

Abstract
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease marked by the presence of pathogenic autoantibodies, immune dysregulation, and chronic inflammation that may lead to increased morbidity and early mortality from end-organ damage. Over half of all SLE patients will develop lupus nephritis. Genetic association studies have identified more than fifty polymorphisms that contribute to lupus nephritis pathogenesis, including genetic variants associated with altered programmed cell death (PCD) and defective immune clearance of PCD debris. These variants may support the generation of autoantibody-containing immune complexes that contribute to lupus nephritis. Genetic variants associated with lupus nephritis also affect the initial phase of innate immunity and the amplifying, adaptive phase of the immune response. Finally, genetic variants associated with the kidney-specific effector response may influence end-organ damage and the progression to end-stage renal disease and death. This review discusses genetic insights of key pathogenic processes and pathways that may lead to lupus nephritis, as well as the clinical implications of these findings as they apply to recent advances in biologic therapies.
Keywords: SLE, lupus nephritis, genetics, immune response
Introduction
The heterogeneous manifestations of systemic lupus erythematosus (SLE) are caused by chronic immune dysregulation and pathogenic autoantibody production that leads to progressive end-organ damage. Kidney damage resulting from lupus nephritis (LN) is among the most severe sequelae of SLE, contributing substantially to SLE-related morbidity and mortality.1 Despite advances in the management of LN, little progress has been made with respect to the adverse outcomes of LN, including chronic kidney disease, end-stage renal disease (ESRD) or mortality. This is particularly problematic in non-Caucasian SLE patients, who are at increased risk of developing LN, with increased disease severity and altered response to treatment protocols.2
Early detection and treatment of LN are imperative to minimize the risk of inflammation-induced irreversible kidney damage and to preserve renal function. In addition, analysis of pathway-specific immune dysregulation may one day enable personalized, precision medicine for LN. The success of such approaches will require methods for identifying individuals at greatest risk of developing LN and for defining measures of pathway-specific immune dysregulation to select the most appropriate LN patients for a given pathway-specific biologic treatment. With the advent of lower-cost genome analysis techniques, both of these goals may be met in part by determining each SLE patient’s individual genetic risk factors for LN. Genetic association studies have identified over fifty SLE disease susceptibility loci.3 Loci associated with LN may influence intra-renal mechanisms of LN that directly produce kidney damage as well as extra-renal mechanisms that promote LN through dysregulation of innate, adaptive, and effector mechanisms of inflammation1 This work reviews genes implicated in the pathogenic mechanisms of LN according to cell types and molecular pathways associated with immune dysregulation (extrarenal etiology) and kidney damage (intrarenal etiology) (Table 1). The clinical implications, shortfalls, and opportunities for future genetic studies linked to LN are briefly discussed.
Table 1.
Disease susceptibility genes associated with lupus nephritis
| Programmed cell death (PCD) | Innate Immunity |
|---|---|
| FAS | IFIH1 |
| DNASE1 | RIG1 |
| ATG5 | TLR3, 7, 9 |
| MTMR3 | MAVS |
| TREX1 | |
| Immune Complex Clearance | MYD88 |
| FCGR2A, 2B, 3A, 3B | TRAF6 |
| C1Q (A,B,C) | IRAK1 |
| C4 (A,B) | IRF5, 7 |
| CRP | TNFAIP3 |
| MBL2 | TNIP1 |
| CR1 | UBE2L3 |
| ITGAM | |
| IKZF1 | Adaptive Immunity |
| HLA-DR | |
| Intra-Renal Pathogenesis | PTPN22 |
| TNFRSF1B | CTLA4 |
| CCL2 | PKCD1 |
| CXCL8 | TNFSF4 |
| CCR5 | STAT1 |
| CXCL12 | STAT4 |
| KLK1, 3 | IFNG |
| ACE | TGFB1 |
| AGT | IL10 |
| APOL1 | BLK CD40 |
Extrarenal etiology
The pathogenesis of LN is largely related to that of SLE: complex dysregulation of immune responses to nuclear autoantigens, including inhibition of regulatory mechanisms, chronic inflammation, accumulation of autoantibody specificities, and formation of pathogenic immune complexes (ICs). Here we discuss the phases of the autoimmune response and those genes which may contribute to the downstream pathogenesis of LN.
Programmed cell death and autoantigens
Numerous studies have shown that nuclear antigens drive SLE pathogenesis, with strong immune responses to nucleic acids, histones, and ribonuclear proteins.4 Sequestered within cellular and nuclear membranes, lupus-specific autoantigens are normally segregated from the immune system. However, enhanced programmed cell death (PCD) mechanisms coupled with alterations in clearance machinery allow for the persistence of antigens that can be modified and perceived by the immune system as non-self (Figure 1A).5
Figure 1.
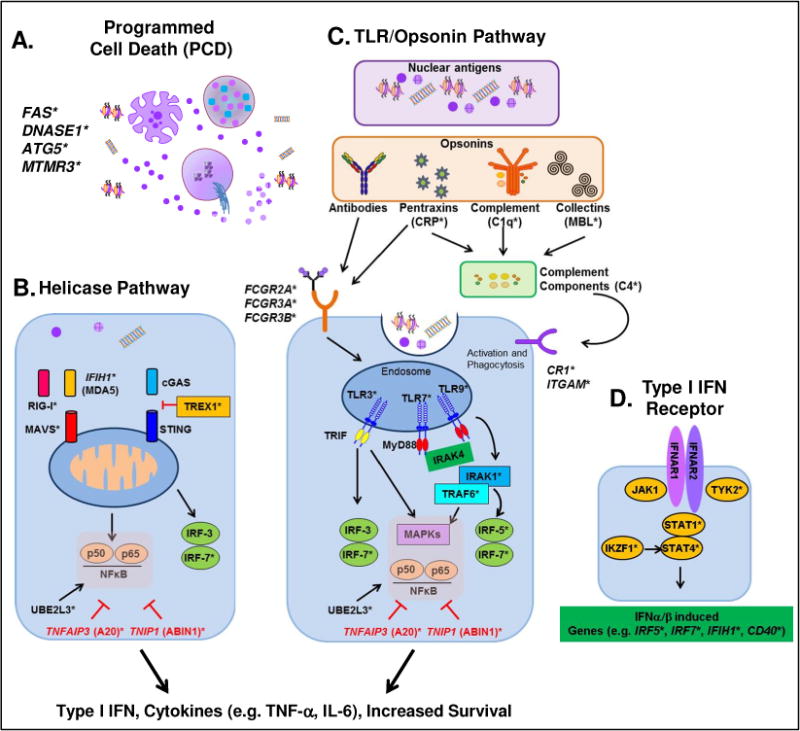
Innate immunity processes contribute to lupus nephritis. A. Enhanced programmed cell death (PCD) coupled with decreased clearance of PCD debris results in the availability of nuclear autoantigens, including nucleosomes, nuclear particles, RNA, and DNA. B. Intracellular RNA (RIGI-I and MDA5) and DNA (cGAS) sensors interact with RNA, DNA, and nuclear particles. C. Nuclear antigens are opsonized by antibodies, complement, pentraxins, and/or collectins (the latter two then interact with complement components), which then interact with Fc (antibody) or complement (CR) receptors to bring antigens into phagocytic endosomes, where RNA and DNA can interact with the TLR receptors TLR3, TLR7 and TLR9, respectively. The helicase (B) and Opsonin/TLR (C) pathways result in prolonged immune cell survival and secretion of pro-inflammatory cytokines. These include Type I IFNs, which then engage the Type I IFN receptor (D). Activation of the Type I IFN receptor results in the production of transcription factors, cytokines, and receptors that prolong and amplify the immune response.
*Genes/molecules with genetic variants associated with lupus nephritis
Perhaps the most extensively studied mechanism of PCD in SLE pathogenesis is apoptosis. Induced by extrinsic (e.g. Fas/FasL) or intrinsic (e.g. DNA damage) factors, downstream activation of caspases leads to changes in the plasma membrane and chromatin structure, causing the cell to disintegrate into apoptotic blebs.4 Variants of genes encoding Fas6 and its ligand, FasL,7 are linked to SLE pathogenesis, with the −670 FAS polymorphism being linked to LN.6 DNase1 activity in the intrinsic apoptotic pathway is increased in SLE patients with nephropathy,8 and polymorphisms in the DNASE1 gene have been linked to LN.9
Clearance of apoptotic cells is altered in SLE patients.5 This results in secondary necrosis, whereby nucleosomes are exposed at the surface of apoptotic blebs and can be proteolytically modified to enhance their immunogenicity.4 Necroptosis leads to rapid plasma membrane permeabilization and the release of nucleosomes and other damage-associated molecular patterns (DAMPs) that serve as lupus-associated autoantigens. Several pro-inflammatory factors linked to LN can trigger necroptosis, including members of the tumor necrosis factor (TNF) superfamily (e.g. TNF and TWEAK), Toll-like receptors (TLRs), and other DNA and RNA sensing receptors.4
Other mechanisms of PCD that may influence LN pathogenesis include autophagy and NETosis.4 Autophagy, an intracellular degradation system where the cell consumes itself for energy, can act as a regulator of both innate and adaptive immune mechanisms. Polymorphisms in the autophagy gene ATG510 that contributes to autophagosome formation and the autophagy initiator and phosphatase gene, MTMR3,11 have been linked to LN. In NETosis, neutrophils (PMNs) release neutrophil extracellular traps (NETs) composed of decondensed chromatin in association with histones, granular proteins, and some cytoplasmic proteins.4 Many receptors that contribute to LN-associated immune dysregulation activate NETosis, including TLRs, Fc receptors, and certain pro-inflammatory cytokine receptors, including IL-8 and TNF-α. NETs released by dying PMNs are normally degraded by DNase1. However, impaired NET degradation, as the result of DNASE1 genetic variants9 and decreased DNase1 activity8 have been associated with LN.
Innate immunity
The primary function of the innate response is the initial recognition of danger signals to facilitate phagocytosis and clearance of infectious pathogens. In SLE, these mechanisms are misdirected to target self, such that endogenous, immunostimulatory nucleic acids, alone or in conjunction with nuclear particles, nucleosomes, or opsonins, stimulate the innate immune response to drive systemic inflammation. Enhanced PCD pathways coupled with decreased clearance of cellular debris increases the availability of pattern recognition receptor (PRR) ligands and opsonized antigens that can activate an enhanced and sustained innate immune response.12
Pattern recognition receptors
Several genetic variants within nucleic acid cytosolic sensor genes have been implicated in LN (Figure 1B). Polymorphisms in the IFIH1 gene, which encodes the dsRNA sensor MDA5, allow for more avid binding of RNA and increased baseline and ligand-induced type I IFN responses. SLE patients carrying IFIH1 risk variants have enhanced responses to type I IFN and are more likely to develop anti-dsDNA antibodies that may contribute to LN.13 Glomeruli of patients with LN exhibit enhanced expression of MDA5.14 Both MDA5 and RIG-I, whose genetic variant is also associated with LN,15 mediate downstream signaling via the adaptor molecule MAVS. The MAVS polymorphism most commonly found in African-American SLE patients has not yet been studied as a modifier of LN risk, but it could feasibly protect against LN because patients with this polymorphism exhibit decreased levels of type I IFN and an absence of autoantibodies to RNA-binding proteins.16 The DNA-specific exonuclease Trex1 inhibits pro-inflammatory responses driven by cytosolic dsDNA sensors. Some genetic variants of TREX1 have been implicated in LN, while others are thought to protect against the development of anti-Ro and anti-dsDNA autoantibodies.17
Endogenous nuclear particles undergoing receptor-mediated endocytosis can reach endosomes and interact with endosomal TLRs (Figure 1C). Genetic variants of TLR3 (dsRNA), TLR7/8 (ssRNA), and TLR9 (DNA) have been associated with LN. Activation of TLR3 on antigen presenting cells (APCs) or renal mesangial cells can aggravate LN15 by upregulating the expression of CXCL1/GROα to recruit PMNs to the site of inflammation, where they can contribute to renal injury.18 TLR7/819 and TLR920 signal through MyD88,21 TRAF6,22 and IRAK1,23,24 genetic variants of which may contribute to severe renal insufficiency in LN. In addition, signaling through particular TLR9 genetic variants has been linked to more severe renal disease at the time of LN presentation.20
Signaling through PRRs leads to type I IFN production through transcriptional activation of interferon regulatory factors (IRF), including IRF3, IRF5, and IRF7. IRF52,25,26 and IRF72,25 polymorphisms are associated with LN. Particular genetic variants of these IRFs are associated with the presence of anti-dsDNA27 and increased type I IFN activity levels,28 which may contribute to LN. In addition, signaling through the NFκB and MAPK pathways results in upregulation of co-stimulatory molecules and other pro-inflammatory mediators that can contribute to LN. Such pathways are regulated in part by ubiquitination and proteasomal degradation. TNFAIP3, which encodes the deubiquitinase A20, has shown strong genetic association with LN.29 In SLE patients, downregulation of A20 allows for enhanced activity of transcription factors that promote inflammation and survival of autoreactive B cells.29,30 A20 also contributes to the regulation of MAPK signaling, most notably through the TNIP-encoded A20-binding inhibitors of NF-κB (ABIN) proteins.31 Dysregulation of NF-κB and MAPK pathways in SLE may be attributed to TNIP1 genetic variants.26,32 Another regulator of ubiquitin-mediated transcriptional control, UBE2L3, is also associated with LN.33 In contrast to A20 and ABINs, UBE2L3 amplifies NF-κB activation via linear ubiquitination, promoting development of autoreactive B-lymphocytes into plasma cells that secrete autoantibodies capable of forming ICs, which may contribute to LN pathogenesis.34,35
Opsonin receptors
Like PRRs, opsonins also recognize nuclear antigens and cellular debris. IgG antibodies, complement, pentraxins, and collectins engage FcR and/or complement receptors (after interacting with complement components) to activate innate immunity and enhance phagocytosis. Genetic variants of opsonins and of their receptors have been associated with LN.12 For example, nuclear antigens opsonized with IgG form ICs that contribute directly to renal pathology in LN, and both ICs and complement components have been found in glomerular biopsies from LN patients.1 In the extra-renal space, IgG-opsonized nuclear antigens engage FcγRs to activate APCs and facilitate phagocytosis, which clears antigen and moves the immune response toward the adaptive phase. Polymorphisms and/or copy number variants in multiple FcγR subtype genes are associated with LN,36–40 including FCGR2A, FCGR2B, FCGR3A, and FCGR3B.
The FcγRIIa R131 polymorphism is associated with LN across multiple ethnicities.40–42 This polymorphism decreases binding affinity for IgG2, thereby reducing clearance of IgG2-containing ICs that contribute to LN pathogenesis, particularly in SLE patients who have C1q IgG2 responses.43 The R131 polymorphism FcγRIIa also increases responsiveness to the pentraxin opsonin, C-reactive protein (CRP), allowing for increased intra-renal inflammation.41 Impairment of the inhibitory receptor FcγRIIb supports the development of SLE by allowing unopposed FcγR signaling. A loss-of-function FcγRIIb variant, T232, excludes FcγRIIb from lipid rafts, leading to increased pro-inflammatory signaling.37
An LN-associated genetic variant of the FcγRIIIa receptor, FCGR3A-158V (also known as 176V/F when the leader sequence is included), increases binding affinity for IgG1, IgG3, and IgG4 relative to the non-risk allele.44 In addition, patients who carry the 158V (176V) allele have increased CD25 expression, Ca2+ flux and NK cell activation.44 Recent evidence from a multi-ethnic inception cohort of SLE patients demonstrated that LN patients carrying the high-binding FCGR3A V176 variant progressed more quickly to ESRD.45 Two different allotypic variants of FCGR3B, NA1 and NA2, have been associated with LN.36 The product of the NA1 allele shows higher binding for IgG1 and IgG3.46 The NA1 and NA2 alleles may also affect interactions with other cell surface receptors, such as the β2-integrin, CD11b/CD1846 (ITGAM), whose genetic variants have also been linked to LN.26,47,48 Low copy number of FCGR3A49 and FCGR3B50 have also been linked to LN.
The complement system includes a series of proteins that opsonize antigens, irreparably damage membranes, and induce inflammatory responses through the three complement pathways: classical (C1q, CRP), alternative, and collectin (e.g. mannose binding lectin [MBL]). In addition, the complement system may affect the ability of innate immune cells to facilitate phagocytosis of nuclear antigens and PCD debris. LN has been genetically associated with the opsonin C1q, as well as complement proteins C2 and C4, and complement inhibitors, which affect the clearance of cellular debris after PCD.51 Genetic variants of pentraxins, such as CRP,36,41 and collectins, such as MBL,52,53 also contribute to LN by disrupting complete clearance of autoantigens, enhancing inflammation, and increasing autoantibodies to C1q.
Multiple complement receptors have been genetically linked to LN. Perhaps the most frequently noted is CR3 (ITGAM/CD11b/Mac-1). The R77H, P1146S, and A858V variants of ITGAM decrease phagocytic clearance of antigens, alter leukocyte adhesion,54,55 and decrease potentially inhibitory interactions with TLR7/8.56 In addition to CR3, CR1 (CD35), CR2 (CD21), and CR4 (ITGAX subunit) have also been linked to LN. Reduced transcription and expression of the CR1 receptor is associated with the presence of anti-dsDNA responses and decreased clearance of ICs that contribute to renal pathology.57 Genetic variants of the CR2 receptor reduce protein expression through decreased transcription and alternative splicing, leading to decreased phagocytosis of apoptotic cells and decreased activation of the alternative complement pathway.51
Activation of FcRs or CR3/ITGAM leads to the secretion of innate cytokines, including type I IFN, which engages the IFNα receptor (IFNAR) (Figure 1D).58 IFNAR activates NF-κB and MAPK pathways in addition to JAK-STAT pathway proteins59 that are genetically linked to LN. Of note are TYK2,60 which binds to the IFNAR-JAK1-STAT4 complex. The gene encoding STAT4 is perhaps the most noted IFNAR-mediated signaling protein associated with LN.2,25,26 LN is also genetically associated with IKZF1, which encodes the Ikaros family zinc finger 1 transcription factor that enhances STAT4 signaling.61
Adaptive Immunity
T-lymphocyte activation
T-lymphocytes are the coordinators of the adaptive immune response. Full T-lymphocyte activation requires: (i) presentation of antigen on the major histocompatibility complex (MHC) of an APC (macrophages, dendritic cells, or B-lymphocytes) to the T-cell receptor (TCR) on the T-lymphocyte, (ii) interactions of cognate co-stimulatory molecules between the APC and T-lymphocyte, and (iii) soluble mediator communication between the APC and T-lymphocyte to determine T-lymphocyte differentiation and downstream effector responses.62 Genetic variants affecting all phases of T-lymphocyte activation have been associated with LN (Figure 2A).
Figure 2.
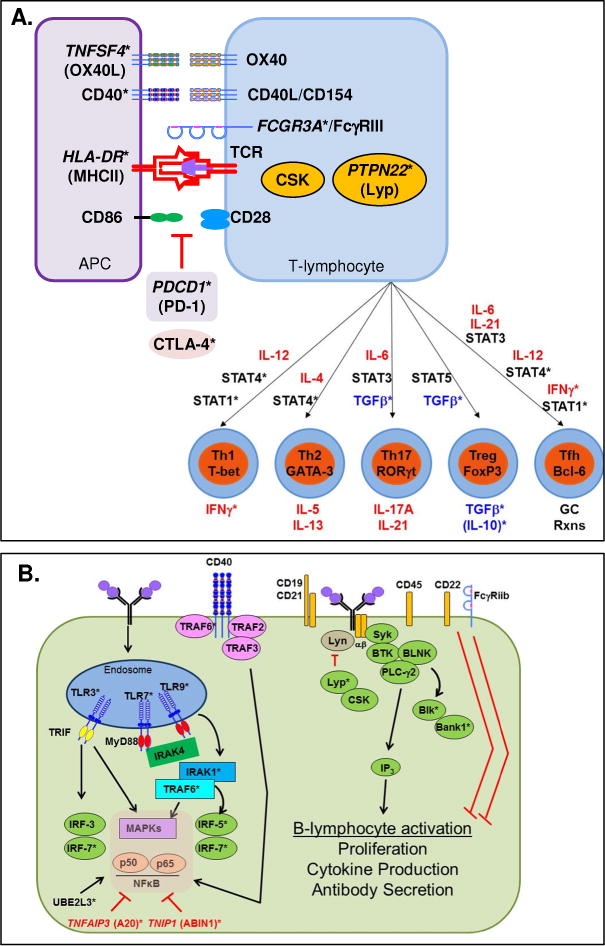
Adaptive immunity processes contribute to lupus nephritis. A. T-lymphocyte activation. Nuclear antigens are phagocytosed, processed, and presented in the context of MHC by APC, which then interacts with the TCR on T-lymphocytes and activates downstream signaling events (Signal 1). Co-stimulatory molecules from the CD28 and TNFR families engage to prevent anergy and amplify the MHCII/TCR response in CD4 T-helper cells (Signal 2). Downstream signals lead to the secretion of cytokines (Signal 3), which then interact with transcriptional regulators to drive T-lymphocyte differentiation pathways. B. B-lymphocyte activation. Nuclear antigens stimulate the BCR, activating the BCR signaling complex that results in downstream signaling events. Co-stimulatory molecules, including CD40 and TLRs, synergize with the BCR to amplify B-lymphocyte activation.
*Genes/molecules with genetic variants associated with lupus nephritis
First, the strongest SLE genetic associations to date have been in the MHC region. Two well-established SLE susceptibility loci, encoded by HLA-DR263,64 and HLA-DR3,26,63 are associated with LN. In addition, a meta-analysis of 25 case-control LN studies suggested that HLA-DR15 increases risk of LN, while HLA-DR4 and HLA-DR11 might be protective.64 The TCR complex is composed of many proteins, including TCRα, β, and ζ chains and CD3. Multiple studies have noted decreased TCRζ chain expression in SLE. Decreased TCRζ chain expression can be caused by a genetic variant in the 3′-UTR that also decreases downstream IL-2 expression and increases IFN-γ expression, which contributes to LN.65 FcγRIII is also part of the TCRζ complex and amplifies TCR-mediated T-lymphocyte activation. As discussed above, genetic variants in FCGR3A are thought to contribute to altered cellular activation44 and are associated with LN36 and progression to ESRD.45 Proximal TCR signaling events are regulated by protein tyrosine phosphatases, including PTPN22-encoded Lyp. Genetic variants of PTPN22 are associated with LN, and C1858T (R620W) is thought to be a gain-of-function variant,66,67 such that TCR-mediated IL-2 is decreased67 while IFN-γ is increased and resistant to control by T-regulatory mechanisms (also see “B-lymphocyte activation”).66
Second, engagement of co-stimulatory molecules is required to prevent anergy, and co-stimulatory molecules are associated with SLE and LN. The classic co-stimulatory molecules are CD28 on the T-lymphocyte, engaged with B7 family members on the APC. Alternatively, engagement of B7 family members by CTLA-4 on the T-lymphocyte negatively regulates T-cell activation. Of particular interest are genetic variants in CTLA4 that lead to decreased expression and potential dysregulation of T-lymphocyte pro-inflammatory signaling that may contribute to LN.3 Genetic variants in PDCD1, which encodes the CD28 family member PD-1 expressed on T-lymphocytes, are associated with LN.68 SLE patients harboring such variants display aberrant expression and negative regulatory function of PD-1 in T-lymphocyte activation.69 Other co-stimulatory molecule partners include members of the TNF receptor (TNFR) superfamily. Genetic variants within the genes encoding APC-expressed CD4070 and T-lymphocyte-expressed CD40L/CD15471 result in altered receptor/ligand expression and cellular activation71 and are associated with LN.70 Genetic variants within TNFSF4 (OX40L, which activates OX40 on T-lymphocytes) are also associated with LN.55,72 One of the strongest LN associations within TNFSF4 is at the upstream variant rs2205960, where alternate alleles are present in the protective and risk haplotypes.72 The risk haplotype is associated with increased OX40L expression, enhanced T-lymphocyte activation with decreased generation of IL-10 producing T-regulatory cells, and increased SLE disease activity with LN.73
Third, the presence of cytokines drives activation and differentiation of T-lymphocytes, and multiple cytokine-driven T-lymphocyte activation pathways are implicated in LN pathogenesis. Associated genetic variants have been identified in each of these pathways, including enhanced effector Th1, Th2, and Th17 pathways, attenuated T-regulatory (Treg) mechanisms, and augmented T-follicular helper (Tfh) cell mechanisms that select high-affinity, antibody-producing B cells for clonal expansion in germinal centers.74 APC-secreted cytokines engage in receptor-mediated phosphorylation of STAT proteins, leading to activation of transcription master regulators that drive the differentiation of pathway-specific T-lymphocytes. In addition, many APC-secreted cytokines are altered during the innate autoimmune response that leads to LN.
The Th2 differentiation pathway is associated with LN and affects B-lymphocyte activation. Within this pathway, IL-4 drives both STAT6 phosphorylation that subsequently activates GATA-3 to drive Th2 differentiation, and STAT4 activation that leads to autoantibody production. Autoantibody-mediated pathology in LN is further supported by genetic variants within the T follicular helper differentiation pathway,75,76 where IL-6 and IFN-γ77 via STAT178 activate Tfh-differentiating Bcl-6. In low IL-2 conditions, as observed in SLE and LN, Bcl-6 is also activated by IL-12 via STAT426,55 (IL-12-STAT4 inhibits Bcl-6 in high IL-2 conditions).
In addition to autoantibody-mediated inflammation, Th17-mediated intra-renal cellular inflammation has been intimately linked to LN, and copy number variation of several genes encoding Th17-type cytokines is associated with SLE pathogenesis.74 With respect to Th1 differentiation, as described above, genetic variants in proximal TCR signaling mediators may result in enhanced IFN-γ production, which contributes to LN pathogenesis. In the Th1 differentiation pathway, IL-12/IL-12 receptor interactions lead to STAT1→T-bet→IFN-γ production or STAT4→IFN-γ production, and components of these pathways are genetically linked to SLE and LN.79 STAT4 genetic variants associate with LN in multiple studies,26,55 and the proximity of some variants in the STAT1-STAT4 region implicates STAT1 as well.78 Finally, genetic variants within IFNG that lead to enhanced IFN-γ secretion77 are also implicated in LN.
Under normal circumstances, activation of inflammatory pathways ultimately results in immune regulation through the differentiation of T regulatory cells. However, in SLE patients (and in LN), many regulatory mechanisms are impaired.80 Two key soluble mediators which negatively regulate inflammation are genetically associated with LN. LN-associated variants of TGFB181 decrease TGFβ secretion and affect the number of circulating T regulatory cells. Polymorphisms in the IL10 gene82 that decrease IL-10 expression and loss-of-function IL10R variants83 contribute to LN and higher SLICC damage scores.84
B-lymphocyte activation
B-lymphocytes act as both APC and effector cells, secreting both (auto)antibody and soluble mediators. Similar to TCR signaling in T-lymphocytes, B-lymphocytes are activated through the antigen-specific B-cell receptor (BCR) (Figure 2B). Genetic variants in the BCR complex and proximal signaling molecules are enriched in SLE patients and may contribute to LN.85 Similar to the TCR, BCR proximal signaling events are controlled by src family protein tyrosine kinases (e.g. Csk, Lyn, and Blk) and protein tyrosine phosphatases (e.g. Lyp, encoded by PTPN22; see “T-lymphocyte activation”). The gain-of-function R620W variant of PTPN22 results in Lyp that does not bind to Csk,67 causing Csk to allow proximal BCR signaling and to mediate inhibitory phosphorylation of Lyn. The inhibitory phosphorylation of Lyn is amplified in SLE patients carrying a genetic variation of CSK that increases Csk expression, thus increasing BCR-mediated activation of mature B cells and higher plasma concentrations of total IgM in the periphery.86 Genetic variants with LYN are also associated with SLE,10,87 and patients carrying the risk allele more frequently carry the lupus-associated autoantibodies that contribute to LN.87
Lyn is a double-edged kinase, acting as both negative and positive regulator of the BCR signaling complex. Phosphorylation of the negative regulators FcγRIIb and CD22 by Lyn inhibits BCR signaling, yet Lyn positively amplifies BCR-mediated B-lymphocyte activation by phosphorylating the CD19/CD21 (CR2) signal transducing complex. Genetic variants in CD19 are associated with LN,88 resulting in decreased expression in naïve B-lymphocytes, but increased expression in memory B-lymphocytes and the presence of lupus-associated autoantibodies.88 The adaptor molecule BANK, encoded by BANK1, binds to Lyn and promotes Lyn-mediated tyrosine phosphorylation of the inositol triphosphate receptor, IP3R, when BANK is bound to it, leading to BCR-induced calcium mobilization. Multiple functional genetic variants of BANK1 are associated with SLE.89,90 The BANK1-61H variant is thought to be protective, while the R61 variant results in protein isoforms that are able to self-multimerize without being phosphorylated by Syk, which may result in enhanced downstream, BCR-mediated calcium mobilization.90 In addition to Lyn, BANK also binds Blk, acting through PLC-γ2 to convert phosphatidylinositol 4,5-bisphosphate into the second messenger inositol 1,4,5-trisphosphate (IP3) to trigger calcium release from the ER and subsequent entry of extracellular calcium. Genetic variants of BLK have been found to associate with LN across multiple ancestral backgrounds,2,26 and two functional BLK promoter variants have been found to modulate transcription of BLK in B-lymphocytes via the use of alternative promoters in immature vs. mature B-lymphocytes.91
Stimulation of B-lymphocytes by co-receptors, such as the TNFR superfamily member CD40, or through TLR activation, synergizes with BCR signaling and leads to B-lymphocyte proliferation, cytokine production, antibody secretion, and isotype switching.92 The P227A variant of CD40 has been shown to enhance transcriptional regulation of pro-inflammatory cytokine secretion and antibody production.93 While a number of CD40 variants affect receptor expression,70 the P227A variant causes the normal CD40-inhibitory adaptor molecule, TRAF3, to become stimulatory and enhance transcriptional signaling pathways.94 CD40 also positively regulates B-lymphocyte activation through the adaptor molecule TRAF6, genetic variants of which are associated with LN.22 CD40 synergizes with TLRs and the BCR to amplify B-lymphocyte activation.93 Both the BCR and the IFN-α receptor95 are able to rescue TLRs from post-activation TLR tolerance on B-lymphocytes, allowing them to further drive immune dysregulation associated with SLE and renal disease in LN. These findings suggest that genetic variants which affect negative regulators of B-lymphocyte activation and plasma cell differentiation allow B-lymphocytes to break tolerance, secrete autoantibodies that contribute to renal disease and exacerbate inflammation that causes kidney damage in LN.
Intrarenal etiology
The development and renal deposition of ICs in LN has been extensively studied, as renal deposits are found in almost all patients with SLE (Figure 3).96 Recent studies indicate that ICs containing anti-nuclear autoantibodies interact with trapped nucleosomes in an antigen-specific manner, particularly in SLE patients with autoantibody specificities to nucleosome components, such as chromatin and dsDNA.1 In addition, a large number of SLE patients carry anti-C1q antibodies, which are able to amplify pathogenic complement activation that results in renal damage when the autoantibodies interact with anti-C1q containing ICs.1 Despite the presence of ICs, many SLE patients never develop clinical kidney disease, although they have histological evidence of intrarenal inflammation. Clinically silent nephritis suggests that the presence and/or deposition of ICs alone is not enough to drive the development of LN and that other pathological factors are involved.97
Figure 3.
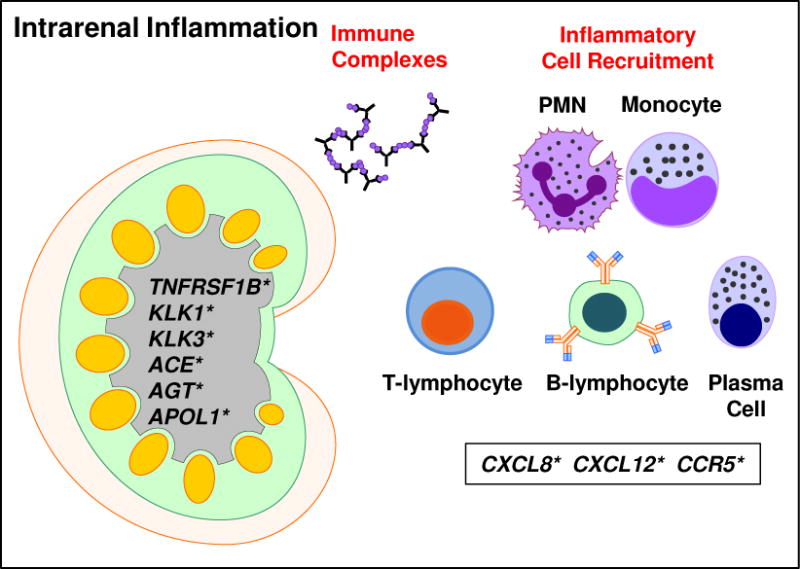
Intrarenal inflammation contributes to lupus nephritis. Autoantibody-containing immune complexes become bound within the glomerulus, leading to inflammation that results in the recruitment of immune cells to the kidney, including PMNs, monocytes, T-lymphocytes, B-lymphocytes, and plasma cells. The immune cells further interact and amplify the immune response (see Figures 1–2). In addition, genetic variants in genes expressed in the kidney (including TNFRSF1B, KLK1, KLK3, ACE, AGT, and APOL1) may result in enhanced susceptibility to kidney damage and lupus nephritis.
*Genes/molecules with genetic variants associated with lupus nephritis
Increasing evidence implicates the TNFR superfamily in the pathogenesis of renal injury in LN by promoting inflammation, apoptosis, and the accumulation of extracellular matrix, which can reduce glomerular blood flow and damage the glomerular permeability barrier.98 Inflammation associated with kidney damage in SLE results includes the production of TNF-α at the site of damage.98 The TNF-α receptor TNFR1 is normally expressed within the glomeruli and by peritubular endothelial cells and is upregulated in response to renal injury in LN,99 primarily associating with the soluble form of TNF-α.100 The second TNF-α receptor, TNFR2, is not expressed in the kidney until inflammation has occurred, and it is the presence of TNFR2 that is essential for the development of glomerulonephritis.101 Polymorphisms of TNFR2 are associated with LN.102 TNFR2 associates with the membrane-bound form of TNF-α,100 promoting the secretion of pro-inflammatory mediators. Perhaps the most promising TNFR superfamily member to contribute to LN is the recently identified ligand, TWEAK. TWEAK interacts with the Fn14 receptor, and expression of genes encoding TWEAK and Fn14 are upregulated in the glomerulus and tubulointerstitium of SLE patients with LN;103 however it is unknown if genetic variants of TNFSF12 (TWEAK) associate with LN.
Histopathological data from kidneys of SLE patients with LN suggest that the inflammation taking place within the kidney is a microcosm of systemic immune dysregulation, with the interaction between parenchymal cells and leukocytes determining the extent of renal damage.1 APC, differentiated Th cells (Th1, Th2, Th3, and Tfh), and B-lymphocytes, as well as ectopic germinal centers, are all present within the LN kidney.1 These cell types must be recruited to the inflamed kidney by chemokines, such as MCP-1/CCL2 (macrophages), 104 IL-8/CXCL8 (T-lymphocytes)105 and CXCR5, which binds BCA-1/CXCL13 (B-lymphocytes),106 all of which are associated with LN.
In addition to enhanced pro-inflammatory processes that lead to LN and kidney damage, renal protective mechanisms that prevent clinical LN in some SLE patients with ICs may be altered, enhancing the risk of developing LN that may be refractory to current treatment regimens and progress to ESRD. As discussed above, immune regulatory mechanisms are altered in SLE patients with LN, including at the genetic level. The chemokine SDF-1/CXCL12 recruits T-regulatory cells and polymorphisms within CXCL12 are associated with LN,107,108 and their effects may be enhanced by the down-regulation of SDF-1 by anti-dsDNA autoantibodies.109 In addition, genetic variants in the Kallikrein genes, KLK1 and KLK3, are associated with immune complex-mediated LN,110 and delivery of the Klk1 gene in mouse models of lupus-like disease downmodulates antibody-mediated glomerulonephritis.111
Genetic variants contributing to systemic co-morbidities may also contribute to the development and severity of LN. For example, hypertension has been associated with LN112 and may be a risk factor for poor outcome.113 Genetic variants within the ACE (encoding angiotensin-converting enzyme)114 and AGT (encoding angiotensinogen)115 genes confer greater susceptibility to the development and severity of LN. In addition, genetic variants in APOL1 have been significantly linked to early development of LN and faster progression to ESRD, particularly in SLE patients of African descent.116 Although the mechanism by which APOL1 facilitates the progression of kidney disease has not been completely elucidated, there is some evidence to suggest that a second hit is required to induce renal disease.117 For SLE patients, immune dysregulation of type I IFN responses and the presence of lupus-associated autoantibodies may provide the second hit required to damage podocytes, leading to the development of LN and progression to ESRD.117
Clinical Implications and Opportunities
Considerable advances have been made in our understanding of the genetic basis of SLE, and novel approaches and studies that explore the relationship between genetics, cellular function, and clinical sequelae are moving forward. Given the renewed interest in personalized, precision medicine, there is a strong push to use what we know about the presence of genetic variants and altered immune pathways to make treatment decisions for SLE patients on an individual basis.118,119 The significant side effects associated with current immunosuppressants and the presence of LN in over half of SLE patients drive home the need for this initiative.
This approach to patient treatment is certainly not without its challenges.118 In addition to cost considerations, there are considerations for the contribution of the identified genetic variations to altered immune pathways leading to LN, potential epistatic interactions of genes, current inability to predict the course of renal disease (which will most likely require genomic, proteomic, and clinical approaches), and the need for improved clinical study designs to permit development of targeted therapy. The good news is that other fields, such as oncology, are beginning to apply pharmacogenomic approaches to patient treatment with some success, using genetic approaches to determine which patients will respond to a selected therapy.120 We also have a treasure trove of recent and upcoming biologic therapies that address dysregulation of pathways that contribute to LN, and growing knowledge of LN-associated genetic risk variants may potentially inform the selection of particular therapies for SLE patients with LN or those who may be at increased genetic risk of developing LN, as outlined in Table 2.121,122
Table 2.
Pathway-specific therapeutic strategies for lupus nephritis
| Pathway | Biologic Therapy | Mechanism of Action | Potential Genes of Interest |
|---|---|---|---|
| Innate Immunity | Sifalimumab Rontazilumab MEDI-546 |
anti-IFNα monclonal antibody (human) anti-IFNα monclonal antibody (humanized) anti-IFNα receptor monclonal antibody (human) |
TYK2, STAT1, STAT4, IKZF1, IRF5, IRF7, IFIH1, CD40 |
| Sirukumab Tocilizumab |
anti-IL-6 monclonal antibody (human) anti-IL-6 receptor monclonal antibody (humanized); blocks IL-6 from binding sIL-6R and mIL-6R |
TNFAIP3, TNIP1, STAT1 | |
| Eculizumab | anti-complement component C5 (humanized); terminal complement inhibitor | TNFAIP3, TNIP1 | |
| SM-101 | Recombinant FcγRIIB (Inhibitory FCγR) agonist | FCGR2B | |
| Adaptive Immunity: T-lymphocytes | Abatacept | Fc:CTLA-4 fusion protein; CD28-B7 blocker | CTLA4 |
| Rigerimod | Synthetic peptide; CD4 modulator | HLA-DR | |
| Adaptive Immunity: B-lymphocytes | Rituximab Ocrelizumab |
Anti-CD20 monoclonal antibody (chimeric mouse human); depletes B-lymphocytes Anti-CD20 monoclonal antibody (humanized); depletes B-lymphocytes |
CD40, TRAF6, PTPN22, BLK, BANK1 |
| Epratuzumab | Anti-CD22 monclonal antibody (humanized); inhibits BCR signaling | PTPN22 | |
| Belumimab Tabalumab Blisibimod |
Anti-sBlyS monoclonal antibody (humanized); Anti-BlyS monoclonal antibody (human); neutralizes sBLyS and mBLyS Peptibody antagonist of BLyS; neutralizes sBLyS and mBLyS | TNFSF13B, TNFRSF13B, TNFAIP3, TNIP1 | |
| Bortezomib | Proteasome inhibitor; depletes plasma cells | TNFAIP3, TNIP1 | |
| Atacicept | Fc-TACI fusion protein; neutralizes BLyS and APRIL | TNFRSF13B, TNFAIP3, TNIP1 | |
| Tissue Injury and Inflammation | Laquinimod | Small molecule; anti-inflammatory | MYD88, TNFAIP3, TNIP1, HLA-DR, CD40, STAT1, IFNG, CCR5 |
| BIIB023 | Anti-TWEAK monoclonal antibody (humanized) | TNFSF12, TNFAIP3, TNIP1 | |
| Fresolimumab | Anti-TGFβ monclonal antibody (human); blocks fibrosis | TGFB1 | |
| Tamibarotene | Small molecule; RAR agonist, anti-inflammatory | MYD88, PTPN22, TNFAIP3, TNIP1, STAT1, IFNG, TGFB1 | |
| GSK-2586184 | Small molecule; JAK inhibitor | MYD88, HLA-DR, STAT1, IFNG, TGFB1 |
For example, utilizing B-lymphocyte depleting therapy with rituximab in refractory LN led to an excellent response in uncontrolled studies, even without the use of cyclophosphamide or steroids. In addition, the mechanism of action was not limited to B-lymphocyte depletion, but also increased the number of T regulatory cells.123,124 In contrast, in the Lupus Nephritis Assessment with Rituximab (LUNAR) trial, rituximab therapy led to increased response rates and greater reductions in anti-dsDNA and C3/C4 levels, yet did not improve clinical outcomes after one year of treatment.125 In addition to other factors, this disparity could be due to genetic differences that influence B-lymphocyte activation or other inflammatory mechanisms involved in LN. Because rituximab primarily acts by depleting CD20-positive B-lymphocytes, LN patients who carry genetic variants within B-lymphocyte activation pathways may benefit the most from rituximab therapy (Table 2). For LN patients (or SLE patients at increased genetic risk of developing LN) who carry genetic variants that influence LN pathogenesis through other mechanisms, rituximab therapy may be less than ideal or may require additional therapeutic modalities to block immune complex-independent mechanisms of LN pathogenesis. For example, patients with genetic risk variants impacting multiple inflammatory pathways and cell types may benefit from a broader spectrum biologic therapy, such as the retinoic acid receptor (RAR) target tamibarotene or the small molecule anti-inflammatory laquinimod.122
Personalized treatment could also benefit from genetic studies for other possible drug targets in LN, including BLyS (encoded by TNFSF13B) and TWEAK (encoded by TNFSF12). The BLyS inhibitor belimumab, the first drug approved for SLE in over 50 years, shows promise for reducing or sustaining disease activity levels, including the renal domain of the BILAG. Patients treated with belimumab showed increased levels of complement components with decreased levels of anti-dsDNA autoantibodies, but studies are needed to specifically evaluate the effects of belimumab treatment on LN.126 Recent studies significantly associated LN with TWEAK, leading to the proof-of-concept Anti-TWEAK in Lupus Nephritis Patient Study (ATLAS), a double-blind controlled trial for patients with Class II and IV LN that is currently underway, with high expectations.127
While many genetic variants have been identified which associate with LN, there is still much work to be done. For example, a number of recent studies have identified proteomic biomarkers which are strongly associated with LN in SLE patients.128 While certain chemokine family members, including MCP-1/CCL2,104 IL-8/CXCL8,105 and CXCR5106 are encoded by genes with known variants associated with LN, others like TWEAK, BLyS, and APRIL have not been explored in detail for the presence of LN associated variants. Given the number of genetic variations in regions that encode proteins which regulate multiple signaling pathways (e.g. IRF/NFκB/MAPK) or regulate multiple receptors (e.g. TCR/BCR and TNFRs), studies addressing epistatic interactions of LN-associated gene variants are warranted. Studies are also needed to determine which epigenetic changes affect expression of associated genes that lead to downstream cellular and clinical sequelae in LN.
On a larger scale, there is a need for opportunities to more adequately address what genetic mechanisms drive LN pathogenesis. Genome wide association studies (GWAS) that not only address SLE cases vs. healthy controls, but SLE patients with and without LN, as well as SLE patients with different classes of LN, would allow us to identify genes that are associated with this specific disease manifestation. Including non-SLE patients with nephritis will allow us to decipher the pathogenesis of autoimmune vs. non-autoimmune mediated renal injury. Alternatively, it is possible that some genes that drive non-autoimmune nephritis (e.g. possibly TNFRSF1B [TNFR2]), may be additionally altered as the result of lupus-associated immune dysregulation and thus contribute to the development of LN. At this time, genetics do not completely explain LN or SLE, and environmental or occupational exposure to a number of infectious and chemical agents may contribute to disease.129 Evaluation of genetic-environmental interacts in the context of LN may provide additional insights into disease pathogenesis.
Building on pharmacogenomic studies in oncology, potential successes using immune pathway-specific biologic therapies in LN, and technological advances in cost-effective genetic screening, it may soon be feasible to sequence patients’ DNA and apply this information to understanding immune system function and selecting ideal treatment. Improved clinical trial designs would also help move this approach forward, bringing the field closer to our goal of decreasing morbidity and mortality in SLE patients with LN.
Acknowledgments
We would like to thank Rebecka Bourn, PhD, and Jennifer Kelly for critical reading of the manuscript.
Financial support and conflict of interest: This work was made possible by funding from the National Institutes of General Medical Sciences (U54GM104938, P30GM103510), Arthritis and Musculoskeletal and Skin Diseases (P30AR053483), and Allergy and Infectious Diseases (U19AI082714, U01AI101934). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borchers AT, Leibushor N, Naguwa SM, Cheema GS, Shoenfeld Y, Gershwin ME. Lupus nephritis: a critical review. Autoimmun Rev. 2012;12(2):174–194. doi: 10.1016/j.autrev.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 2.Richman IB, Taylor KE, Chung SA, et al. European genetic ancestry is associated with a decreased risk of lupus nephritis. Arthritis Rheum. 2012;64(10):3374–3382. doi: 10.1002/art.34567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis. 2013;72(Suppl 2):ii56–61. doi: 10.1136/annrheumdis-2012-202351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fenton K. The effect of cell death in the initiation of lupus nephritis. Clin Exp Immunol. 2015;179(1):11–16. doi: 10.1111/cei.12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6(5):280–289. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 6.Bollain YGJJ, Arellano-Rodriguez M, Torres-Del-Muro Fde J, et al. Soluble fas and the -670 polymorphism of fas in lupus nephritis. International journal of nephrology. 2014;2014:780406. doi: 10.1155/2014/780406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moudi B, Salimi S, Farajian Mashhadi F, Sandoughi M, Zakeri Z. Association of FAS and FAS ligand genes polymorphism and risk of systemic lupus erythematosus. The Scientific World Journal. 2013;2013:176741. doi: 10.1155/2013/176741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez-Valle F, Balada E, Ordi-Ros J, Bujan-Rivas S, Sellas-Fernandez A, Vilardell-Tarres M. DNase1 activity in systemic lupus erythematosus patients with and without nephropathy. Rheumatol Int. 2010;30(12):1601–1604. doi: 10.1007/s00296-009-1199-6. [DOI] [PubMed] [Google Scholar]
- 9.Panneer D, Antony PT, Negi VS. Q222R polymorphism in DNAse I gene is a risk factor for nephritis in South Indian SLE patients. Lupus. 2013;22(10):996–1000. doi: 10.1177/0961203313498801. [DOI] [PubMed] [Google Scholar]
- 10.Harley JB, Alarcon-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature genetics. 2008;40(2):204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou XJ, Nath SK, Qi YY, et al. Brief Report: identification of MTMR3 as a novel susceptibility gene for lupus nephritis in northern Han Chinese by shared-gene analysis with IgA nephropathy. Arthritis & rheumatology. 2014;66(10):2842–2848. doi: 10.1002/art.38749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelley JM, Edberg JC, Kimberly RP. Pathways: Strategies for susceptibility genes in SLE. Autoimmun Rev. 2010;9(7):473–476. doi: 10.1016/j.autrev.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson T, Kariuki SN, Franek BS, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-alpha and serologic autoimmunity in lupus patients. J Immunol. 2011;187(3):1298–1303. doi: 10.4049/jimmunol.1100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imaizumi T, Aizawa-Yashiro T, Tsuruga K, et al. Melanoma differentiation-associated gene 5 regulates the expression of a chemokine CXCL10 in human mesangial cells: implications for chronic inflammatory renal diseases. The Tohoku journal of experimental medicine. 2012;228(1):17–26. doi: 10.1620/tjem.228.17. [DOI] [PubMed] [Google Scholar]
- 15.Enevold C, Kjaer L, Nielsen CH, et al. Genetic polymorphisms of dsRNA ligating pattern recognition receptors TLR3, MDA5, and RIG-I. Association with systemic lupus erythematosus and clinical phenotypes. Rheumatol Int. 2014 doi: 10.1007/s00296-014-3012-4. [DOI] [PubMed] [Google Scholar]
- 16.Pothlichet J, Niewold TB, Vitour D, Solhonne B, Crow MK, Si-Tahar M. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO molecular medicine. 2011;3(3):142–152. doi: 10.1002/emmm.201000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Namjou B, Kothari PH, Kelly JA, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011;12(4):270–279. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imaizumi T, Aizawa T, Segawa C, et al. Toll-like receptor 3 signaling contributes to the expression of a neutrophil chemoattractant, CXCL1 in human mesangial cells. Clinical and experimental nephrology. 2014 doi: 10.1007/s10157-014-1060-4. [DOI] [PubMed] [Google Scholar]
- 19.Tian J, Ma Y, Li J, et al. The TLR7 7926A>G polymorphism is associated with susceptibility to systemic lupus erythematosus. Molecular medicine reports. 2012;6(1):105–110. doi: 10.3892/mmr.2012.865. [DOI] [PubMed] [Google Scholar]
- 20.Ramachandran R, Sharma V, Rathi M, et al. Association between -1486 T>C and +1174 G>A single nucleotide polymorphisms in TLR9 gene and severity of lupus nephritis. Indian journal of nephrology. 2012;22(2):125–129. doi: 10.4103/0971-4065.97133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38(3):528–540. doi: 10.1016/j.immuni.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Namjou B, Choi CB, Harley IT, et al. Evaluation of TRAF6 in a large multiancestral lupus cohort. Arthritis Rheum. 2012;64(6):1960–1969. doi: 10.1002/art.34361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacob CO, Zhu J, Armstrong DL, et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2009;106(15):6256–6261. doi: 10.1073/pnas.0901181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufman KM, Zhao J, Kelly JA, et al. Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Ann Rheum Dis. 2013;72(3):437–444. doi: 10.1136/annrheumdis-2012-201851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P, Cao C, Luan H, et al. Association of genetic variations in the STAT4 and IRF7/KIAA1542 regions with systemic lupus erythematosus in a Northern Han Chinese population. Hum Immunol. 2011;72(3):249–255. doi: 10.1016/j.humimm.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Bolin K, Sandling JK, Zickert A, et al. Association of STAT4 polymorphism with severe renal insufficiency in lupus nephritis. PLoS One. 2013;8(12):e84450. doi: 10.1371/journal.pone.0084450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salloum R, Franek BS, Kariuki SN, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum. 2010;62(2):553–561. doi: 10.1002/art.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58(8):2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bates JS, Lessard CJ, Leon JM, et al. Meta-analysis and imputation identifies a 109 kb risk haplotype spanning TNFAIP3 associated with lupus nephritis and hematologic manifestations. Genes Immun. 2009;10(5):470–477. doi: 10.1038/gene.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tavares RM, Turer EE, Liu CL, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33(2):181–191. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verstrepen L, Carpentier I, Beyaert R. The biology of A20-binding inhibitors of NF-kappaB activation (ABINs) Adv Exp Med Biol. 2014;809:13–31. doi: 10.1007/978-1-4939-0398-6_2. [DOI] [PubMed] [Google Scholar]
- 32.Caster DJ, Korte EA, Nanda SK, et al. ABIN1 dysfunction as a genetic basis for lupus nephritis. Journal of the American Society of Nephrology: JASN. 2013;24(11):1743–1754. doi: 10.1681/ASN.2013020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou XJ, Cheng FJ, Zhu L, et al. Association of systemic lupus erythematosus susceptibility genes with IgA nephropathy in a Chinese cohort. Clinical journal of the American Society of Nephrology: CJASN. 2014;9(4):788–797. doi: 10.2215/CJN.01860213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis MJ, Vyse S, Shields AM, et al. UBE2L3 polymorphism amplifies NF-kappaB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. American journal of human genetics. 2015;96(2):221–234. doi: 10.1016/j.ajhg.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chung SA, Taylor KE, Graham RR, et al. Differential genetic associations for systemic lupus erythematosus based on anti-dsDNA autoantibody production. PLoS genetics. 2011;7(3):e1001323. doi: 10.1371/journal.pgen.1001323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jonsen A, Gunnarsson I, Gullstrand B, et al. Association between SLE nephritis and polymorphic variants of the CRP and FcgammaRIIIa genes. Rheumatology (Oxford) 2007;46(9):1417–1421. doi: 10.1093/rheumatology/kem167. [DOI] [PubMed] [Google Scholar]
- 37.Floto RA, Clatworthy MR, Heilbronn KR, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med. 2005;11(10):1056–1058. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 38.Karassa FB, Trikalinos TA, Ioannidis JP, Fcgamma R-SLEM-AI Role of the Fcgamma receptor IIa polymorphism in susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Arthritis Rheum. 2002;46(6):1563–1571. doi: 10.1002/art.10306. [DOI] [PubMed] [Google Scholar]
- 39.Karassa FB, Trikalinos TA, Ioannidis JP, Fc gamma R-SLEm-ai The Fc gamma RIIIA-F158 allele is a risk factor for the development of lupus nephritis: a meta-analysis. Kidney Int. 2003;63(4):1475–1482. doi: 10.1046/j.1523-1755.2003.00873.x. [DOI] [PubMed] [Google Scholar]
- 40.Gelmetti AP, Freitas AC, Woronik V, Barros RT, Bonfa E, Monteiro RC. Polymorphism of the FcgammaRIIalpha IgG receptor in patients with lupus nephritis and glomerulopathy. J Rheumatol. 2006;33(3):523–530. [PubMed] [Google Scholar]
- 41.Zuniga R, Markowitz GS, Arkachaisri T, Imperatore EA, D’Agati VD, Salmon JE. Identification of IgG subclasses and C-reactive protein in lupus nephritis: the relationship between the composition of immune deposits and FCgamma receptor type IIA alleles. Arthritis Rheum. 2003;48(2):460–470. doi: 10.1002/art.10930. [DOI] [PubMed] [Google Scholar]
- 42.Salmon JE, Millard S, Schachter LA, et al. Fc gamma RIIA alleles are heritable risk factors for lupus nephritis in African Americans. J Clin Invest. 1996;97(5):1348–1354. doi: 10.1172/JCI118552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haseley LA, Wisnieski JJ, Denburg MR, et al. Antibodies to C1q in systemic lupus erythematosus: characteristics and relation to Fc gamma RIIA alleles. Kidney Int. 1997;52(5):1375–1380. doi: 10.1038/ki.1997.464. [DOI] [PubMed] [Google Scholar]
- 44.Wu J, Edberg JC, Redecha PB, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100(5):1059–1070. doi: 10.1172/JCI119616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alarcon GS, McGwin G, Jr, Petri M, et al. Time to renal disease and end-stage renal disease in PROFILE: a multiethnic lupus cohort. PLoS Med. 2006;3(10):e396. doi: 10.1371/journal.pmed.0030396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salmon JE, Edberg JC, Kimberly RP. Fc gamma receptor III on human neutrophils. Allelic variants have functionally distinct capacities. J Clin Invest. 1990;85(4):1287–1295. doi: 10.1172/JCI114566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee YH, Bae SC. Association between the functional ITGAM rs1143679 G/A polymorphism and systemic lupus erythematosus/lupus nephritis or rheumatoid arthritis: an update meta-analysis. Rheumatol Int. 2014 doi: 10.1007/s00296-014-3156-2. [DOI] [PubMed] [Google Scholar]
- 48.Kim-Howard X, Maiti AK, Anaya JM, et al. ITGAM coding variant (rs1143679) influences the risk of renal disease, discoid rash and immunological manifestations in patients with systemic lupus erythematosus with European ancestry. Ann Rheum Dis. 2010;69(7):1329–1332. doi: 10.1136/ard.2009.120543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439(7078):851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 50.Nossent JC, Becker-Merok A, Rischmueller M, Lester S. Susceptibility for Lupus Nephritis by Low Copy Number of the FCGR3B Gene Is Linked to Increased Levels of Pathogenic Autoantibodies. Autoimmune diseases. 2013;2013:750814. doi: 10.1155/2013/750814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis. 2014;73(9):1601–1606. doi: 10.1136/annrheumdis-2014-205287. [DOI] [PubMed] [Google Scholar]
- 52.Villarreal J, Crosdale D, Ollier W, et al. Mannose binding lectin and FcgammaRIIa (CD32) polymorphism in Spanish systemic lupus erythematosus patients. Rheumatology (Oxford) 2001;40(9):1009–1012. doi: 10.1093/rheumatology/40.9.1009. [DOI] [PubMed] [Google Scholar]
- 53.Seelen MA, van der Bijl EA, Trouw LA, et al. A role for mannose-binding lectin dysfunction in generation of autoantibodies in systemic lupus erythematosus. Rheumatology (Oxford) 2005;44(1):111–119. doi: 10.1093/rheumatology/keh417. [DOI] [PubMed] [Google Scholar]
- 54.MacPherson M, Lek HS, Prescott A, Fagerholm SC. A systemic lupus erythematosus-associated R77H substitution in the CD11b chain of the Mac-1 integrin compromises leukocyte adhesion and phagocytosis. J Biol Chem. 2011;286(19):17303–17310. doi: 10.1074/jbc.M110.182998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanchez E, Nadig A, Richardson BC, et al. Phenotypic associations of genetic susceptibility loci in systemic lupus erythematosus. Ann Rheum Dis. 2011;70(10):1752–1757. doi: 10.1136/ard.2011.154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rhodes B, Furnrohr BG, Roberts AL, et al. The rs1143679 (R77H) lupus associated variant of ITGAM (CD11b) impairs complement receptor 3 mediated functions in human monocytes. Ann Rheum Dis. 2012;71(12):2028–2034. doi: 10.1136/annrheumdis-2012-201390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verma J, Arora V, Marwaha V, Kumar A, Das N. Association of leukocyte CR1 gene transcription with the disease severity and renal involvement in systemic lupus erythematosus. Lupus. 2005;14(4):273–279. doi: 10.1191/0961203305lu2074oa. [DOI] [PubMed] [Google Scholar]
- 58.Ivashkiv LB. A signal-switch hypothesis for cross-regulation of cytokine and TLR signalling pathways. Nat Rev Immunol. 2008;8(10):816–822. doi: 10.1038/nri2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res. 2011;17(9):2619–2627. doi: 10.1158/1078-0432.CCR-10-1114. [DOI] [PubMed] [Google Scholar]
- 60.Sigurdsson S, Nordmark G, Goring HH, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. American journal of human genetics. 2005;76(3):528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He CF, Liu YS, Cheng YL, et al. TNIP1, SLC15A4, ETS1, RasGRP3 and IKZF1 are associated with clinical features of systemic lupus erythematosus in a Chinese Han population. Lupus. 2010;19(10):1181–1186. doi: 10.1177/0961203310367918. [DOI] [PubMed] [Google Scholar]
- 62.Couser WG. Basic and translational concepts of immune-mediated glomerular diseases. Journal of the American Society of Nephrology: JASN. 2012;23(3):381–399. doi: 10.1681/ASN.2011030304. [DOI] [PubMed] [Google Scholar]
- 63.Chung SA, Brown EE, Williams AH, et al. Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. Journal of the American Society of Nephrology: JASN. 2014;25(12):2859–2870. doi: 10.1681/ASN.2013050446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niu Z, Zhang P, Tong Y. Value of HLA-DR genotype in systemic lupus erythematosus and lupus nephritis: a meta-analysis. International journal of rheumatic diseases. 2014 doi: 10.1111/1756-185X.12528. [DOI] [PubMed] [Google Scholar]
- 65.Masutani K, Akahoshi M, Tsuruya K, et al. Predominance of Th1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheum. 2001;44(9):2097–2106. doi: 10.1002/1529-0131(200109)44:9<2097::AID-ART360>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 66.Vang T, Landskron J, Viken MK, et al. The autoimmune-predisposing variant of lymphoid tyrosine phosphatase favors T helper 1 responses. Hum Immunol. 2013;74(5):574–585. doi: 10.1016/j.humimm.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vang T, Congia M, Macis MD, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nature genetics. 2005;37(12):1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 68.Prokunina L, Gunnarsson I, Sturfelt G, et al. The systemic lupus erythematosus-associated PDCD1 polymorphism PD1.3A in lupus nephritis. Arthritis Rheum. 2004;50(1):327–328. doi: 10.1002/art.11442. [DOI] [PubMed] [Google Scholar]
- 69.Bertsias GK, Nakou M, Choulaki C, et al. Genetic, immunologic, and immunohistochemical analysis of the programmed death 1/programmed death ligand 1 pathway in human systemic lupus erythematosus. Arthritis Rheum. 2009;60(1):207–218. doi: 10.1002/art.24227. [DOI] [PubMed] [Google Scholar]
- 70.Joo YB, Park BL, Shin HD, Park SY, Kim I, Bae SC. Association of genetic polymorphisms in CD40 with susceptibility to SLE in the Korean population. Rheumatology (Oxford) 2013;52(4):623–630. doi: 10.1093/rheumatology/kes339. [DOI] [PubMed] [Google Scholar]
- 71.Citores MJ, Rua-Figueroa I, Rodriguez-Gallego C, et al. The dinucleotide repeat polymorphism in the 3′UTR of the CD154 gene has a functional role on protein expression and is associated with systemic lupus erythematosus. Ann Rheum Dis. 2004;63(3):310–317. doi: 10.1136/ard.2003.006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou XJ, Cheng FJ, Qi YY, Zhao MH, Zhang H. A replication study from Chinese supports association between lupus-risk allele in TNFSF4 and renal disorder. BioMed research international. 2013;2013:597921. doi: 10.1155/2013/597921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patschan S, Dolff S, Kribben A, et al. CD134 expression on CD4+ T cells is associated with nephritis and disease activity in patients with systemic lupus erythematosus. Clin Exp Immunol. 2006;145(2):235–242. doi: 10.1111/j.1365-2249.2006.03141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yap DY, Lai KN. Pathogenesis of Renal Disease in Systemic Lupus Erythematosus-The Role of Autoantibodies and Lymphocytes Subset Abnormalities. International journal of molecular sciences. 2015;16(4):7917–7931. doi: 10.3390/ijms16047917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liarski VM, Kaverina N, Chang A, et al. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Science translational medicine. 2014;6(230):230ra246. doi: 10.1126/scitranslmed.3008146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee SK, Silva DG, Martin JL, et al. Interferon-gamma excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity. 2012;37(5):880–892. doi: 10.1016/j.immuni.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 77.Miyake K, Nakashima H, Akahoshi M, et al. Genetically determined interferon-gamma production influences the histological phenotype of lupus nephritis. Rheumatology (Oxford) 2002;41(5):518–524. doi: 10.1093/rheumatology/41.5.518. [DOI] [PubMed] [Google Scholar]
- 78.Namjou B, Sestak AL, Armstrong DL, et al. High-density genotyping of STAT4 reveals multiple haplotypic associations with systemic lupus erythematosus in different racial groups. Arthritis Rheum. 2009;60(4):1085–1095. doi: 10.1002/art.24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lessard CJ, Adrianto I, Ice JA, et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. American journal of human genetics. 2012;90(4):648–660. doi: 10.1016/j.ajhg.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tucci M, Stucci S, Strippoli S, Silvestris F. Cytokine overproduction, T-cell activation, and defective T-regulatory functions promote nephritis in systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010:457146. doi: 10.1155/2010/457146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Susianti H, Handono K, Purnomo BB, Widodo N, Gunawan A, Kalim H. Changes to signal peptide and the level of transforming growth factor- beta1 due to T869C polymorphism of TGF beta1 associated with lupus renal fibrosis. SpringerPlus. 2014;3:514. doi: 10.1186/2193-1801-3-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu LJ, Liu ZH, Zeng CH, Chen ZH, Yu C, Li LS. Association of interleukin-10 gene-592 A/C polymorphism with the clinical and pathological diversity of lupus nephritis. Clin Exp Rheumatol. 2005;23(6):854–860. [PubMed] [Google Scholar]
- 83.Hermann J, Gruber S, Neufeld JB, et al. IL10R1 loss-of-function alleles in rheumatoid arthritis and systemic lupus erythematosus. Clin Exp Rheumatol. 2009;27(4):603–608. [PubMed] [Google Scholar]
- 84.Sung YK, Park BL, Shin HD, Kim LH, Kim SY, Bae SC. Interleukin-10 gene polymorphisms are associated with the SLICC/ACR Damage Index in systemic lupus erythematosus. Rheumatology (Oxford) 2006;45(4):400–404. doi: 10.1093/rheumatology/kei184. [DOI] [PubMed] [Google Scholar]
- 85.Packard TA, Cambier JC. B lymphocyte antigen receptor signaling: initiation, amplification, and regulation. F1000prime reports. 2013;5:40. doi: 10.12703/P5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Manjarrez-Orduno N, Marasco E, Chung SA, et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nature genetics. 2012;44(11):1227–1230. doi: 10.1038/ng.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu R, Vidal GS, Kelly JA, et al. Genetic associations of LYN with systemic lupus erythematosus. Genes Immun. 2009;10(5):397–403. doi: 10.1038/gene.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Culton DA, Nicholas MW, Bunch DO, et al. Similar CD19 dysregulation in two autoantibody-associated autoimmune diseases suggests a shared mechanism of B-cell tolerance loss. J Clin Immunol. 2007;27(1):53–68. doi: 10.1007/s10875-006-9051-1. [DOI] [PubMed] [Google Scholar]
- 89.Guo L, Deshmukh H, Lu R, et al. Replication of the BANK1 genetic association with systemic lupus erythematosus in a European-derived population. Genes Immun. 2009;10(5):531–538. doi: 10.1038/gene.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kozyrev SV, Bernal-Quiros M, Alarcon-Riquelme ME, Castillejo-Lopez C. The dual effect of the lupus-associated polymorphism rs10516487 on BANK1 gene expression and protein localization. Genes Immun. 2012;13(2):129–138. doi: 10.1038/gene.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guthridge JM, Lu R, Sun H, et al. Two functional lupus-associated BLK promoter variants control cell-type- and developmental-stage-specific transcription. American journal of human genetics. 2014;94(4):586–598. doi: 10.1016/j.ajhg.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Buchta CM, Bishop GA. Toll-like receptors and B cells: functions and mechanisms. Immunol Res. 2014;59(1–3):12–22. doi: 10.1007/s12026-014-8523-2. [DOI] [PubMed] [Google Scholar]
- 93.Peters AL, Plenge RM, Graham RR, et al. A novel polymorphism of the human CD40 receptor with enhanced function. Blood. 2008;112(5):1863–1871. doi: 10.1182/blood-2008-02-138925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peters AL, Bishop GA. Differential TRAF3 Utilization by a Variant Human CD40 Receptor with Enhanced Signaling. J Immunol. 2010 doi: 10.4049/jimmunol.1000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Poovassery JS, Bishop GA. Type I IFN receptor and the B cell antigen receptor regulate TLR7 responses via distinct molecular mechanisms. J Immunol. 2012;189(4):1757–1764. doi: 10.4049/jimmunol.1200624. [DOI] [PubMed] [Google Scholar]
- 96.Cervera R, Khamashta MA, Font J, et al. Morbidity and mortality in systemic lupus erythematosus during a 10-year period: a comparison of early and late manifestations in a cohort of 1,000 patients. Medicine. 2003;82(5):299–308. doi: 10.1097/01.md.0000091181.93122.55. [DOI] [PubMed] [Google Scholar]
- 97.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279(5353):1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 98.Sanchez-Nino MD, Benito-Martin A, Goncalves S, et al. TNF superfamily: a growing saga of kidney injury modulators. Mediators of inflammation. 2010;2010 doi: 10.1155/2010/182958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aten J, Roos A, Claessen N, Schilder-Tol EJ, Ten Berge IJ, Weening JJ. Strong and selective glomerular localization of CD134 ligand and TNF receptor-1 in proliferative lupus nephritis. Journal of the American Society of Nephrology: JASN. 2000;11(8):1426–1438. doi: 10.1681/ASN.V1181426. [DOI] [PubMed] [Google Scholar]
- 100.Richter C, Messerschmidt S, Holeiter G, et al. The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mol Cell Biol. 2012;32(13):2515–2529. doi: 10.1128/MCB.06458-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vielhauer V, Stavrakis G, Mayadas TN. Renal cell-expressed TNF receptor 2, not receptor 1, is essential for the development of glomerulonephritis. J Clin Invest. 2005;115(5):1199–1209. doi: 10.1172/JCI23348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tsuchiya N, Komata T, Matsushita M, Ohashi J, Tokunaga K. New single nucleotide polymorphisms in the coding region of human TNFR2: association with systemic lupus erythematosus. Genes Immun. 2000;1(8):501–503. doi: 10.1038/sj.gene.6363700. [DOI] [PubMed] [Google Scholar]
- 103.Lu J, Kwan BC, Lai FM, et al. Gene expression of TWEAK/Fn14 and IP-10/CXCR3 in glomerulus and tubulointerstitium of patients with lupus nephritis. Nephrology (Carlton) 2011;16(4):426–432. doi: 10.1111/j.1440-1797.2011.01449.x. [DOI] [PubMed] [Google Scholar]
- 104.Tucci M, Barnes EV, Sobel ES, et al. Strong association of a functional polymorphism in the monocyte chemoattractant protein 1 promoter gene with lupus nephritis. Arthritis Rheum. 2004;50(6):1842–1849. doi: 10.1002/art.20266. [DOI] [PubMed] [Google Scholar]
- 105.Rovin BH, Lu L, Zhang X. A novel interleukin-8 polymorphism is associated with severe systemic lupus erythematosus nephritis. Kidney Int. 2002;62(1):261–265. doi: 10.1046/j.1523-1755.2002.00438.x. [DOI] [PubMed] [Google Scholar]
- 106.Zhang J, Zhang Y, Yang J, et al. Three SNPs in chromosome 11q23.3 are independently associated with systemic lupus erythematosus in Asians. Human molecular genetics. 2014;23(2):524–533. doi: 10.1093/hmg/ddt424. [DOI] [PubMed] [Google Scholar]
- 107.Warchol T, Lianeri M, Lacki JK, Jagodzinski PP. SDF1-3′ G801A polymorphisms in Polish patients with systemic lupus erythematosus. Mol Biol Rep. 2010;37(7):3121–3125. doi: 10.1007/s11033-009-9890-y. [DOI] [PubMed] [Google Scholar]
- 108.Wu FX, Luo XY, Wu LJ, et al. Association of chemokine CXCL12–3′G801A polymorphism with systemic lupus erythematosus in a Han Chinese population. Lupus. 2012;21(6):604–610. doi: 10.1177/0961203311435266. [DOI] [PubMed] [Google Scholar]
- 109.Sela U, Hershkoviz R, Cahalon L, Lider O, Mozes E. Down-regulation of stromal cell-derived factor-1alpha-induced T cell chemotaxis by a peptide based on the complementarity-determining region 1 of an anti-DNA autoantibody via up-regulation of TGF-beta secretion. J Immunol. 2005;174(1):302–309. doi: 10.4049/jimmunol.174.1.302. [DOI] [PubMed] [Google Scholar]
- 110.Liu K, Li QZ, Delgado-Vega AM, et al. Kallikrein genes are associated with lupus and glomerular basement membrane-specific antibody-induced nephritis in mice and humans. J Clin Invest. 2009;119(4):911–923. doi: 10.1172/JCI36728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li QZ, Zhou J, Yang R, et al. The lupus-susceptibility gene kallikrein downmodulates antibody-mediated glomerulonephritis. Genes Immun. 2009;10(5):503–508. doi: 10.1038/gene.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pogorevici A, Gluhovschi C, Gluhovschi G, Velciov SM, Trandafirescu V, Secrii RV. Hypertension in lupus nephritis. Romanian journal of internal medicine = Revue roumaine de medecine interne. 2006;44(3):295–316. [PubMed] [Google Scholar]
- 113.Kammoun K, Jarraya F, Bouhamed L, et al. Poor prognostic factors of lupus nephritis. Saudi J Kidney Dis Transpl. 2011;22(4):727–732. [PubMed] [Google Scholar]
- 114.Sprovieri SR, Sens YA, Martini Filho D. Association between polymorphisms of the renin-angiotensin system and more severe histological forms of lupus nephritis. Clinical nephrology. 2005;64(1):20–27. doi: 10.5414/cnp64020. [DOI] [PubMed] [Google Scholar]
- 115.Parsa A, Lovett DH, Peden EA, Zhu L, Seldin MF, Criswell LA. Renin-angiotensin system gene polymorphisms predict the progression to renal insufficiency among Asians with lupus nephritis. Genes Immun. 2005;6(3):217–224. doi: 10.1038/sj.gene.6364179. [DOI] [PubMed] [Google Scholar]
- 116.Lin CP, Adrianto I, Lessard CJ, et al. Role of MYH9 and APOL1 in African and non-African populations with lupus nephritis. Genes Immun. 2012;13(3):232–238. doi: 10.1038/gene.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kruzel-Davila E, Wasser WG, Aviram S, Skorecki K. APOL1 nephropathy: from gene to mechanisms of kidney injury. Nephrol Dial Transplant. 2015 doi: 10.1093/ndt/gfu391. [DOI] [PubMed] [Google Scholar]
- 118.Rhodes B, Vyse TJ. Using genetics to deliver personalized SLE therapy-a realistic prospect? Nat Rev Rheumatol. 2010;6(6):373–377. doi: 10.1038/nrrheum.2010.67. [DOI] [PubMed] [Google Scholar]
- 119.Rovin BH, McKinley AM, Birmingham DJ. Can we personalize treatment for kidney diseases? Clinical journal of the American Society of Nephrology: CJASN. 2009;4(10):1670–1676. doi: 10.2215/CJN.04140609. [DOI] [PubMed] [Google Scholar]
- 120.Filipski KK, Mechanic LE, Long R, Freedman AN. Pharmacogenomics in oncology care. Frontiers in genetics. 2014;5:73. doi: 10.3389/fgene.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Koutsokeras T, Healy T. Systemic lupus erythematosus and lupus nephritis. Nat Rev Drug Discov. 2014;13(3):173–174. doi: 10.1038/nrd4227. [DOI] [PubMed] [Google Scholar]
- 122.Rovin BH, Parikh SV. Lupus nephritis: the evolving role of novel therapeutics. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2014;63(4):677–690. doi: 10.1053/j.ajkd.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Diaz-Lagares C, Croca S, Sangle S, et al. Efficacy of rituximab in 164 patients with biopsy-proven lupus nephritis: pooled data from European cohorts. Autoimmun Rev. 2012;11(5):357–364. doi: 10.1016/j.autrev.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 124.Vigna-Perez M, Hernandez-Castro B, Paredes-Saharopulos O, et al. Clinical and immunological effects of Rituximab in patients with lupus nephritis refractory to conventional therapy: a pilot study. Arthritis Res Ther. 2006;8(3):R83. doi: 10.1186/ar1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64(4):1215–1226. doi: 10.1002/art.34359. [DOI] [PubMed] [Google Scholar]
- 126.Manzi S, Sanchez-Guerrero J, Merrill JT, et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: combined results from two phase III trials. Ann Rheum Dis. 2012;71(11):1833–1838. doi: 10.1136/annrheumdis-2011-200831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Michaelson JS, Wisniacki N, Burkly LC, Putterman C. Role of TWEAK in lupus nephritis: a bench-to-bedside review. J Autoimmun. 2012;39(3):130–142. doi: 10.1016/j.jaut.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li Y, Fang X, Li QZ. Biomarker profiling for lupus nephritis. Genomics, proteomics & bioinformatics. 2013;11(3):158–165. doi: 10.1016/j.gpb.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sarzi-Puttini P, Atzeni F, Iaccarino L, Doria A. Environment and systemic lupus erythematosus: an overview. Autoimmunity. 2005;38(7):465–472. doi: 10.1080/08916930500285394. [DOI] [PubMed] [Google Scholar]