

Significance
The previously unidentified virus-mimetic nanovesicles (VMVs) described in this manuscript consist of phospholipid derived from mammalian cell plasma membrane, recombinant protein anchored to cell membrane via the route of signal peptide sorting, and surfactants capable of controlling the VMV size and strength, which allows the VMVs to display functional polypeptides or maintain the correct conformation of protein antigen. The protein integrated into VMV by its hydrophobic transmembrane peptide has more modifications, such as glycosylation, than proteins in conventional subunit vaccines. Moreover, many viral envelope glycoproteins can be genetically engineered onto VMV liposomal surface so as to mimic the properties and conformational epitopes of natural virus. VMV provides an effective, straightforward, and tunable approach against a wide range of emerging enveloped viruses.
Keywords: virus-mimetic vesicle, vaccine, nanobiotechnology, antigen delivery system, cell membrane
Abstract
It is a critically important challenge to rapidly design effective vaccines to reduce the morbidity and mortality of unexpected pandemics. Inspired from the way that most enveloped viruses hijack a host cell membrane and subsequently release by a budding process that requires cell membrane scission, we genetically engineered viral antigen to harbor into cell membrane, then form uniform spherical virus-mimetic nanovesicles (VMVs) that resemble natural virus in size, shape, and specific immunogenicity with the help of surfactants. Incubation of major cell membrane vesicles with surfactants generates a large amount of nano-sized uniform VMVs displaying the native conformational epitopes. With the diverse display of epitopes and viral envelope glycoproteins that can be functionally anchored onto VMVs, we demonstrate VMVs to be straightforward, robust and tunable nanobiotechnology platforms for fabricating antigen delivery systems against a wide range of enveloped viruses.
Enveloped viruses, such as avian influenza virus, Ebola, and SARS virus, pose a great threat to public health, and new generations of vaccines are needed to combat these, as well as future, viruses (1). However, it is very difficult to rapidly design new, safe, effective vaccines to reduce the morbidity and mortality of potential or ongoing pandemics (2, 3). Conventional vaccines produced by live or killed organisms have the potential disadvantages of safety; their packed viral nucleic acids may have risk of replication (4). Thus, more efforts have been made to develop safer recombinant subunit vaccines based on known protective antigens. However, there are at least two major challenges in developing subunit vaccines against enveloped virus: (i) the native conformational epitope of viral envelope protein is difficult to produce because the glycoproteins of enveloped viruses are usually subjected to posttranslational protein modifications in eukaryotic cells; (ii) because viral envelope glycoproteins often need to be incorporated in lipid membrane environment to maintain correct conformation of multimer, recombinant purified subunit antigens often elicit anti-incorrect conformation antibody and have poor immunogenicity mostly due to their great differences from “native” conformations on viral envelope. The key to success of effective vaccine against enveloped virus depends on complete simulation of natural virions. Therefore, studies about display of viral envelope glycoprotein have gradually become a hot spot in the fields of enveloped virus vaccine (5, 6).
New advances (6–12) in nanotechnology and biomaterial sciences have created the possibility of improving vaccine products. Biological nanovaccines from various natural sources, such as virus-like particles (10), bacterially-derived membrane vesicles (6) and mammalian cell-derived exosomes (11), have emerged and attracted much attention in developing vaccine. With inspiration from the way that most enveloped viruses hijack a host cell membrane and subsequently release from the infected cells by a budding process that requires cell membrane scission, we reasoned that we could genetically engineer viral antigen to harbor into cell membrane, followed by the formation of uniform spherical virus-mimetic nanovesicles (VMVs) that resemble natural viruses in size, shape, and specific immunogenicity (Fig. 1) with the help of surfactants. Incubation of major cell membrane vesicles with surfactants would help generate a large amount of nano-sized uniform VMVs displaying the native conformational epitopes.
Fig. 1.

Schematic showing the preparation process of virus-mimetic nanovesicles (VMV) for anchoring anti-tumor epitopes or enveloped-virus glycoprotein to VMV surface. (A) Viral antigen genes were subcloned into lentivirus PLV vector and then transfected into eukaryotic cells to express the viral antigen. (B) HEK 293T cells stably expressing viral antigen on cell membrane were acquired after selection of puromycin. (C) The antigenic proteins were located in endoplasmic reticulum by the recognition of signal peptide. (D) The sorting signal peptide guided antigenic proteins to plasma membrane or Golgi complex with the help of transport vesicles. (E) Transport vesicles carrying antigenic proteins fused with cell plasma membrane, anchoring the viral antigen to cellular surface. (F) HEK 293T cells expressing viral antigen were incubated with sodium deoxycholate to generate major cell membrane vesicles (MCV, 500–2,500 nm). (G) The purified MCV, treated by ultrasonication, were then mixed with 0.045% sodium deoxycholate and 0.05% triton-X100 to produce uniform VMV (50–150 nm). (H) The vaccinated mice were challenged intranasally with 50 times lethal doses of mouse-adapted influenza viruses to verify the in vivo immune protection of VMV-HA.
Herein, we describe a new type of VMVs (Fig. 1) consisting of a complex of phospholipids derived from mammalian cell plasma membrane, recombinant protein anchored to cell membrane via the route of signal peptide sorting (13), and surfactants capable of controlling VMV size and strength (14–16), thereby allowing the VMVs to display functional polypeptides (e.g., anti-cancer epitopes) or correctly conformational protein antigen (e.g., influenza hemagglutinin proteins). These proteins, integrated into VMVs by their hydrophobic-rich transmembrane peptide, can have more posttranslation modifications, such as glycosylation, than ones in conventional subunit vaccines expressed by the Escherichia coli expression system. Moreover, many viral envelope glycoproteins can be genetically engineered onto VMV liposomal surfaces so as to mimic the properties and conformational epitopes of natural viruses. Importantly, VMVs are generated without the need of propagating potentially dangerous pathogens in cells or egg culture, and they allow for additional modifications that, due to unique structure and properties, enhance vaccine immunogenicity. Therefore, VMVs provide an effective, straightforward, and tunable approach to combating a wide range of emerging enveloped viruses.
Results and Discussion
VMVs Expressing HPV L2 Epitope Peptide on the Exterior of the Vesicles (VMV-16L2).
As a proof of principle, we established both HEK 293T and HeLa cell lines that stably express an epitope of L2 protein of human papilloma virus 16 (HPV16) on the surface of cellular membrane. To image and guide the epitope into cell plasma membrane, a signal peptide sequence (20 amino acids) from membrane-target integrin protein (17), an epitope sequence (24 amino acids) from HPV16 L2 protein (18), and a linker of transmembrane peptide sequence (17) (22 amino acids) (Table S1) were genetically fused to the N-terminal of enhanced green fluorescent protein (eGFP), forming sig-16L2-eGFP recombinant protein (Fig. 2A and Fig. S1). Based on an understanding of membrane protein transport mechanism via the route of signal peptide sorting (19), we were able to target the location of cargo protein from cytoplasm to cell plasma membrane. As shown in Fig. 2 B–F, abundant sig-16L2-eGFPs were embedded in both nuclear membrane and plasma membrane of HEK 293T and HeLa cells expressing sig-16L2-eGFP, whereas a 16L2-eGFP group without signal peptide only existed in the cytoplasm, as demonstrated by the green fluorescence of eGFP.
Table S1.
The sequence information of sig-16L2-eGFP proteins
| Peptides | Sequences |
| Signal peptide | MNLQPIFWIG LISSVCCVFA |
| HPV16 L2 epitope | TQLYKTCKQAGTCPPDIIPKVGP |
| Transmembrane peptide | LWVILLSAFAGLLLLMLLILAL |
Fig. 2.

The identification of eukaryotic cell displaying Sig-L2-eGFP. (A) Design of membrane-targeted Sig-L2-eGFP in eukaryotic cells and schematic representation of VMV displaying Sig-L2-eGFP. (B–F) The location of Sig-16L2-eGFP in HeLa and HEK 293T cells. Both the cells transfected with lentivirus PLV vectors encoding the sig-16L2-Egfp or PLV-L2-Egfp gene were observed using confocal laser scanning microscopy. There were abundant sig-16L2-eGFPs embedded in both nuclear membrane and plasma membrane of HeLa cells, whereas L2-eGFP without signal peptide only existed in the cytoplasm, as demonstrated by the green fluorescence from eGFP. The process of the protein sorting and vesicle transport (the white arrows) can be clearly seen. (Scale bar: 10 μm.) (G) Determination of Sig-16L2-eGFP sorting and transportation by Golgi body and ER staining. The yellow light resulting from coincidence of green light (Sig-16L2-eGFP) and red light (Golgi body) were observed, suggesting that Sig-16L2-eGFP was located in Golgi body via signal peptide sorting route.
Fig. S1.

Schematic representation of the lentivirus PLV-sig-16L2-eGFP construct.
Interestingly, the process of protein sorting and vesicle transport (Fig. 2F) can be clearly observed. Sig-16L2-eGFP sorting and transportation were verified by carrying out Golgi body and endoplasmic reticulum (ER) staining in HEK 293T cells (Fig. 2G). As expected, the yellow light, produced from coincidence of green light (Sig-16L2-eGFP) and red light (Golgi body) was observed, suggesting the Sig-16L2-eGFP was located in Golgi body via signal peptide sorting route and then guided to plasma membrane with the help of transport vesicles. The close connection of membrane system between cell nucleus and organelles may account for the fluorescence emission from both types of membranes. Obviously, it is very important to select appropriate transmembrane peptide and signal peptide to target cargo proteins to cell membrane. The signal peptide we used in this study with great potential for surface functionalization is generalizable for different cell lines and ensures a high yield of displayed protein, because it is derived from integrin, which is abundant in most human cells.
Next, the engineered cells were incubated with 0.015% sodium deoxycholate in PBS for 15 min to generate major cell membrane vesicles (MCVs, 500–2,500 nm, Fig. 3 A, i), and then the MCVs were purified by sequential density sucrose gradient ultracentrifugation (20). To prepare VMVs displaying sig-16L2-eGFP (Fig. 3A), the purified MCVs were further mixed with 0.045% sodium deoxycholate and 0.05% triton-X100, which played a crucial role in generating nano-sized VMVs (50–150 nm), as demonstrated by cryo-electron microscopy (cryo-EM) (Fig. 3 A, iii) and transmission electron microscopy (TEM) (Fig. 3C), and in maintaining adequate strength of the materials (Fig. S2i). The entrapment of triton-X100 and sodium deoxycholate into VMV membrane ensured the size distribution, homogeneity and stability of the liposomes, as shown by dynamic light scattering analysis (DLS, Fig. 3B), phase contrast microscopy and confocal laser scanning microscopy (Fig. 3D). The confocal images illustrated the existence of sig-16L2-eGFP in VMV (<250 nm), as shown by the green light spots. The size distribution of VMVs was not changed by at least three freezing/defrosting cycles. It is of note that natural vesicles without surfactants (Fig. S2i) are more prone to be disrupted in the process of drying in negative stain than nano-sized VMVs, indicating that the surfactants could maintain the strength and integrity of VMV as well as uniformity. Impressively, we could obtain 64 ± 27 μg of VMV (measured based on protein concentration by BCA assay) per 106 cells. Out of 100 μg of total protein in VMV samples, 13.49 ± 0.61 μg of eGFP and 1.33 μg of L2 peptide at equimolar concentration of sig-L2-eGFP were detected by anti-eGFP ELISA kit (Fig. S3). The percentage of target antigen, sig-L2-eGFP, in total protein of VMV sample reached ∼14.82%. In addition, VMV-L2 antigen purity was also assessed by Western blot (WB) using anti-plasma membrane-protein (Na+/K+-ATPase) antibody, anti-cell nucleus (Histone H3a) antibody, and anti-cytoplasm protein (actin and GAPDH) antibody. Fig. S4 showed that intracellular contamination was virtually negligible, whereas the fact that Na+/K+-ATPase could be detected by WB assay further demonstrated that the VMVs were indeed generated from cell plasma membrane.
Fig. 3.

The preparation and characterization of VMV displaying Sig-L2-eGFP. (A) The cryo-EM images of major cell membrane vesicles (MCV-L2) incubated with different concentrations of sodium deoxycholate and triton-X100: (i) MCV-L2 resulting from HEK 293T cell incubated with 0.015% sodium deoxycholate; (ii) MCV-L2 mixed with 0.045% sodium deoxycholate; (iii) MCV-L2 mixed with 0.045% sodium deoxycholate and 0.05% triton-X100; (iv) MCV mixed with 0.045% sodium deoxycholate and 0.1% triton-X100, indicating that the entrapping of surfactants into vesicular membrane ensures the shape integrity and homogeneity of VMV. (Scale bar: 100 nm.) (B) The size distribution of VMV-L2 under different concentrations of surfactants measured by DLS. (C) The TEM images revealed similar shape and size of VMV-L2 to natural enveloped virus (<200 nm), which were produced from MCV-L2 mixed with 0.045% sodium deoxycholate and 0.05% triton-X100. (Scale bar: 200 nm.) (D) The imaging of VMV-L2using phase contrast microscope (Left) and confocal laser scanning microscopy (Right). The results verify the existence of sig-16L2-eGFP in VMV (<200 nm), as shown by the green light spots. (Scale bar: 500 nm.) (E) Co-IP and WB analysis of VMV-sig-16L2-eGFP. Co-IP assay confirmed the presence of the epitope from 16L2 protein on the exterior of VMV. In contrast, there was no VMV in supernatant after Co-IP. (F) Schematic representation of the Co-IP beads capturing the VMV-sig-16L2-eGFP by the strong affinity of protein A/G on beads to anti-L2 antibodies because of the existence of sig-16L2 epitope on VMV surface, rather than in vesicular interiors.
Fig. S2.
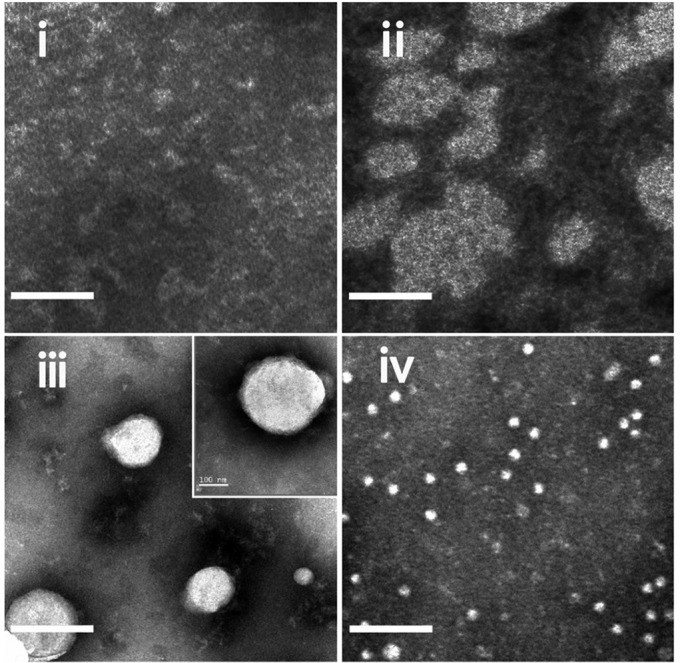
The TEM images revealed distinct morphology of major cell membrane vesicles (MCV-L2) incubated with different concentrations of sodium deoxycholate and triton-X100: (i) Natural vesicles without surfactants; (ii) MCV-L2 mixed with 0.045% sodium deoxycholate; (iii) MCV-L2 mixed with 0.045% sodium deoxycholate and 0.05% triton-X100; (iv) MCV mixed with 0.045% sodium deoxycholate and 0.1% triton-X100, indicating that the entrapping of surfactants into vesicular membrane ensures the shape integrity and homogeneity of VMVs. (Scale bar: 200 nm.) It is of note that natural vesicles without surfactants (i) are more prone to be disrupted in the process of drying in negative stain than nano-sized VMVs.
Fig. S3.

To determine the content of sig-16L2-eGFP in the total amount of protein, the anti-eGFP ELISA kit was used to quantify the amount of sig-16L2-eGFP after the VMV-L2 samples (100 μg of total protein in 1 mL of normal saline) were diluted 10,000 times. As seen from the figure, the concentration of eGFP in diluted sample could reach 1,349 pg/mL and thus 100 μg of total protein in 1 mL of normal saline contained 13.49 ± 0.61 μg of eGFP and 1.33 μg of L2 peptide at equimolar concentration of sig-L2-eGFP.
Fig. S4.

Western blot assay using anti-plasma membrane-protein (Na+/K+-ATPase) antibody, anti-cell nucleus (Histone H3a) antibody, and anti-cytoplasm protein (actin and GAPDH) antibody. The intracellular contamination was virtually negligible, whereas the fact that Na+/K+-ATPase could be detected further confirmed that the VMVs were indeed generated from cell plasma membrane.
Our next challenge was to determine whether the epitope (21) from 16L2 protein was presented on the exterior of the VMVs, rather than their interior (Fig. 2A). Hence, the VMVs were incubated, washed, and immobilized on protein A/G conjugated beads via anti-16L2 epitope antibodies in a coimmunoprecipitation (co-IP) assay. Co-IP and WB analysis (Fig. 3E) of the complex, using 14H6 antibodies, also confirmed the expression of sig-16L2 epitope on the surface of VMV. The schematic (Fig. 3F) shows that VMV displaying the epitope of VMV-L2 was captured by anti-16L2 epitope antibody (14H6), followed by binding of protein A/G beads to the antibody to form a complex of beads and VMV.
As VMV resembles natural virus in size, shape, and specific activity, we next investigated the in vivo immunogenicity of VMV-L2. BALB/c mice were vaccinated with VMV-L2, VMVblank+ free L2 peptide, VMVblank or free L2 peptide via different routes of administration with or without adjuvants. Sera from immunized mice were collected 2 wk after the last boost and tested by enzyme linked immunosorbent assay (ELISA) for anti-HPV L2 IgG. As shown in Fig. 4 A and B, vaccination of mice with VMV-L2 could not only induce high level of antibody titers to the L2 polypeptide, but also elicit 7–8 times higher total antibody titers (Fig. 4A) and ∼10.5-fold higher neutralizing antibody titers (Fig. 4B), in contrast with free L2 peptide group in the presence of Alum adjuvant via intramuscular (i.m.) route. Moreover, similar antibody titers induced by VMV-L2 alone via i.v. administration with VMV-L2 in Alum adjuvant groups via i.m. route were detected.
Fig. 4.

The immunogenicity of VMV-L2. (A) Mice (n = 5 per group) were immunized three times with 100 μg of VMV-l2 or 1.33 μg of L2 peptide with incomplete Freund’s adjuvant (IFA) or Alhydrogel (alum) adjuvant via i.v. , i.m., or s.c. administration. Serum anti-L2 epitope antibody titer was determined by end-point dilution ELISA. (B) A comparison of neutralizing antibody response of mice vaccinated thrice with VMV-L2 with IFA or Alhydrogel (alum) adjuvant via i.v., i.m., or s.c. administration. In vitro neutralization assays were performed using HPV16 pseudovirus. All data indicated that the antiserum from mice immunized with VMV-L2 could effectively inhibit the invasion of HPV16 pseudovirus into the host cells by neutralizing response and VMV-L2 was significantly superior to L2 peptide or VMVblank+ L2 peptide in eliciting the immune protection. Native mouse serum and antiserum in mice vaccinated with VMVblank were used as negative controls. (C) The exposure time of Cy5.5-labeled L2 peptide and VMV-L2 to immune system following i.m. injection into left leg in mice, as visualized by fluorescence imaging. The change in fluorescence intensity at different time points postinjection suggested longer exposure time of VMV-L2 over free L2 peptide to immune cell because of the larger molecular weight of VMV-L2 (∼30 μg of Cy5.5 each mouse). (D) SPET/CT images at 1, 3, and 5 h after i.v. injection of 99mTc labeled L2 peptide and VMV-L2 (15 MBq each mouse). The spleen and bladder are indicated by white arrow and red circle, respectively. The imaging data showed that the VMV can effectively accumulate into liver, spleen and other parts of the reticuloendothelial system by passive targeting to the immune system via i.v. administration route, whereas free L2 peptide was rapidly cleared.
In accordance with the trend of total IgG antibody titers, neutralization activities of antibodies triggered by VMV-L2 in mice were verified in vitro against HPV16. Fig. 4B shows that neutralization activities of anti-sera vaccinated with VMV-L2 against HPV16 pseudotyped virus, the highest value of IC50, could reach 1,024, which was much higher than ones elicited by free L2 peptide in Alum adjuvant. In contrast, the anti-sera from mice treated with VMVblank derived from HEK 293T cells expressing 16L2-eGFP in cytoplasm exhibited no neutralization activities against HPV16 pseudotyped virus. It is concluded that VMV-L2 as a subunit vaccine delivery vehicle elicits neutralization antibodies specific for the epitope of L2 protein, which is essential to inhibit the entry of HPV into cells.
Biological Behavior of VMVs.
Notably, even the group of VMV-L2 without adjuvant also exhibited strong humoral immune response, so we speculated that satisfactory immunogenicity of VMV-L2 was most likely due to the larger molecular weight and exogenous properties of VMV that would can be recognized and swallowed by immune cell. To test this hypothesis, the kinetic clearance of VMV-L2 via different administration routes was assessed through molecular imaging methods. We first performed near-infrared (NIR) fluorescence imaging to analyze the antigen exposure time of VMVs in mice treated by i.m. injection. Labeling of VMVs with a NHS-Cy5.5 dye did not affect the size distribution and activity of VMVs. Antigen exposure at the injection site was monitored by ex vivo NIR imaging at different time points. As a result, the VMVs (mostly due to their unique virus-like structure) maintained the integrity of VMV-L2 and retained the high molecular weight antigen in the muscle tissue, resulting in longer antigen stimulation time than the L2 epitope peptide alone (Fig. 4C). Next, we used SPECT/CT (22) to quantify the distribution of 99mTc-labeled VMV-L2 and L2 epitope peptide in mice vaccinated via i.v. administration route. Normal ICR mice injected with the same amount (15 MBq) of 99mTc-labeled L2 peptide or VMV-L2 were monitored at 1, 3, and 5 h after injection. Quantitative analysis of SPECT/CT images (Fig. 4D) showed that VMVs tend to accumulate in the liver, spleen, and kidneys, as well as other portions of reticuloendothelial system (RES) within 5 h of i.v. administration, whereas the L2 epitope peptide had rapid renal clearance within 1 h (Fig. S5).
Fig. S5.

(A) Representative SPECT images of normal ICR mice at 5 h after i.v. injection of 15 MBq VMV-L2. 1, liver; 2, spleen; 3, kidneys. The images confirmed accumulation of VMV-L2 into liver and spleen (immune organ) as well as the lymphatic system because of its passive targeting to lymphatic and reticuloendothelial system. (B and C) Biodistribution of 99mTc-labeled VMV-L2 (B) and L2 peptide (C) in normal mice at different time points after i.v. injection. Data were calculated from regions of interest analysis of SPECT images.
Given the aluminum hydroxide particles in the micrometer size range and the conventional way of vaccination, the alum-adjuvanted formulation was administrated via the i.m. route to ensure the vaccine safety and long-term antigen exposure. As mentioned above, the antibody and neutralization titers induced by VMV-L2 alone via i.v. administration were comparable with VMV-L2 in Alum adjuvant groups via i.m. route, owing largely to the nanosize and exogenous property of VMV-L2, which allows VMV to effectively deliver antigen into immune system through blood circulation. These results about immunogenicity of VMV-L2 are nicely associated with the biodistribution studies performed by molecular imaging methods. Taken together, we conclude that the VMV vaccine not only offers long-term antigen exposure but also confers excellent target recognition by the immune system, including the spleen–which acts as important immune organ (23, 24) to remove pathogen and senescent blood cells.
VMV Displaying Integral Hemagglutinin (HA) Glycoprotein from Influenza A (H1N1) Virus.
Motivated by the goal of developing a method to use VMVs as a broad vaccine delivery vehicle for rapid response to emerging catastrophic pandemics, we further prepared VMVs to display integral membrane hemagglutinin (HA) glycoprotein (570 amino acids) from influenza A (H1N1) virus, rather than an HPV L2 epitope peptide (24 amino acids). The HA glycoprotein can be anchored through its hydrophobic transmembrane region to vesicular membrane, which provides a befitting lipid environment analogous to the one in enveloped viruses. Because the recombinant HA protein produced in eukaryotic cells is subjected to posttranslational modifications, including glycosylation essential for trimerization of influenza hemagglutinin, as well as correct folding and transport to cell plasma membrane (19, 25), we presumed that the protein molecules displayed on VMVs resemble native viral trimeric spike proteins so that they can induce neutralizing antibodies targeting the correctly conformational epitopes.
In this study, the preparation process of VMVs carrying HA (VMV-HA) involved a procedure similar to that mentioned above. Briefly, the HA gene was subcloned into lentiviral PLV vector and then transfected into HEK 293T cells to express HA protein in cell plasma membrane. HEK 293T cells transfected with PLV-HA construct was incubated with 0.01–0.015% sodium deoxycholate in PBS buffer for 15 min to generate major cell membrane vesicles (MCVs, 500–2,500 nm), which were separated by multiple centrifugation and ultracentrifugation for 4 h at 4 °C. Then, the pellets were resuspended and fractionated through a sucrose density gradient centrifugation (21). Finally, the purified MCVs were mixed with 0.045% (wt/vol) sodium deoxycholate and 0.05% (wt/vol) triton-X100 in PBS to produce uniform sized VMVs (50–150 nm), as demonstrated by cryo-EM (Fig. 5B).
Fig. 5.

Functional characterization of VMV displaying influenza HA protein (VMV-HA). (A) Immunofluorescence assay of HEK 293T cells transfected with lentivirus PLV-HA vector after adding 0.015% sodium deoxycholate. Laser confocal images show location of the recombinant HA protein (green) in cytoplasmic membrane. Incubation of these transfected 293T cells with 0.015% sodium deoxycholate facilitated generation of MCV-HA on cellular surface. (Scale bar: 10 μm.) (B) Cryo-EM images of VMV-HA showed similar size as well as shape to native virus. (Scale bar: 100 nm.) (C) Agglutination of chicken red blood cells (RBCs) by VMV-HA, suggesting that HA protein coupled to MCV is capable of binding to cellular surfaces similar to those on influenza virions. (D) Schematic of agglutination and cross-linking of chicken RBCs by VMV-HA. (E) Co-IP and WB analysis of VMV-HA verified the existence of HA glycoprotein on the exterior of VMV similar to real influenza virus, as demonstrated by the band in Co-IP pellet lane. (F) VMV-HA derived from 5 × 105 HEK293T cells for hemagglutination assay, which were treated with TPCK-trypsin at 28 °C for 0, 5, 10, or 15 min and neutralized with protease inhibitors. The comparison of hemagglutination capacity between VMV-HA and inactive influenza virus (H1N1) indicated that TPCK-trypsin–treated VMV-HA had similar hemagglutination function and HA protein conformation to inactive influenza virus and that trypsin contributes to maturing VMV-HA via a protein cleavage mechanism similar to that of influenza virions because it has been confirmed that TPCK-trypsin treatment can improve cleavage and maturation of HA0 protein in the same cleavage site as HA0 protein expressed in cell infected by virus. The H1N1 influenza viruses resulting from 5 × 105 cells were harvested 48 h after seeding in MDCK cells.
As seen in Fig. 5A, MCV-HA binds to the surfaces of 293T host cells after several washings and the green fluorescence from MCVs (green spotlights) confirms the existence of viral antigen HA, suggesting that HA protein coupled to MCV is capable of binding to cellular surfaces similar to those on influenza virions. Furthermore, co-IP and WB assay (Fig. 5E) also demonstrated that the beads can bridge the VMVs, mediated by anti-HA epitope antibodies, due to the presence of HA glycoproteins on VMV surface.
To verify the antigenicity of HA protein on VMVs produced by 293T cells, we analyzed its reactivity with six conformation-dependent neutralizing antibodies (J3F11, 13G7, 2H3, 18B10, J8A1, 16D5) by sandwich ELISA, which recognizes different specific sites on live influenza virus. Table S2 shows that the HA glycoprotein on VMVs binds to all conformation-dependent neutralizing antibodies with affinities similar to those of inactivated influenza viruses, except for the control antibody 14H6. The results suggest that HA molecules on VMVs antigenically resemble the trimeric HA viral spikes on native virions.
Table S2.
Antigenicity of HA protein on VMV using sandwich ELISA and WB assays with conformation-dependent neutralizing antibodies
| VMV-HA | H1N1 virus | |||
| Neutralizing antibodies | ELISA | WB | ELSIA | WB |
| J3F11 | +++ | — | +++ | — |
| 13G7 | +++ | — | +++ | — |
| 2H3 | ++ | — | ++ | — |
| 18B10 | +++ | — | ++ | — |
| J8A1 | ++ | — | +++ | — |
| 16D5 | +++ | — | +++ | — |
| 14H6 | — | — | — | — |
The reactivity of HA protein on VMV was analyzed with six conformation-dependent neutralizing antibodies (J3F11, 13G7, 2H3, 18B10, J8A1, and 16D5) by sandwich ELISA, which recognized specific sites on live influenza virus. The results suggest that HA molecules on VMV antigenically resemble the trimeric HA viral spikes of native virions. (+: 0.1 < OD450nm < 1.0, ++: 1.0 < OD450nm < 2.0, +++: OD450nm > 2.0, -: OD450nm < 0.1 for WB detection at exposure time of 30 s).
We next studied whether VMV-HA was conferred the capacities of receptor binding by hemagglutination assay (Fig. 5 C and D), which is a conventional assay to measure the functionality and amount of HA in live influenza virus. As seen in Fig. 5C, VMV-HA can efficiently agglutinate red blood cells (RBC) like active influenza virus. This result reflects the presence of conformationally correct HA glycoprotein on the nanovesicular surfaces. Interestingly, VMV-HA treated by trypsin (TPCK treated) induced higher HA titers than ones without TPCK-trypsin treatment (Fig. 5F). On the contrary, nanovesicles without HA protein (VMVblank) exhibited no HA activities. The comparison of hemagglutination capacity (Fig. 5F) between VMV-HA and inactive influenza virus (H1N1) indicated that TPCK-trypsin–treated VMV-HA had similar hemagglutination function and conformation to inactive influenza virus and that trypsin contributes to maturing VMV-HA via a protein cleavage mechanism in similar way to that of influenza virions, because it has been proven (19, 25) that TPCK-trypsin treatment can improve cleavage and maturation of HA0 protein at the same cleavage site as HA0 protein expressed in cells infected by virus. All of the data showed that VMV-HA resembled live influenza virus in shape, size as well as specific function.
Immunogenicity of VMV-HA.
To evaluate the immunogenicity of VMV-HA, the humoral immune response and protective efficacy of VMV-HA were assessed in mice. Quantification of HA antigen in the VMV-HA (Fig. S6) was performed by WB assay and sandwich ELISA to normalize the antigen dose. The mice were immunized thrice with 100 μg per injection of VMV-HA (containing 6.5 μg of HA antigen), the same HA content of inactive influenza virus or purified HA glycoprotein in 10 μL of Alum adjuvant via i.m. administration route. Sera from immunized mice were collected 2 wk after the last boost by standard methods and ELISA was performed on 96-microwell plates coated with inactive influenza virus (H1N1) rather than purified HA protein. For comparison, the preimmune sera and the VMVblank group were pooled as control samples. The data (Fig. 6A) showed that VMV-HA could trigger high level of serum IgG titers via i.m. administration route with or without Alum adjuvant. The total antibody titers induced by VMV-L2 in Alum adjuvant was comparable to that elicited by standard HA glycoproteins recombinant from Baculovirus. The inactive influenza virus in Alum adjuvant led to significant increase in total antibody level relative to other groups because of coating ELISA plate with whole inactive influenza virus. VMVs as a class of subunit vaccine indeed mimicked the important immunogenic part of enveloped virus.
Fig. S6.

Quantification of HA antigen in VMV-HA by Western blot assay. The data suggested that VMV-HA samples (5 μg of total protein) contained 325 ± 33 ng of HA protein, the percentage of HA protein in total protein of VMV could reach 6.5 ± 0.66%. The results in accordance with ones by sandwich ELISA using purchased HA protein sample.
Fig. 6.

Evaluation of the immunogenicity of VMV-HA. (A) Total antibodies titers in mice (n = 5 per group) immunized with inactive influenza virus, HA protein, or VMV-HA in the same amount of HA antigen (6.5 μg of HA each mouse) with or without Alum adjuvant via i.m. administration routes were measured by ELISA. Native mouse serum and anti-serum in mice vaccinated with VMVblank were used as negative controls. (B) Neutralization capacities of antisera from mice vaccinated with inactive influenza virus, HA protein, or VMV-HA with or without adjuvant via i.m. administration routes was assessed by incubation of mouse sera with live H1N1 influenza virus. Neutralizing titers of pre- and postvaccination sera were determined by a microneutralization assay. (C) Hemagglutinin inhibition (HAI) titers after three immunizations with the same HA amount of inactive influenza virus, HA protein, VMV-HA with or without Alum adjuvant via i.m. administration route. (D and E) Decrease in body weight (D) and survival (E) of mice challenged with 50 times lethal dose of mouse-adapted H1N1 (A/California/04/2009) viruses. All of the data confirmed the ability of VMV-HA to elicit a protective immune response in vivo and suggested the VMV vaccine could match purified HA subunit vaccines in immunogenicity.
The neutralization activities and HA inhibition response (HAI) of antibodies elicited by VMV-HA in mice immunized via i.m. routes were verified in vitro against live H1N1 influenza viruses. Fig. 6 B and C showed that the neutralization activities in mice induced by VMV-HA were similar to ones elicited by the same HA amount of inactive influenza virus and standard purchased HA protein via i.m. route in the presence of Alum adjuvant. Moreover, although the HAI titers were significantly higher in all vaccinated groups in contrast to the nonvaccinated groups, the changes among the immunized groups using Alum as adjuvant did not reach statistical significance, suggesting that the VMV vaccine could match purified HA subunit vaccines in immunogenicity.
We further examined the protective immune response of VMV-HA in vivo. Four weeks after the last immunization, mice were challenged intranasally with 50 times lethal dose of mouse-adapted H1N1 virus, and body weight of treated mice was monitored daily (Fig. 6D). Mice immunized intramuscularly with VMV-HA in the absence of Alum adjuvant exhibited good in vivo protection against challenges of lethal dose of influenza virus, with 80% survival (Fig. 6E). Importantly, all of the mice vaccinated with inactive virus, HA protein, VMV-HA in Alum adjuvant finally survived, whereas the mice in VMVblank groups died 9 d after challenges of mouse-adapted influenza viruses. Body weight data (Fig. 6D) showed that the initial weights of VMV-HA-treated mice were recovered and most of the treated mice had parallel weight loss with the groups vaccinated with HA proteins, whereas the group immunized by inactive influenza virus exhibited significantly less weight loss than those vaccinated with VMV-HA or HA protein, which may be due to the fact that inactive virus as whole pathogen consisted of many immune stimulating antigen such as M1, M2, and NA protein inducing broader protection.
In this study, we did not notice obvious signs of toxic side effects in the VMV-HA–treated cells (Fig. S7), and neither death nor significant body weight drop was observed in the treated mice. The results of blood chemistry (26) for the mice vaccinated thrice with VMV-HA fluctuated in normal range (Fig. S8) and no obvious abnormalities were found by hematoxylin and eosin (HE) staining of the major organs of the treated mice (Fig. S9), both suggest safety of VMV.
Fig. S7.

Cellular viability (by MTT assay) after different period incubation with VMV-HA (400 μg of total protein per mL).
Fig. S8.

The blood chemistry of the mice vaccinated thrice with 100 μg of VMV-HA (6.5 μg of HA antigen) with or without Alum adjuvant after i.m. administration to test (A) inflammation-associated cell counts, (B) blood cell counts, (C) hemoglobin levels, and (D) renal toxicity. Although some results (A) between VMV-HA groups and PBS groups have some difference, the changes fluctuated in the normal range. (BNU, urine nitrogen content; EON, eosinophils; HGB, hemoglobin; LYMPHN, lymphocyte; MCHC, mean corpuscular hemoglobin concentration; MONON, mononuclear macrophages; NEUT, neutrophile granulocyte). All results suggest that VMV-HA is biodegradable and a safe vaccine candidate.
Fig. S9.

HE staining of major organs from mice treated with PBS (control) and VMV-HA, respectively. No obvious abnormalities were observed in the heart, liver, spleen, and kidneys.
However, attention should be paid to the potential toxicity and biocompatibility of VMVs before this strategy may be translated into clinic. The additives such as synthetic surfactants in a vaccine formulation may cause unwanted immune reactions and lead to the induction of autoimmunity. Some vesicles from organelles other than the plasma membrane may harbor immature constructs. Although the intracellular contamination in VMVs was virtually negligible in the present study, more systematic investigations including screening biosurfactants with less toxicity and higher biodegradability than the currently used synthetic surfactants will be a subject of our future studies.
Because VMV platform not only presents foreign epitope or other important antigen to immune cell, but also displays whole viral enveloped glycoprotein in native conformation by mimicking the budding process of natural enveloped virus and being subjected to posttranslation modification in eukaryocyte. In addition, VMV as attractive vaccine candidate could induce good immune protection as well as subunit protein vaccine. All these traits make VMV possible to design a wide range of vaccines against newly emerging enveloped virus through incorporating the viral enveloped glycoprotein into nanovesicles.
In this work, we have designed VMVs to serve as broad vaccine delivery vehicles and developed a straightforward, robust and tunable method of functionalizing the VMV surface with a variety of protein probe for targeted delivery (27). The VMV technology is also rapidly scalable to mass production by bioreactors, whenever cell lines/patients' own cells [e.g., MSCs, DC cells or engineered red blood cells (27) as nanocarriers] stably expressing a virus antigen on their cell plasma membrane surfaces become available. Due to the tolerance of large protein insertion in natural forms by VMVs, the VMV nanotechnology has the potential to be adapted to the design of vaccines against a wide array of enveloped viruses, including ones with pandemic potential.
Materials and Methods
Vector Construction.
The HPV L2 epitope with a C-terminal transmembrane sequence was placed in the upstream of enhanced green fluorescent protein (eGFP) at EcoR1 and BamH1 restriction sites, forming 16L2-eGFP recombinant protein. Next, the human integrin signal sequence was fused to the 16L2-eGFP gene fragment encoding sig-16L2-eGFP to functionalize cell plasma membrane, using PCR with long primers which include integrin signal sequence. The gene encoding sig-16L2-eGFP or HA gene from H1N1 (A/California/04/2009) was finally subcloned into lentiviral PLV vector to express cargo protein in mammalian cells (Fig. S1 and Table S1).
Intracellular Localization of Sig-16L2-eGFP, 16L2-eGFP, and HA Protein.
Briefly, HEK293 and HeLa cells in DMEM containing 10% FBS and antibiotics were cultured on gelatin-coated coverslips in six-well dishes. The cells were transfected with PLV-sig-16L2-eGFP, PLV-16L2-eGFP or PLV-HA construct using Lipofectamine 2000, in accordance with the manufacturer’s instructions. At 48 h after the transfection with these plasmids, the cells on coverslips were washed twice with PBS and stained with DAPI (Roche), followed by fixation with 4% paraformaldehyde for 15 min. Finally, the cell-coated coverslips were evaluated using a laser confocal microscope (LSM780) equipped with 60× oil-immersion lenses. Sig-16L2-eGFP sorting and transportation were determined by using Golgi body staining (Beyotime) and ER staining (Thermo) for live cells in confocal dish.
Preparation of VMV-L2 or VMV-HA.
The 293T cells transfected with PLV-sig-16L2-eGFP or PLV-HA constructs were collected and incubated with 0.01–0.015% (wt/vol) sodium deoxycholate in PBS buffer for 15 min to generate major cell membrane vesicles (MCV, 500–2500nm) under the condition of strenuous vibration, followed by the addition of protease inhibitors (Roche). Engineered MCV carrying sig-16L2-eGFP or HA protein were centrifuged at 2,000 × g and 4,000 × g, respectively, for 5 min at 4 °C to remove cell nuclei and organelles, filtered through a 0.45 µm filter at least 20 times, ultracentrifuged at 25,000 rpm for 4 h at 4 °C (Beckman Coulter). The pellets were resuspended and purified through a 5–65% sucrose density gradient at 25,000 rpm for 16 h at 4 °C. Each separated fraction was carefully collected from the top to the bottom of the gradient. Finally, the purified MCV was mixed with 0.045% (wt/vol) sodium deoxycholate and 0.05% (wt/vol) triton-X100 in PBS to produce size-controlled and uniform VMV displaying 16L2-eGFP or HA protein.
Western Blot Analysis and Coimmunoprecipitation Assay.
To test the incorporation and configuration of sig-16L2-eGFP onto VMV, the purified samples (VMV-L2 or VMV-HA) were incubated with beads (Santa Cruz Biotechnology) conjugated to protein A/G at 30 °C after addition of 14H6 antibody or anti-HA antibody (10 μg) that were specific for the antigen protein. The beads were then centrifuged at 1,000 × g for 4 min at 4 °C to determinate whether the beads were capable of capturing the VMV by the binding of protein A/G on beads to the antibodies. The pellets were washed thrice in PBS and analyzed by Western blot assay. Briefly, the pellets were mixed with cell lysis solution in the presence of loading buffer, boiled for 15 min, separated by 8% SDS/PAGE, and transferred onto PVDF membranes. The PVDF membranes were then blocked with a TBST solution containing 7% nonfat milk power and subsequently probed with an anti-L2 14H6 antibody. Sig-L2-eGFP was visualized with a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (Southern Biotech) and ECL substrate (Thermo). To determine the content of sig-16L2-eGFP in the total amount of protein, the eGFP ELISA kit (Abcam) was used for quantifying the amount of sig-16L2-eGFP. VMV-L2 antigen purity was assessed by Western blot assay using anti-plasma membrane-protein (Na+/K+-ATPase) antibody, anti-cell nucleus (Histone H3a) antibody, and anti-cytoplasm protein (actin and GAPDH) antibody (Santa Cruz Biotechnology). All the antibodies used in these experiments, except as specifically noted, were produced by the National Institute of Diagnostics and Vaccine Development in Infectious Diseases, Xiamen University.
Characterization of VMV.
The VMV-L2 or VMV-HA was analyzed by dynamic light scattering (DLS, Zetasizer Nano) for the size distribution. VMVs were suspended in PBS at a protein concentration of 2 μg/μL. All measurements were conducted in triplicate. To observe VMV-L2 and VMV-HA by cryo-EM, 5 μL of samples were loaded onto Quantitoil Holey grid for 3 min, swept with a filter paper, and then quickly dipped into liquid ethane by use of FEI Vitrobot Marker. The samples were transferred and observed on FEI Tecnai G2 electron microscope under the condition of 300 kV and a defocus of approximately 2.4 μm. Transmission electron microscopy (TEM) imaging was performed to measure the VMV size. VMVs at 1 μg/μL were dropped on freshly discharged 400 mesh carbon parlodion-coated copper grids. The grids were then negatively stained with 1% phosphotungstic acid and naturally dried for 1 h. The VMV was finally imaged on a JEM-2100 transmission electron microscope at 90 kV and digitally captured with a CCD camera at 1k×1k resolution.
Immunization and Animal Studies.
All animal protocols were in accordance with the instructions of and approved by the Institutional Animal Care and Use Committee of Xiamen University. The VMV-L2 (50–150 nm) was generated from MCV mixed with 0.045% (wt/vol) sodium deoxycholate and 0.05% (wt/vol) triton-X100. BALB/c mice were vaccinated with VMV-L2 samples (100 μg of total protein containing 13.49 ± 0.61 μg of eGFP and 1.33 μg of L2 peptide at equimolar concentration), VMV-L2 + free L2 peptide (1.33 μg), VMVblank, or free L2 peptide (1.33 μg) via different routes of administration with or without adjuvants (n = 5 per group). Immunizations were performed thrice at a 2-wk interval with or without IFA (Sigma) or Alhydrogel adjuvant (InvivoGen). Lastly, sera from immunized mice were collected two weeks after the last boost by standard methods and stored at −20 °C until use. Protein concentrations of the purified VMV were measured with a BCA protein assay (Pierce), using BSA as a standard.
Determination of Serum Antibody Titers by ELISA.
To estimate the antigenicity of VMV-HA, a sandwich ELISA (28) was performed. The purified VMV-HA was added to anti-HA antibody-coated 96-well plates, followed by incubation at 37 °C for 1.5 h. The treated plates were then washed five times with PBST buffer (0.05% Tween-20 in PBS). Anti-HA monoclonal antibody (J3F11, 13G7, 2H3, 18B10, J8A1, 16D5) and control antibody (14H6) at a 1:1,000 dilution were then seeded, followed by incubation for another 1.5 h. HRP-conjugated goat anti-mouse Ig (H+L) at a 1:2,000 dilution as a secondary antibody (Southern Biotech) was added. Colorimetric analysis was carried out using a TMB substrate kit (Pierce), and the absorbance was obtained at 450 nm by a spectrophotometer.
To determine the total serum IgG antibody response against L2 protein or H1N1 influenza virus, 96-well plates were coated with 300 ng of purified L2 protein/well or 500 ng of inactive H1N1 influenza virus per well at 4 °C overnight. The plates were blocked with PBS containing 5% (wt/vol) BSA at 37 °C for 1.5 h, followed by addition of serially diluted mouse anti-serum at 37 °C for 1.5 h. Plates were washed four times with PBST buffer (0.05% Tween-20 in PBS) and incubated with HRP-conjugated goat anti-mouse IgG (Southern Biotech, 1:4,000) for 1 h. Colorimetric analysis was performed as described above. Endpoint titers were detectable as the last reciprocal serial serum dilution at which the absorbance at 450 nm was twofold greater than the mean value of the background signals from preimmune serum and VMVblank-treated groups.
An L2-based ELISA was conducted to assess the titer of anti-16L2 antibodies. To generate hexahistidine (His)-labeled 16L2 polypeptide, the L2 gene encompassing amino acid residues 13–105 was attained by PCR and subcloned into the pET28a vector. ELISA plates were coated with 300 ng of bacterially expressed 16L2 polypeptide containing amino acid residues 13–105, which were purified by affinity to a nickel-nitrilotriacetic acid column in 7 M urea. Antibody titer was determined as the highest sera dilution factors within OD value twofold greater than control sera at the same dilution.
Neutralization Assay (HPV).
The human papillomavirus (HPV16) pseudovirions loaded with secreted alkaline phosphatase (SEAP) were produced by cotransfection of HEK 293T cells with plasmids encoding a SEAP reporter genes and HPV16 L1 and L2 gene. HEK 293T cells were added into 96-well plates at 1.5 × 104 cells per well, cultured overnight, and then used for pseudovirion neutralization assays. Briefly, the serial dilutions of immune sera and the HPV16 pseudovirus were incubated for 2 h and the mixture was used to infect HEK 293T cells. Seventy hours after infection, the supernatants in wells were carefully collected and SEAP activity in cell-free supernatants by centrifugation was analyzed using p-nitrophenyl phosphate (Sigma). Anti-serum IgG neutralizationtiter was defined as the highest dilution that result into a greater than 50% reduction in absorbance, compared with control serum samples. Titers less than 50 were considered as undetectable.
In Vivo Imaging of Cy5.5 or 99mTc-Labeled VMVs.
The labeling of VMVs with NHS-Cy5.5 dye or 99mTc was performed according to our previous report (23). Cy5.5-labeled VMVs (100 μg of total protein) were injected into mice (male, 6∼8 wk old). Cy5.5 fluorescence in the whole body of the mice was acquired by IVIS Lumina II (Caliper Life Sciences). Radiant efficiency was measured using Living Image 3.1 software. To quantify the distribution of VMV, 99mTc-labeled VMV and L2 peptide (15 MBq per mouse) were injected, and tracer in the whole body of the mice was acquired by SPECT/CT (22).
Immunization and Animal Studies for VMV-HA.
The quantification of HA antigen in VMV-HA was performed by Western blot assay and sandwich ELISA (28) using purchased HA protein as a standard to normalize the antigen dose. The female mice (n = 5 per group) were immunized three times with 100 μg per injection of VMV-HA (containing 6.5 μg of HA antigen), the same HA content of inactive influenza virus or purified HA protein in 10 μL of Alum adjuvant via i.m. administration route. Control group was treated with VMVblank from the HEK 293T cells. The HA glycoprotein recombinant from Baculovirus was from BEIResources. H1N1 influenza virus (A/California/04/2009) was generated by reverse genetics technique and expanded in MDCK cells.
Hemagglutination Assay (29).
VMV-HA, which was treated with TPCK-trypsin at 28 °C for 0, 5, 10, or 15 min and neutralized with protease inhibitors, was serially diluted in 50-μL volumes in a 96-well plate. To each treated VMV-HA dilution, 50 μL of 0.5% chicken red blood cells (RBCs) solution in normal saline was seeded; mixtures of the red blood cells and VMV-HA were gently shaken, and the plates were incubated at 20–30 °C for 40 min before examination. Negative hemagglutination results were observed as dots in the center of the wells. The hemagglutination titer was determined as the highest dilution factor that exhibited an absolute positive result. Serial dilutions of VMV-HA derived from 5 × 105 HEK 293T cells were mixed with washed RBCs and incubated to analyze the cross-linking and receptor binding of chicken RBCs.
Hemagglutinin Inhibition Assay (HAI).
The H1N1 influenza viruses were expanded in Madin–Darby canine kidney (MDCK) cells. Immune mouse sera were pretreated with receptor-destroying enzyme (RDE) and the assays were conducted using 0.5% chicken red blood cells and influenza viruses measured as four hemagglutinating units per well. HAI titers were calculated by the mean value of the last dilution factors of sera which completely inhibited the cross-linking and hemagglutination of chicken red blood cells (30).
Neutralization Assay (H1N1).
BALB/c mice were divided into five groups (n = 5 per group) and immunized thrice with 100 μg of VMV-HA via different routes of administration in the presence of Alum adjuvant. All of the VMV-HA (50–150 nm) was produced from MCV mixed with 0.045% surfactant A and 0.05% surfactant B, aimed at mimicking real virions. Neutralizing antibody titers of pre- and postvaccination sera were calculated in a microneutralization assay on the basis of the standard methods of pandemic influenza reference laboratories. Anti-serum neutralization titers were verified as the highest dilution factors that caused a greater than 50% reduction in OD450nm compared with control groups. Low pathogenic H1N1 influenza virus (A/California/04/2009) was generated by reverse genetics technique and expanded in MDCK cells. The neutralization assays were performed with three replicates for each serum sample.
H1N1 Virus Challenge.
Four weeks after the last immunization, the mice were challenged intranasally with 50 times lethal dose of mouse-adapted H1N1 (A/California/04/2009) viruses, and body weights of challenged mice were observed daily. The influenza virus was expanded in MDCK cells. The mice were monitored and body weights were recorded daily. Results depict the average of five mice per group.
Biosafety Assays.
The cytotoxicity of the VMVs was evaluated by measuring the HEK293T and HeLa cell growth using the MTT assay (31). The cells were cocultured with VMVs for different time. After incubation with the MTT for 4 h, the media were removed and 100 µL of dimethyl sulfoxide (DMSO) was added. The absorbance was monitored at a wavelength of 450 nm, and the cell viability was expressed as the ratio of the number of treated cells to the number of untreated control cells. The blood routine examination and histology studies were performed to evaluate the in vivo biocompatibility. Two groups were injected with 100 μg of VMVs through the tail vein or leg muscle, and the other was the untreated control group. The blood and the tissues (heart, liver, spleen, lung, and kidney) were collected for the routine analysis (26). Hematoxylin and eosin (HE) staining (32) was used to stain the tissue slices. For HE staining, tissues from each group were harvested, fixed in 10% neutral buffered formalin, processed into paraffin, sectioned at 5-µm thickness, stained with HE, and examined by a digital microscope.
Statistical Analysis.
Statistical analyses were performed using the GraphPad Prism 5 software. P value less than 0.05 was considered statistically significant. Data plotted with error bars are expressed as means with SD, unless otherwise indicated. The P values were calculated by analyzing data with a two-tailed unpaired t test.
Acknowledgments
We thank Dr. Henry S. Eden for careful proofreading of the manuscript. This work was supported by the Ministry of Science and Technology of China (MOST) (2014CB744503, 2013CB733802, and 2012DFH30020); the National Natural Science Foundation of China (NSFC) Grants 81422023, 51273165, 81101101, 81371596, and 81371817, Program for New Century Excellent Talents in University (NCET-13-0502); Fundamental Research Funds for the Central Universities (2013121039 and 20720150141); and the Intramural Research Program, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. O.C.F. is a Guest Editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505799112/-/DCSupplemental.
References
- 1.Smith DM, Simon JK, Baker JR., Jr Applications of nanotechnology for immunology. Nat Rev Immunol. 2013;13(8):592–605. doi: 10.1038/nri3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 3.Hu CMJ, Fang RH, Luk BT, Zhang L. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8(12):933–938. doi: 10.1038/nnano.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ulmer JB, Valley U, Rappuoli R. Vaccine manufacturing: Challenges and solutions. Nat Biotechnol. 2006;24(11):1377–1383. doi: 10.1038/nbt1261. [DOI] [PubMed] [Google Scholar]
- 5.Pitoiset F, Vazquez T, Bellier B. Enveloped virus-like particle platforms: Vaccines of the future? Expert Rev Vaccines. 2015;14(7):913–915. doi: 10.1586/14760584.2015.1046440. [DOI] [PubMed] [Google Scholar]
- 6.Chen DJ, et al. Delivery of foreign antigens by engineered outer membrane vesicle vaccines. Proc Natl Acad Sci USA. 2010;107(7):3099–3104. doi: 10.1073/pnas.0805532107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10(7):521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- 8.Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat Nanotechnol. 2007;2(8):469–478. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 9.Fang RH, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Q, Li S, Yu H, Xia N, Modis Y. Virus-like particle-based human vaccines: Quality assessment based on structural and functional properties. Trends Biotechnol. 2013;31(11):654–663. doi: 10.1016/j.tibtech.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Wolfers J, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7(3):297–303. doi: 10.1038/85438. [DOI] [PubMed] [Google Scholar]
- 12.Parodi A, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hegde RS, Bernstein HD. The surprising complexity of signal sequences. Trends Biochem Sci. 2006;31(10):563–571. doi: 10.1016/j.tibs.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Hildebrand A, Neubert R, Garidel P, Blume A. Bile salt induced solubilization of synthetic phosphatidylcholine vesicles studied by isothermal titration calorimetry. Langmuir. 2002;18(7):2836–2847. [Google Scholar]
- 15.Ross BP, et al. Micellar aggregation and membrane partitioning of bile salts, fatty acids, sodium dodecyl sulfate, and sugar-conjugated fatty acids: Correlation with hemolytic potency and implications for drug delivery. Mol Pharm. 2004;1(3):233–245. doi: 10.1021/mp049964d. [DOI] [PubMed] [Google Scholar]
- 16.Garidel P, Hildebrand A, Knauf K, Blume A. Membranolytic activity of bile salts: Influence of biological membrane properties and composition. Molecules. 2007;12(10):2292–2326. doi: 10.3390/12102292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balzac F, et al. Expression and functional analysis of a cytoplasmic domain variant of the beta 1 integrin subunit. J Cell Biol. 1993;121(1):171–178. doi: 10.1083/jcb.121.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gambhira R, et al. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J Virol. 2007;81(24):13927–13931. doi: 10.1128/JVI.00936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Copeland CS, Doms RW, Bolzau EM, Webster RG, Helenius A. Assembly of influenza hemagglutinin trimers and its role in intracellular transport. J Cell Biol. 1986;103(4):1179–1191. doi: 10.1083/jcb.103.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang L, et al. HIV-1 virus-like particles produced by stably transfected Drosophila S2 cells: A desirable vaccine component. J Virol. 2012;86(14):7662–7676. doi: 10.1128/JVI.07164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jagu S, et al. Concatenated multitype L2 fusion proteins as candidate prophylactic pan-human papillomavirus vaccines. J Natl Cancer Inst. 2009;101(11):782–792. doi: 10.1093/jnci/djp106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li ZB, Chen X. MicroPET, MicroSPECT, and NIR fluorescence imaging of biomolecules in vivo. Methods Mol Biol. 2009;544:461–481. doi: 10.1007/978-1-59745-483-4_31. [DOI] [PubMed] [Google Scholar]
- 23.Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity. 2013;39(5):806–818. doi: 10.1016/j.immuni.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barral P, Sánchez-Niño MD, van Rooijen N, Cerundolo V, Batista FD. The location of splenic NKT cells favours their rapid activation by blood-borne antigen. EMBO J. 2012;31(10):2378–2390. doi: 10.1038/emboj.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ceriotti A, Colman A. Trimer formation determines the rate of influenza virus haemagglutinin transport in the early stages of secretion in Xenopus oocytes. J Cell Biol. 1990;111(2):409–420. doi: 10.1083/jcb.111.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaaijk P, et al. Preclinical safety and immunogenicity evaluation of a nonavalent PorA native outer membrane vesicle vaccine against serogroup B meningococcal disease. Vaccine. 2013;31(7):1065–1071. doi: 10.1016/j.vaccine.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 27.Shi J, et al. Engineered red blood cells as carriers for systemic delivery of a wide array of functional probes. Proc Natl Acad Sci USA. 2014;111(28):10131–10136. doi: 10.1073/pnas.1409861111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan Q, et al. Differential diagnosis of pandemic (H1N1) 2009 infection by detection of haemagglutinin with an enzyme-linked immunoassay. Clin Microbiol Infect. 2011;17(10):1574–1580. doi: 10.1111/j.1469-0691.2010.03413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, et al. Broad cross-protection against H5N1 avian influenza virus infection by means of monoclonal antibodies that map to conserved viral epitopes. J Infect Dis. 2009;199(1):49–58. doi: 10.1086/594374. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, et al. Serological survey of antibodies to influenza A viruses in a group of people without a history of influenza vaccination. Clin Microbiol Infect. 2011;17(9):1347–1349. doi: 10.1111/j.1469-0691.2011.03538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He R, et al. Facile synthesis of pentacle gold-copper alloy nanocrystals and their plasmonic and catalytic properties. Nat Commun. 2014;5:4327. doi: 10.1038/ncomms5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song XR, et al. Co9 se8 nanoplates as a new theranostic platform for photoacoustic/magnetic resonance dual-modal-imaging-guided chemo-photothermal combination therapy. Adv Mater. 2015;27(21):3285–3291. doi: 10.1002/adma.201405634. [DOI] [PMC free article] [PubMed] [Google Scholar]