

Abstract
Nephritis is a common complication of systemic lupus erythematosus (SLE) for which current therapies often prove inadequate. Current lupus nephritis classification systems emphasize glomerular acuity and scarring. However, tubulointerstitial inflammation (TII) and scarring are much better predictors of progression to renal failure. It is now becoming clear that the immunological features, and probable underlying mechanisms, are very different in lupus glomerulonephritis (GN) and TII at time of biopsy. While GN is a manifestation of systemic autoimmunity, TII is associated with local, in situ adaptive immune cell networks predicted to amplify local inflammation and tissue damage. In addition, poorly defined networks of innate immune cells and effectors likely contribute to the severity of local inflammation. Understanding these in situ immune mechanisms should lead to a better understanding of prognostically meaningful lupus nephritis subsets and reveal novel therapeutic opportunities.
Keywords: Lupus nephritis, Tubulointerstitial inflammation, Adaptive immunity, Innate immunity
Introduction
The most common and severe manifestation of systemic lupus erythematosus (SLE) is certainly lupus nephritis1, 2, 3, 4, 5. Up to 60 % of SLE patients develop lupus nephritis with most of these requiring major immunosuppressive therapies such as cyclophosphamide or mycophenolate mofetil6, 7, 8, 9. Yet, despite aggressive treatment, up to 50% of lupus nephritis patients progress to renal failure within 5 years of diagnosis 10, 11, 12.
Ethnicity is a major determinant of renal failure risk, with African-Americans and Hispanics having a worse prognosis than Caucasians11, 13. Reflecting their worse prognosis, and possibly differing responses to therapies, the treatment recommendations for African-Americans and Hispanics are different than those for Caucasians and Asians 9. It is not entirely clear however if African-Americans and Hispanics have a higher ultimate risk of renal failure or if they just progress to renal failure more quickly. Most studies demonstrating the risk associated with ethnicity are five years or less in duration. However, at least one study suggests that patients continue to progress to renal failure beyond five years14. In this Danish study, less than 20% progressed in five years while about 50% were in renal failure 25 years after diagnosis.
These more recent epidemiological studies have all been done in the modern era of treatment in which cyclophosphamide and/or mycophenolate mofetil were the standards of care. While these drugs are clearly effective in some patients, short-term response rates have not appreciably improved since the introduction of cyclophosphamide for lupus nephritis in the 1980s14, 15, 16. Therefore, either rapidly or eventually, half of lupus nephritis patients fail these modalities and progress to end-stage kidney disease.
The need for both more effective and less toxic therapies in lupus nephritis is obvious and pressing. However, it is unclear which therapies to pursue and in which sub-populations of lupus they might be efficacious. We suggest that this uncertainty in how to proceed reflects limitations in both our understanding of lupus nephritis and in how we classify patients and assign prognosis.
Prognostic value of renal biopsies
The current standard is to biopsy all SLE patients who present with an active urinary sediment and/or greater than 500 mg/protein in 24 hours9, 17. Lupus patients are then broadly categorized as having either proliferative or nonproliferative nephritis based on the activity and frequency of glomerular lesions with therapeutic decisions being based upon this classification. However, current histologic measures of disease activity, which emphasize glomerular involvement, perform poorly in identifying those patients at risk for subsequent renal failure.
The most commonly used classification system reflects this focus on glomerular inflammation. The 2003 International Society of Nephrology/Renal Pathology Society (ISN/RPS)18 lupus nephritis classification focuses exclusively on histologic changes of the glomerulus. Similarly, the NIH activity index quantifies the severity of lupus nephritis and is scored using six pathologic features, of which five involve the glomerular compartment, with 21 of the 24 activity points awarded based on glomerular findings13, 19. However, the prognostic value of glomerular inflammation, at best, remains unclear.
Several studies have demonstrated that glomerular measures of disease activity do not accurately predict subsequent clinical course13,12, 19, 20, 21, 22, 23. For the most part, these studies were performed during the modern era when all patients received cytotoxic therapies. Earlier studies clearly demonstrated that patients with proliferative nephritis have a worse prognosis than non-proliferative nephritis and that this group does better with immunosuppressives24. However, in these earlier studies, other features of the biopsy, such as tubulointerstitial inflammation were not systematically assessed. Furthermore, features predictive of resistance to immunosuppressive therapy were not analyzed.
Rather, several studies in the immunosuppressive era of lupus nephritis treatment, extending back to the 1980s, have indicated that tubulointerstitial inflammation is prognostically more meaningful than glomerular inflammation and more likely to be correlated with elevated creatinine at time of biopsy and with risk for subsequent renal failure13, 22, 25, 26, 27, 28. Many of these studies noted that more active TII tended to be associated with active GN. However, multivariate analysis demonstrated that TII was an independent predictor of progression to renal failure 13 and correlated with serum creatinine at time of biopsy13, 26. Furthermore, TII is not associated with low complement levels, or high titers of dsDNA antibodies13, 26, factors epidemiologically and mechanistically tied to GN. Therefore, TII is an independent and important predictor of renal failure in lupus nephritis.
The current assessments of TII are largely qualitative with severity scored as the fraction of the tubulointerstitium infiltrated with inflammatory cells on PAS stained paraffin embedded sections. By simply staining with anti-CD45 antibodies, and assessing the fraction of the tubulointerstitium infiltrated with CD45+ cells, intermediate grades of TII can be more accurately assessed which are prognostically significant13.
While the degree of TII is prognostically more important than GN activity, it is not clear how this information should inform therapy. Clinical trials have not been stratified by TII and therefore it is not clear if one therapy is relatively more effective in TII. However, the fact that severe TII predicts renal failure in all lupus patients suggests that all current therapies are relatively ineffective for this manifestation.
In contrast to commonly used indices of active glomerular inflammation, indices of scarring (glomerulosclerosis, interstitial fibrosis and tubular atrophy) are strongly predictive of subsequent renal failure13, 22, 26, 29, 30. The NIH chronicity index is a composite score that equally reflects scarring in both the glomeruli and the tubulointerstitium. However, prognostic value of the chronicity index lies primarily in those components that capture interstitial scarring13. Measures of glomerular scarring do not provide independent prognostic information to the chronicity index. In other renal diseases, interstitial scarring also identifies patients with a poor prognosis31. In IgA nephropathy, which is primarily considered to be a glomerulonephritis, tubular atrophy and interstitial fibrosis are more predictive of subsequent renal insufficiency than segmental glomerulosclerosis32.
There is substantial evidence that inflammation leads to fibrosis. This central idea is an extension of the known roles of both inflammation and fibrosis, in the normal processes critical for organ repair following injury. Macrophages play a role in both processes33 and ablation of macrophages mitigates fibrosis34, 35, 36. Furthermore, the extent of macrophage infiltration correlates with the extent of fibrosis37. Therefore, the overall effect of macrophages in these model systems appears to be to promote fibrosis. However, infusion of M2 macrophages, which act to limit inflammation, attenuate renal fibrosis in mice38. Adaptive immunity appears important as deletion of Rag, thereby eliminating both B and T cells, can protect against renal fibrosis but not, interestingly, GN39. Furthermore, T cells are required for fibrosis following ischemia-reperfusion injury40, 41. However, it is not clear that monotherapy targeting adaptive immunity, or inflammation, will be sufficient to prevent fibrosis in most patients.
The pathogenesis of tubulointerstitial inflammation
While GN is a manifestation of systemic autoimmunity42, 43, 44, lupus TII has histological features suggesting that local, in situ immunity might contribute to, and propagate, local tubuloinflammation and organ damage45, 46. What is most striking is how different the inflammatory infiltrates are in glomerular inflammation and TII. In lupus glomeruli, the degree and type of involvement varies with ISN/RPS class. In the non-proliferative lupus nephritis (classes I and II), patients have immune complex deposits in the mesangium which can be associated with mesangial hypercellularity (class II). In class V (membranous), immune complex deposition is subepithelial and is associated with thickening of the glomerular basement membrane. None of these lesions are associated with a significant influx of inflammatory cells into the kidneys. In contrast, the proliferative forms of lupus nephritis (classes III and IV) are characterized by inflammation. Active glomerular lesions have prominent subendothelial immune complexes that sometimes fill the glomerular capillary loops (hyaline thrombi). T cells and macrophages accumulate at sites of subendothelial immune complexes. This is associated with ruptured glomerular basement membranes, fibrinoid necrosis and cellular crescents. Neutrophils are not prominent within the glomerular capillaries except when fibrinoid necrosis and crescent formation are present. B cells and plasma cells are rare in glomeruli, and are usually confined to intravascular spaces.
In contrast, B cells and plasma cells are a common, almost invariant, feature of TII. Likewise, T follicular helper-like cells (described below) are only found in the inflamed tubulointerstitium. In about 50% of patients, immune complexes are deposited throughout the tubulointerstitium with characteristic accumulations of immune complexes in the tubular basement membranes46, 47. These immune complexes are often associated with C3c and C1q deposition. Immune complexes in the tubulointerstitium can be associated with more severe inflammation13 although this point is controversial47, 48, 49. The distribution of immune complex deposits within the tubulointerstitum speaks against the immune complexes having a purely hematogenous origin. Interestingly, the isotypes of antibodies deposited in the glomeruli and tubulointerstitium can be quite divergent in the same patient47. These observations suggest that different mechanisms underlie immune complex deposition in GN and TII.
Furthermore, many renal biopsies with TII have features of lymphoid organization45, 46. In up to 8% of clinical biopsies, germinal center-like structures are observed. More commonly, well-formed aggregates of B and T cells are seen in up to 50% of biopsies. Lymphoid-like structures were associated with both more severe inflammation and with the presence of tubular basement membrane immune complexes46. These histological features suggest that interstitial B and T cell infiltrates are being selected in situ by locally occurring antigen. Consistent with this, sampling of expressed immunoglobulin repertoires from the tubulointerstitum has revealed local clonal expansion and ongoing somatic hypermutation46, 50. Analysis of the distribution of mutations in those regions of expressed antibodies containing the variable regions (Complementarity determining regions or CDRs) indicted that mutations arising from somatic hypermutation were being subsequently selected by antigen. Furthermore, T cell populations in approximation with tubulointerstitial B cells exhibit clonality51. Such characteristics are defining features of in situ adaptive immunity or tertiary lymphoid neogenesis.
To identify the antigens driving in situ B cell selection, variable region heavy and light chain immunoglobulin pairs from clonally expanded B cell populations were cloned and expressed50. Remarkably, most of these antibodies, expressed from seven patients, were reactive with cytoplasmic, and not nuclear, antigens in HEp-2 cells. Subsequent studies identified vimentin as the most common antigen targeted by these anti-cytoplasmic antibodies. This result was not totally expected, because vimentin is generally considered to be an intermediate filament, and therefore has been assumed to primarily play a structural role. However, mice deficient in vimentin are relatively normal52. Furthermore vimentin is highly expressed in activated T cells and macrophages53. In the latter cells, vimentin is expressed on the cell surface. Consistent with these observations, vimentin was highly expressed in TII and many of our anti-cytoplasmic antibodies bound inflamed, but not normal, tubulointerstitium50. Vimentin is a large, complex and charged protein that might be expected to be very immunogenic. Furthermore, vimentin can bind dectin 1, a C-type lectin receptor expressed on dendritic cells, macrophages and B cells54. Therefore, vimentin might be an immunodominant pattern of inflammation and the in situ adaptive immune response we have observed is against inflammation.
Interestingly, most anti-vimentin antibodies were also reactive with other antigens on large protein arrays and were reactive with other cytoplasmic structures50. Such polyreactivity could be an intrinsic property of anti-vimentin antibodies. Alternatively, this polyreactivity could be a consequence of the unique environment in which antibodies are selected. In contrast to germinal centers, antigen is not limiting in TII and therefore there might not be sufficiently stringent selection to diminish polyreactivity. Polyreactivity could also arise from poly-selection in which clonal populations are selected on different antigens over time. Persistent polyreactivity might reflect the fact that many antibodies arose from T:B aggregates or plasmablast foci in which selection might not be as stringent. Alternatively, it could be that the inflammatory milieu, and inflammatory cytokines alter the stringency with which repertoire is selected.
Remarkably, in serum, high-titer antibodies were almost exclusively restricted to those patients with severe TII50. Therefore, anti-vimentin antibodies appear to serve as biomarker of a specific disease manifestation. Mechanistically, this correlation suggests that in situ anti-vimentin antibody responses might function as a local amplification or feed-forward mechanism to drive severe inflammation. It remains to be determined how anti-vimentin antibodies vary with disease activity or if high anti-vimentin antibody titers identity specific therapeutic opportunities.
It is likely that anti-vimentin antibodies are not a unique feature of lupus. In allograft transplantation, high titers of anti-vimentin antibodies are associated with rejection55, 56, 57 and immunization with vimentin can accelerate cardiac rejection in a mouse model58. Therefore, anti-vimentin immune responses might arise when tolerance to inflammation is broken in a variety of disease contexts.
Antigen is usually insufficient to drive B cells to secrete antibodies; second signals are required. In lupus, much work has focused on the role of toll-like receptors (TLRs) and the role of TLR7 and TLR9 contributing to anti-RNP and and-dsDNA responses respectively59, 60. However, remarkably, these specificities were not found to be selected in the tubulointerstitium suggesting that other mechanisms are likely driving in situ selection.
In secondary lymphoid organs, necessary second signals for B cell selection are usually provided by T follicular helper cells (TFH cells), a subset of CD4+ T cells that express ICOS and high levels of IL-21 which contribute to B cell activation61. While in vitro studies of circulating putative TFH populations have given insights into potential TFH cell capacities, there have been no methods to assess if TFH cells were contributing to B cell activation in situ. To address this experimental limitation, we developed computational tools to analyze the spatial relationships between different lymphocyte subsets in clinical biopsies imaged by multicolor confocal microscopy62. Interestingly, only competent TFH cells came within a short critical distance (less than 0.5 Pm) of B cells. Three-dimensional imaging and other modalities indicated that these tight juxtapositions of cells represented the formation of complex supramolecular activation complexes (SMACs) and the delivery of cognate help63.
Quantifying the prevalence of cell-cell interactions allowed us to determine how commonly one type of interaction occurred between two different cell populations. Remarkably, when TFH cells were present on biopsy, almost all of the B cells in that biopsy were organized around them with predictable geometries and stochastic relationships reminiscent of those found in germinal center light zones62. However, none of these biopsies had discernable germinal centers histologically. These observations provide insights into why T:B aggregates form in the TII and reveal mechanistic links between these histological features and frank germinal centers. Furthermore, for those with more severe TII, one general mechanism, TFH help, primarily maintained B cell within the interstitium. This is consistent with the remarkably restricted repertoire of B cells present in TII for a protein antigen, vimentin50.
Ultimately, many studies in humans are unavoidably descriptive with the only true in vivo mechanistic experiments being clinical trials. Therefore, studies in mice are very attractive. Furthermore, murine models have been critical to understanding many fundamental mechanisms of lymphocyte tolerance and systemic autoimmunity42. However, it appears that most available murine models of lupus nephritis do not accurately mimic key pathological features of the human disease. Lymphocytic infiltrates in the kidneys of both NZB/NZW and MRL/Mplpr/lpr mice contain B cells and/or plasma cells that can express antibodies with broad repertoires64, 65, 66, 67. However, NZB/NZW and MRL/Mplpr/lpr mice have diffuse or perivascular intrarenal lymphocytic infiltrations. The in situ organization of B and T cells into lymphoid-like structures appears to be a unique feature of human lupus nephritis. Furthermore, tubular basement membrane immune complexes are not found in common murine models. Therefore, it is not reasonable to assume that any murine model will provide universal insights into the specifics of the human disease. Rather what is needed is a quantitative and comprehensive understanding of human lupus nephritis. This will then allow aspects of the human disease to be modeled in rational murine models that accurately reflect specific and relevant pathogenic processes.
Beyond T:B collaboration in tubulointerstitial inflammation
Our initial studies have focused on B cells and their interplay with T cells. In part, this reflects both technical limitations and the central role B cells play in the autoimmunity associated with SLE. Furthermore, recent clinical trials suggest that B cells might not be the critical therapeutic target for many SLE patients9. However, there are many more T cell and antigen presenting cell populations resident in lupus TII. The cortex of normal human kidneys contains a network BDCA-1+DC-SIGN+ and DC-SIGN− myeloid DCs (mDCs) as well as fewer numbers of BDCA-2+DC-SIGN− plasmacytoid DCs (pDCs)68. The cortex also contains macrophages that have similar surface phenotypes, and possibly functions, as DCs69. In murine models, these populations appear to actively maintain tolerance in normal tissue, limit inflammation in response to tissue damage70, 71, 72, 73, drive inflammation in response to ischemia, infection and ureteral obstruction70, 74, 75, 76 and resolve inflammation to allow repair77, 78. In NZB/W mice, kidneys are infiltrated with pro-inflammatory macrophages and DCs79 and both human and murine lupus is associated with a pattern of in situ mRNA expression indicative of activated macrophages and dendritic cells80. Histologically, human lupus is associated with increased DC infiltration81 and increased chemokine expression82. However, the specific phenotype and activation state of these DCs remains unclear81.
Other inflammatory pathways are clearly active within TII with tumor necrosis factor (TNF) being one of the most interesting and potentially relevant. TNF can play a central role in inflammatory cascades83 and has been successfully targeted in other inflammatory diseases including rheumatoid arthritis and inflammatory bowel disease. MRL/lpr mice have high levels of TNF in both the serum and kidneys which correlates with disease activity84, 85. Renal TNF is also elevated in NZB/W mice86. In human lupus, class III and IV lupus nephritis is associated with elevated TNF in inflamed glomeruli and tubulointerstitium, especially in infiltrating mononuclear cells87, 88.
Clinical trials suggest that targeting TNF might be efficacious for some SLE manifestations. In a small trial of nine patients, Infliximab diminished SLEDAI scores 89, while in a trial including four lupus nephritis patients 83 proteinuria was reduced and this benefit lasted as long as four years in some patients90. These four patients did experience an increase in dsDNA titers but this did not correspond to increased disease activity. Anti-TNF therapies are well known to be associated with the development of autoantibodies and SLE-like manifestations including pericarditis, neuritis, and nephritis91, 92. Therefore, more clinical studies are needed to define which lupus patients might benefit from anti-TNF therapy.
A possibly more specific target than TNF is TWEAK (TNF-related weak inducer of apoptosis), which is a member of the TNF superfamily. TWEAK and its receptor FN14, have also been found to promote inflammation in SLE93, 94. In the graft versus host murine model of lupus, gene targeting of FN14 diminished proteinuria, glomerular immune complexes and intrarenal inflammatory cytokines95. In these studies, anti-TWEAK antibodies were also beneficial. In human lupus nephritis, TWEAK is elevated in the glomeruli, tubulointerstitium and urine96, 97. Furthermore, high urinary TWEAK levels are associated with renal flares and correlated more closely with lupus nephritis than serum anti-dsDNA titers and complement levels98. A clinical trial with a monoclonal anti-TWEAK antibody (BIIB023) is underway in lupus nephritis.
Possible mechanistic relationships between GN and TII
By the time most patients go to renal biopsy, the immunological processes apparent in inflamed glomeruli and inflamed tubulointerstitium are very distinct. As discussed above this is consistent with different mechanisms driving inflammation in each renal compartment. However, there are at least three mechanisms by which inflammation initiated in glomeruli could, in turn, initiate TII (Figure 1).
Figure 1. Potential mechanisms for how glomerulonephritis might initiate tubulointerstitial inflammation.
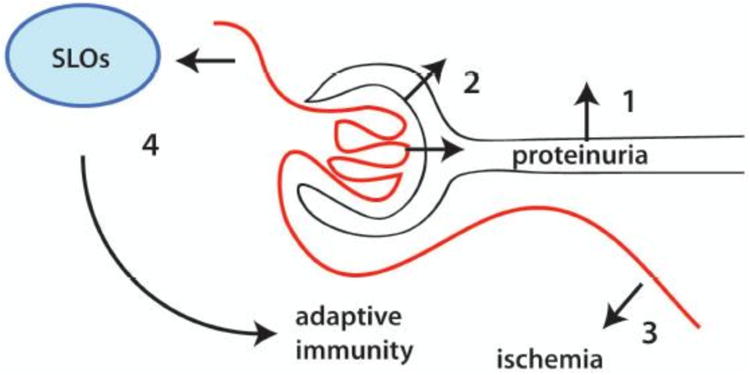
1. GN leads to proteinuria that both activates and damages tubular epithelial cells leading to the release of inflammatory mediators in the tubulointerstitium and inflammation. 2. Inflammation can also result when severe GN ruptures Bowman's capsule. 3. Severe GN can induce tubulointerstitial ischemia, damage and inflammation. 4. Adaptive immune responses which begin with a break in tolerance in glomeruli can be established in secondary lymphoid organs (SLOs) and amplified in the tubulointerstitium.
One possible relationship is ischemia. The glomerular efferent arteriole feeds the peritubular vascular bed. Therefore, it has been postulated that severe GN results in tubulointerstitial ischemia, damage and secondary inflammation. Interestingly, proximal tubules are likely more susceptible to hypoxia as they are dependent on aerobic oxidative metabolism99. Severe glomerular and tubulointerstitial inflammation are both associated with hypoxia. Indeed, the transcriptional signature of hypoxia has been observed in both murine models of lupus and human lupus nephritis100, 101, 102. Furthermore, in experimental models of nephritis, strategies targeting hypoxia have shown promise in impeding progressive fibrosis. These include inhibiting Angiotensin II, calcium channel blockers, inhibiting endothelin and activation of hypoxia-inducible factors (HIFs)103, 104, 105, 106, 107. In renal ischemia-reperfusion models, ischemia induces endogenous ligands that directly activate complement and therefore targeting complement might mitigate both renal injury and inflammation108.
Progression to tubulointerstitial fibrosis evokes additional mechanisms that feed-forward to worsen renal disease and accelerate progression to renal failure. Fibrosis attenuates peritubular vessels and, through exuberant matrix deposition, creates barriers to the diffusion of oxygen. Increasing hypoxia then feeds forward to drive further fibrosis and worsening hypoxia and renal failure109.
The tubulointerstitium also lies downstream of glomerular eluent and there is a well-described relationship between proteinuria and tubulointerstitial injury110, 111. Loss of glomerular integrity allows proteins to pass through and come into direct contact with proximal tubules. Albumin and transferrin are among the best studied and they can induce multiple inflammatory and fibrogenic mediators including monocyte chemoattractant protein 1 (MCP-1), IL-8, RANTES and TGFβ110, 112, 113, 114. In vitro studies of tubular cells showed that apical presentation of proteins leads to the release of inflammatory mediators across the basolateral membrane110, 115. Albumin might also be directly toxic to tubular cells and induce apoptosis. Similar direct toxicity might be mediated by other filtered proteins including complement and IgG116. Interestingly, in crescentic glomerular diseases, breaks are induced in Bowman's capsule that can lead to leakage of protein containing glomerular ultrafiltrate directly into the tublointerstitium110. This mechanism is consistent with our own observations that TII can be severe at the Bowman's capsule border.
In contrast to these potential physiological links between GN and TII, an immunological link has been described117. In this study, model antigens were expressed in the glomeruli and antigen specific effector CD4+ and CD8+ cells repeatedly transferred. In this defined system, dendritic cells in both draining lymph nodes and in the tubulointerstitium amplified immune responses in the tubulointerstitium including production of intrarenal cytokines, chemokines and infiltration by monocyte-derived DCs and macrophages. These data provide a mechanism whereby breaking of tolerance in glomeruli leads to TII. This mechanism might be very relevant to the anti-vimentin immune response described above as vimentin is highly expressed in normal glomeruli and is only abundant in the tubulointerstitium after injury and inflammation.
While there is likely a strong mechanistic interplay between GN and the initiation of TII, it is unclear if these same mechanisms are propagating TII at time of biopsy. Rather, it is likely that many of the additional in situ mechanisms described above (Figure 2), worsen local inflammation and tissue damage. In human lupus nephritis, the severity of interstitial nephritis does not necessarily correlate with glomerular activity. Could we add some histopath here?Cases in which very active glomerulonephritis is associated with little tubulointerstitial inflammation are common. Conversely, there are rare cases of lupus nephritis in which severe interstitial nephritis occurs in the absence of appreciable glomerulonephritis118, 119. Therefore, glomerulonephritis and interstitial nephritis can occur independently. Finally, as described above, the immunological milieus resident in the glomeruli and TI of lupus nephritis are very different.
Figure 2. Propagation of tubulointerstitial inflammation.
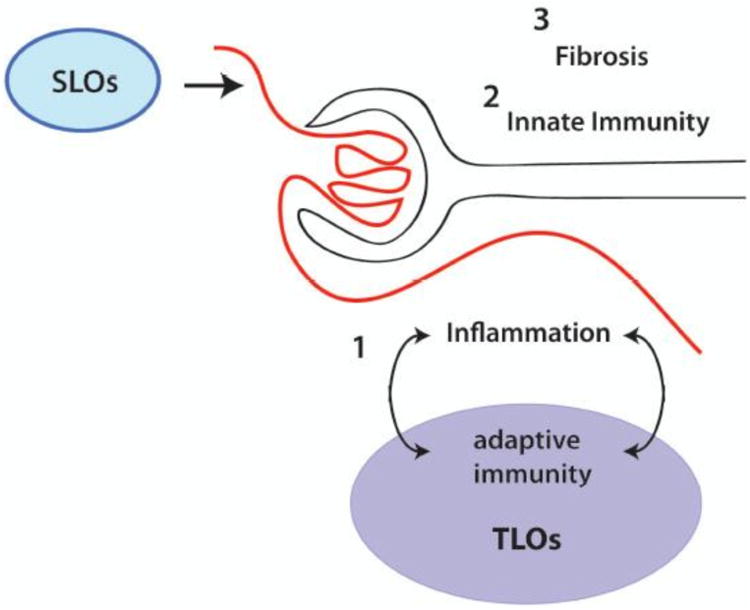
While many mechanisms might initiate tubulointerstitial inflammation, additional in situ mechanisms likely propagate and amplify local inflammation and tissue damage. 1. Within tertiary-like organ (TLO) structures, including T:B aggregates and plasmablast foci, local adaptive immune responses to antigenic features of inflammation are propagated. This feeds forward to worsen inflammation and tissue damage. In addition, networks of innate cells and mediators (2) and fibrogenic pathways (3) lead to progressive functional loss and ultimate renal failure.
This seeming disparity between animal models of pathogenesis and observations of the human disease might be resolved in one of two ways. First, simply, the pathogenic mechanisms of lupus nephritis are almost certainly many and complex. Therefore, it is likely that TII arises in different patients through different mechanisms that have variable relationships to GN. Second, while the mechanisms that initiate TII might be linked to GN, the mechanisms that propagate each process, and that are present at time of biopsy, might be very different.
Therapeutic Implications
Current therapies are either non-specific or are predicated on the idea that SLE is a systemic autoimmune disease in which adaptive immune responses, arising in secondary lymphoid organs, play a central role. However, it is becoming apparent that in those lupus nephritis patients with a poor prognosis, additional in situ adaptive immune cell networks are present that likely contribute to disease severity. We do not know if our current B and T cell targeted therapies disrupt these networks. Furthermore, there are additional innate immune and fibrogenic renal intrinsic processes that are poorly understood and for which relevant clinical trial data is largely lacking. Defining these renal intrinsic mechanisms of disease, and targeting them effectively, should provide a path towards better therapies for the most severe cases of lupus nephritis.
Acknowledgments
Funding: University of Chicago Autoimmunity Center of Excellence (U19AI082724) and RO1-AR55646.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mok C, Tang SS. Incidence and predictors of renal disease in Chinese patients with systemic lupus erythematosus. Am J Med. 2004;117(10):791–795. doi: 10.1016/j.amjmed.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 2.Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, Mejia JC, Aydingtug AO, Chwalinska-Sadowska H, de Ramon E, FernandezNebro A, Galeazzi M, Valen M, Mathieu A, Houssiau F, Caro N, Alba P, Ramos-Casals M, Ingelmo M, Hughes GR. European Working Party on Systemic Lupus Erythematosus. Morbidity and mortality in systemic lupus erythematosus during a 10-year period: a comparison of early and late manifestations in a cohort of 1,000 patients. Medicine. 2003;82(5):299–308. doi: 10.1097/01.md.0000091181.93122.55. [DOI] [PubMed] [Google Scholar]
- 3.Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, Urowitz M, Fortin PR, Petri M, Barr S, Gordon C, Bae SC, Isenberg D, Zoma A, Aranow C, Dooley MA, Nived O, Sturfelt G, Steinsson K, Alarcon G, Senecal JL, Zummer M, Hanly J, Ensworth S, Pope J, Edworthy S, Rahman A, Sibley J, El-Gabalawy H, McCarthy T, St Pierre Y, Clarke A, Ramsey-Goldman R. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54(8):2550–2557. doi: 10.1002/art.21955. [DOI] [PubMed] [Google Scholar]
- 4.McLaughlin J, Bombardier C, Farewell VT, Gladman DD, Urowitz MB. Kidney biopsy in systemic lupus erythematosus. III. Survival analysis controlling for clinical and laboratory variables. Arthritis Rheum. 1994;37(4):559–567. doi: 10.1002/art.1780370417. [DOI] [PubMed] [Google Scholar]
- 5.Cook R, Gladman DD, Pericak D, Urositz MB. Prediction of short term mortality in systemic lupus erythematosus with time dependent measures of disease activity. J Rheumatol. 2000;27(8):1892–1895. [PubMed] [Google Scholar]
- 6.Ginzler E, Dooley MA, Aranow C, Kim MY, Buyon J, Merrill JT, Petri, Gilkeson S, Wallace DJ, Weisman MH, Appel GB. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med. 2005;353(21):2219–2228. doi: 10.1056/NEJMoa043731. [DOI] [PubMed] [Google Scholar]
- 7.Appel G, Contreras G, Dooley MA, Ginzler EM, Isenberg D, Jayne D, Li LS, Mysler E, Sanchez-Guerrero J, Solomons N, Wofsy D Aspreva Lupus Management Study Group. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol. 2009;20(5):1103–1112. doi: 10.1681/ASN.2008101028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flanc R, Roberts MA, Strippoli GF, Chadban SJ, Kerr PG, Atkins RC. Treatment for lupus nephritis. Cochrane Database Syst Rev. 2004;2004(1):CD0002922. doi: 10.1002/14651858.CD002922.pub2. [DOI] [PubMed] [Google Scholar]
- 9.Chan TM. Treatment of severe lupus nephritis: the new horizon. Nat Rev Nephrol. 2015;11:46–56. doi: 10.1038/nrneph.2014.215. [DOI] [PubMed] [Google Scholar]
- 10.Korbet S, Schwartz MM, Evans J, Lewis EJ. Severe lupus nephritis:racial differences in presentation and outcome. J Am Soc Nephrol. 2007;18:244–254. doi: 10.1681/ASN.2006090992. [DOI] [PubMed] [Google Scholar]
- 11.Donadio JJ, Hart GM, Bergstralh EJ, Holley KE. Prognostic determinants in lupus nephritis: a long-term clinicopathologic study. Lupus. 1995;4(2):109–115. doi: 10.1177/096120339500400206. [DOI] [PubMed] [Google Scholar]
- 12.Neumann K, Wallace DJ, Azen C, Nessim S, Fichman M, Metzger AL, Klinenberg JR. Lupus in the 1980s: III. Influence of clinical variables, biopsy, and treatment on the outcome in 150 patients with lupus nephritis seen at a single center. Semin Arthritis Rheum. 1995;25(1):47–55. doi: 10.1016/s0049-0172(95)80017-4. [DOI] [PubMed] [Google Scholar]
- 13.Hsieh C, Chang A, Brandt D, Guttikonda R, Utset TO, Clark MR. Predicting outcomes of lupus nephritis with tubulointerstitial inflammation and scarring. Arthritis care & research. 2011;63(6):865–874. doi: 10.1002/acr.20441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faurschou M, Dreyer L, Kamper AL, Starklint H, Jacobsen S. Long-term mortality and renal outcome in a cohort of 100 patients with lupus nephritis. Arthritis care & research. 2010;62(6):873–880. doi: 10.1002/acr.20116. [DOI] [PubMed] [Google Scholar]
- 15.Austin Hr, Klippel JH, Balow JE, le Riche NG, Steinberg AD, Plotz PH, Decker JL. Therapy of lupus nephritis. Controlled trial of prednisone and cytotoxic drugs. N Engl J Med. 1986;314(10):614–619. doi: 10.1056/NEJM198603063141004. [DOI] [PubMed] [Google Scholar]
- 16.Ong L, Hooi LS, Lim TO, Goh BL, Ahmad G, Ghazalli R, Teo SM, Wong HS, Tan SY, Shaariah W, Tan CC, Morad Z. Randomized controlled trial of pulse intravenous cyclophosphamide versus mycophenolate mofetil in the induction therapy of proliferative lupus nephritis. Nephrology (Carlton) 2005;10(5):504–510. doi: 10.1111/j.1440-1797.2005.00444.x. [DOI] [PubMed] [Google Scholar]
- 17.Hahn BH, McMahon MA, Wilkinson A, Wallace WD, Daikh DI, Fitzgerald JD, et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis care & research. 2012;64(6):797–808. doi: 10.1002/acr.21664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weening JJ, D'Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65(2):521–530. doi: 10.1111/j.1523-1755.2004.00443.x. [DOI] [PubMed] [Google Scholar]
- 19.Austin HA, 3rd, Muenz LR, Joyce KM, Antonovych TA, Kullick ME, Klippel JH, et al. Prognostic factors in lupus nephritis. Contribution of renal histologic data. Am J Med. 1983;75(3):382–391. doi: 10.1016/0002-9343(83)90338-8. [DOI] [PubMed] [Google Scholar]
- 20.Appel G, Cohen DJ, Pirani CL, Meltzer JI, Estes D. Long-term follow-up of patients with lupus nephritis. A study based on the classfication of the World Health Organization. Am J Med. 1987;83(5):877–885. doi: 10.1016/0002-9343(87)90645-0. [DOI] [PubMed] [Google Scholar]
- 21.Parichatikanond P, Francis ND, Malasit P, Laohapand T, Nimmannit S, Singchoovong L, Nilwarangkur S, Chrirawong P, Vanichakarn S. Lupus nephritis: clinicopathological study of 162 cases in Thailand. J Clin Pathol. 1986;39(2):160–166. doi: 10.1136/jcp.39.2.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esdaile JM, Levinton C, Federgreen W, Hayslett JP, Kashgarian M. The clinical and renal biopsy predictors of long-term outcome in lupus nephritis: a study of 87 patients and review of the literature. Q J Med. 1989;72(269):779–833. [PubMed] [Google Scholar]
- 23.Williams W, Sargeant LA, Smilkle M, Smith R, Edwards H, Shah D. The outcome of lupus nephritis in Jamaican patients. Am J Med Sci. 2007;334(6):426–430. doi: 10.1097/MAJ.0b013e3180de4997. [DOI] [PubMed] [Google Scholar]
- 24.Dubois EL. Lupus Erythematosus. Second. W.B Saunders; London: 1974. [Google Scholar]
- 25.Park MH, D'Agati V, Appel GB, Pirani CL. Tubulointerstitial disease in lupus nephritis: relationship to immune deposits, interstitial inflammation, glomerular changes, renal function, and prognosis. Nephron. 1986;44:309–319. doi: 10.1159/000184012. [DOI] [PubMed] [Google Scholar]
- 26.Hill GS, Delahousse M, Nochy D, Tomkiewicz E, Remy P, Mignon F, et al. A new morphologic index for the evaluation of renal biopsies in lupus nephritis. Kid Int. 2000;58:1160–1173. doi: 10.1046/j.1523-1755.2000.00272.x. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz M, Fennell JS, Lewis EJ. Pathologic changes in the renal tubule in systemic lupus erythematosus. Hum Pathol. 1982;13:534–547. doi: 10.1016/s0046-8177(82)80268-2. [DOI] [PubMed] [Google Scholar]
- 28.Bohle A, Wehrmann M, Bogenschutz O, Batz C, Vogl W, Schmitt H, et al. The long-term prognosis of the primary glomerulonephritides. A morphological and clinical analysis of 1747 cases. Pathol Res Pract. 1992;188:908–924. doi: 10.1016/s0344-0338(11)80252-9. [DOI] [PubMed] [Google Scholar]
- 29.Yu F, Wu LH, Tan Y, Li LH, Wang CL, Wang CL, et al. Tubulointerstitial lesions of patients with lupus nephritis classified by the 2003 international society of nephrology and renal pathology society system. Kid Int. 2010;77:820–829. doi: 10.1038/ki.2010.13. [DOI] [PubMed] [Google Scholar]
- 30.Austin HA, Boumpas DT, Vaughan EM, Balow JE. Predicting renal outcomes in severe lupus nephritis: contributions of clinical and histologic data. Kid Int. 1994;45:544–450. doi: 10.1038/ki.1994.70. [DOI] [PubMed] [Google Scholar]
- 31.Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis. 1992;20:1–17. doi: 10.1016/s0272-6386(12)80312-x. [DOI] [PubMed] [Google Scholar]
- 32.Coppo R, D'Amico G. Factors predicting progression of IgA nephropathies. J Nephrol. 2005;18(5):503–512. [PubMed] [Google Scholar]
- 33.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin SL, Castano AP, Nowlin BT, Lupher ML, Jr, Duffield JS. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol. 2009;183(10):6733–6743. doi: 10.4049/jimmunol.0901473. [DOI] [PubMed] [Google Scholar]
- 35.Ko GJ, Boo CS, Jo SK, Cho WY, Kim HK. Macrophages contribute to the development of renal fibrosis following ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant. 2008;23(3):842–852. doi: 10.1093/ndt/gfm694. [DOI] [PubMed] [Google Scholar]
- 36.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115(1):56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishida M, Hamaoka K. Macrophage phenotype and renal fibrosis in obstructive nephropathy. Nephron Exp Nephrol. 2008;110(1):e31–36. doi: 10.1159/000151561. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Wang YP, Zheng G, Lee VW, Ouyang L, Chang DH, et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007;72(3):290–299. doi: 10.1038/sj.ki.5002275. [DOI] [PubMed] [Google Scholar]
- 39.Lebleu VS, Sugimoto H, Miller CA, Gattone VH, 2nd, Kalluri R. Lymphocytes are dispensable for glomerulonephritis but required for renal interstitial fibrosis in matrix defect-induced Alport renal disease. Lab Invest. 2008;88(3):284–292. doi: 10.1038/labinvest.3700715. [DOI] [PubMed] [Google Scholar]
- 40.Ascon M, Ascon DB, Liu M, Cheadle C, Sarkar C, Racusen L, et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009;75(5):526–535. doi: 10.1038/ki.2008.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burne-Taney MJ, Liu M, Ascon D, Molls RR, Racusen L, Rabb H. Transfer of lymphocytes from mice with renal ischemia can induce albuminuria in naive mice: a possible mechanism linking early injury and progressive renal disease? Am J Physiol Renal Physiol. 2006;291(5):F981–986. doi: 10.1152/ajprenal.00229.2005. [DOI] [PubMed] [Google Scholar]
- 42.Davidson A, Aranow C. Lupus nephritis: lessons from murine models. Nat Rev Immunol. 2010;6:13–20. doi: 10.1038/nrrheum.2009.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waldman M, Madaio MP. Pathogenic autoantibodies in lupus nephritis. Lupus. 2005;14:19–24. doi: 10.1191/0961203305lu2054oa. [DOI] [PubMed] [Google Scholar]
- 44.Kalaaji M, Fenton KA, Mortensen ES, Olsen R, Sturfelt G, Alm P, et al. Glomerular apoptotic nucleosomes are central target structures for nephritogenic antibodies in human SLE nephritis. Kid Int. 2007;71:664–672. doi: 10.1038/sj.ki.5002133. [DOI] [PubMed] [Google Scholar]
- 45.Steinmetz OM, Velden J, Kneissler U, Marx M, Klein A, Helmchen U, et al. Analysis and classification of B-cell infiltrates in lupus and ANCA-associated vasculitis. Kid Int. 2008;74:448–457. doi: 10.1038/ki.2008.191. [DOI] [PubMed] [Google Scholar]
- 46.Chang A, Henderson SG, Brandt D, Liu N, Guttikonda R, Hsieh C, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol. 2011;186(3):1849–1860. doi: 10.4049/jimmunol.1001983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satoskar AA, Brodsky SV, Nadasdy G, Bott C, Rovin B, Hebert L, et al. Discrepancies in glomerular and tubulointerstitial/vascular immune complex IgG subclasses in lupus nephritis. Lupus. 2011;20(13):1396–1403. doi: 10.1177/0961203311416533. [DOI] [PubMed] [Google Scholar]
- 48.Jeruc J, Jurcic V, Vizjak A, Hvala A, Babic N, Kveder R, et al. Tubulointerstitial involvement in lupus nephritis with emphasis on pathogenesis. Wien Klin Wochenschr. 2000;112(15-16):702–706. [PubMed] [Google Scholar]
- 49.Park MH, D'Agati V, Appel GB, Pirani CL. Tubulointerstitial disease in lupus nephritis: relationship to immune deposits, interstitial inflammation, glomerular changes, renal function, and prognosis. Nephron. 1986;44(4):309–319. doi: 10.1159/000184012. [DOI] [PubMed] [Google Scholar]
- 50.Kinloch AJ, Chang A, Ko K, Henry Dunand CJ, Henderson S, Maienschein-Cline M, et al. Vimentin is a dominant target of in situ humoral immunity in human lupus tubulointerstitial nephritis. Arthritis Rheumatol. 2014;66(12):3359–3370. doi: 10.1002/art.38888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winchester R, Wiesendanger M, Zhang HZ, Steshenko V, Peterson K, Geraldino-Pardilla L, et al. Immunologic characteristics of intrarenal T cells: trafficking of expanded CD8+ T cell beta-chain clonotypes in progressive lupus nephritis. Arthritis Rheum. 2012;64(5):1589–1600. doi: 10.1002/art.33488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eckes B, Colucci-Guyon E, Smola H, Nodder S, Babinet C, Krieg T, et al. Impaired wound healing in embryonic and adult mice lacking vimentin. J Cell Sci. 2000;113:2455–2462. doi: 10.1242/jcs.113.13.2455. [DOI] [PubMed] [Google Scholar]
- 53.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol. 2003;5:59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 54.Thiagarajan PS, Yakubenko VP, Elsori DH, Yadav SP, Willard B, Tan CD, et al. Vimentin is an endogenous ligand for the pattern recognition receptor Dectin-1. Cardiovasc Res. 2013;99(3):494–504. doi: 10.1093/cvr/cvt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jurcevic S, Ainsworth ME, Pomerance A, Smith JD, Robinson DR, Dunn MJ, et al. Antivimentin antibodies are an independent predictor of transplant-associated coronary artery disease after cardiac transplantation. Transplantation. 2001;71:886–892. doi: 10.1097/00007890-200104150-00011. [DOI] [PubMed] [Google Scholar]
- 56.Nath DS, Ilias BG, Tiriveedhi V, Alur C, Phelan D, Eward GA, et al. Characterization of immune responses to cardiac self-antigens myosin and vimentin in human cardiac allograft recipients with antibody-mediated rejection and cardiac allograft vasculopathy. J Heart Lung Transplant. 2010;29:1277–1285. doi: 10.1016/j.healun.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carter V, Shenton BK, Jacques B, Turner D, Talbot D, Gupta A. Anti-vimentin antibody detection in recipients of heart-beating and non-heart beating donor kidneys. Transplant Proc. 2005;8:3269–3271. doi: 10.1016/j.transproceed.2005.09.062. [DOI] [PubMed] [Google Scholar]
- 58.Mahesh B, Leong JS, McCormak A, Sarathchandra P, Holder A, Rose ML. Autoantibodies to vimentin cause accelerated rejection of cardiac allografts. Am J Pathol. 2007;170:1415–1427. doi: 10.2353/ajpath.2007.060728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 60.Avalos AM, Busconi L, Marshak-Rothstein A. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity. 2010;43(1):76–83. doi: 10.3109/08916930903374618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crotty S. Follicular helper CD4 T cells (TFH) Ann Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 62.Liarski V, Kaverina N, Chang A, Brand D, Carlesso G, Utset TO, et al. Quantitative cell distance mapping in human nephritis reveals organization of in situ adaptive immune responses. Sci Trans Med. 2014;6(230):230ra46. doi: 10.1126/scitranslmed.3008146. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, et al. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adalid-Peralta L, Mathian A, Tran T, Delbos L, Durand-Gasselin I, Berrebi D, et al. Leukocytes and the kidney contribute to interstitial inflammation in lupus nephritis. Kid Int. 2008;73:172–180. doi: 10.1038/sj.ki.5002625. [DOI] [PubMed] [Google Scholar]
- 65.Odegard JM, DiPlacido LD, Greenwald L, Kashgarian M, Kono DH, Dong C, et al. ICOS controls effector function but not trafficking receptor expression of kidney-infiltrating effector T cells in murine lupus. J Immunol. 2009;182:4076–4084. doi: 10.4049/jimmunol.0800758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cassese G, Lindenau S, de Boer B, Arce S, Hauser A, Riemekasten G, et al. Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur J Immunol. 2001;31:2726–2732. doi: 10.1002/1521-4141(200109)31:9<2726::aid-immu2726>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 67.Sekine H, Watanabe H, Gilkeson GS. Enrichment of anti-glomerular antigen antibody-producing cells in the kidneys of MRL/MpJ-Fas(lpr) mice. J Immunol. 2004;172:3913–3921. doi: 10.4049/jimmunol.172.6.3913. [DOI] [PubMed] [Google Scholar]
- 68.Woltman AM, de Fijter JW, Zuidwijk K, Vlug AG, Bajema IM, van der Kooij SW, et al. Quantification of dendritic cell subsets in human renal tissue under normal and pathological conditions. Kid Int. 2007;71:1001–1008. doi: 10.1038/sj.ki.5002187. [DOI] [PubMed] [Google Scholar]
- 69.Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J. The renal mononuclear phagocytic system. J Am Soc Nephrol. 2012;23:194–203. doi: 10.1681/ASN.2011070680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. 2005;68:1096–1108. doi: 10.1111/j.1523-1755.2005.00502.x. [DOI] [PubMed] [Google Scholar]
- 71.Lukacs-Kornek V, Burgdorf S, Diehl L, Specht S, Kornek M, Kurts C. The kidney-renal lymph node-system contributes to cross-tolerance against innocuous circulating antigen. J Immunol. 2008;180:706–715. doi: 10.4049/jimmunol.180.2.706. [DOI] [PubMed] [Google Scholar]
- 72.Coates PT, Duncan FJ, Colvin GL, Wang Z, Zhahorchak AF, Shufesky WJ, et al. In vivo-mobilized kidney dendritic cells are functionally immature, subvert alloreactive T-cell responses, and prolong organ allograft survival. Transplantation. 2004;77:1080–1089. doi: 10.1097/01.tp.0000122183.60680.c9. [DOI] [PubMed] [Google Scholar]
- 73.Scholz JL, Lukas-Kornek V, Engel DR, Specht S, Kiss E, Eitner F, et al. Renal dendritic cells stimulate IL-10 production and attenuate nephrotoxic nephritis. J Am Soc Nephrol. 2008;19:527–537. doi: 10.1681/ASN.2007060684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dong X, Bachman LA, Miller MN, Nath KA, Griffin MD. Dendritic cells facilitate accumulation of IL-17 T cells in the kidney following acute renal obstruction. Kidney Int. 2008;74:1294–1309. doi: 10.1038/ki.2008.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dong X, Swarminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kid Int. 2007;71:619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- 76.Tittel AP, Heuser C, Ohliger C, Knoller PA, Engel DR, Kurts C. Kideny dendritic cells induce innate immunity against bacterial pyelonephritis. J Am Soc Nephrol. 2011;22:1435–1441. doi: 10.1681/ASN.2010101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cao Q, Wang Y, Zheng D, Sun Y, Wang Y, Lee VWS, et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol. 2010;21:933–942. doi: 10.1681/ASN.2009060592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22:317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bethunaickan R, Berthier CC, Ramanujam M, Sahu R, Zhang W, Sun Y, et al. A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus nephritis. J Immunol. 2011;186:4994–5003. doi: 10.4049/jimmunol.1003010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berthier CC, Bethunaickan R, Gonzalez-Riveria T, Nair V, Ramanujam M, Zhang W, et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol. 2012;189:988–1001. doi: 10.4049/jimmunol.1103031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fiore N, Castellano G, Blasi A, Capobianco C, Loverre A, Montinaro V, et al. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol Immunol. 2008;45:259–265. doi: 10.1016/j.molimm.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 82.De Palma G, Castellano G, Del Prete A, Sozzani S, Fiore N, Loverre A, et al. The possible role of ChemR23/Chemerin axis in the recruitment of dendritic cells in lupus nephritis. Kid Int. 2011;79:1228–1235. doi: 10.1038/ki.2011.32. [DOI] [PubMed] [Google Scholar]
- 83.Aringer M, Graninger WB, Steiner G, Smolen JS. Safety and efficacy of tumor necrosis factor alpha blockade in systemic lupus erythematosus: an open-label study. Arthritis Rheum. 2004;50(10):3161–3169. doi: 10.1002/art.20576. [DOI] [PubMed] [Google Scholar]
- 84.Boswell JM, Yui MA, Burt DW, Kelley VE. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J Immunol. 1988;141(9):3050–3054. [PubMed] [Google Scholar]
- 85.Yokoyama H, Kreft B, Kelley VR. Biphasic increase in circulating and renal TNF-alpha in MRL-lpr mice with differing regulatory mechanisms. Kidney Int. 1995;47(1):122–130. doi: 10.1038/ki.1995.14. [DOI] [PubMed] [Google Scholar]
- 86.Brennan DC, Yui MA, Wuthrich RP, Kelley VE. Tumor necrosis factor and IL-1 in New Zealand Black/White mice. Enhanced gene expression and acceleration of renal injury. J Immunol. 1989;143(11):3470–3475. [PubMed] [Google Scholar]
- 87.Takemura T, Yoshioka K, Murakami K, Akano N, Okada M, Aya N, et al. Cellular localization of inflammatory cytokines in human glomerulonephritis. Virchows Arch. 1994;424(5):459–464. doi: 10.1007/BF00191429. [DOI] [PubMed] [Google Scholar]
- 88.Herrera-Esparza R, Barbosa-Cisneros O, Villalobos-Hurtado R, AvalosDiaz E. Renal expression of IL-6 and TNFalpha genes in lupus nephritis. Lupus. 1998;7(3):154–158. doi: 10.1191/096120398678919949. [DOI] [PubMed] [Google Scholar]
- 89.Uppal SS, Hayat SJ, Raghupathy R. Efficacy and safety of infliximab in active SLE: a pilot study. Lupus. 2009;18(8):690–697. doi: 10.1177/0961203309102557. [DOI] [PubMed] [Google Scholar]
- 90.Aringer M, Houssiau F, Gordon C, Graninger WB, Voll RE, Rath E, et al. Adverse events and efficacy of TNF-alpha blockade with infliximab in patients with systemic lupus erythematosus: long-term follow-up of 13 patients. Rheumatology (Oxford) 2009;48(11):1451–1454. doi: 10.1093/rheumatology/kep270. [DOI] [PubMed] [Google Scholar]
- 91.De Bandt M, Sibilia J, Le Loet X, Prouzeau S, Fautrel B, Marcelli C, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7(3):R545–551. doi: 10.1186/ar1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Soforo E, Baumgartner M, Francis L, Allam F, Phillips PE, Perl A. Induction of systemic lupus erythematosus with tumor necrosis factor blockers. J Rheumatol. 2010;37(1):204–205. doi: 10.3899/jrheum.081312. [DOI] [PubMed] [Google Scholar]
- 93.Campbell S, Burkly LC, Gao HX, Berman JW, Su L, Browning B, et al. Proinflammatory effects of TWEAK/Fn14 interactions in glomerular mesangial cells. J Immunol. 2006;176(3):1889–1898. doi: 10.4049/jimmunol.176.3.1889. [DOI] [PubMed] [Google Scholar]
- 94.Chicheportiche Y, Chicheportiche R, Sizing I, Thompson J, Benjamin CB, Ambrose C, et al. Proinflammatory activity of TWEAK on human dermal fibroblasts and synoviocytes: blocking and enhancing effects of anti-TWEAK monoclonal antibodies. Arthritis Res. 2002;4(2):126–133. doi: 10.1186/ar388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao Z, Burkly LC, Campbell S, Schwartz N, Molano A, Choudhury A, et al. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol. 2007;179(11):7949–7958. doi: 10.4049/jimmunol.179.11.7949. [DOI] [PubMed] [Google Scholar]
- 96.Lu J, Kwan BC, Lai FM, Choi PC, Tam LS, Li EK, et al. Gene expression of TWEAK/Fn14 and IP-10/CXCR3 in glomerulus and tubulointerstitium of patients with lupus nephritis. Nephrology (Carlton) 2011;16(4):426–432. doi: 10.1111/j.1440-1797.2011.01449.x. [DOI] [PubMed] [Google Scholar]
- 97.Schwartz N, Rubinstein T, Burkly LC, Collins CE, Blanco I, Su L, et al. Urinary TWEAK as a biomarker of lupus nephritis: a multicenter cohort study. Arthritis Res Ther. 2009;11(5):R143. doi: 10.1186/ar2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.El-Shehaby A, Darweesh H, El-Khatib M, Momtaz M, Marzouk S, ElShaarawy N, et al. Correlations of urinary biomarkers, TNF-like weak inducer of apoptosis (TWEAK), osteoprotegerin (OPG), monocyte chemoattractant protein-1 (MCP-1), and IL-8 with lupus nephritis. J Clin Immunol. 2011;31(5):848–856. doi: 10.1007/s10875-011-9555-1. [DOI] [PubMed] [Google Scholar]
- 99.Epstein FH. Oxygen and renal metabolism. Kidney Int. 1997;51(2):381–385. doi: 10.1038/ki.1997.50. [DOI] [PubMed] [Google Scholar]
- 100.Bethunaickan R, Berthier CC, Zhang W, Eksi R, Li HD, Guan Y, et al. Identification of stage-specific genes associated with lupus nephritis and response to remission induction in (NZB × NZW)F1 and NZM2410 mice. Arthritis Rheumatol. 2014;66(8):2246–2258. doi: 10.1002/art.38679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bethunaickan R, Berthier CC, Zhang W, Kretzler M, Davidson A. Comparative transcriptional profiling of 3 murine models of SLE nephritis reveals both unique and shared regulatory networks. PLoS One. 2013;8(10):e77489. doi: 10.1371/journal.pone.0077489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Berthier CC, Bethunaickan R, Gonzalez-Rivera T, Nair V, Ramanujam M, Zhang W, et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol. 2012;189(2):988–1001. doi: 10.4049/jimmunol.1103031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gunaratnam L, Bonventre JV. HIF in kidney disease and development. J Am Soc Nephrol. 2009;20(9):1877–1887. doi: 10.1681/ASN.2008070804. [DOI] [PubMed] [Google Scholar]
- 104.Nangaku M, Eckardt KU. Hypoxia and the HIF system in kidney disease. J Mol Med (Berl) 2007;85(12):1325–1330. doi: 10.1007/s00109-007-0278-y. [DOI] [PubMed] [Google Scholar]
- 105.Norman JT, Stidwill R, Singer M, Fine LG. Angiotensin II blockade augments renal cortical microvascular pO2 indicating a novel, potentially renoprotective action. Nephron Physiol. 2003;94(2):39–46. doi: 10.1159/000071289. [DOI] [PubMed] [Google Scholar]
- 106.Gagliardini E, Corna D, Zoja C, Sangalli F, Carrara F, Rossi M, et al. Unlike each drug alone, lisinopril if combined with avosentan promotes regression of renal lesions in experimental diabetes. Am J Physiol Renal Physiol. 2009;297(5):F1448–1456. doi: 10.1152/ajprenal.00340.2009. [DOI] [PubMed] [Google Scholar]
- 107.Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, et al. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295(4):F1023–1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Danobeitia JS, Djamali A, Fernandez LA. The role of complement in the pathogenesis of renal ischemia-reperfusion injury and fibrosis. Fibrogenesis Tissue Repair. 2014;7:16. doi: 10.1186/1755-1536-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21(11):1819–1834. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 110.Eddy AA. Proteinuria and interstitial injury. Nephrol Dial Transplant. 2004;19(2):277–281. doi: 10.1093/ndt/gfg533. [DOI] [PubMed] [Google Scholar]
- 111.Theilig F. Spread of glomerular to tubulointerstitial disease with a focus on proteinuria. Ann Anat. 2010;192(3):125–132. doi: 10.1016/j.aanat.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 112.Tang S, Leung JC, Abe K, Chan KW, Chan LY, Chan TM, et al. Albumin stimulates interleukin-8 expression in proximal tubular epithelial cells in vitro and in vivo. J Clin Invest. 2003;111(4):515–527. doi: 10.1172/JCI16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shimizu H, Maruyama S, Yuzawa Y, Kato T, Miki Y, Suzuki S, et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates renal injury induced by protein-overload proteinuria. J Am Soc Nephrol. 2003;14(6):1496–1505. doi: 10.1097/01.asn.0000069223.98703.8e. [DOI] [PubMed] [Google Scholar]
- 114.Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. J Exp Med. 1999;190(12):1813–1824. doi: 10.1084/jem.190.12.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339(20):1448–1456. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- 116.Vernon KA, Cook HT. Complement in glomerular disease. Adv Chronic Kidney Dis. 2012;19(2):84–92. doi: 10.1053/j.ackd.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 117.Heymann F, Meyer-Schwesinger C, Hamilton-Williams EE, Hammerich L, Panzer U, Kaden S, et al. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest. 2009;119(5):1286–1297. doi: 10.1172/JCI38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mori Y, Kishimoto N, Yamahara H, Kijima Y, Nose A, Uchiyama-Tanaka Y, et al. Predominant tubulointerstitial nephritis in a patient with systemic lupus nephritis. Clin Exp Nephrol. 2005;9:79–84. doi: 10.1007/s10157-004-0338-3. [DOI] [PubMed] [Google Scholar]
- 119.Omokawa A, Wakui H, Okuyama S, Togashi M, Ohtani H, Komatsuda A, et al. Predominant tubulointerstitial nephritis in a patient with systemic lupus erythematosus: phenotype of infiltrating cells. Clin Nephrol. 2008;69(6):436–444. doi: 10.5414/cnp69436. [DOI] [PubMed] [Google Scholar]