

Abstract
To elucidate the transcriptional landscape that regulates human lymphoid commitment during postnatal life, we used RNA sequencing to assemble the long non-coding transcriptome across human bone marrow and thymic progenitors spanning the earliest stages of B and T lymphoid specification. Over 3000 novel long non-coding RNA genes (lncRNAs) were revealed through the analysis of these rare populations. Lymphoid commitment was characterized by lncRNA expression patterns that were highly stage-specific and more lineage-specific than protein coding patterns. Protein-coding genes co-expressed with neighboring lncRNA genes were enriched for ontologies related to lymphoid differentiation. The exquisite cell-type specificity of global lncRNA expression patterns independently revealed new developmental relationships between the earliest progenitors in the human bone marrow and thymus.
INTRODUCTION
Long non-coding RNAs (lncRNAs) comprise a key component of the repertoire of regulatory elements that control cell differentiation1. However, the lncRNA landscape of human lymphoid commitment is unknown; thus knowledge of the transcriptional programs that launch human lymphopoiesis and regulate the subsequent fate decision between the T and B cell lineages is incomplete. We assembled a lncRNA catalog through whole transcriptome sequencing of rare hematopoietic stem cell (HSC) and lymphoid progenitor populations that encompass the earliest stages of B and T lineage commitment in the human bone marrow (BM) and thymus.
HSCs and lymphoid progenitors in both the BM and thymus express the surface antigen CD34 (refs.2,3). Although various immunophenotypic subsets of the CD34+ progenitors have been identified, the exact developmental relationships between these populations remain undefined. In particular, the identity of the dominant BM progenitor that migrates to the human thymus and initiates T cell differentiation remains an unanswered question due to the inherent limitations of functional assays of human progenitors. Expression of the cell surface antigen CD10 has long been used to define so-called “common lymphoid progenitors” (CLPs), which have been thought to represent the first stage of human lymphoid commitment in the BM and cord blood because of their broad ability to generate all lymphoid lineages2. The CD10+ progenitors have also been postulated to be the BM-derived precursors that seed the thymus because of their ability to generate T cells in culture2,4,5. Recently, we identified a lymphoid progenitor population that lacks CD10 expression but nonetheless possesses robust B, T and natural killer (NK) lymphoid potential; these CD34+CD38+CD45RA+CD10− CD62Lhi lineage-negative (lin−) cells were termed lymphoid-primed multipotent progenitors (LMPPs) because of functional similarity to the previously described Flt3+ murine LMPPs which lack erythroid and megakaryocytic potential6.
B lymphoid commitment begins in the BM; the cell surface co-expression of CD19 and CD34 defines a fully B-committed progenitor (BCP) that lacks the potential for any other lymphoid (NK or T) lineages7. T lymphoid commitment is initiated by the arrival of BM-derived precursors in the thymus. CD34+ thymic progenitors constitute <1% of all thymocytes8 and can be further fractionated based on CD7 and CD1a expression8,9. Two progenitor populations in the thymus with both lymphoid and myeloid potential have been described; the CD34+CD7−CD1a− progenitors (~5% of CD34+ thymocytes), and the CD34+CD7+CD1a− progenitors (~30–50% of CD34+ thymocytes)8,9. The degree of myeloid potential varies between these two progenitors, and the developmental relationship between them is unknown8,9. Upregulation of CD1a expression marks the third thymic progenitor population, the CD34+CD7+CD1a+ cells that represent the earliest fully T lineage-committed progenitor stage9. These latter cells generate CD4+CD8+ (double-positive, DP) thymocytes, which comprise the predominant population in the thymus, and further differentiate into single-positive (either CD4+ or CD8+) T cells that egress from the thymus to constitute mature peripheral T lymphocytes3,9.
We here demonstrate that the transcriptional repertoire of the lymphoid progenitors in BM and thymus, and the HSCs from which they are generated, is comprised of at least 9,400 lncRNA genes, over a third of which represent previously undiscovered genes. Sample clustering analysis and Bayesian polytomous model selection revealed distinct global lncRNA expression patterns that corresponded to early lymphoid commitment or lineage (B or T) specification. Global lncRNA expression patterns independently revealed developmental relationships between BM and thymic progenitors, which supported the conclusions that uncommitted thymic progenitors directly arise from BM-derived HSCs and/or LMPPs while CD10+ CLPs and subsequent B cell differentiation proceed through a separate pathway, and that CD34+CD7−CD1a− progenitors represent the earliest developmental stage of thymopoiesis. Our data constitute a resource for the elucidation of lncRNA mechanisms that regulate the earliest stages of human lymphopoiesis.
RESULTS
Novel lncRNAs revealed in HSC and lymphoid progenitors
To annotate novel lncRNAs during human lymphoid commitment, we performed RNA-Seq of 10 distinct cell types isolated by flow cytometry. From BM, we isolated CD34+CD38−lin− cells, a population highly enriched for HSCs, as well as three lymphoid progenitor populations; LMPPs, CLPs (CD34+CD38+CD10+CD45RA+lin−) and fully B cell-committed progenitors (BCPs, CD34+CD38+CD19+). From thymus we isolated three CD34+CD4−CD8− subsets; Thy1 (CD34+CD7−CD1a−), Thy2 (CD34+CD7+CD1a−) and Thy 3 (CD34+CD7+CD1a+), as well as fully T cell-committed populations; CD4+CD8+ (Thy 4), CD3+CD4+CD8− (Thy5) and CD3+CD4−CD8+ (Thy6). (Fig. 1a, Supplementary Fig.1a,b). We assembled the transcriptome by aligning and pooling reads from twenty samples (n = 2 biological replicates per cell type) to identify individual transcripts. Expression patterns for protein-coding genes known to be specifically expressed in hematopoietic stem/progenitor cells (HSPCs) or lymphoid lineages (B, T, or both) (Supplementary Fig.1c) were consistent with published studies7,10,11,12, validating our isolation and sequencing methods.
Figure 1. Human HSC and lymphoid transcriptomes are characterized by novel lncRNAs.

(a) Schema of HSC and 9 lymphoid cell types in human bone marrow (BM) and thymus that were analyzed by RNA-Seq (n=20 samples [two biological replicates per population]). * Lineage cocktail included CD19 except in the case of BCP) (b) Bioinformatic analysis pipeline for annotating novel lncRNAs. (c) Number of expressed protein coding and long non-coding RNA genes (>1 FPKM in at least one sample). (d) Violin plot showing expression levels of protein coding and lncRNA genes (FPKM mean, standard deviation and range are depicted). For each gene the maximum expression value (of the 20 samples) was used to generate the plot. (e) Expression levels of novel lncRNA genes in BM and thymus samples, and 16 other cell types from the Human Body Map project (adipose, adrenal, breast, brain, colon, heart, kidney, leucocyte, liver, lung, lymph node, ovary, prostate, skeletal muscle, testis, thyroid, supplementary table 3). Shown are 357 genes differentially expressed in at least one pairwise comparison of the 10 populations. HSC-hematopoietic stem cell, LMPP-lymphoid primed multipotent progenitor, CLP-common lymphoid progenitor, BCP- B committed progenitor. “Annotated lncRNA genes” were defined as genes annotated in GencodeV19 and/or lncipedia databases. “Novel lncRNA genes” were defined as lncRNA genes that have not been annotated in these databases. Hybrid lncRNA genes represent a subset of annotated lncRNA genes for which we discovered novel transcripts.
LncRNAs were identified using an ab-initio assembly pipeline (Fig. 1b). The final lncRNA annotation (Supplementary Table 1) used throughout the paper was comprised of long (> 200 bp), spliced transcripts with no sense exonic overlap with protein-coding genes. Analysis by CPAT13and PhlyoCSF14 confirmed these transcripts to be largely devoid of protein-coding potential (Supplementary Fig.1d). This comprehensive annotation comprised a total of 18,268 lncRNA genes, with 3,880 novel lncRNA genes not annotated in existing lncRNA databases15,16. Owing to the generation of multiple transcripts from each gene through alternative splicing, a total of 6,851 novel lncRNAs transcripts were detected, of which 4,661 were transcribed from previously unannotated gene loci while 2,190 represented novel isoforms generated from previously annotated loci.
Using unique whole-genome alignments of our RNA-Seq libraries to estimate expression levels on the final merged annotation (protein-coding and lncRNA genes), we found that 9,444 lncRNA genes and 3,348 novel lncRNA genes were expressed at >1 FKPM in at least one sample (Fig.1c, gene expression estimates listed at NCBI’s Gene Expression Omnibus [GSE69239]). Supplementary Table 2 lists the number of genes expressed in each cell type. Consistent with the strong correlation of gene expression between replicate samples (mean correlation coefficient = 0.95, standard deviation = 0.02, Supplementary Fig. 2a), 80% (2688 /3348) of novel lncRNA genes were expressed (FPKM>1) in both replicates of at least one cell type.
To validate the RNA-Seq expression data, a subset of B cell-specific lncRNAs was assessed by qPCR, (n = 2 biological replicates per cell type, including a replicate that was separate from the samples used for RNA-Seq). High concordance was observed between qPCR and RNA-Seq expression results across the ten cell types, for each of these genes (mean correlation = 0.92, standard deviation = 0.07, Supplementary Fig. 2b).
Among the novel lncRNA genes expressed at >1 FKPM in at least one sample, 2,652 were intergenic while 696 flanked a protein-coding gene in a divergent orientation (overlapping with a protein-coding gene on the antisense strand). The size distribution of novel lncRNA transcripts was similar to that of previously annotated lncRNAs15,16 (Supplementary Fig. 2c). Of note, novel lncRNA transcripts, irrespective of whether they were transcribed from previously unnannotated or annotated gene loci, were more abundantly expressed in these hematopoietic populations than were previously annotated lncRNAs (Fig. 1d, Supplementary Fig.2d).
Analysis of RNA-Seq data from Illumina’s Human Body Map (Supplementary Table 3), revealed that the novel lncRNA genes identified from our data were highly specific to the HSC and/or lymphoid populations studied in this current report (Fig. 1e). Interestingly, among the 3,109 novel lncRNA genes expressed in the six thymic populations, less than a third overlapped with a recently published catalog of lncRNAs derived from transcriptional profiles of unfractionated thymocytes and T cell acute lymphoblastic leukemia cells17.
To further characterize the lncRNA expression signature obtained from our RNA-Seq analysis, we analyzed the transcription start sites (TSS) of the novel lncRNAs for the presence of histone modifications. Actively transcribed lncRNA genes have been shown to be associated with, among others, promoter- (histone 3 lysine 4 trimethylation [H3K4me3]) or enhancer-associated (histone 3 lysine 4 monomethylation [H3K4me1]) histone modifications18,19. Although it has been previously shown that histone modifications are regulated during differentiation, only a fraction is subjected to significant dynamic changes20. Therefore, as low cell numbers render ChIP-sequencing of the rare populations used in this study largely unfeasible, we used publically available H3K4me1 and H3K4me3 ChIP-Seq data for hematopoietic progenitor (CD34+) and lymphoid cell populations (Supplementary Table 3)21. In addition, RNA-Seq libraries for these same populations were downloaded and re-analyzed to identify the populations that were transcriptionally similar to the specific progenitors used to generate our RNA-Seq dataset (Supplementary Fig. 3). ChIP-Seq data from three populations were thus chosen for analysis: whole thymic tissue, CD19+ primary B lymphocytes, and mobilized peripheral blood HSPCs (defined as either CD34+ or CD133+) (Supplementary Table 3).
Over a third of the novel lncRNA TSSs (37%) showed overlap with statistically significant peaks (Supplementary Methods) for at least one type of histone modification, a proportion similar to that of previously annotated lncRNAs associated with such histone modifications in these datasets (37.5%) as well as in a published analysis of epigenetic modifications22. H3K4me1 modifications showed a broader density distribution around TSSs when compared with H3K4me3 modifications (Fig. 2a,b). Protein-coding gene TSSs tended to show high density of H3K4me3 modifications (low H3K4me1/H3K4me3 ratio) in HSPCs and lymphoid cells, a feature commonly associated with promoter regions. On the other hand, lncRNA TSSs tended to show high H3K4me1/H3K4me3 ratios, a feature commonly associated with enhancer regions18,19. H3K4me1/H3K4me3 ratios for TSSs with overlapping histone modification peaks are depicted in Fig. 2c (data for individual TSSs listed in Supplementary Table 4).
Figure 2. LncRNAs transcription start sites (TSS) show cell type specific active chromatin profiles.

(a) Histone modification profiles at TSSs of novel lncRNAs (ChiP-Seq data from hematopoietic stem and progenitor [CD133+] cells). (b) UCSC overlay tracks depicting representative novel lncRNAs overlapping histone modifications typically associated with enhancer (high H3K4Me1/ H3K4Me3 signal ratio) or promoter- (low H3K4Me1/ H3K4Me3 signal ratio) elements. Overlay tracks depict the normalized Chip-Seq signal for HSPC (CD34+ and CD133+), CD19+ primary cells, and thymic cells (color code not shown). (c) H3K4Me1/ H3K4Me3 signal intensity ratios for protein coding genes and novel lncRNAs for ChIP-Seq data from CD34+ mobilized peripheral blood cells, CD19+ primary B lymphocytes, and thymic cells. (d) ChIP-Seq metaplots (histone mark density) at TSSs of protein coding and lncRNA genes, stratified by gene expression level are depicted for CD34+ HSPC, CD19+ B lymphocytes, and unfractionated thymocytes; HSC, BCP, and Thy4 respectively represent the closest related cell types in the RNA-Seq dataset (see Supplementary Fig.3). “Highly expressed” was defined as the top 2,000 transcripts when all the transcripts are ranked by expression level in that cell type. Cell type “specific” transcripts were defined as those showing peak expression in that cell type, and the peak value exceeds twice the mean of the expression in all other cell types. Publically available ChIP-Seq datasets (Supplementary table 3) were used for the analysis of histone profiles.
In the case of protein-coding genes, highly expressed transcripts were associated with a higher level of histone modifications compared to transcripts that were not expressed, or a set of randomly chosen transcripts (Fig. 2d). In contrast, in the case of lncRNA genes, cell type-specific transcripts were associated with the highest density of histone modifications (Fig. 2d). In summary, our results indicate that the transcriptional landscapes of human HSCs and lymphoid progenitors are characterized by numerous previously undescribed lncRNAs that are unique to these cell types.
Correlation between lncRNA and coding gene expression
To investigate co-expression patterns between lncRNA and protein-coding genes during lymphoid differentiation, we computed pairwise expression correlations between genes across all RNA-Seq samples. We first analyzed trans correlations of expression (defined as pairs consisting of genes separated by a distance >1 Mb, or located in different chromosomes). Expression of lncRNA genes tended to be more positively than negatively correlated with protein-coding genes in trans (4.0% of pairs had Spearman correlation coefficient |rho| (rs) >0.5, vs. 2.7% rs < −0.05, out of a total of 215 million trans-correlations tested). Consistent with previous reports2, the same tendency toward positive correlation was observed for pairs of protein coding-protein coding genes in trans (Supplementary Table 5). In all cases, the bias to positive correlations was significantly higher than that obtained from a control set of trans correlations, where the expression of protein-coding genes was randomly shuffled (Fig. 3a).
Figure 3. LncRNA genes are co-expressed with protein coding genes involved in hematopoiesis and immune function, during lymphoid differentiation.

(a) Density histograms of pairwise Spearman expression correlations between genes from different classes, in trans or cis. (b) Gene ontology enrichment for protein coding genes in cis and positively correlated with lncRNA genes (mRNA – lncRNA rs > 0.8), other protein coding genes (mRNA – mRNA rs > 0.8), or negatively correlated (mRNA – lncRNA rs < −0.5, mRNA – mRNA rs < −0.5). Colormap indicates the –log10 hypergeometric p value for enrichment as provided by DAVID functional annotation tools. (c) Model-based expression profiles (Profiles 1–20, Group I–VII) of differentially expressed genes (protein coding and lncRNA genes) during lymphoid differentiation. Numbers above each plot indicate: Profile identifier number (Total number of genes in the profile; number of lncRNA genes in the profile). Group VII contains profiles that could not be assigned to a specific differentiation related pattern. * Profiles enriched for lncRNA genes (p<0.05 when compared with the proportion of lncRNAs among all differe;ntially expressed genes). HSPC: hematopoietic stem/progenitor cells.
We then analyzed cis correlations of expression (defined as pairs consisting of genes located within a genomic window of 100 kb). Interestingly, we found a higher proportion of positive correlations among cis correlations than among trans correlations, in the cases of both lncRNA-protein coding and protein-coding–protein-coding pairs (13.3% and 15.0% respectively for cis vs 4.0% and 7.3% respectively for trans, for rs >0.5). In all sets of gene pairs (protein-coding–protein-coding, protein-coding–lncRNA, and protein-coding–novel lncRNA), positive but not negative cis-correlations were higher than those obtained from random controls (Fig. 3a). The highest proportion of positive and extreme positive correlations were found when we restricted the analysis to novel lncRNA genes (Supplementary Table 5).
Gene Ontology analysis of the protein coding genes that showed strong positive cis-correlations with lncRNA genes revealed enrichment for genes involved in the regulation of lymphocyte development and proliferation, hematopoiesis and immune processes (Fig. 3b). In contrast, genes showing negative cis-correlations were not statistically associated with these functional annotations (Fig. 3b).
To gain further insight into the co-expression signature of genes under strong transcriptional regulation, we delineated the expression profiles of 6,793 genes (protein-coding and lncRNA), which were differentially expressed in at least one pairwise comparison among the ten cell types (fold change > 2, false discovery rate <5%). Model-based gene clustering23 was used to obtain twenty non-redundant expression profiles (Profiles 1–20), which could be classified into seven broad groups (Groups I–VII, Fig. 3c, Supplementary Table 6). Interestingly, four specific profiles showed significant enrichment for lncRNA genes (P < 0.05 when compared with the proportion of lncRNA genes among all differentially expressed genes): those that peaked with B lineage commitment (Profile 7); peaked at the Thy3 stage of T cell commitment (Profile 13); and were upregulated in both B and T lymphoid populations relative to HSCs (Profiles 14 and 15).
Differentially expressed lncRNA genes were significantly enriched for loci that neighbored protein-coding genes involved in transcriptional regulatory mechanisms, T and B cell immune responses, and HSC migration (analysis based on the GREAT24 tool, Supplementary Fig. 4, Supplementary Tables 7,8). Moreover, lncRNA genes with cell type-specific expression patterns were enriched for loci that neighbored protein-coding genes with similar cell type specificity (Supplementary Fig. 4). For instance, HSPC (Group I) lncRNA genes were enriched for loci that neighbored HSPC specific protein-coding genes. In summary, global co-expression analysis and gene-expression profiling suggest an important and previously unappreciated role for lncRNAs in lymphoid commitment from HSC, and T and B cell specification.
LncRNA-based classification of BM and thymic progenitors
Sample clustering of transcriptomes was next used to investigate developmental relationships between lymphoid progenitors in the BM and thymus. Samples were clustered based on genes (both protein-coding and lncRNA genes) that were differentially expressed in at least one pairwise comparison of the ten cell types (“Tree 1”, Fig. 4a). CD34+ thymic progenitors (Thy1, Thy2, and Thy3) segregated with all four CD34+ populations in the BM (HSCs, LMPPs, CLPs and BCPs) rather than late (CD34−) thymic populations (Thy4, Thy5, and Thy6). Moreover, within the branch containing CD34+ progenitors, early thymic progenitors (Thy 1 and Thy 2) sub-clustered with HSCs and LMPPs while CLPs and BCPs formed a separate group. This phylogeny of samples was not affected by variations in FDR and fold change criteria for gene selection, or the inclusion of publically available RNA-Seq datasets from a wide range of tissues, indicating the robustness and validity of this clustering approach (Online methods, Supplementary Fig. 5a,b).
Figure 4. Lymphoid commitment and differentiation are characterized by stage and lineage specific global lncRNA expression patterns.
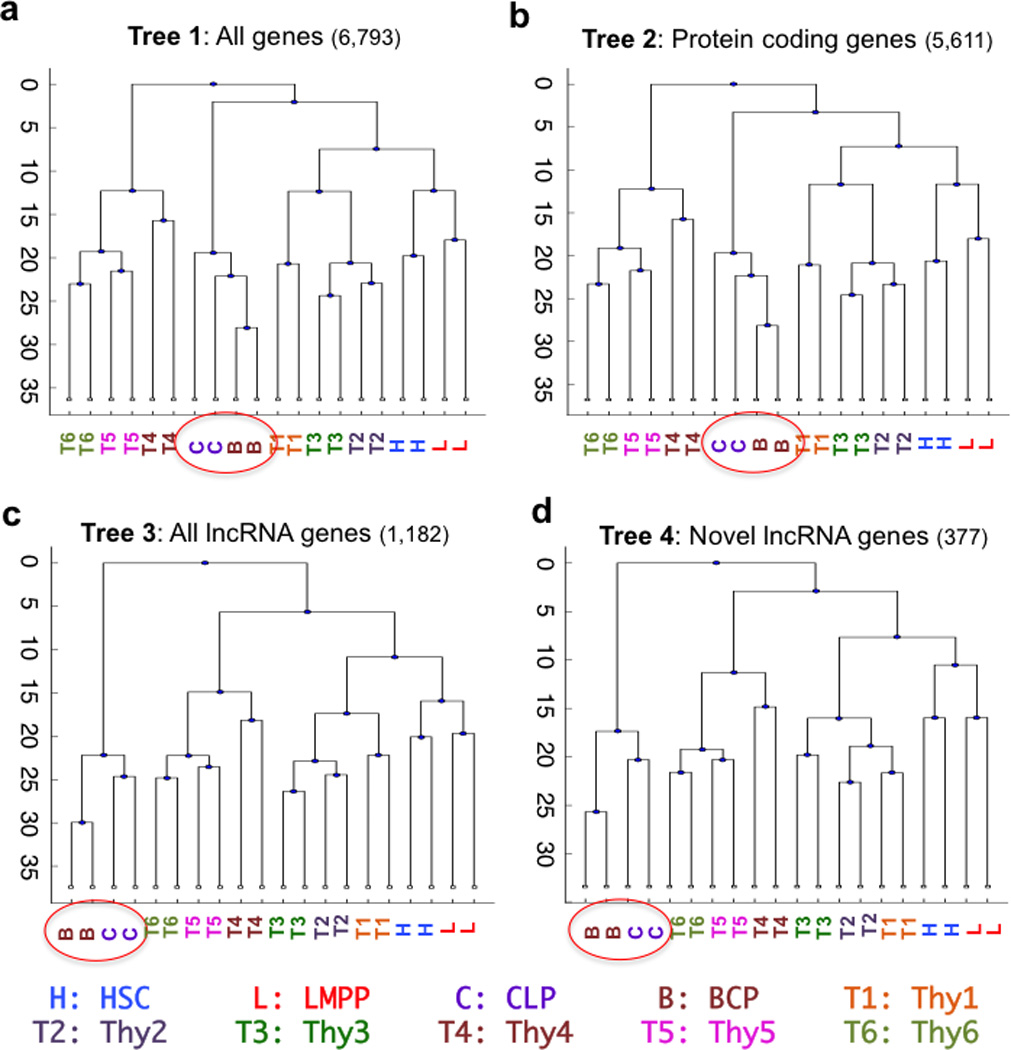
Sample clustering analysis based on differentially expressed genes (fold change >2 and FDR<5% for at least one pairwise comparison of the ten cell types). (a) All genes (protein coding and lncRNA genes); (b) Protein coding genes; (c) All lncRNA genes; and (d) Novel lncRNA genes. Profiles of protein coding genes cluster all CD34+ cells (HSC, LMPP, Thy 1–3, CLP and BCP) separately from CD34neg (Thy4–6). In all analyses, CLP cluster with BCP (circled). LncRNA expression levels completely segregate B (CLP and BCP) and T lineage (Thy 1–6) lineages. Two biological replicates of each cell type are shown.
We next constructed sample trees based on differentially expressed genes from the following classes: Tree 2, protein coding genes (Fig. 4b); Tree 3, all lncRNA genes (novel and previously annotated lncRNAs, Fig. 4c); and Tree 4, novel lncRNA genes (Fig. 4d). Of note, the clustering of B lineage-skewed (CLP and BCP) progenitors varied among the trees, whereas CD34+ thymic progenitors (Thy1, 2 and 3) clustered with HSCs and LMPPs in all four trees. Protein-coding gene expression profiles clustered CLPs and BCPs with the other CD34+ cells in the BM (HSCs and LMPPs) and the thymus (Thy1, Thy2, and Thy3). In contrast, lncRNA gene expression profiles (Trees 3 and 4) segregated B lineage-skewed cells in a cluster distinct from all thymic populations (Thy 1–6), indicating that lncRNAs are expressed in a highly lineage (B versus T) specific manner (Fig. 4c,d). To test whether the difference between trees based on protein-coding and lncRNA genes was due either to a difference in the number of protein-coding (5,611) and lncRNA (1,182) genes, or to differences in expression levels of these two types of genes, we generated trees using randomly selected sets of differentially expressed genes of the same size and similar expression levels. In all cases, the observed trees remained unchanged, indicating that the differences between protein-coding and lncRNA trees reflected actual differences in the lineage specificity of expression of the two classes of genes. Overall, the clear proximity of early thymic progenitor transcriptomes (Thy1, Thy2) to HSCs and LMPPs rather than to CLPs suggests the pre-commitment thymic progenitors that give rise to the T lineage directly arise from BM-derived multipotent HSCs and/ or LMPPs, and that the CLP stage marks the launch of a transcriptionally separate program, which largely overlaps with that of fully B lineage committed CD19+ lymphoid progenitors.
To further dissect the transcriptional relationships between the earliest BM and thymic progenitors, we used Bayesian polytomous model selection, an algorithm that has previously been used to study transcriptional changes at known differentiation branch points among cord blood progenitors25. This approach enabled transcriptional proximities between selected early progenitor cell types to be scrutinized in a highly stringent statistical context, avoiding the influence of transcriptomes of fully lineage committed populations (BCP, Thy3, Thy4, Thy5, and Thy6). Model selection was first performed using both protein-coding and lncRNA genes (n = 38613 genes) (Fig. 5a). For a given combination of any three cell types, each gene was assigned to either the null model (expression similar in all 3 cell types) or one of the alternative (non-null) models (expression different in at least one, and possibly all, cell types) (Fig. 5a). The total number of genes classified in non-null models (“classified genes”) for a given combination of samples can be interpreted as an inverse measure of the transcriptional similarity between the cell types in the combination. As predicted, a markedly lower number of classified genes (5.5 fold lower) was seen with the Thy4-Thy5-Thy6 combination (closely related differentiated T lineage populations) than with the HSC-BCP-Thy4 combination (members from distinct lineages).
Figure 5. LncRNA gene expression defines developmental relationships between bone marrow and thymic progenitors prior to complete lineage commitment, independent of protein coding gene expression.
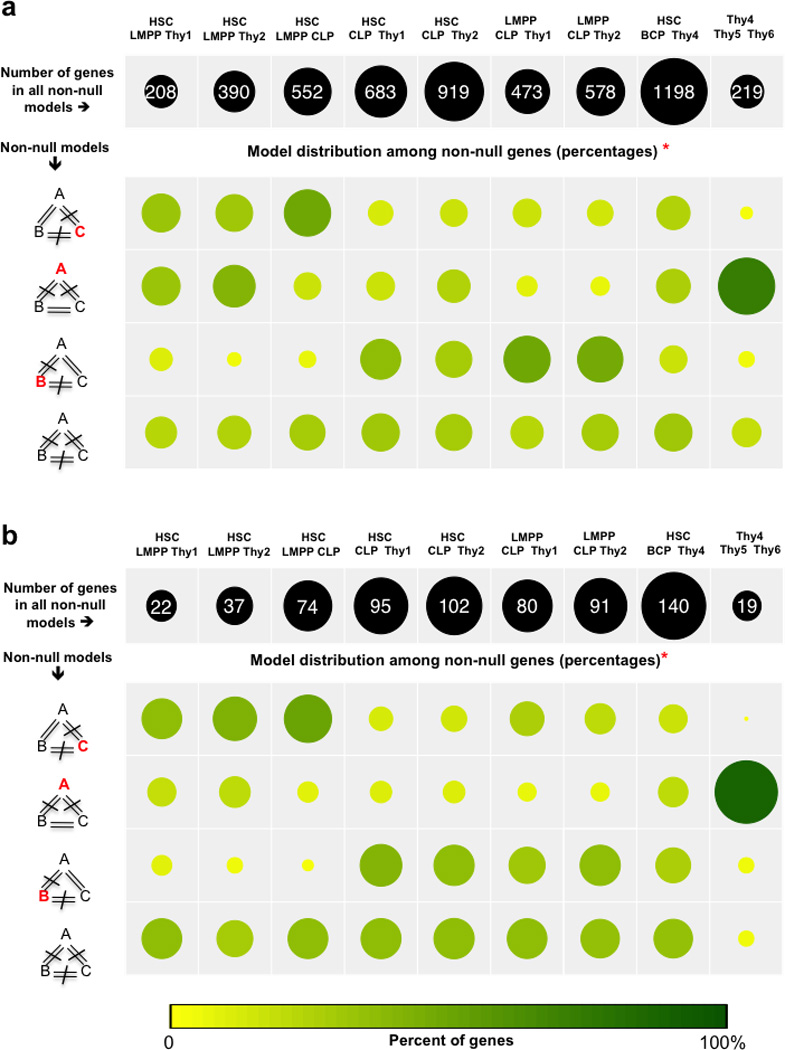
Bayesian polytomous model selection of (a) all genes (protein coding and lncRNA genes), and (b) lncRNA genes only, to analyze transcriptional differences between the least committed BM (HSC, LMPP, CLP) and thymic (Thy1, Thy2) progenitors. For a given combination of three cell types (depicted in column headers), each gene was assigned to either the null model (expression similar in all 3 cell types) or one of the alternative (non-null) models (expression different in at least one, and possibly all, cell types, depicted to left of rows 2–5). A, B and C represent the average expression of the gene in each of the three cell types. The null model is defined as A=B=C. The total number of genes classified in non-null models (shown in the black circles along the top row both numerically and by relative size) for a given combination represents an inverse measure of the transcriptional proximity between the cell types in the combination. *For each combination, the proportion of classified genes assigned to each non-null model is indicated by both circle size and depth of color (yellow-green scale). Model selection for combinations containing cell types known to be either from distinct (HSC, BCP, and Thy4), or closely related lineages (Thy4, Thy5, and Thy6) was performed to estimate, within our dataset, the upper and lower bounds for the number of genes in the null model.
We then applied model selection to the least committed BM (HSC, LMPP, and CLP) and thymic (Thy1 and Thy2) cell types. Gene ontology analysis of classified genes confirmed the ability of this approach to appropriately identify the core transcriptional differences between cell types within a given combination, for example, T cell differentiation genes were significantly enriched among the genes expressed uniquely in thymic progenitors (Supplementary Fig. 6).
Less than 3% of all genes were found to be classified in non-null models when the earliest progenitors were studied (HSC, LMPP, CLP, Thy1 and Thy2), demonstrating the close biological similarities between these cell populations, and the stringent model assignment criteria used. However, combinations that included the CLP cell type contained markedly higher numbers of classified genes than those that did not include this cell type (Fig. 5a, Supplementary Table 9), confirming that CLPs have a transcriptional program that is distinct from that of uncommitted thymic progenitors (Thy1 and Thy2), HSCs, and LMPPs. Interestingly, the least number of classified genes were seen for the HSC-LMPP-Thy1 combination. The striking similarity between these three cell types strongly suggests close developmental relationships for LMPPs and Thy1 to HSCs, and points toward Thy1 as the most primitive progenitor in the human thymus.
When the input set of genes for Bayesian polytomous analysis was restricted only to lncRNA genes (n = 18,268 genes) (Fig. 5b, Supplementary Table 9), the transcriptional relationships seen between the progenitors were recapitulated; differences in lncRNA gene expression were dominated by the presence of CLP. These data demonstrate that the exquisitely cell-type specific nature of lncRNA expression in BM and thymic progenitors can be used to define developmental relationships, independent of protein coding gene expression.
Co-expression modules of LncRNA and coding genes
Correlation based approaches are widely used to infer the function of lncRNAs. These include co-expression analysis (i.e. putative functions of a given lncRNA are inferred based on those protein-coding genes that are highly co-expressed with the lncRNA - guilt by association), and functional associations based on the cell type specificity of lncRNAs26. Here, we employed weighted gene co-expression network analysis (WGCNA), which allows combining these two strategies27. WGCNA first identifies modules of highly co-expressed genes, from which modules with cell type-specific expression profiles can be easily selected. Additionally, individual genes within these modules can be ranked based on module membership (a measure of how similar the expression profile of a gene is to that of the module). Screening for genes with high module membership has been shown to be a useful strategy to identify genes of biological interest28.
Of the 45 modules identified by WGCNA in our samples, five were chosen for further analysis as they demonstrated significant lineage or differentiation stage-specific profiles (P < 0.05) (Fig. 6a,b): HSPC module (all CD34+ cells), B lineage module (CLP and BCP), T lineage module (Thy1–6), lymphoid module (both B lineage [CLP, BCP] and T lineage [Thy2–Thy4] populations), and early thymic progenitor module (Thy1–3). These modules contained protein-coding genes known to be specific to each of the cell types; these include, respectively, HSPCs (CD34, ERG), B lineage (PAX5, EBF1), T lineage (BCL11B, CD3), lymphoid (RAG1, RAG2), and early thymic progenitor cells29 (NOTCH1). The individual genes in each module were then ranked based on module membership (Supplementary Table 10).
Figure 6. Identifying lineage or differentiation stage specific, lncRNA-protein coding gene co-expression modules.

Weighted gene co-expression network analysis (WGCNA) across all samples (n=2 biological replicates per cell type) was used to identify modules containing genes with highly correlated expression. a) Shown is WGCNA’s Topological Overlap Matrix (red: high expression correlation, yellow: low expression correlation) for genes in the 5 modules shown in b). (b) Lineage or differentiation stage specific (p<0.05 for expression specificity) modules with the depicted expression profiles were then selected. (c, d) Two suggested screening strategies for the identification of potentially interesting candidate lncRNA genes and lncRNA-protein coding gene co-expression associations within specific modules. Analyses of the hematopoietic stem and progenitor cell (HSPC) module are depicted in the form of circos plots (broken lines in the circumference indicate individual chromosomes) as illustrative examples to demonstrate these strategies. (c) Coding to non-coding association: candidate lncRNA genes were identified based on high co-expression with protein coding genes in the HSPC module that belong to the functional annotation “proto-oncogenes” (includes genes known to be important for HSPC maintenance) (black circles). The three most positively (yellow circles, correlation coefficient >0) and negatively (orange circles, correlation coefficient <0) correlated lncRNA genes for each coding gene are shown. (d) Non-coding to coding association: Among genes with high module membership (module membership adjusted p value<0.01), the 10 most highly expressed lncRNA genes (black circles), and the 3 most positively (yellow circles) and negatively (orange circles) correlated protein coding genes for each of these lncRNA genes are shown.
To aid the utilization of our database as a resource, we provide two examples of how the above approach can be used to identify interesting candidate lncRNA and lncRNA-protein coding gene co-expression associations for further functional studies (circos plots in Fig. 6c,d). The first strategy (coding to non-coding association, Fig. 6c) involves selection, from a module of interest, of protein-coding genes known to be important in a specific functional process. Interesting lncRNA gene candidates are then identified based on how strongly their expression profile correlates with that of the selected protein-coding genes. The second strategy (non-coding to coding association, Fig. 6d) involves selection of candidate lncRNA genes based on module membership and expression strength (since high expression may facilitate functional characterization). Associated protein-coding genes are then identified based on how strongly their expression profile correlates with that of the selected lncRNA genes. Supplementary Table 10 lists the lncRNA-protein coding gene associations identified when the five chosen modules were analyzed using these two strategies. Overall, the WGCNA results provide a searchable resource to identify lncRNA candidates and generate hypotheses for functional studies to elucidate the role of lncRNAs in lymphopoiesis.
DISCUSSION
To the best of our knowledge, this report represents the first integrated transcriptional map of protein coding and lncRNA elements underlying the initial phases of post-natal lymphoid commitment in the human BM and thymus.
A recent evolutionary study showed significant differences between the repertoires of murine and human lncRNAs30. Of the few studies performed in human hematopoiesis, most have analyzed cord blood progenitors or fully differentiated peripheral blood lymphocytes25,31, which are functionally and immunophenotypically different from the BM and thymic progenitors underlying steady state postnatal lymphoid commitment6. Furthermore our conceptual framework of lymphoid development in the BM and thymus is largely based on the lineage potential of isolated progenitors using in vitro or xenotransplantation assays. However, the non-physiological nature of these assays create significant challenges when interpreting such studies of human lymphopoiesis. Thus, in the current report we examined the relationship between BM and thymic progenitors through the use of global gene expression profile analyses of purified unperturbed cells. Even among these closely related cell types, analysis of global lncRNA expression profiles uncovered new relationships, suggesting an important role for lncRNAs during the initial stages of lymphopoiesis.
Two recent studies annotated lncRNAs in unfractionated thymocytes17, and thymic progenitors stimulated ex vivo with Notch ligands32. While approximately one fourth of the lncRNAs expressed in thymic progenitors in our dataset overlapped with those reported in these studies, we identified over two thousand unique, previously undescribed lncRNA genes in the thymic transcriptome. The use of purified subpopulations of CD34+ progenitors (which are extremely rare in whole thymic tissue) in our study, the predominance of leukemic samples in the datasets used for defining lncRNAs in other studies, and the transcriptional changes related to non-physiological conditions in culture may account for the differences in the repertoires of lncRNA genes described in the current and previous reports. Our results underscore the importance of defining the lncRNAs de novo in unmanipulated purified rare progenitor populations.
To date, the unmapped nature of lncRNAs underlying human lymphoid commitment has represented a substantial obstacle to the dissection of transcriptional circuits regulating lymphopoiesis. The database presented here of annotated lncRNAs expressed in the rare progenitors that mark the initiation of B and T lineage divergence has allowed the cataloging of protein-coding–lncRNA gene associations, and the identification of developmental relationships between these progenitor stages, thus providing a resource for parsing the lncRNA circuitry in normal lymphopoiesis, and understanding aberrations of these mechanisms in immune deficiencies and lymphoid malignancies.
Online METHODS
Isolation and RNA extraction from bone marrow and thymic progenitors
Anonymous normal human thymi were obtained via the UCLA Translational Pathology Core Laboratory and the CHLA cardiovascular thoracic surgery department. Bone marrow (BM) was obtained from donors via ALLCELLS, Alameda, CA. All tissues were collected and used in accordance with UCLA and CHLA Institutional Review Board guidelines. The magnetic activated cell sorting (MACS) system (Miltenyi Biotec) was used to enrich CD34+ cells from BM and thymus prior to isolation of CD34+ subsets by flow cytometry. BM CD34+ cells were incubated with the following antibodies: CD34-APC-Cy7 (581), CD38-APC (HIT2), CD10-PE-Cy7 (HI10a), CD62L-PE (DREG-56), CD45RA Percp-cy5.5 (HI100) (all from Biolegend), as well as the following FITC-labeled lineage depletion antibodies: CD3 (SK7), CD14 (M5E2), CD19 (4G7), CD56 (MY31), and CD235a (GA-R2)(Becton Dickinson). CD19 was not included in the lineage depletion cocktail used for sorting BCPs. An unstained control was used to set gates. CD34+ enriched thymic cells were incubated with CD34 Percp-cy5.5 (8G12), CD7 FITC (4H9), CD1a PE (HI149) and APC-labeled CD4 (RPA-T4) and CD8 (SK1). To isolate DP and SP thymocytes, non-CD34 enriched thymic cells were incubated with CD3 APC (UCHT1), CD4 FITC (RPA-T4), and CD8 Percp (SK1) (all from BD). Dead cells were gated out using 4′,6-diamidino-2-phenylindole (DAPI). FMO (fluorescence minus one) controls were used to set gates for isolating thymic cells. The following immunophenotypic definitions were used to isolate progenitors from BM CD34+ cells: CD34+CD38−lin− (HSCs), CD34+CD45RA+CD38+CD10− CD62Lhi lin− (LMPPs), CD34+CD38+CD10+CD45RA+ lin− (CLPs) and CD34+CD38+CD19+lin− (BCPs); thymic CD34+ cells: CD34+CD7−CD1a−CD4−CD8− (Thy1), CD34+CD7+CD1a−CD4−CD8− (Thy2), CD34+CD7+CD1a+CD4−CD8− (Thy3); and thymic CD34− cells: CD4+CD8+ (Thy4), CD3+CD4+CD8− (Thy5), and CD3+CD4−CD8+ (Thy6) (Supplementary Fig.1a, b). All populations were isolated on a FACSAria (355, 405, 488, 561 and 633 nm lasers) (BD Immunocytometry Systems).
Progenitor populations were isolated for RNA-Seq from 2 donors (2 biological replicates) for bone marrow and thymus (separate donors for bone marrow and thymus). Fresh samples were used for both replicates of the bone marrow and for one replicate of the thymus. Cells cryopreserved post MACS were thawed and used for isolation of subsets by flow cytometry for the other replicate of the thymus. Progenitors were isolated from fresh samples for qPCR from additional donors for bone marrow and thymus (separate donors for bone marrow and thymus). The Trizol method (Mirneasy RNA extraction kit, Qiagen) was used to extract RNA from all samples. The RNA 6000 Pico kit (Agilent technologies) was used to assess RNA integrity prior to library preparation.
RNA-Seq library preparation and sequencing protocol
The Ovation RNA- Seq System V2 (NuGen), which uses oligo dT as well as random primers for reverse transcription, was used to convert 6–10 ng of total RNA (not selected for polyadenylation) into amplified cDNA (linear amplification). The cDNA was sheared using a S220 focused ultrasonicator (Covaris) to generate an average fragment size of 350 base pairs. 900 ng of sheared cDNA was converted into sequencing libraries using the Encore Rapid DR Multiplex kit (NUGen), which were then sequenced on an Illumina Hiseq 2000 sequencer (paired end 100 base pair sequencing). A total of 541 million paired-end reads were generated (on average 27 million reads for each sample). Raw sequence files have been deposited at NCBI’s Gene Expression Omnibus (GSE69239).
RNA-Seq and ChIP-Seq data analysis
The STAR33 aligner was used to align paired end reads to the human genome. Reference Annotation Based Transcript (RABT) assembly34 was used to assemble the transcriptome. Alignment files for each of our samples as well as those derived from publicly available RNA-Seq datasets were analyzed with HTSeq35, using our gene annotation file to generate gene-level counts for each sample. Pairwise expression correlations between protein coding and lncRNA genes were computed using a strategy that was previously described for the characterization of the Gencode lncRNA catalog15. Gene clustering of differentially expressed genes was performed by means of MBCluster.Seq23. GREAT24 was used to analyze functional annotations of protein-coding genes whose regulatory domains overlapped lncRNA gene loci. Pair-wise differential expression was performed with DESeq36. Bayesian model polytomous selection and WGCNA were done as previously described25,27. Peak detection and signal intensity analyses for ChIP-seq data were done using MACS237 and HOMER38 respectively. A detailed description of the RNA-Seq and ChIP-Seq analysis methods is provided in the supplementary methods section.
Quantitative Polymerase Chain Reaction (PCR)
Predesigned primers (Qiagen) were used for lnc-UBE2N-2 (catalog no. LPH15913A-200), lnc-FBXO31-1 (catalog no. LPH23574A), AC006129.1 (catalog no. LPH25998A), RP11-301G19.1 (catalog no. LPH06790A), and ACTB (catalog no. PPH00073G). Primers for LINC00544, BMThy_chr2_0447, BMThy_chr2_1049, and BMThy_chrX_ were designed using Primer3Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) and synthesized by Life Technologies (Supplementary Table 11 lists primer sequences). The RT2 SYBR Green ROX qPCR Mastermix (Qiagen) was used for reactions involving predesigned primers (RT2 lncRNA qPCR assay). The Power SYBR Green PCR master mix (Life Technologies) was used for reactions involving primers designed with Primer3. cDNA prepared using the Ovation RNA- Seq System V2 or the Superscript Vilo CDNA synthesis kit (Life Technologies) was used as input for all PCR reactions. All reactions were run in triplicate using the Applied Biosystems 7900 HT Real Time PCR system (Life Technologies). Thermocycling parameters: 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 60 s at 60 °C. Gene expression was calculated relative to ACTB expression using the ΔCt method. A Pearson correlation coefficient between qPCR and RNA-Seq gene expression measurements was calculated for each gene.
Supplementary Material
ACKNOWLEDGEMENTS
We thank F. Codrea (BSCRC flow cytometry core), A. George (CHLA flow cytometry core), and S. Feng (BSCRC Highthroughput Sequencing Core) for technical assistance; and M. Pellegrini and R. D’Auria for computational support. C.P. was supported by St. Baldrick’s Foundation Scholar and NIH CHLA K12 Child Health Research (HD052954) Career Development awards, and Nautica, Tower Cancer Research, Couples against Leukemia, and Joseph Drown Foundations. This work was also supported by NIH P01 HL073104 (G.M.C.), UCLA Broad Stem Cell Research Center (BSCRC) (G.M.C., D.C.), NIH T32HL066992 (C.S.S.), and NIH T32 HL086345 (S.S.). S.S. also acknowledges support from California Institute for Regenerative Medicine (CIRM) Training Grant TG2-01169 for this work. We would like to thank the Center for AIDS Research Virology Core Lab that is supported by the National Institutes of Health award AI-28697 and by the UCLA AIDS Institute and the UCLA Council of Bioscience Resources, for providing reagents.
Footnotes
Accession codes
Raw sequence files (RNA-Seq) have been deposited at NCBI’s Gene Expression Omnibus (GSE69239).
Author Contributions
D. C., conception and design, developed bioinformatics analysis pipeline, bioinformatic analysis and interpretation, manuscript writing. S.S., conception and design, collection and assembly of data (performed experiments), data analysis and interpretation. C.S.S., data analysis and interpretation. J. S., conception and design. Y.Z., collection and assembly of data (assisted in performance of experiments). V.L.H. and A.L., collection and assembly of data (performed experiments). C.P., conception and design, collection and assembly of data (performed experiments), data analysis and interpretation, manuscript writing, final approval of manuscript. G.M.C., conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 2.Galy A, Travis M, Cen D, Chen B, Human TB. natural killer, and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity. 1995;3:459–473. doi: 10.1016/1074-7613(95)90175-2. [DOI] [PubMed] [Google Scholar]
- 3.Plum J, et al. Human intrathymic development: a selective approach. Semin Immunopathol. 2008;30:411–423. doi: 10.1007/s00281-008-0135-2. [DOI] [PubMed] [Google Scholar]
- 4.Doulatov S, et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585–593. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 5.Six EM, et al. A human postnatal lymphoid progenitor capable of circulating and seeding the thymus. J Exp Med. 2007;204:3085–3093. doi: 10.1084/jem.20071003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohn LA, et al. Lymphoid priming in human bone marrow begins before expression of CD10 with upregulation of L-selectin. Nat Immunol. 2012;13:963–971. doi: 10.1038/ni.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blom B, Spits H. Development of human lymphoid cells. Annu Rev Immunol. 2006;24:287–320. doi: 10.1146/annurev.immunol.24.021605.090612. [DOI] [PubMed] [Google Scholar]
- 8.Hao QL, et al. Human intrathymic lineage commitment is marked by differential CD7 expression: identification of CD7- lympho-myeloid thymic progenitors. Blood. 2008;111:1318–1326. doi: 10.1182/blood-2007-08-106294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weerkamp F, et al. Human thymus contains multipotent progenitors with T/B lymphoid, myeloid, and erythroid lineage potential. Blood. 2006;107:3131–3137. doi: 10.1182/blood-2005-08-3412. [DOI] [PubMed] [Google Scholar]
- 10.Novershtern N, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dik WA, et al. New insights on human T cell development by quantitative T cell receptor gene rearrangement studies and gene expression profiling. J Exp Med. 2005;201:1715–1723. doi: 10.1084/jem.20042524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tydell CC, et al. Molecular dissection of prethymic progenitor entry into the T lymphocyte developmental pathway. J Immunol. 2007;179:421–438. doi: 10.4049/jimmunol.179.1.421. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–i282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volders PJ, et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013;41:D246–D251. doi: 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trimarchi T, et al. Genome-wide Mapping and Characterization of Notch-Regulated Long Noncoding RNAs in Acute Leukemia. Cell. 2014;158:593–606. doi: 10.1016/j.cell.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marques AC, et al. Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs. Genome Biol. 2013;14:R131. doi: 10.1186/gb-2013-14-11-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.NE II, et al. Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat Commun. 2014;5:3979. doi: 10.1038/ncomms4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saeed S, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chadwick LH. The NIH Roadmap Epigenomics Program data resource. Epigenomics. 2012;4:317–324. doi: 10.2217/epi.12.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sati S, Ghosh S, Jain V, Scaria V, Sengupta S. Genome-wide analysis reveals distinct patterns of epigenetic features in long non-coding RNA loci. Nucleic Acids Res. 2012;40:10018–10031. doi: 10.1093/nar/gks776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Si Y, Liu P, Li P, Brutnell TP. Model-based clustering for RNA-Seq data. Bioinformatics. 2014;30:197–205. doi: 10.1093/bioinformatics/btt632. [DOI] [PubMed] [Google Scholar]
- 24.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science. 2014;345:1251033. doi: 10.1126/science.1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horvath S, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A. 2006;103:17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taghon T, et al. Notch signaling is required for proliferation but not for differentiation at a well-defined beta-selection checkpoint during human T-cell development. Blood. 2009;113:3254–3263. doi: 10.1182/blood-2008-07-168906. [DOI] [PubMed] [Google Scholar]
- 30.Necsulea A, et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505:635–640. doi: 10.1038/nature12943. [DOI] [PubMed] [Google Scholar]
- 31.Ranzani V, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol. 2015;16:318–325. doi: 10.1038/ni.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durinck K, et al. The Notch driven long non-coding RNA repertoire in T-cell acute lymphoblastic leukemia. Hematologica. 2014 doi: 10.3324/haematol.2014.115683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 35.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.