

Abstract
Talin (tln) binds and activates integrins to couple extracellular matrix–bound integrins to the cytoskeleton; however, its role in heart development is not well characterized. We identified the defective gene and the resulting cardiovascular phenotypes in zebrafish tln1fl02k mutants. The ethylnitrosourea-induced fl02k mutant showed heart failure, brain hemorrhage, and diminished cardiac and vessel lumens at 52 h post fertilization. Positional cloning revealed a nonsense mutation of tln1 in this mutant. tln1, but neither tln2 nor -2a, was dominantly expressed in the heart and vessels. Unlike tln1 and -2 in the mouse heart, the unique tln1 expression in the heart enabled us, for the first time, to determine the critical roles of Tln1 in the maintenance of cardiac sarcomeric Z-disks and endothelial/endocardial cell integrity, partly through regulating F-actin networks in zebrafish. The similar expression profiles of tln1 and integrin β1b (itgb1b) and synergistic function of the 2 genes revealed that itgb1b is a potential partner for tln1 in the stabilization of cardiac Z-disks and vessel lumens. Taken together, the results of this work suggest that Tln1-mediated Itgβ1b plays a crucial role in maintaining cardiac sarcomeric Z-disks and endothelial/endocardial cell integrity in zebrafish and may also help to gain molecular insights into congenital heart diseases.—Wu, Q., Zhang, J., Koh, W., Yu, Q., Zhu, X., Amsterdam, A., Davis, G. E., Arnaout, M. A., Xiong, J.-W. Talin1 is required for cardiac Z-disk stabilization and endothelial integrity in zebrafish.
Keywords: tln1, itgb1b, cardiovascular development, cardiac sarcomere
Cell–matrix adhesion is critical for normal cardiac function and cardiomyopathy (1); it is mainly mediated by the integrin α/β heterodimeric adhesion receptors, which connect the extracellular matrix (ECM) with the intracellular actin cytoskeleton and form integrin adhesion complexes (2). These structures localize in the costameres and circumferentially align with the Z-disks in the cardiomyocytes (CMs) (3). Many studies have demonstrated the essential roles of integrins and adhesion-complex proteins in heart development (4), cardiac function (5–7), and stress responses of the heart (8–10). However, an understanding of how the integrin adhesion-complex proteins regulate cardiac sarcomeric structure in heart development and subsequently affect cardiac function remains elusive.
It is well known that integrin activation by the cytoskeletal protein talin (Tln) results in the assembly of multicomponent, force-generating signals and adhesion complexes at focal adhesions (FAs) that are necessary for sustaining cell spreading and migration (11, 12), as well as for generating FA-like costameres, intercalated disks, and myotendinous junctions in skeletal and cardiac striated muscles (13, 14). Tln is cytoskeletal a protein of ∼270 kDa with an N-terminal 47 kDa globular head domain and a C-terminal ∼220 kDa elongated rod domain, which links integrins to the actin cytoskeleton. The head domain comprises an F0 subdomain followed by a 4.1, ezrin, radixin, moesin (FERM) domain consisting of 3 subdomains, F1, F2, and F3 (15, 16). Binding of the F3 subdomain to the cytoplasmic tail of the integrin β-subunit converts the integrin into a high-affinity state by disrupting the salt bridge between the integrin α- and β-cytoplasmic tails that normally locks the integrin in the default low-affinity state (15, 17). The F0, F1, and F2 subdomains participate in integrin activation by stabilizing the association of the Tln head with the plasma membrane, through electrostatic interactions with PIP2-rich membrane microdomains and by reorienting the Tln head–β-cytoplasmic tail complex, which disrupts the integrin α/β transmembrane helical interface (15). The Tln rod domain comprises a series of helical bundles and contains a second integrin binding site, multiple vinculin binding sites, and a dimerization and actin-binding domain (18). The Tln rod domain is necessary for the clustering of ligand-bound integrins, interactive signaling, and the assembly of cytoskeletal proteins at FAs.
Studies in Caenorhabditis elegans and Drosophila have confirmed that Tln is essential for integrin function in embryogenesis and in organ function (19–21). Unlike the lower eukaryotes, vertebrates contain 2 Tln genes encoding closely related homologs, Tln1 and -2, both of which are expressed in the heart of mouse embryos and adults (10). Global ablation of Tln1 results in embryonic lethality because of failure of cell migration during gastrulation, preventing further evaluation of its role in heart development (22). Conditional knockout of Tln1 and -2 in mice has shown that Tln plays a role in the cardiovascular system (23). Tln2 mutant mice are viable and fertile and show a mildly dystrophic phenotype resulting from defects in the myotendinous junctions (24). Conditional knockout of Tln1 in mouse endothelial cells causes embryonic lethality, primarily by affecting angiogenesis and endothelial cell spreading in vivo, but heart development appears unaffected (25). Myocardium-specific knockout of Tln1 has shown that the gene is not necessary for heart development, but it acts as a mechanotransducer in the stress response of the adult heart (10). However, a compensatory role of Tln2 in Tln1-mutant mice during heart development has not been excluded.
In this study, we first identified the mutated gene tln1 by positional cloning in fl02k zebrafish mutant that was isolated during an ethylnitrosourea-induced mutagenesis screen for genes involved in cardiovascular development. Unlike in mice, only tln1, but neither tln2 nor tln2a, was expressed in the zebrafish cardiovascular system. This unique tln1 expression allowed us to determine, for the first time, that tln1 is necessary for cardiac Z-disk stabilization, endocardial/endothelial cell integrity, and cardiac function. Therefore, this work provides novel insights into the roles of tln1 in normal embryonic cardiovascular development and suggested its potential roles in the pathogenesis of human congenital heart disease.
MATERIALS AND METHODS
Zebrafish lines and ethics statement
The fl02k zebrafish, an ethylnitrosourea-induced mutant, was isolated in Mark Fishman’s laboratory (Massachusetts General Hospital, Boston, MA, USA). The tln1hi3093 zebrafish was isolated from a retroviral-based insertional mutagenesis screen in Nancy Hopkins’ laboratory (Massachusetts Institute of Technology, Cambridge, MA, USA) (26, 27). The Tg[kinase insert domain receptor like (kdrl):enhanced green fluorescent protein (EGFP)] zebrafish was a gift from Dr. Shuo Lin (University of California, Los Angeles, CA, USA) (28). The Tg(myl7:EGFP) zebrafish was a gift from Dr. C. Geoffrey Burns (Massachusetts General Hospital) (29). Tg(myl7:EGFP-UtrCH)pku316, Tg(myl7:tln1H-EGFP)pku317, and Tg(myl7:EGFP-tln1)pku318 were generated in wild-type (WT) TL embryos by using Tol2-based transgenesis (30). The above mutant and transgenic lines, as well as the WT TL, WIK, and AB zebrafish lines, were used according to an animal protocol approved by the Institutional Animal Care and Use Committees at Peking University and Massachusetts General Hospital.
Positional cloning of fl02k and genotyping of tln1hi3093
The fl02k mutant embryos (n = 5377) were sorted for positional cloning based on defects in the cardiac lumen and cardiac contractility. Genomic DNA was extracted from each embryo for PCR-based genotyping for recombinants by using various genetic markers (Table 1). The fl02k locus was mapped to link marker Z8146 on chromosome 10 by using bulked segregant analysis and was further narrowed to a region demarcated by markers s5097ca4 and z66779.2. Markers developed during the chromosomal walk include sequence-length polymorphism (SLP) markers (nos. 2, 31, 44, and 46) and single nucleotide polymorphism (SNP) markers (nos. 26 and 410). SLP markers were assayed by agarose gel electrophoresis of the PCR products. SNP markers were assayed by sequencing PCR products. Marker 2 is in intron 6 of tln1, which gave 0 recombinants in 10,754 meioses. The transcript isoforms of tln1 was amplified by RT-PCR with the primers: Exon6-forward (F) (5′-AAAGTTCTTCTACTCAGACC-3′) and Exon10-reverse (R) (5′-TAGTGATGCCCAACAAACGC-3′).
TABLE 1.
Genetic markers for positional cloning of tln1
| Markers | Primer A | Primer B |
|---|---|---|
| s5097ca4 | GCTCAAACTGTGGGCTTGTT | TGAGCGTGTCATGGATTGAT |
| 2 | AGCTCAACCTGCTGTATGTGCAG | CCGTTCAAAATGTCATCACGGG |
| 46 | TTTTCTGAAAATAAAAAGTAAGGGTG | GAAATGGTCTATATATCTATGTACTA |
| 44 | GGAACGCTGAAAAACACTCC | CCCCATTGCGTTCTATTGTT |
| 410 | GCTCGCCGAACCCTGGATCAAG | TGCAGCGTTTTGCTGTAGCGTGG |
| 26 | GTATGTGTGTAAATGAGAGTTTATGGG | CAACAACAGCCAATGAGTGCTTCG |
| 31 | TGAACATGTAGAGGGCCAGTC | ACCCTTGGCAAAACAAACAA |
Genotyping of tln1hi3093 was performed by PCR with 2 pairs of primers simultaneously (tln1hi3093-P1: 5′-CAAGGGTGGGAAGTTTCCTT-3′ and 5′-TGCTGTCGCCTCTAAACCTT-3′; and tln1hi3093-P2: 5′-ATATCGACGGTTTCCATATGGG-3′ and 5′-GTACTCTATAGGCTTCAGCTGG-3′). The tln1hi3093-P1 primers were designed on 2 flanking genomic regions of the retroviral insertion site, which were expected to amplify a 200 bp fragment from the WT allele, but failed to amplify the insertional allele because of its large size (6 kb). The tln1hi3093-P2 primers were designed to amplify a 300 bp fragment of the mutagenic retroviral vector.
Whole-mount in situ hybridization and morpholino analysis
RNA in situ probes were made from cDNA templates generated by RT-PCR with the primers Tln1-11-F (5′-ATGGTACGGGGGCTGGAGAG-3′) and Tln1-11-R (5′-ACCGCGCGAGCAGCAGCAGC-3′) (for tln1 probe); Tln2a-F (5′-TCCGGTATGTCAGGAGCAGC-3′) and Tln2a-R (5′-GGTTTCAACTGTCCCTCAGA-3′) (for tln2a probe); Tln2-F (5′-TCGACTCCGCTCTCAGTGCT-3′) and Tln2R (5′-TGACAGCTCGTGCGGCGG-3′) (for tln2 probe); and Itgβ1b-52-F (5′-AAACAGGGAGAGCAGAAATCCACA-3′) and Itgβ1b-297-R (5′-GCATTTCCCGTCATTAGGAAGCAC-3′) (for itgb1b probe). Whole-mount RNA in situ hybridization was performed as has been described (31).
Antisense morpholinos (MOs): tln1MO (5′-TTCCTGTTGAGTAAATGCTGGTATC-3′), itgb1bMO (5′-AATCAGGAGCAGCCTTACGTCCATC-3′), itgb1bmisMO (5′-AATaAGGAcCAGCaTTAaGTCaATC-3′), and tnnt2aMO (5′-CATGTTTGCTCTGATCTGACACGCA-3′) were synthesized (Gene Tools, Philomath, OR, USA); itgb1bMO and tnnt2aMO were the same as previously reported (32, 33). Embryos were injected with MOs at the 1-cell stage and allowed to grow to the desired stages for analysis. Images were captured with a DFC 300 FX camera on an MZ 165F fluorescence microscope (Leica, Bannockburn, IL, USA).
Quantification of the lumens of the dorsal aorta and posterior cardinal vein and of heart function
To evaluate the dorsal aorta (DA) and posterior cardinal vein (PCV) lumens, we mounted embryos in 1.0% low-melting-point agarose for imaging. Images were acquired with a laser scanning confocal microscope (LSM 700; Zeiss, Oberkochen, Germany). ImageJ [National Institutes of Health (NIH), Bethesda, MD, USA] and ZenLightEdition software (Zeiss) were used to analyze the lumen diameters of the DA and PCV.
To determine heart rate and the ventricular shortening fraction (VSF), embryos were loaded in a recording chamber filled with E3 solution at the desired stage. Heart rate was calculated by counting the number of sequential contractions in 30 s intervals under a dissecting microscope (S8APO; Leica). Cardiac contractility was assessed as VSF. The method of calculating VSF has been described (34). In brief, movies of cardiac contractions were acquired under a compound microscope (DM5000B; Leica) with a video camera (DFC500; Leica) at a frame rate of 20 ms/frame (680 × 512 pixels). The lengths of ventricles in end-diastolic and -systolic conditions were measured to calculate the VSF (Eq. 1):
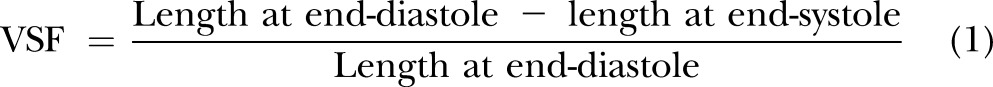 |
To measure the ventricular chamber volume at end-diastole, fluorescent images of Tg(myl7:nuDsRED) embryos were used, and the heartbeat was stopped by treatment with 20 μM 2,3-butanedione monoxime (BDM) before image acquisition. The heart images were analyzed with Imaris software (Bitplane, Concord, MA, USA).
Antibody generation and Western blot analysis
The peptide TMVRGLERDMQEAKASAAEGKLR, from aa 1100 to 1122 in Tln1, and the peptide QCRAFGTGDKKDTCEEQCSYFTM from aa 645 to 667 in Itgβ1b were used to generate affinity-purified antibodies (Genemed Synthesis, San Antonio, TX, USA). For Western blot analysis, zebrafish embryos were manually dechorionated, deyolked in 0.5× Ginzburg fish Ringer buffer, and lysed in Laemmli SDS-sample buffer (Boston BioProducts, Boston, MA, USA). Thirty to 50 embryonic zebrafish hearts per experimental group were dissected and homogenized in Laemmli SDS-sample buffer. Protein samples were heated and then resolved on polyacrylamide gels, blotted onto PVDF membranes, and probed with anti-Tln1, anti-vinculin, or anti-pFAK antibodies. α-Tubulin antibody (Easybio, Seongnam, China) and GAPDH antibody (GenTex, Zeeland, MI, USA) were used for protein loading controls. The signals were developed with ECL Western blot detection reagents and an analysis system (GE Healthcare, Pittsburgh, PA, USA). For Western blot analysis of cultured HUVEC extracts, the anti-human TLN1 antibody was used (Sigma-Aldrich, St. Louis, MO, USA).
Immunostaining of agarose-embedded tissue sections
For antibody staining, all embryos were immersed in 0.12% tricaine (Sigma-Aldrich) to stop and relax the hearts before fixation. Embryos were fixed in 4% paraformaldehyde (PFA) at room temperature for 2 h, embedded in 3% low-melting-point agarose gel, and cut at 200 μm on a vibratome (VT 1000s; Leica). For anti-laminin antibody staining, embryo sections were treated with 0.5% papain (Sigma-Aldrich) in phosphate buffer (pH 4.7) at 37°C for 10 min and refixed in 4% PFA for 20 min. They were blocked with blocking solution [10% normal goat serum (NGS) plus 2% blocking reagent (BR) (Roche Diagnostics, Indianapolis, IN, USA) in maleic acid buffer (MAB; 150 mM maleic acid and 100 mM NaCl, pH 7.5)]. Staining with anti-laminin (1:300; Sigma-Aldrich), anti-vinculin (1:300; Sigma-Aldrich), and anti-zonula occludens (ZO)-1 (1:100; Life Technologies-Invitrogen, Eugene, OR, USA) antibody was performed in incubation solution (2% NGS plus 2% BR in MAB) at 4°C overnight. For anti-laminin staining, peroxidase-labeled goat anti-rabbit IgG(H+L) (1:2000; Vector Laboratories, Burlingame, CA, USA) was used as the secondary antibody. The signal was developed with the TSA Plus Cyanine 3 System (PerkinElmer, Waltham, MA, USA). For anti-vinculin and anti-ZO1 staining, Alexa Fluor 555-conjugated goat anti-mouse IgG(H+L) (Life Technologies-Invitrogen) was used as the secondary antibody. Images were captured with a confocal microscope (LSM 5 Pascal; Zeiss).
Immunostaining of dissected embryonic hearts
The zebrafish hearts were dissected, stained, and imaged as previously described (35). The following antibodies were used: anti–β-catenin (1:200; Sigma-Aldrich); anti–α-actinin (clone EA 53, 1:300; Sigma-Aldrich); tropomyosin antibody (CH1) (1:100; Developmental Studies Hybridoma Bank, Iowa City, IA, USA); myosin antibody (F59) (1:10; Developmental Studies Hybridoma Bank); Alexa-conjugated anti-mouse IgG secondary antibodies (1:500; Life Technologies-Invitrogen); and DAPI (1:1000; Sigma-Aldrich). Alexa-conjugated phalloidin (1:20; Life Technologies-Invitrogen) was used to show F-actin structures. Heart samples were imaged with an LSM 510 Meta Confocal microscope (Zeiss), and ImageJ (NIH) and ZenLightEdition software (Zeiss) were used to process and analyze the immunostained images.
Immunostaining of paraffin-embedded tissue sections
Ten to 15 embryos/sample were fixed in 4% PFA at room temperature for 2 h, embedded in paraffin, and cut at 5 μm on a microtome (RM2265; Leica). The sections were then mounted on gelatin-coated slides and blocked with blocking solution. Staining with anti-Itgβ1b (1:200; Genemed Synthesis, San Antonio, TX, USA), anti-pFAK (Y576) (1:300; Life Technologies-Invitrogen), anti-vinculin (1:300; Sigma-Aldrich), or anti-ZO1 (1:100; Life Technologies-Invitrogen) antibodies and with Alexa-conjugated anti-mouse IgG or anti-rabbit IgG secondary antibody (Life Technologies-Invitrogen) was performed in incubation solution (5% fetal bovine serum plus 5% bovine serum albumin in PBS) at 4°C overnight. DAPI (1:1000; Sigma-Aldrich) was used to label nuclei after incubation with secondary antibodies. Images were captured by confocal microscope (LSM 510 Meta; Zeiss).
Molecular cloning and injection
To form the Tol2-based construct expressing the fusion gene of EGFP and calponin homology domain of utrophin (UtrCH), we amplified the 3.1 kb myl7 enhancer (GenBank: BX248505.9) by PCR of myl7-F (5′-GAAGGATCCATGATTAAGCAACTCCACAA-3′) and myl7-R (5′-TCCTCGAGACGTTCACTGTCTGCTTTGCTG-3′), from zebrafish genomic DNA, and then cloned it into a pT2K-EGFP plasmid to generate pT2K-myl7-EGFP. The cDNA for UtrCH was amplified by the PCR primers UtrCH-F (5′-CAAGTCCGGAACCATGGCCAAGTAT-3′) and UtrCH-R (5′-ACGCGTTTAGTCTATGGTGACTTGC-3′), from plasmid mCherry-UtrCH (26740; Addgene, Boston, MA, USA) and then cloned into the pT2K-myl7-EGFP vector to generate the final construct pT2K-myl7-EGFP-UtrCH.
The cypher coding sequence (GenBank: NM_201505.1), tln1 head domain (1-470 aa) sequence, and tln1 coding sequence (GenBank: NM_001009560.1) were amplified by the PCR primers cypher-F (5′-CCGGTCGACCGCCACCATGACTTCGTACAACGTG-3′) and cypher-R (5′-GCGGGATCCCCTGAGGCACGTTGAGTATTTGTG-3′); Tln1H-F (5′-CCGCTCGAGCGCCACCATGGTGGCGCTGTCGCTG-3′), and Tln1H-R (5′-CGCGGATCCCGTGCTGTTTGGCCTGTGG-3′); and Tln1cds-F (5′-GTCGACATGGTGGCGCTGTCGCTGAA-3′), and Tln1cds-R (5′-ACGCGTTCACTGCTCCTGTCCGTCCT-3′), respectively. These cDNAs were then cloned into the pT2K-myl7-EGFP plasmid to generate the constructs pT2K-myl7-cypher-EGFP, pT2K-myl7-tln1H-EGFP, and pT2K-myl7-EGFP-tln1.
For the itgb1bMO rescue experiment, we created MO-resistance itgb1b mRNA by changing 5 nucleotides at the itgb1bMO binding site to prevent direct interaction with the itgb1bMO, while keeping the Itgβ1b amino acids unchanged. To generate this construct, we amplified the itgb1b coding sequence (GenBank: NM_001034987.1) using the following primers: Itgβ1bcds- F (5′- ATGGACGTtAGcCTcCTgCTcATTTCAGTTCTGCTTGGAC -3′) and Itgβ1bcds-R (5′- CTATTTGCCCTCATATTTAGGGTTG-3′). The sequence was then cloned into the pXT7 plasmid.
To construct the 5′UTR tln1-EGFP and 5′UTR itgb1b-EGFP reporters, the EGFP coding sequence was amplified from pEGFP-N1 and cloned into pCS2+ vector, then −141 to +33 bp of tln1 and −257 to +33 bp of itgb1b were cloned into the modified pCS2+ vector. The corresponding primers were: egfp-F (5′-CCGGAATTCGCGGTGAGCAAGGGC-3′) and egfp-R (5′-CCGCTCGAGTTACTTGTACAGCTCGT-3′); Tln1-F (5′-CGCGGATCCGGGATACCAGCATTTACTC-3′) and Tln1-R (5′-CCGGAATTCCCCCACCCCGATCTTCAG-3′); and Itgβ1b-F (5′-CGCGGATCCACCCGGACTGGGGACACGCC-3′) and Itgβ1b-R (5′-CCGGAATTCCAGAACTGAAATCAGGAGCAG-3′). Capped sense Tol2 transposed, 5′UTR tln1-EGFP, and 5′UTR itgb1b-EGFP mRNAs were synthesized with the mMessage mMachine in vitro transcription kit (Life Technologies-Ambion, Austin, TX, USA) and were purified with an RNeasy mini kit (Qiagen, Hilden, Germany). The plasmid vector and purified mRNA were injected separately or together into zebrafish embryos at the 1-cell stage.
Isolating the 5′UTR of itgb1b cDNA by 5′RACE and RT-PCR
The total RNA was isolated from zebrafish embryos at 52 h post fertilization (hpf). 5′ rapid amplification of cDNA ends (RACE) was performed with the Smarter RACE cDNA amplification kit (Clontech Corp., San Diego, CA, USA). The sequences of 2 antisense primers against itgb1b for the 5′ RACE PCR were 5′-CATTAGGAAGCACGATGCCGCCCAGTT-3′ and 5′-TCACAAACTCTGCCAATGCGCCCCTC-3′. For RT-PCR, adult zebrafish were anesthetized with 0.016% tricaine (Sigma-Aldrich) and then euthanized on ice based on an animal protocol approved by the Peking University Institutional Animal Care and Use Committees. Adult heart ventricles were then dissected and washed several times in PBS to remove debris. Total RNA from 50 zebrafish embryos, 35 embryonic hearts, or 10 adult ventricles was extracted and purified with Trizol reagent (Sigma-Aldrich) and an RNeasy mini kit (Qiagen). The cDNA was synthesized using the SuperScript II First-Strand Synthesis System (Life Technologies-Invitrogen). To test the expression of tln1, -2, and -2a, RT-PCR was performed in a Master PCR system (Eppendorf, Bochum, Germany) and the following primer pairs were used: Tln1-rt-F (5′-GGTCTCATTTCAGCTGCTCG-3′) and Tln1-rt-R (5′-CGCCCTCTTGACAGCATTAC-3′) (for tln1); Tln2-rt-F (5′-GAAAAGTGCTGCCAAGGTCA-3′) and Tln2-rt-R (5′-CTCTTCTGGGGTGGTGGATT-3′) (for tln2); and Tln2a-rt-F (5′-CATGGGTACAATGCCTGCTC-3′) and Tln2a-rt-R (5′-AGAAGCCATGTCATCCCCAA-3′) (for tln2a).
Transmission electron microscopy
Zebrafish embryos at 52 hpf were bathed in E3 solution with 20 mM BDM for 10 min, to completely relax the cardiac muscles. Five to 10 dissected embryonic zebrafish hearts per sample were collected and fixed in 2% glutaraldehyde, 2% PFA, and 0.1 M PBS overnight at 4°C before transfer to the Electron Microscopy Core Facility (Peking University, Peking, China). Stained sections were imaged under a Tecnai T20 (LaB6, 200KV) transmission electron microscope (FEI, Hillsboro, OR, USA).
RNA interference and HUVECs for vessel formation in 3-dimensional collagen gel cultures
HUVECs (Clontech Corp.) were treated with siRNAs targeting either human TLN1 or control luciferase and then resuspended in 3-dimensional (3D) collagen matrices. HUVEC preparation, collagen matrix preparation, cell culture, fixation, and analysis were as published elsewhere (36). The data was derived from triplicate wells and 8 different fields from each of the triplicate wells. These fields were quantitated (24 total fields) to obtain lumen area and standard deviation.
Statistics
The values shown in the figures are means ± sem, unless otherwise stated. The number of samples is given in each of the figures. Unpaired Student’s t test was used to assess the difference between 2 groups, and significance was set at P < 0.05.
RESULTS
Altered cardiac and vascular development and function in zebrafish mutant fl02k
We isolated the zebrafish mutant fl02k in a large-scale ethylnitrosourea-induced mutagenesis screening for recessive lethal mutations in the cardiovascular system. Homozygous fl02k mutant embryos displayed progressive reduction of cardiac contractility. They were indistinguishable from their WT and heterozygous fl02k siblings at 36 hpf, with a well-patterned cardiovascular system and normal blood circulation (Table 2; Supplemental Movie S1). However, by 52 hpf, ventricular contractility and function deteriorated in the mutant embryos (Fig. 1A, B); the VSF declined from 24.0 ± 0.7 (48 hpf) to 4.9 ± 0.4% (60 hpf) (Fig. 1C) and the heart rate declined (Fig. 1D). Cardiac growth and chamber formation were significantly reduced because of the diastolic dysfunction in the fl02k mutant embryos at 52 hpf (Fig. 1F, G; Supplemental Movie S1. The endocardium was labeled with the Tg(kdrl:EGFP) transgene and the myocardial nuclei with the Tg(myl7:nuDsRed) transgene (Fig. 1E, F). Since the size and number of CMs are critical for chamber enlargement in developing zebrafish embryos (37), we then examined the size, proliferation, and apoptosis of mutant CMs. Consistent with the smaller heart (Fig. 1G), both the size and number of CMs were reduced in fl02k mutants at 52 hpf (Fig. 1H; Supplemental Fig. S4B, C) compared with WT controls (Fig. 1H; Supplemental Fig. S4A, C). These defects in cardiac growth in the mutants were primarily caused by the inhibition of myocardial proliferation (as assayed by either proliferating cell nuclear antigen or pH3 immunostaining; Supplemental Fig. S4D–I), not by myocardial apoptosis (as revealed by TUNEL assay; Supplemental Fig. S4J–L). Therefore, these results suggested that cardiac function and growth/expansion are compromised in fl02k mutants.
TABLE 2.
Statistics of fl02k phenotype at 36 hpf
| Pairs | Mutants |
WT siblings | ||
|---|---|---|---|---|
| Normal | Cardiac dysfunction without brain hemorrhage | Cardiac dysfunction with brain hemorrhage | ||
| 1 | 19 | 0 | 2 | 67 |
| 2 | 17 | 0 | 0 | 48 |
Phenotypes were scored from 2 pairs of offspring. Genotypes were identified by PCR and Sanger sequencing. All WT siblings were normal.
Figure 1.

Cardiovascular defects are evident in fl02k mutant embryos. A and B) Live images of a WT sibling and fl02k mutant embryo at 52 hpf. Arrow: interstitial hemorrhage in the mutant. C) VSF gradually declined from 48 to 60 hpf in fl02k embryos compared with their WT siblings. D) The heart rate was reduced in fl02k embryos at 52 hpf. E and F) Cardiac growth and expansion were compromised in fl02k mutants. The heart was stopped at end-diastole by 20 µM BDM treatment. Endocardial cells were labeled by Tg(kdrl:EGFP) (green), and myocardial nuclei by Tg(myl7:nuDsRED) (red). a, atrium; v, ventricle. G) Ventricular volume was reduced in fl02k mutants. The heart was stopped at end-diastole by 20 µM BDM. H) The number of CMs labeled by Tg(myl7:nuDsRED) decreased in fl02k mutants. I–N) The lumen of the aorta and PCV were formed at 36 hpf (J) but were not maintained at 54 hpf (L) in fl02k mutants compared with their WT siblings (I, K). I–L) Lateral views of trunk vessels of WT siblings and fl02k mutants in Tg(kdrl:EGFP) transgenic zebrafish. Lumens of the DA (L, M) and PCV (L, N) were narrower in fl02k embryos at 54 hpf, although the lumens were comparable in WT and fl02k mutant embryos at 36 hpf (I, J, M, N). The anterior is to the left in (I–L). The number of zebrafish embryos/group is shown in the histograms. *P < 0.05; **P < 0.01. Scale bars, 50 μm (E, F); 100 μm (I–L).
In addition to cardiac dysfunction, a brain hemorrhage was found in a proportion of fl02k embryos at 52 hpf (Fig. 1B; Table 3), but was never detected at 36 hpf (Table 2). To assess vascular defects in fl02k mutants, we used the Tg(kdrl:EGFP) transgene to label vascular endothelial cells (28). The patterning of trunk vessels appeared comparable in WT siblings and fl02k mutants at 36 and 54 hpf (Fig. 1I–L). However, the diameters of the DA and PCV were normal at 36 hpf, but decreased at 54 hpf in the mutants (Fig. 1M, N), suggesting that the vascular lumens are initially formed but lose integrity later during zebrafish development. Although mutant embryos had normal morphology and swam normally until 5 d post fertilization, they died of severe pericardial edema and heart dysfunction.
TABLE 3.
Statistics of fl02k phenotype at 52 hpf
| Pairs | Mutants |
WT siblings | |
|---|---|---|---|
| Cardiac dysfunction with brain hemorrhage | Normal cardiac function with brain hemorrhage | ||
| 1 | 32 | 13 | 136 |
| 2 | 35 | 28 | 172 |
| 3 | 20 | 15 | 124 |
Phenotypes were scored from 3 pairs of offspring. All WT siblings were normal.
The tln1 gene is mutated in fl02k mutants
We mapped fl02k to chromosome 10 between marker s5097ca4 (15 recombinants/7600 meioses) and marker z66779.2 (9 recombinants/1200 meioses) (Fig. 2A). Subsequent fine-mapping and Sanger sequencing identified tln1 as the defective gene responsible for the fl02k phenotype. The splice donor site at intron 9 in the fl02k allele was abolished by a T-to-A transition, which led to the retention of intron 9 in the mutant tln1fl02k transcript (Fig. 2B). Intron 9 contains 7 stop codons in all 3 frames. By RT-PCR with a forward primer in exon 6 and a reverse primer in exon 10, we showed that the 416 bp WT band (Fig. 2C, lane 1) was missing, whereas a 503 bp band with intron 9 was present in the tln1fl02k mutant (Fig. 2C, lane 2). The major band (311 bp) in the tln1fl02k mutant was generated by in-frame deletion of exon 9 (Fig. 2C, lane 2). Exon 9 encodes 35 amino acids and includes parts of the F2 and F3 domains (Fig. 2D, underlined sequence), comprising the middle of the α3- and α4-helices of the F2 subdomain and the linker and the first β1 strand of the F3 subdomain in the FERM domain of the Tln head. The Tln1fl02k protein lacking the 35 amino acids encoded by exon 9 led to disruption of the integrin binding site of the Tln1 head domain. The relatively low abundance of the 503 bp band suggests that the transcript containing intron 9 is unstable and probably is subject to nonsense-mediated decay. Bands immediately below the 503 bp band sometimes appeared and also contained intron 9, but with other variants. The WT Tln1 protein was ∼270 kDa (Fig. 2E, lanes 1–3). Of note, the maternal Tln1 protein was present at the 256-cell stage (Fig. 2E, lane 1), a stage when zygotic transcription has not yet started. In the tln1fl02k mutant, the Tln1fl02k protein level was markedly low (Fig. 2E, lane 4), suggesting that Tln1fl02k protein predicted to be translated from the mRNA of tln1fl02k with exon 9 deleted is unstable. These genetic data showed that tln1 is mutated in fl02k mutants.
Figure 2.

Positional cloning reveals that tln1 is mutated in fl02k mutants. A–E) The zebrafish fl02k locus encoded tln1. A) A regional fine map of the fl02k locus on chromosome 10, determined to lie between genetic marker s5097ca4 (15 recombinants/7600 meioses) and marker 46 (4 recombinants/3154 meioses). B) Partial cDNA sequence amplified from tln1fl02k mutants by RT-PCR. A thymine-to-adenine transition (highlighted in yellow) at the splice donor site of intron 9 leads to its retention (between the 2 arrows) in the tln1fl02k transcript. C) RT-PCR products amplified with a forward primer in exon 6 and a reverse primer in exon 10 in the tln1 gene. mRNA templates: a fin clip from a WT TL zebrafish (lane 1); 35 tln1fl02k mutant embryos (lane 2); 35 tln1fl02k WT siblings that contain WT and tln1fl02k heterozygous embryos (lane 3); and 100 bp DNA marker (lane 4). There was a dominant exon 9–depleted transcript and a less abundant intron 9–included transcript in tln1fl02k mutants (lane 2). D) Primary sequence of the N-terminal F0–F3 subdomains of the zebrafish Tln1 head domain. Subdomains are: F0, black; F1, blue; F2, red; and F3, green. The amino acids encoded by exon 9 and deleted in the major transcript (C, lane 2) in the tln1fl02k mutant are underlined. E) Western blots of maternal Tln1 with anti-Tln1 antibody in 256-cell embryos (lane 1), 52 hpf WT siblings of tln1fl02k mutant embryos (lane 2), 52 hpf WT siblings of tln1hi3093 mutant embryos (lane 3), 52 hpf tln1fl02k mutant embryos (lane 4), and 52 hpf tln1hi3093mutant embryos (lane 5). Very little Tln1 was detected in tln1fl02k mutants (lane 4), whereas Tln1 was not detectable in tln1hi3093 mutants (lane 5). α-Tubulin was used as a loading control. F–I) tln1fl02k is allelic to tln1hi3093. F) Ventricular volume was reduced in tln1hi3093 mutants at 52 hpf as in tln1fl02k mutants (Fig. 1G). G) The number of CMs decreased in tln1hi3093 mutants at 52 hpf as in tln1fl02k mutants (Fig. 1H). H, I) VSF was reduced in tln1hi3093/tln1hi3093 mutants at 52 hpf from heterozygous tln1hi3093 crosses (H), or tln1hi3093/tln1fl02k mutants from transheterozygous tln1hi3093 and tln1fl02k crosses (I), suggesting that tln1hi3093 failed to complement tln1fl02k. The numbers of zebrafish embryos/group are shown in the histograms; *P < 0.05; **P < 0.01. J–R) The tln1 gene is expressed in the cardiovascular system. Whole-mount in situ hybridization of embryos with an antisense tln1 RNA probe at 6 somites (s) (J) and 18s (K) and at 24 (L, Q), 36 (R), 48 (M), and 72 (N) hpf; transverse sections of the trunk region at 24 (O) and 72 (P) hpf. Arrows: (L) axial vessels; (Q, R) the heart tube. (P) 1, Ventricle of neural tube; 2, myoseptum; and 3, wall of body cavity. S–U) The Tln1 head domain localizes to the Z-disk structure in CMs. Expression of the Tg(myl7:tln1H-EGFP) transgene (S) colocalized with the Z-disk marker α-actinin (T) in isolated ventricles at 52 hpf. The tln1H-EGFP is a fusion gene of the Tln1 head domain (470 amino acids) and EGFP. U) The merged images of (S) and (T) show the overlapping signals of the tln1H-EGFP fusion protein and α-actinin (arrows). Scale bars, 25 μm (O, P); 4 μm (S–U).
Previous studies have demonstrated that the Tln1 rod domain has partial function (18, 38, 39). To further evaluate whether the Tln1fl02k protein carrying the defective Tln1 head domain retains partial function, we obtained the tln1hi3093 zebrafish line from the laboratory of Dr. Nancy Hopkins (Massachusetts Institute of Technology, Cambridge, MA, USA). This mutant has a retroviral insertion at the first tln1 intron (26, 27). The Tln1 protein was undetectable in 2-d-old tln1hi3093 mutants (Fig. 2E, lane 5) compared with that in their WT siblings (Fig. 2E, lane 3). This tln1hi3093 mutant exhibited phenotypes similar to tln1fl02k, including reduced size of the ventricular chamber, a decreased number of CMs, and reduced VSF (Fig. 2F–H). Just as in tln1fl02k mutants, reduced size and proliferation but not apoptosis of CMs contributed to the compromised cardiac growth and function in tln1hi3093 mutants (Supplemental Fig. S4C, F, I, L). Furthermore, tln1hi3093 failed to complement tln1fl02k (Fig. 2I), suggesting that the 2 mutants are allelic to tln1 and the truncated Tln1fl02k protein has null function.
To further confirm that the tln1 gene is defective in tln1fl02k mutants, we designed an antisense MO targeting the tln1 5′-UTR (tln1MO) to examine tln1 morphant phenotypes. Knockdown efficiency was demonstrated by showing effective suppression of the 5′UTR tln1-EGFP reporter (Supplemental Fig. S1C) and Tln1 protein expression (Supplemental Fig. S1F) by tln1MO. We found that tln1 morphants induced by tln1MO had phenotypes similar to those of the tln1fl02k mutants (Supplemental Fig. S1E). Most embryos injected with 4 ng of tln1MO had decreased cardiac contractility, reduced cardiovascular lumen diameter, sluggish or absent blood circulation, and hemorrhages at 52 hpf (Table 4). Together with the genetic mapping and splice donor mutation in tln1fl02k and depletion of tln1 in tln1hi309, these results substantiated the conclusion that tln1 is responsible for the tln1fl02k mutant phenotype.
TABLE 4.
tln1 morphants phenocopy tln1fl02k mutants
| Dosage (ng/embryo) | tln1fl02k-like | Milder tln1fl02k-like | Normal | Deformed |
|---|---|---|---|---|
| 6 | 85 | 0 | 0 | 3 |
| 4 | 105 | 5 | 20 | 4 |
One typical experiment is shown. Noninjected control embryos were all normal.
The tln1 gene, but not tln2 and -2a, is expressed in the zebrafish heart
To identify the zebrafish Tln orthologs, we Blast-searched (NIH) the zebrafish Ensembl genomic database (www.ensembl.org) using the human and mouse Tln1 and -2 protein sequences, and found 3 genes, tln1, -2, and -2a. Synteny analysis suggested that zebrafish tln1 is located on chromosome 10 and tln2 on chromosome 25, whereas tln2a is a new tln homolog on chromosome 7 (data not shown). In situ hybridization was used to assess the tln gene expression profiles during zebrafish embryogenesis. The tln1 gene was expressed in the notochord at the somitogenesis stage (Fig. 2J, K), in the vasculature at 24 (Fig. 2L, O) and 72 (Fig. 2P) hpf, and in the heart at 24 and 36 hpf (Fig. 2Q, R). From 48 to 72 hpf, tln1 was expressed in the vessels, brain ventricles, gill arches, gut, skin, and cells lining the body cavity (Fig. 2M, N, P). A feature of the cells expressing tln1 is that they must stretch extensively and maintain polarity for their function. The tln2 and -2a expression patterns were similar, but tln2 was expressed at a higher level in the somites (Fig. 3A–H). Both genes were expressed during somitogenesis in the somites, eyes, and brain ventricles, similar to the expression pattern of human and mouse Tln2 (40). However, tln2 and -2a were undetectable in the heart by in situ hybridization. RT-PCR also confirmed that tln1, but neither tln2 nor -2a, was expressed in zebrafish embryonic and adult hearts (Fig. 3I). Therefore, unlike in the mouse, tln1 was the only Tln gene present in the cardiovascular system during zebrafish development. This feature allowed us to isolate the tln1fl02k and tln1hi3093 alleles as cardiovascular mutants without tln2 gene redundancy in zebrafish. We then determined the Tln1 protein localization in CMs by using a fusion gene of the Tln1 head domain and EGFP (tln1H-EGFP) that was driven by the zebrafish cardiac-specific myl7 promoter (Fig. 2S). Immunostaining assays showed that the strongest tln1H-EGFP fluorescence signals colocalized well with anti–α-actinin signals (Fig. 2T, U), suggesting that zebrafish Tln1 is a structural protein in the cardiac Z-disks.
Figure 3.

The tln2 and -2a genes are expressed in the head and somites, but not in the cardiovascular system. A–H) Both tln2 and -2a are expressed in the ectoderm and paraxial mesoderm at 13 somites, as well as the brain somites, from 24 to 45 hpf. Whole-mount in situ hybridization in embryos at 13 somites (A, B), at 24 (C–F) and 45 (G, H) hpf, with antisense tln2 (A, C, E, G) or antisense tln2a (B, D, F, H) probes. The anterior is to the left in all embryos. (A, B, E, F) Dorsal views; (C, D, G, H) lateral views. I) The tln1 gene, but neither tln2 nor -2a, is expressed in the embryonic and adult heart. RT-PCR revealed that tln2 and -2a were expressed in the whole embryo but not in embryonic and adult hearts; gapdh was the loading control. DNA marker, 100 bp ladder; mpf, months post fertilization.
Tln1 is critical for the stabilization of cardiac sarcomeric Z-disks
Although tln1 deficiency had no effect on the key steps of early heart morphogenesis in zebrafish (heart tube jogging, looping, and heart chamber demarcation) (Supplemental Movie S1), cardiac dysfunction was evident in tln1 mutants by 52 hpf (Fig. 1C; Supplemental Movie S1). Because of the increased load on the heart at this stage, the sarcomeric structures undergo a critical maturation and remodeling (41). Because tln1H-EGFP was located in the cardiac Z-disks, as assayed by anti–α-actinin antibody staining (Fig. 2U), we hypothesized that Tln1 regulates maturation or stabilization of the Z-disks. We then compared the expression levels of several Z-disk proteins in WT siblings and tln1 mutant embryos by immunostaining. Consistently, the Z-disk protein α-actinin was barely detected in the tln1fl02k and tln1hi3093 mutants at 52 hpf (Fig. 4E, F, arrows) although it had been evident just 4 h earlier (Fig. 4B, C). The thin filament CH1 was slightly reduced (Fig. 4H, I), and the thick filament F59 appeared unchanged (Fig. 4K, L) in both mutants. On the other hand, quantitative RT-PCR analysis showed that the mRNA expression of 6 major structural Z-disk genes [actinin2α (actn2), myotilin, lasp2, tcapa, mlp, and cypher] was not affected in tln1fl02k mutant embryos (data not shown). These data suggest, for the first time, that tln1 is essential for the stabilization, but not the early assembly, of sarcomeric Z-disks during embryonic heart development.
Figure 4.

Tln1 is critical for the stabilization of sarcomeric structures during heart development. Confocal images from the ventricles at 48 (A–C) and 52 (D–L) hpf with the indicated genotypes and antibody staining. The Z-disk staining by α-actinin was normal in tln1 mutants (B, C) compared with WT controls (A) at 48 hpf, but was disrupted in tln1 mutants (E, F) compared with WT controls (D) at 52 hpf; arrows pointed to a few weak Z-disks in mutants. The thin filament staining by CH1 was slightly affected in tln1 mutants (H, I) compared with WT controls (G) at 52 hpf, whereas the thick filament staining by F59 was comparable in WT (J) and tln1 mutants (K, L) at 52 hpf. M and N) The Z-disks in ventricular CMs, stained by anti–α-actinin, were rescued in tln1 morphants with the Tg(myl7:EGFP-tln1) transgene (M) compared with their nontransgenic siblings (N). n, hearts per group. O) VSF decreased in tln1 morphants (tln1-MO) compared with untreated WT controls, which was fully rescued in tln1 morphants by overexpression of the fusion gene EGFP-tln1 in CMs. **P < 0.01; number of hearts assayed is shown in the histograms. Scale bars, 4 μm (A–N).
To demonstrate the intrinsic role of tln1 in CMs, we constructed a transgenic zebrafish with cardiac-specific expression of the fusion coding sequence of EGFP and tln1, driven by the zebrafish myosin light chain promoter (myl7). Inhibition of tln1 by tln1MO, which targets only the endogenous 5′-UTR sequences of tln1, but not the transgenic EGFP-tln1, caused cardiac dysfunction with evident disruption of α-actinin–positive sarcomeres and decreased VSF (Fig. 4N, O), which were fully rescued by transgenic overexpression of the EGFP-tln1 fusion gene (Fig. 4M) compared with noninjected controls. On the other hand, transgenic overexpression of the EGFP-tln1 fusion gene failed to restore blood circulation along with rescued cardiac function in tln1 morphants, of which 103 of 147 showed no circulation. These data suggest that tln1 has intrinsic function in CMs that is independent of its vascular function.
To substantiate the role of Tln1 in stabilization of Z-disks, we used transmission electron microscopy to examine cardiac sarcomeric Z-disks in tln1 mutants and WT siblings by the cypher-EGFP reporter for sarcomeric Z-disks, and the F-actin biosensor EGFP-UtrCH. First, the Z-disks were disrupted in both tln1fl02k and tln1hi3093 mutants (Fig. 5B, C), compared with the well-formed Z-disks in WT siblings (Fig. 5A). Second, cypher is a critical Z-disk protein and the cypher-EGFP fusion protein is known to label cardiac Z-disk structures (35). Compared with the Z-disks well-labeled by transient expression of the cypher-EGFP fusion protein driven by the myl7 promoter (Fig. 5E), the cypher-EGFP signals were disrupted in the tln1fl02k and tln1hi3093 mutants (Fig. 5F, G). Third, it is known that the F-actin network is essential for cardiac sarcomeric structure (42) and is regulated by the integrin–talin complex (43). To visualize F-actin networks in CMs, we used the F-actin biosensor EGFP-UtrCH, which contains the calponin homology domain of utrophin fused to EGFP (44) for cardiac-specific transgenic expression of EGFP-UtrCH. Unlike the main phalloidin labeling of the F-actin in thin filaments, the strongest fluorescence signal of this biosensor revealed much higher resolution in Z-disks that colocalized with α-actinin (Supplemental Fig. S5A–C). As expected, the expression pattern of EGFP-UtrCH was disturbed in tln1 morphants at 52 hpf (Supplemental Fig. S5E), compared with the well-formed Z-disks in control embryos (Supplemental Fig. S5D). Together, these data further suggest that Tln1/F-actin have essential functions in the stabilization of cardiac sarcomeric Z-disks in the developing zebrafish heart.
Figure 5.

Z-disks are disrupted in the absence of either tln1 or itgb1b. A–D) Transmission electron micrographs reveal that the Z-disks of cardiac sarcomeres were disrupted in tln1 mutants (B, C) and itgb1b morphants (D) compared with WT controls (A) at 52 hpf. Arrows: disrupted Z-disks. Z-disks labeled by transient expression of the cypher-EGFP fusion protein in CMs were disrupted in tln1 mutants (F, G) and itgb1b morphants (H) compared with WT controls (E) at 52 hpf. n, number of hearts. Scale bars, 200 nm (A–D); 4 μm (E–H).
Tln1 is crucial for endocardial/endothelial cell integrity
In addition to cardiac dysfunction, we showed that the lumens of the DA and PCV were severely reduced in fl02k mutants by 52 hpf (Fig. 1M, N). In an intriguing observation, the cardiac lumen also appeared narrower or disrupted in tln1fl02k mutants at 52 hpf. In contrast to the flattened appearance of endocardial cells in WT embryos at 52 hpf (Fig. 6A–E), Tg(kdrl:EGFP)-labeled endocardial cells were cuboidal and aggregated, thus surrounding a narrow lumen (Fig. 6F–J). Both endocardial and myocardial cells labeled by Tg(myl7:EGFP) in tln1fl02k mutants remained associated with their respective basement membranes, but the distance between these cell layers increased (Fig. 6I, J). Eventually, the endocardium was ruptured (Fig. 6J, arrowhead) and blood cells leaked into the interstitium (Fig. 6F, arrows). Anti-ZO1 staining showed comparable tight junctions in tln1 mutants and WT siblings (Supplemental Fig. S6A–F). Together, these data show that tln1 is critical for endocardial cell integrity in zebrafish heart development.
Figure 6.

Tln1 is essential for endothelial/endocardial cell integrity. A–J) The endocardium became square shaped and detached from the myocardium in tln1fl02k embryos (F–J) compared with spindle-like endocardial cells in WT siblings (A–E) at 52 hpf. The extracellular matrix was labeled with anti-laminin antibody (red) (B, G), and Tg(myl7:EGFP) was used to outline the myocardium (C, H, white arrows) and Tg(kdrl:EGFP) was used to label the endocardium (C, H, red arrows). (F) Arrows: blood cells that abnormally entered and formed a hemorrhage in the space between the myocardium and endocardium. A and F) Lower laser intensity was used for imaging so that the basement membrane of the endocardium was not shown. D and I) Superimposed images of (B, C) and (G, H), respectively. E and J) High-magnification images of the respective regions outlined by white rectangles in (D) and (I). J) Asterisk: the abnormal separation of endocardial and myocardial basement membranes in the tln1fl02k embryo; arrowhead: the deformed endocardium. K–Q) The tln1 gene is essential for formation of the F-actin networks in vascular endothelial cells. The LDA and DA of WT siblings (L–N) and tln1fl02k mutant embryos (O–Q) in Tg(kdrl:EGFP) transgenic embryos at 52 hpf stained with Alexa-conjugated phalloidin (red). K) Blue frame: scanning area of (L–Q). Vascular endothelial cells were labeled by the Tg(kdrl:EGFP) transgene (green). The density of F-actin networks in tln1fl02k mutants (P, Q) decreased compared with that in WT embryos (M, N). a, atrium; v, ventricle; n, number of zebrafish embryos/group. Scale bars, 25 μm (A–D, F–I); 50 μm (K); 4 μm (L–Q).
We then addressed whether tln1 deficiency affects vascular endothelial cell integrity. We set out to examine endothelial tight junctions by using phalloidin staining for F-actin networks, anti-ZO1 antibody staining, and recording of endothelial tube formation in tln1 mutant embryos and TLN1-deficient HUVECs. Tight junctions detected with anti-ZO1 antibody appeared unaffected in the axial vessels of tln1 mutants (Supplemental Fig. S6L–N), suggesting that mutant endothelial cells maintain cell–cell adhesion. However, the density of phalloidin-visualized F-actin networks decreased in the lateral dorsal aortae (LDA) and DA of the tln1fl02k mutants (Fig. 6P, Q), compared with the WT embryos (Fig. 6M, N). Because F-actin is essential for cell spreading, these data partially explain why tln1 deficiency leads to cardiovascular lumen reduction and blood cell leakage in the brain and heart. In addition, the vascular tube formation of the subintestinal vessel (SIV) plexus was defective in the tln1fl02k mutants at 72 hpf (Fig. 7B), consistent with that in the DA and PCV (Fig. 1K–N). Furthermore, knockdown of Tln1 in HUVECs resulted in impaired vascular tube formation in 3D collagen matrices (Fig. 7D, E; areas of the lumens traced with Metamorph software; Molecular Devices, Sunnyvale, CA, USA), a phenotype known to be integrin mediated (45). These data showed that Tln1 acts through regulating F-actin networks and cell spreading to control vascular endothelial cell integrity in zebrafish and human cells.
Figure 7.

The tln1 gene is essential for endothelial tube formation in both the SIV plexus of zebrafish embryos and HUVECs. A–C) The vessel lumen of the SIV plexus (red arrows) was hardly detectable in tln1fl02k mutants (B) compared with WT siblings (A) and tnnt2a morphants (C) in Tg(kdrl:EGFP) transgenic embryos at 72 hpf. n, number of hearts. D–G) Vessels with lumens were barely formed by HUVECs treated with TLN1 siRNAs (E) compared with control siRNA (D) after culture in 3D collagen matrices for 24 h. D) Arrows: representative endothelial tube structures with an open lumen lined by flat endothelial cells, which are lacking in TLN1-deficient HUVECs (E). F) Statistics for endothelial cell (EC) tubes in (D, E), where the areas of the lumens were traced. Data are mean luminal areas ± sd (n = 3). **P < 0.01 vs. luciferase control. G) HUVEC extracts were prepared for Western blot analysis and probed for anti-Tln1 and anti-actin control. siRNA efficiently knocked down Tln1. Scale bars, 100 μm (A–C); 50 μm (D, E).
The itgb1b gene has a function similar to tln1 in zebrafish cardiovascular development
The major function of Tln1 is activating integrins by interacting with the Itgβ subunit (11, 17). The Itgβ1 subunit is involved in angiogenesis, vascular remodeling, and cardiac contractility in mice (46, 47). The zebrafish genome contains multiple homologs of this subunit: itgb1a, -1b, -b.1, and -b.2 (48). To determine which of these homologs mediates the function of tln1 in cardiovascular development, we performed a candidate screen with RNA in situ hybridization and found a remarkable similarity in the expression patterns of tln1 (Fig. 2J–R) and itgb1b (Fig. 8A–G). The itgb1b gene was expressed in the notochord, somites, and progenitor cells of the cardiovascular system in the somitogenesis stages (Fig. 8A, B); in the vasculature at 24 hpf (Fig. 8C; arrow); in the brain ventricles, gill arches, and gut at 48 hpf and later (Fig. 8D, E); and in the heart at 24 and 36 hpf (Fig. 8F, G; arrows). It was also expressed in the somites and at the border between the neural plate and epidermal ectoderm (Fig. 8A–C), overlapping with tln2 and -2a expression (Fig. 3E, F).
Figure 8.

itgb1b has functions and expression patterns overlapping with tln1 in cardiac growth and stabilization of the vessel lumen and sarcomeric structures. A–G) Whole-mount in situ hybridization of embryos with antisense itgb1b RNA probe at 12 somites (A, B), 24 (C, F), 36 (G), 48 (D), and 72 (E) hpf. A) Black arrow: developing heart field; white arrow: border between neural ectoderm and epidermal ectoderm. Arrows indicate progenitors of axial vessels (B), axial vessels (C), and heart tube (F, G). The itgb1b morphant (I) and noninjected control embryo (H) at 72 hpf; arrow: brain hemorrhage. J) Ventricular volume was reduced in itgb1b morphants at 52 hpf, and the heart was stopped at end diastole by 20 µM BDM as in fl02k mutants (Fig. 1G). K) The number of CMs labeled by Tg(myl7:nuDsRed) was reduced at 52 hpf, in itgb1b morphants as in fl02k mutants (Fig. 1H). L–N) The itgb1b gene is necessary for maintenance of the blood vessel lumen. L and M) Lateral views of trunk vessels of a WT sibling (L) and an itgb1b morphant (M) in Tg(kdrl:EGFP) zebrafish. The DA and PCV (M, N) were narrower in the itgb1b morphants at 52 hpf. O–T) The itgb1b morphants showed cardiac sarcomere defects similar to those in tln1 mutants during heart development. Confocal images of ventricles from itgb1b morphants (R–T) and WT siblings (O–Q) at 52 hpf after antibody staining. In itgb1b morphants, the Z-disks immunostained by α-actinin were hardly detectable (R), and the thin filaments stained by CH1 were only slightly affected (S), whereas the thick filaments stained by F59 were normal (T) compared with those in noninjected controls (O-Q). R) Arrows: residual signals of sarcomeric structures in itgb1b morphants. The number of embryos/group is shown in the histograms; n, number of hearts analyzed. Immunostaining of isolated embryonic hearts at 52 hpf with anti–α-actinin antibody (red). The Z-disks were disrupted in morphants by coinjecting low-dose tln1MO (2 ng) and itgb1bMO (3 ng) (X) compared with noninjected embryos (U), low-dose tln1MO (2 ng) only (V), or low-dose itgb1bMO (3 ng) only (W). n, number of hearts. H) VSF decreased in embryos with coinjection of both low-dose tln1MO (2 ng) and itgb1bMO (3 ng) compared with noninjected embryos, low-dose tln1MO (2 ng) only, or itgb1bMO (3 ng) only. The number of hearts is shown in the histograms. *P < 0.05; **P < 0.01. Scale bars, 40 μm (L, M); 4 μm (O–X).
To confirm the role of igb1b in cardiovascular development, we used an antisense MO targeting the itgb1b ATG (itgb1bMO) to knockdown itgb1b expression. These itgb1b morphants showed phenotypes similar to those of tln1 mutants at 52 hpf compared with noninjected control siblings or mismatch MO (itgb1bmisMO)-injected embryos (Fig. 8H, I; Table 5; Supplemental Fig. S2E, H). Although all noninjected control siblings showed normal circulation at 52 hpf, most of the embryos injected with 8 ng of itgb1bMO displayed tln1fl02k-like phenotypes (Table 5). The effectiveness and specificity of the itgb1bMO were confirmed, as it suppressed the 5′UTR itgb1b-EGFP reporter (Supplemental Fig. S2) and Itgβ1b expression (Supplemental Fig. S3C, D) as well as morphant phenotypes were rescued by injection of MO-resistance itgb1b mRNA (Supplemental Fig. S2F, H). We then evaluated how itgb1b regulates heart function by using the same methods as were used for tln1 mutants. The ventricular chamber size and number of CMs were reduced in itgb1b morphants (Fig. 8J, K), and the reduced size and proliferation, as well as increased apoptosis of CMs, contributed to compromised cardiac growth (Supplemental Fig. S4C, F, I, L). The lumens of major axial vessels were closed or diminished in the itgb1b morphants (Fig. 8M, N) compared with the well-formed lumens in WT siblings (Fig. 8L, N). Furthermore, anti–α-actinin staining showed that the Z-disks were disrupted (Fig. 8R), whereas CH1 staining showed that the thin filament CH1 was only slightly affected (Fig. 8S), and with F59 staining, the thick filament F59 appeared normal (Fig. 8T) in itgb1b morphants. The Z-disk abnormalities in the itgb1b morphants were further confirmed by transmission electron microscopy (Fig. 5D), as were the cypher-EGFP reporter for sarcomeric Z-disks (Fig. 5H), and the F-actin biosensor EGFP-UtrCH (Supplemental Fig. S5F). To further explore whether Tln1 functionally interacts with Itgβ1b during heart development, we simultaneously knocked down both tln1 and itgb1b and found synergistic effects on the formation of Z-disks and cardiac function in morphants injected with low doses of tln1MO (2 ng) and itgb1bMO (3 ng) (Fig. 8X, Y), whereas we found no evident phenotypes in embryos injected with either tln1MO (2 ng) or itgb1bMO (3 ng) (Fig. 8V, W, Y). Integrin activation by Tln results in assembling multicomponent force-generating cell adhesion complexes at FAs that are necessary for sustaining cell spreading (11, 12).The expression patterns of the FA complex proteins vinculin and pFAK(Y576) were unexpectedly normal in itgb1b morphants (Fig. 9J–M), as were those in tln1 mutants (Fig. 9D, I, M; Supplemental Fig. S3E–H). In addition, the tight junctions stained with anti-ZO1 were intact in itgb1b morphants (Supplemental Fig. S6G, H). Together with the tln1 mutant phenotypes, these results support the notion that Itgβ1b is a potential partner for Tln1 in the stabilization of cardiac Z-disks and vessel lumens, as well as in cardiac growth and function, and the formation of FAs and tight junctions is independent of Tln1-Itgβ1b in the embryonic heart in zebrafish.
TABLE 5.
itgb1b is required for cardiovascular development
| Dosage (ng/embryo) | Embryos (52 hpf) in each phenotypic group (n) |
Total embryos (n) | ||||
|---|---|---|---|---|---|---|
| a | b | c | d | e | ||
| 8 | 0 | 4 | 15 | 108 | 4 | 131 |
| 6 | 6 | 15 | 31 | 86 | 0 | 138 |
A typical experiment is shown. Noninjected control embryos were all normal. a, With circulation; b, with circulation, reduced ventricular lumen; c, milder tln1f02k-like, some with brain hemorrhage; d, tln1f02k-like, some with brain hemorrhage; and e, deformed.
Figure 9.

Focal adhesion proteins vinculin and pFAK are normally expressed in tln1 mutants and itgb1b morphants. A–L) Immunostaining of paraffin-embedded sections of embryonic hearts at 52 hpf with anti-vinculin (red) and anti-pFAK (green) antibodies, with DAPI costaining (blue) for nuclei. Expression of vinculin (A, D, G, J), pFAK (B, E, H, K), and merged images (C, F, I, L) in the heart of tln1 mutants (D–I) and itgb1b morphants (J–L) was comparable with WT controls (A–C), suggesting that Itgβ1b-Tln1 plays almost no role in the formation of FAs in zebrafish embryos. C, F, I, L) Arrows: myocardium (my) and endocardium (en). v, ventricle; a, atrium; avc, atrioventricular canal; n, number of hearts. M) Western blot analysis showing that vinculin and pFAK were normally expressed in dissected embryonic hearts from tln1 mutants and itgb1b morphants compared with their sibling controls at 52 hpf. The blot is representative of 3 independent experiments. Scale bars, 20 μm.
DISCUSSION
In this study, we identified tln1 as the defective gene in tln1fl02k mutants by positional cloning, found tln1 to be the only cardiac tln in the developing cardiovascular system, showed that its deficiency leads to abnormal cardiovascular morphogenesis and function by affecting F-actin networks and endocardial/endothelial cell spreading, as well as destabilizing cardiac Z-disks and endothelial cell integrity, and found that Tln1 interacts with Itgβ1b during zebrafish cardiovascular development. Unlike in mice, the unique tln1 expression pattern and its mutant phenotype enabled us, for the first time, to demonstrate the essential function of tln1 in the stabilization of cardiac sarcomeres and endocardial/endothelial cell integrity in the developing cardiovascular system. This study has important implications in our further understanding of the molecular mechanisms underlying cardiomyopathy and congenital heart disease.
Constitutive ablation of Tln1 causes embryonic arrest during mouse gastrulation, precluding investigations into its role in organogenesis (22). However, no differences in gross morphology or cardiovascular patterning were evident between WT siblings and tln1 mutants or between control embryos and morphants targeting tln1 ATG, which inhibited translation of both maternal and zygotic tln1 mRNA. A probable explanation is that maternal Tln1 proteins, which are detectable by Western blot analysis at the 256-cell stage, are sufficient for mediating morphogenesis during gastrulation in the tln1 mutants and morphants. Western blot analysis also showed the presence of Tln1fl02k proteins lacking the 35 amino acids encoded by exon 9, which led to disruption of the integrin binding site of the Tln1 head domain, but another integrin binding site localized at the Tln1 rod domain was intact. Studies of Drosophila development have demonstrated that each of the 2 integrin binding sites has a specialized role (49, 50). However, tln1hi3093 mutants, tln1hi3093/tln1fl02k mutants, and tln1 morphants all had the same phenotypes as the tln1fl02k mutants, suggesting that the truncated Tln1fl02k proteins have a null function. Both Tln1 and -2 are expressed in the mouse heart (10), but tln1 is the only Tln gene expressed in the zebrafish embryonic and adult heart. Therefore, this zebrafish tln1 mutant provides an ideal genetic model for determining the role of the tln1 gene in heart development in the absence of other Tln gene redundancy.
Unlike tln1 mutant zebrafish embryos, myocardium-specific Tln1 knockout (Tln1c-KO) mice have normal life spans and show normal cardiac sarcomere structure and function (10). The probable reason is Tln2 compensation, given that mouse Tln2 is expressed in both embryonic and adult CMs and is normally expressed in the Tln1-deficient heart (10, 40). In vitro studies have shown that Tln2 has a higher affinity for F-actin than Tln1 (51). Furthermore, Tln2 plays a major structural role in connecting integrins with the cytoskeleton/sarcomeres, whereas Tln1 dominantly acts as a mechanotransducer under stress to trigger integrin-dependent hypertrophic response signaling in mice (10). In the zebrafish heart, only tln1 undertakes both roles: formation of structural connections between integrin receptors and cytoskeleton/sarcomeres, and a mechanotransducer for stress response. Our data suggest that tln1 does not contribute to early heart development and function, but is essential for the stabilization of sarcomere structure.
Cardiac Z-disks, the lateral borders of the sarcomere unit, withstand mechanical forces during every heartbeat. Actin-containing thin filaments from neighboring sarcomeres overlap at the Z-disks and are cross-linked by tight interactions with α-actinin (42). In zebrafish cardiogenesis, Z-disk assembly starts at the 26-somite stage and matures at 48 hpf (41). The Z-disks, detected by α-actinin antibody staining, appeared almost normal in most of the tln1 mutant embryos at 48 hpf, but were hardly detectable at 52 hpf, and their absence led to heart failure. These results suggested that tln1 is critical for stabilizing the Z-disks to meet the increased mechanical force of heartbeats after 52 hpf. Similar cardiac sarcomeric defects have been reported in zebrafish and human mutants of actn2, cypher, and nexilin, which are also Z-disk components (35, 52, 53). The defective ventricular chamber in tln1 mutants is notably similar to that in actn2 mutants, but differs from the dilated hearts of morphants that are depleted of either cypher or nexilin. Both tln1 and actn2 are consistent components of FAs and bind to another FA component, vinculin (54), which is critical for heart function and cardiomyopathy (55). The genetic interactions among tln1, actn2, and vinculin in cardiac development remain to be determined. Furthermore, the Z-disk is not only an important structure for mechanical stability, but also functions in response to mechanical stress and subsequently initiates signaling pathways (42). Therefore, understanding how Tln1 affects Z-disk structure will help to elucidate the function of the integrin adhesion complex proteins under stress responses in cardiac physiology during cardiomyopathy.
In contrast to the requirement for tln1 in angiogenesis in mice (25), cardiovascular lumens and angiogenesis were normal in zebrafish tln1 mutants before 36 hpf. Endocardial and endothelial cells began to become rounded, with splitting of the endocardial and myocardial layers, resulting in progressive narrowing of cardiovascular lumens and extravascular leakage of blood cells at ∼52 hpf. Endothelial cells in the tln1 mutant embryos unexpectedly remained attached to their basement membranes, suggesting the presence of compensatory proteins for angiogenesis and mediating integrin–ECM interactions in zebrafish endothelial/endocardial cells. One such candidate could be kindlin 2, which also binds and activates β1 integrins (56) and is vital for angiogenesis in mice and zebrafish (57). Constitutive ablation of kindlin 2 causes a similar but more severe phenotype than that of Tln1 mutant mice, leading to peri-implantation lethality because of severe detachment of the endoderm and epiblasts from the basement membrane (58). Several studies have demonstrated that blood flow and shear stress are 2 essential factors for vascular lumen formation and maintenance (33, 59, 60). However, the lack of blood circulation may partly contribute to, but not fully explain, the impaired vessel lumen in tln1 mutants by comparing with those in silent heart (sih) mutants (61). We found that the fully connected SIV lumen appear in sih morphants but not in tln1 mutants. In addition, myocyte-specific expression of EGFP-tln1 restores cardiac function, but still fails to improve blood circulation in tln1 morphants. Therefore, our data suggest that tln1 is essential for both cardiac and vascular development.
The inability of vascular endothelial cells to sustain cell spreading and F-actin networks in the face of the mechanical forces of muscle contraction and shear stress most likely results from loss or weakening of FA and FA-like structures, which normally must have the Tln rod domain to mechanically link ligand-bound β1 integrins to the contractile actin network (11, 43, 62). Increased actomyosin contraction in the absence of mature FA may account for the eventual rounding of endocardial and endothelial cells in tln1 mutants. This interpretation is consistent with previous findings in Drosophila, where in the absence of Tln and β1 integrins remain associated with their ECM ligands, but the ECM-bound integrin is unable to connect stably to the cytoskeleton, leading to defective muscle adhesion and splitting of the 2 cell layers of the wing, resulting in blisters (19, 21).
Both the expression pattern and functional analysis of itgb1b support the notion that tln1 mediates itgb1b during zebrafish cardiovascular development. The β1 integrins are critical for heart function under normal conditions and in the pathologic response to stress (5, 63) and are also essential for CM differentiation and proliferation (4, 64). However, their role in vivo in sarcomere structure during heart development is not clear. Our results provide the first in vivo evidence that, like tln1, itgb1b plays an essential role in the stabilization of cardiac sarcomeric Z-disks and major axial vessel lumens, at least in part by regulating F-actin networks. The similar cardiac and vascular defects found in itgb1b morphants and tln1 mutants reflected the impaired interaction of truncated Tln1fl02k mutant products with the integrin β1 cytoplasmic tail (17), which is necessary for switching the integrin from the default low- to the high-affinity state (11, 43, 62). The in-frame deletion of key segments of the Tln1fl02k FERM domain impaired its ability to switch to high-affinity β1 integrin. On the other hand, expression of the integrin-dependent FA proteins Itgβ1b, vinculin, and pFAK(Y576) was unaffected in the tln1 mutant heart, which is different from other studies in Drosophila (19, 65). The findings in our study suggest that either the molecular nature of the formation and stabilization of integrin clustering is different or another redundant protein (e.g., kindling 2) plays a parallel role in this process in the zebrafish.
Acknowledgments
The authors thank Dr. W. Shou (Indiana University, Indianapolis, IN, USA) and members of the laboratory of J.-W.X. for providing critical comments on the manuscript, Dr. I. C. Bruce (Zhejiang University, Zheijang, China) for reading the manuscript, and Dr. Y. Hu (Peking University, Peking, China) for help with transmission electron microscopy. This work was funded by U.S. National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases Training Grant T32-DK07540 (to Q.Y.) and Grants DK088327 and DK48549 (to M.A.A.); March of Dimes Birth Defects Foundation Grant 1-FY2007-471 (to J.-W.X.); the Milton Fund from Harvard University (to J.-W.X.); the National Basic Research Program of China Grants 2012CB944501 and 2010CB529503 (to J.-W.X.); the National Natural Science Foundation of China Grants 31430059, 81470399, 31221002, 31271549, and 81270164 (to J.-W.X.); NIH National Heart, Lung, and Blood Institute Grant HL59373 (to G.E.D.); and NIH National Center for Research Resources Grant RR012589 (to M.A.A).
Glossary
- 3D
3-dimensional
- actn2
actinin 2 α
- BDM
2,3-butanedione monoxime
- BR
blocking reagent
- CH1
tropomyosin antibody
- CM
cardiomyocyte
- DA
dorsal aorta
- ECM
extracellular matrix
- EGFP
enhanced green fluorescent protein
- F59
myosin antibody
- FA
focal adhesion
- FERM
4.1, ezrin, radixin, moesin
- hpf
hours post fertilization
- itgb
integrin β
- kdrl
kinase insert domain receptor like
- LDA
lateral dorsal aortae
- MAB
maleic acid buffer
- MO
morpholino
- myl
myosin light chain
- NGS
normal goat serum
- PCV
posterior cardinal vein
- PFA
paraformaldehyde
- RACE
rapid amplification of cDNA ends
- siRNA
small interfering RNA
- sih
silent heart
- SIV
subintestinal vessel
- SLP
sequence length polymorphism
- SNP
single nucleotide polymorphism
- Tln
talin
- tnnt2a
troponin t2a
- UtrCH
calponin homology domain of utrophin
- VSF
ventricular shortening fraction
- WT
wild-type
- ZO
zonula occuldens
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Harston R. K., Kuppuswamy D. (2011) Integrins are the necessary links to hypertrophic growth in cardiomyocytes. J. Signal Transduct. 2011, 521742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 3.Samarel A. M. (2005) Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am. J. Physiol. Heart Circ. Physiol. 289, H2291–H2301 [DOI] [PubMed] [Google Scholar]
- 4.Ieda M., Tsuchihashi T., Ivey K. N., Ross R. S., Hong T. T., Shaw R. M., Srivastava D. (2009) Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell 16, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shai S. Y., Harpf A. E., Babbitt C. J., Jordan M. C., Fishbein M. C., Chen J., Omura M., Leil T. A., Becker K. D., Jiang M., Smith D. J., Cherry S. R., Loftus J. C., Ross R. S. (2002) Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ. Res. 90, 458–464 [DOI] [PubMed] [Google Scholar]
- 6.Bendig G., Grimmler M., Huttner I. G., Wessels G., Dahme T., Just S., Trano N., Katus H. A., Fishman M. C., Rottbauer W. (2006) Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 20, 2361–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang X., Sun Y., Ye M., Scimia M. C., Cheng H., Martin J., Wang G., Rearden A., Wu C., Peterson K. L., Powell H. C., Evans S. M., Chen J. (2009) Targeted ablation of PINCH1 and PINCH2 from murine myocardium results in dilated cardiomyopathy and early postnatal lethality. Circulation 120, 568–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brancaccio M., Fratta L., Notte A., Hirsch E., Poulet R., Guazzone S., De Acetis M., Vecchione C., Marino G., Altruda F., Silengo L., Tarone G., Lembo G. (2003) Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat. Med. 9, 68–75 [DOI] [PubMed] [Google Scholar]
- 9.Johnston R. K., Balasubramanian S., Kasiganesan H., Baicu C. F., Zile M. R., Kuppuswamy D. (2009) Beta3 integrin-mediated ubiquitination activates survival signaling during myocardial hypertrophy. FASEB J. 23, 2759–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manso A. M., Li R., Monkley S. J., Cruz N. M., Ong S., Lao D. H., Koshman Y. E., Gu Y., Peterson K. L., Chen J., Abel E. D., Samarel A. M., Critchley D. R., Ross R. S. (2013) Talin1 has unique expression versus talin 2 in the heart and modifies the hypertrophic response to pressure overload. J. Biol. Chem. 288, 4252–4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Critchley D. R. (2009) Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu. Rev. Biophys. 38, 235–254 [DOI] [PubMed] [Google Scholar]
- 12.Puklin-Faucher E., Sheetz M. P. (2009) The mechanical integrin cycle. J. Cell Sci. 122, 179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tidball J. G., O’Halloran T., Burridge K. (1986) Talin at myotendinous junctions. J. Cell Biol. 103, 1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belkin A. M., Zhidkova N. I., Koteliansky V. E. (1986) Localization of talin in skeletal and cardiac muscles. FEBS Lett. 200, 32–36 [DOI] [PubMed] [Google Scholar]
- 15.Anthis N. J., Wegener K. L., Ye F., Kim C., Goult B. T., Lowe E. D., Vakonakis I., Bate N., Critchley D. R., Ginsberg M. H., Campbell I. D. (2009) The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 28, 3623–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goult B. T., Bouaouina M., Elliott P. R., Bate N., Patel B., Gingras A. R., Grossmann J. G., Roberts G. C., Calderwood D. A., Critchley D. R., Barsukov I. L. (2010) Structure of a double ubiquitin-like domain in the talin head: a role in integrin activation. EMBO J. 29, 1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wegener K. L., Partridge A. W., Han J., Pickford A. R., Liddington R. C., Ginsberg M. H., Campbell I. D. (2007) Structural basis of integrin activation by talin. Cell 128, 171–182 [DOI] [PubMed] [Google Scholar]
- 18.Moes M., Rodius S., Coleman S. J., Monkley S. J., Goormaghtigh E., Tremuth L., Kox C., van der Holst P. P., Critchley D. R., Kieffer N. (2007) The integrin binding site 2 (IBS2) in the talin rod domain is essential for linking integrin beta subunits to the cytoskeleton. J. Biol. Chem. 282, 17280–17288 [DOI] [PubMed] [Google Scholar]
- 19.Brown N. H., Gregory S. L., Rickoll W. L., Fessler L. I., Prout M., White R. A., Fristrom J. W. (2002) Talin is essential for integrin function in Drosophila. Dev. Cell 3, 569–579 [DOI] [PubMed] [Google Scholar]
- 20.Cram E. J., Clark S. G., Schwarzbauer J. E. (2003) Talin loss-of-function uncovers roles in cell contractility and migration in C. elegans. J. Cell Sci. 116, 3871–3878 [DOI] [PubMed] [Google Scholar]
- 21.Prout M., Damania Z., Soong J., Fristrom D., Fristrom J. W. (1997) Autosomal mutations affecting adhesion between wing surfaces in Drosophila melanogaster. Genetics 146, 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monkley S. J., Zhou X. H., Kinston S. J., Giblett S. M., Hemmings L., Priddle H., Brown J. E., Pritchard C. A., Critchley D. R., Fassler R. (2000) Disruption of the talin gene arrests mouse development at the gastrulation stage. Dev. Dyn. 219, 560–574 [DOI] [PubMed] [Google Scholar]
- 23.Calderwood D. A., Campbell I. D., Critchley D. R. (2013) Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Debrand E., Conti F. J., Bate N., Spence L., Mazzeo D., Pritchard C. A., Monkley S. J., Critchley D. R. (2012) Mice carrying a complete deletion of the talin2 coding sequence are viable and fertile. Biochem. Biophys. Res. Commun. 426, 190–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monkley S. J., Kostourou V., Spence L., Petrich B., Coleman S., Ginsberg M. H., Pritchard C. A., Critchley D. R. (2011) Endothelial cell talin1 is essential for embryonic angiogenesis. Dev. Biol. 349, 494–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amsterdam A., Burgess S., Golling G., Chen W., Sun Z., Townsend K., Farrington S., Haldi M., Hopkins N. (1999) A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 13, 2713–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amsterdam A., Nissen R. M., Sun Z., Swindell E. C., Farrington S., Hopkins N. (2004) Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA 101, 12792–12797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi J., Dong L., Ahn J., Dao D., Hammerschmidt M., Chen J. N. (2007) FoxH1 negatively modulates flk1 gene expression and vascular formation in zebrafish. Dev. Biol. 304, 735–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burns C. G., Milan D. J., Grande E. J., Rottbauer W., MacRae C. A., Fishman M. C. (2005) High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat. Chem. Biol. 1, 263–264 [DOI] [PubMed] [Google Scholar]
- 30.Kawakami K., Takeda H., Kawakami N., Kobayashi M., Matsuda N., Mishina M. (2004) A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell 7, 133–144 [DOI] [PubMed] [Google Scholar]
- 31.Thisse C., Thisse B., Schilling T. F., Postlethwait J. H. (1993) Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119, 1203–1215 [DOI] [PubMed] [Google Scholar]
- 32.Xiao T., Baier H. (2007) Lamina-specific axonal projections in the zebrafish tectum require the type IV collagen Dragnet. Nat. Neurosci. 10, 1529–1537 [DOI] [PubMed] [Google Scholar]
- 33.Sehnert A. J., Huq A., Weinstein B. M., Walker C., Fishman M., Stainier D. Y. (2002) Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Genet. 31, 106–110 [DOI] [PubMed] [Google Scholar]
- 34.Shu X., Cheng K., Patel N., Chen F., Joseph E., Tsai H. J., Chen J. N. (2003) Na,K-ATPase is essential for embryonic heart development in the zebrafish. Development 130, 6165–6173 [DOI] [PubMed] [Google Scholar]
- 35.Yang J., Xu X. (2012) α-Actinin2 is required for the lateral alignment of Z discs and ventricular chamber enlargement during zebrafish cardiogenesis. FASEB J. 26, 4230–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koh W., Stratman A. N., Sacharidou A., Davis G. E. (2008) In vitro three dimensional collagen matrix models of endothelial lumen formation during vasculogenesis and angiogenesis. Methods Enzymol. 443, 83–101 [DOI] [PubMed] [Google Scholar]
- 37.Bakkers J. (2011) Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 91, 279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tremuth L., Kreis S., Melchior C., Hoebeke J., Rondé P., Plançon S., Takeda K., Kieffer N. (2004) A fluorescence cell biology approach to map the second integrin-binding site of talin to a 130-amino acid sequence within the rod domain. J. Biol. Chem. 279, 22258–22266 [DOI] [PubMed] [Google Scholar]
- 39.Cheung T. Y., Fairchild M. J., Zarivach R., Tanentzapf G., Van Petegem F. (2009) Crystal structure of the talin integrin binding domain 2. J. Mol. Biol. 387, 787–793 [DOI] [PubMed] [Google Scholar]
- 40.Monkley S. J., Pritchard C. A., Critchley D. R. (2001) Analysis of the mammalian talin2 gene TLN2. Biochem. Biophys. Res. Commun. 286, 880–885 [DOI] [PubMed] [Google Scholar]
- 41.Huang W., Zhang R., Xu X. (2009) Myofibrillogenesis in the developing zebrafish heart: A functional study of tnnt2. Dev. Biol. 331, 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knöll R., Buyandelger B., Lab M. (2011) The sarcomeric Z-disc and Z-discopathies. J. Biomed. Biotechnol. 2011, 569628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harburger D. S., Calderwood D. A. (2009) Integrin signalling at a glance. J. Cell Sci. 122, 159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burkel B. M., von Dassow G., Bement W. M. (2007) Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 64, 822–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davis G. E., Koh W., Stratman A. N. (2007) Mechanisms controlling human endothelial lumen formation and tube assembly in three-dimensional extracellular matrices. Birth Defects Res. C Embryo Today 81, 270–285 [DOI] [PubMed] [Google Scholar]
- 46.Krishnamurthy P., Subramanian V., Singh M., Singh K. (2007) Beta1 integrins modulate beta-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Hypertension 49, 865–872 [DOI] [PubMed] [Google Scholar]
- 47.Carlson T. R., Hu H., Braren R., Kim Y. H., Wang R. A. (2008) Cell-autonomous requirement for beta1 integrin in endothelial cell adhesion, migration and survival during angiogenesis in mice. Development 135, 2193–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mould A. P., McLeish J. A., Huxley-Jones J., Goonesinghe A. C., Hurlstone A. F., Boot-Handford R. P., Humphries M. J. (2006) Identification of multiple integrin beta1 homologs in zebrafish (Danio rerio). BMC Cell Biol. 7, 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanentzapf G., Brown N. H. (2006) An interaction between integrin and the talin FERM domain mediates integrin activation but not linkage to the cytoskeleton. Nat. Cell Biol. 8, 601–606 [DOI] [PubMed] [Google Scholar]
- 50.Ellis S. J., Pines M., Fairchild M. J., Tanentzapf G. (2011) In vivo functional analysis reveals specific roles for the integrin-binding sites of talin. J. Cell Sci. 124, 1844–1856 [DOI] [PubMed] [Google Scholar]
- 51.Senetar M. A., Foster S. J., McCann R. O. (2004) Intrasteric inhibition mediates the interaction of the I/LWEQ module proteins Talin1, Talin2, Hip1, and Hip12 with actin. Biochemistry 43, 15418–15428 [DOI] [PubMed] [Google Scholar]
- 52.Hassel D., Dahme T., Erdmann J., Meder B., Huge A., Stoll M., Just S., Hess A., Ehlermann P., Weichenhan D., Grimmler M., Liptau H., Hetzer R., Regitz-Zagrosek V., Fischer C., Nürnberg P., Schunkert H., Katus H. A., Rottbauer W. (2009) Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat. Med. 15, 1281–1288 [DOI] [PubMed] [Google Scholar]
- 53.Zheng M., Cheng H., Li X., Zhang J., Cui L., Ouyang K., Han L., Zhao T., Gu Y., Dalton N. D., Bang M. L., Peterson K. L., Chen J. (2009) Cardiac-specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Hum. Mol. Genet. 18, 701–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bois P. R., O’Hara B. P., Nietlispach D., Kirkpatrick J., Izard T. (2006) The vinculin binding sites of talin and alpha-actinin are sufficient to activate vinculin. J. Biol. Chem. 281, 7228–7236 [DOI] [PubMed] [Google Scholar]
- 55.Zemljic-Harpf A., Manso A. M., Ross R. S. (2009) Vinculin and talin: focus on the myocardium. J. Investig. Med. 57, 849–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moser M., Legate K. R., Zent R., Fässler R. (2009) The tail of integrins, talin, and kindlins. Science 324, 895–899 [DOI] [PubMed] [Google Scholar]
- 57.Pluskota E., Dowling J. J., Gordon N., Golden J. A., Szpak D., West X. Z., Nestor C., Ma Y. Q., Bialkowska K., Byzova T., Plow E. F. (2011) The integrin coactivator kindlin-2 plays a critical role in angiogenesis in mice and zebrafish. Blood 117, 4978–4987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Montanez E., Ussar S., Schifferer M., Bösl M., Zent R., Moser M., Fässler R. (2008) Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 22, 1325–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellertsdóttir E., Lenard A., Blum Y., Krudewig A., Herwig L., Affolter M., Belting H. G. (2010) Vascular morphogenesis in the zebrafish embryo. Dev. Biol. 341, 56–65 [DOI] [PubMed] [Google Scholar]
- 60.Li Y. S., Haga J. H., Chien S. (2005) Molecular basis of the effects of shear stress on vascular endothelial cells. J. Biomech. 38, 1949–1971 [DOI] [PubMed] [Google Scholar]
- 61.Montero-Balaguer M., Swirsding K., Orsenigo F., Cotelli F., Mione M., Dejana E. (2009) Stable vascular connections and remodeling require full expression of VE-cadherin in zebrafish embryos. PLoS One 4, e5772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arnaout M. A., Goodman S. L., Xiong J. P. (2007) Structure and mechanics of integrin-based cell adhesion. Curr. Opin. Cell Biol. 19, 495–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krishnamurthy P., Subramanian V., Singh M., Singh K. (2006) Deficiency of beta1 integrins results in increased myocardial dysfunction after myocardial infarction. Heart 92, 1309–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guan K., Fürst D. O., Wobus A. M. (1999) Modulation of sarcomere organization during embryonic stem cell-derived cardiomyocyte differentiation. Eur. J. Cell Biol. 78, 813–823 [DOI] [PubMed] [Google Scholar]
- 65.Ellis S. J., Lostchuck E., Goult B. T., Bouaouina M., Fairchild M. J., López-Ceballos P., Calderwood D. A., Tanentzapf G. (2014) The talin head domain reinforces integrin-mediated adhesion by promoting adhesion complex stability and clustering. PLoS Genet. 10, e1004756 [DOI] [PMC free article] [PubMed] [Google Scholar]