

Significance
Hydrogen has been recognized as one of the most promising energy carriers for the future, because it can generate enormous energy by clean combustion chemistry without any greenhouse gas emissions. Water splitting under visible light irradiation is an ideal route to cost-effective, large-scale, and sustainable hydrogen production, but it is challenging, because it requires a rare photocatalyst that carries a combination of suitable band gap energy, appropriate band positions, and photochemical stability. To create this rare photocatalyst, we engineered the band edges of BiVO4 by simultaneously substituting In3+ for Bi3+ and Mo6+ for V5+ in the host lattice of monoclinic BiVO4, which induced partial phase transformation from pure monoclinic BiVO4 to a mixture of monoclinic BiVO4 and tetragonal BiVO4.
Keywords: photocatalysis, water splitting, band edge engineering, bismuth vanadate, dual doping
Abstract
Through phase transition-induced band edge engineering by dual doping with In and Mo, a new greenish BiVO4 (Bi1-XInXV1-XMoXO4) is developed that has a larger band gap energy than the usual yellow scheelite monoclinic BiVO4 as well as a higher (more negative) conduction band than H+/H2 potential [0 VRHE (reversible hydrogen electrode) at pH 7]. Hence, it can extract H2 from pure water by visible light-driven overall water splitting without using any sacrificial reagents. The density functional theory calculation indicates that In3+/Mo6+ dual doping triggers partial phase transformation from pure monoclinic BiVO4 to a mixture of monoclinic BiVO4 and tetragonal BiVO4, which sequentially leads to unit cell volume growth, compressive lattice strain increase, conduction band edge uplift, and band gap widening.
Photocatalytic water splitting with a particulate semiconductor powered by sunlight is an ideal route to cost-effective, large-scale, and sustainable hydrogen production because of its extreme simplicity. However, it is challenging, because it requires a rare photocatalyst that carries a combination of suitable band gap energy, appropriate band positions, and photochemical stability (1–5). Thus, reproducible photocatalytic systems for visible light-driven overall water splitting (OWS) by one-step photoexcitation are also rare, although there were several reports of such systems (4–6). In the best-known successful case, Domen and coworkers (5) reported in 2005 that a solid solution of GaN and ZnO [(Ga1−xZnx)(N1−xOx)] was a stable photocatalyst that could split water into H2 and O2 under visible light when modified with a cocatalyst. This system remains the most active and reproducible one-step OWS photocatalyst responsive to visible light so far (4).
Scheelite monoclinic (m-) BiVO4 is a well-documented photocatalyst having suitable band gap energy (Eg ∼ 2.4 eV) for absorbing visible light (7–10). Also, it is chemically stable in aqueous solution under light irradiation. Thus, it functions as an excellent photocatalyst for O2 evolution under visible light in the presence of an appropriate electron acceptor (e.g., AgNO3). However, because the bottom of its conduction band is located at a more positive potential than the potential of water reduction [0 VRHE (reversible hydrogen electrode) at pH 7], it is incapable of evolving H2. In addition, it shows poor charge transport characteristics (11) and weak surface adsorption properties (12), causing low photocatalytic activity. To overcome these weaknesses, a variety of strategies, such as heterojunction structure formation (11, 13, 14), loading cocatalysts (8, 15–17), and impurity doping (1, 7, 12, 18–23), has been attempted. These strategies were successful in improving BiVO4’s oxidation capability for photoelectrochemical water oxidation (1, 11, 13, 15, 19, 24–28) as well as the Z scheme (two-photon excitation) water splitting system (29). Also, cocatalyzed BixY1-xVO4 (x ∼ 0.5) (7, 8) and cocatalyzed Bi0.5La0.5VO4 (23) promoted OWS by raising the conduction band edge (CBE) position, but OWS under visible light irradiation over BiVO4-based photocatalysts has not been fully shown.
To meet this challenge, we developed greenish BiVO4 (GBVOx; x = atom ratio of In and Mo), Bi1-xInxV1-xMoxO4, by simultaneously substituting In3+ for Bi3+ and Mo6+ for V5+ in the host lattice of m-BiVO4. The new GBVOx photocatalyst has a slightly larger band gap energy than the usual yellow scheelite m-BiVO4 as supported by the unique color change to green and a higher (more negative) conduction band than H+/H2 potential (0 VRHE at pH 7). Consequently, as depicted in Fig. 1, GBVOx is able to split water into H2 and O2 under visible light irradiation without using any sacrificial reagents (e.g., CH3OH or AgNO3). Herein, we report the dual-metal doping effects on the optical absorption behavior, crystal structure, and electronic band structure of BiVO4, which led to one-photon OWS under visible light irradiation. We elucidate the physical origin of the augmented photoresponse behaviors of GBVOx through density functional theory (DFT) calculation of electronic structure as well as a variety of physical and electrochemical characterizations.
Fig. 1.

OWS reaction mechanism by GBVOx.
Results and Discussion
Physical Properties of GBVOx.
The magnified X-ray diffraction (XRD) patterns of pristine BiVO4 and four GBVOx samples with different target atom ratios (x = 0.02, 0.05, 0.10, and 0.15) are shown in Fig. 2A (more details are in SI Appendix, Fig. S1 A–D) to identify the evolution of the crystal structure by doping. Split peaks around 35° are merged into one peak as the dopant ratio (x) increases, indicating phase change of BiVO4 from m to tetragonal (t) (11). Furthermore, Rietveld analysis was performed using X'pert Plus 3.0 software (PANalytical) to determine the fraction of m- and t-phases as illustrated in Fig. 2B and SI Appendix, Fig. S2. As the dopant ratio (x in GBVOx) increases, the crystal structure evolves from almost pure m-BiVO4 (clinobisvanite; space group: 12/a/(15); JCPDS card no. 014–0688) to a mixture of m- and t-BiVO4, where t-BiVO4 fraction reaches a maximum of 60% at x = 0.10. This doping-induced crystal structure evolution results in unit cell volume growth (Fig. 2B) that generates compressive lattice strain, which is discussed in SI Appendix, section S4 and visualized in SI Appendix, Fig. S1 E and F. In particular, the compressive lattice strain is the key physical driving force to lift the conduction band of GBVOx over H+/H2 potential (0 VRHE at pH 7), taking collectively both experimental (SI Appendix, Fig. S1 E and F) and theoretical findings (as discussed later).
Fig. 2.

Photophysical characterization of GBVOx. (A) Magnified view of powder XRD patterns of pristine BiVO4 and all GBVOx samples. (B) Crystal-phase diagram with calculated unit cell volume for different x values of GBVOx. (C) UV/Vis absorption spectra of pristine BiVO4 and all GBVOx samples. Inset shows color change of the samples. (D) Mott–Schottky plot of pristine BiVO4, GBVO0.05, GBVO0.10, and GBVO0.15.
The UV/Vis (visible) absorption spectra of all samples are shown in Fig. 2C. With higher x values of GBVOx, the greater blue shift is observed, indicating that the optical band gap energy of GBVOx increases with dopant level, whereas the blue shift is not caused by the single Mo doping into BiVO4 (SI Appendix, Fig. S4F). Actually, the blue shift trend is consistent with the band gap energy transition estimated by extrapolating the linear part of the (αhυ)2- vs. hυ-plot to the energy axis (1) as shown in SI Appendix, Fig. S3. According to the Tauc plot, the band gap energy increases from 2.43 eV of pristine BiVO4 to 2.50 eV of GBVO0.10. Furthermore, the alteration in the optical band gap energy corresponds to the color evolution of GBVOx powder samples from yellow to greenish as shown in Fig. 2C, Inset and SI Appendix, Fig. S4. Thus, the ultimate physical driving force for the apparent color evolution is also the compressive lattice strain that widens the band gap by lifting the conduction band. The Mott–Schottky plot in Fig. 2D that displays the conduction band of GBVO0.10 is elevated to higher than 0 V (vs. RHE at pH 7) by 97.2 mV or ∼151 mV from that of pristine BiVO4 (a more clear visualization of the contrast between schematic band structures of pristine BiVO4 and GBVO0.10 is in SI Appendix, Fig. S5). This finding indicates that photocatalytic water reduction, impossible with pristine BiVO4, becomes possible with GBVO0.10, because the dual doping lifts the CBE above the proton reduction potential of 0 VRHE (at pH 7). For the valance band of GBVO0.10, it is 81 mV higher than that of pristine BiVO4, but it is still far below the water oxidation potential of 1.23 VRHE (at pH 7) as shown in SI Appendix, Fig. S5.
The field emission SEM images in SI Appendix, Fig. S6 reveal that all samples consist of irregular polyhedrons of unsystematic size distribution, typical of powder samples synthesized by solid-state reactions. The real atomic ratios in all GBVOx samples determined by the inductively coupled plasma analysis in SI Appendix, Table S1 are close enough to the target values. The high-resolution transmission EM (HR-TEM) and energy-dispersive X-ray spectroscopy elemental mapping in SI Appendix, Figs. S6–S10 show that Bi, V, In, and Mo are homogeneously distributed within the GBVOx particle. All of these characterization results univocally prove that In3+ and Mo6+ have been effectively inserted into the BiVO4 lattice. The HR-TEM image in SI Appendix, Fig. S9F illustrates the lattice spacing of 0.450 nm corresponding to the interplanar spacing of the (040) plane of GBVO0.10. XPS binding energies in SI Appendix, Fig. S11 indicate that the involved elements are all in their stable oxidation states of Bi3+, V5+, Mo6+, and In3+.
OWS Under Visible Light.
The OWS reaction was carried out under visible light irradiation (λ ≥ 420 nm; 500-W Hg-arc lamp) with 0.3 g photocatalyst powder dispersed in 100 mL distilled water at pH 7. In phase I shown in Fig. 3, GBVO0.10 (the most active GBVOx) showed stoichiometric H2/O2 evolution even without modification by any cocatalyst. Hence, the GBVOx can split pure water by visible light-driven OWS without any sacrificial reagents or additives. In particular, this phenomenon represents the first example, to our knowledge, of a pure water-splitting photocatalyst responding to visible light without any metal cocatalyst. Next, we purged the system with N2 and tested the OWS reaction after photodepositing 3 wt% RuO2 on GBVO0.10 as a cocatalyst. The photocatalytic activity was significantly improved by adding an RuO2 cocatalyst that collects electrons/holes and provides active sites for catalytic water reduction and/or oxidation. The other common cocatalysts, such as Pt, Rh, RhxCryO3, and PtxCryO3 (4–6), were much less effective on GBVO0.10. Phases II–IV present repeated runs of the same catalysts to show the stability of GBVO0.10. In phase V, heat treatment (623 K in air for 1 h) further intensified the photocatalytic activity of RuO2-cocatalyzed GBVO0.10. This treatment was aimed at converting the photodeposited RuO2•xH2O on GBVO0.10 into the more stable and active form of cocatalyst (i.e., RuO2) (30).
Fig. 3.
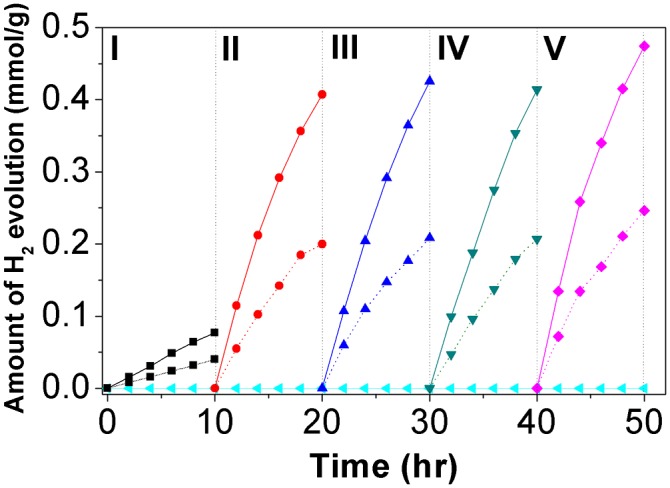
OWS by GBVO0.10 under the visible light (λ ≥ 420 nm) irradiation. Solid and dashed lines indicate evolved H2 and O2, respectively. Baseline represents a control experiment by pristine BiVO4. Phase I, unmodified GBVO0.10; phases II–IV, 3 wt% RuO2/GBVO0.10; phase V, heat treated 3 wt% RuO2/GBVO0.10.
The rate of hydrogen evolution (∼17 μmol h−1) from this 3 wt% RuO2 on GBVO0.10 could be compared with that of the best-known example [∼56 μmol h−1; i.e., 5 wt% RuO2-loaded (Ga1−xZnx)(N1−xOx) under visible light irradiation (λ ≥ 400 nm; 450-W Hg-arc lamp)] (5). The activity of our photocatalyst corresponds to an apparent quantum yield of 3.2% under overall visible light (420–800 nm) irradiation, which was calculated from the amount of evolved hydrogen and photon flux measured by chemical actinometry with ferrioxalate (SI Appendix, section S3). The value is also compared with 5.9% at 420–440 nm for (Ga1−xZnx)(N1−xOx) photocatalysts (31). Considering that we used a longer wavelength filter (λ ≥ 420 nm), we can state that the photocatalytic activity of GBVO0.10 is comparable with that of the best-known (Ga1−xZnx)(N1−xOx) photocatalyst. However, this work represents the first report, to our knowledge, of visible light-driven OWS over modified BiVO4 in pure water of pH 7 without cocatalyst. This work also represents the first, to our knowledge, OWS photocatalyst based on d(0) electron configuration (V5+), whereas (Ga1−xZnx)(N1−xOx) is made of d(10) configuration (Zn2+ and Ga3+). The incident photon to current conversion efficiency was measured for unmodified GBVO0.10 film (details are in SI Appendix, section S2). As shown in SI Appendix, Fig. S12, incident photon to current conversion efficiency of 0.5–2% is observed in 350–480 nm, with the shape of its curve well reflecting the absorption spectrum in Fig. 2C. It confirms that the photoresponse of the material corresponds to light absorption by the band to band transition (Fig. 3).
To understand the nature of the photocatalytic water splitting with GBVOx in more detail, its half reactions were studied in the presence of sacrificial reagents. Namely, Ag+ ions from AgNO3 act as electron scavengers, and CH3OH acts as a hole scavenger:
In SI Appendix, Fig. S13A, O2 evolution rates increased greatly with the single Mo doping (BiV0.98Mo0.02O4). This increase is because of the improved charge transfer properties of BiVO4 as reported earlier (21). However, the dual-metal doping improved the water oxidation activity much further, showing a maximum at the low doping level of GBVO0.02. According to the numerical data from SI Appendix, Fig. S13A, GBVO0.02 has dramatically increased photocatalytic water oxidation activity by factors of about 20 and 5 relative to pristine BiVO4 and single-doped BiV0.98Mo0.02O4, respectively. SI Appendix, Fig. S14 shows that the single In doping did not induce any t-phase formation. However, it increased the rate of O2 evolution in Bi0.99In0.01VO4 and reduced the rate at the higher In doping level of Bi0.98In0.02VO4.
As SI Appendix, Fig. S13B exhibits, the effects of doping on H2 evolution were quite different from those of doping on O2 evolution. Neither the single doping of Mo6+ or In3+ nor the low level of dual doping (x = 0.02) produced H2. The optimum doping level for H2 evolution was found to be x = 0.10. This trend indicates that, when 10% of In3+/Mo6+ is inserted into the BiVO4 lattice, the conduction band gains more negative (higher) potential than 0 VRHE (at pH 7) to achieve photocatalytic water reduction. However, even with the appropriate In3+/Mo6+ dopant concentration (x = 0.10), the H2 evolution rate was two orders of magnitude smaller than the O2 evolution rate. This comparison reveals that water reduction using photogenerated electrons is the rate-determining step in the OWS reaction by GBVO0.10. In this sense, the main role of RuO2 is most likely to provide augmented reaction sites for H2 evolution (water reduction).
The stability of GBVO0.10 is supported by SI Appendix, Fig. S15C, revealing no difference in the XRD patterns of GBVO0.10 before and after the 40-h OWS reaction. The stability is also confirmed by the turnover number of RuO2 loaded on GBVO0.10 (number of hydrogen atoms produced/number of Ru atoms in RuO2 as discussed in SI Appendix, section S4). The turnover number is estimated to be 11 during the OWS reaction phases II–IV shown in Fig. 3, guaranteeing sustainable catalytic cycles on GBVO0.10. However, at 15% In3+/Mo6+ dopant concentration (GBVO0.15), the crystal structure is damaged after a 40-h OWS reaction, which is shown in SI Appendix, Fig. S15C. This inferior crystal structure stability of GBVO0.15 is most likely caused by the too high In3+/Mo6+ dopant concentration of 15%. According to the volumetric strain method and the Williamson–Hall method discussed in SI Appendix, Figs. S1 E and F and S5, the CBE position is proportional to the compressive (minus) lattice stain up to 10% dopant concentration. However, the proportional relation is not valid anymore at 15% dopant concentration because of the inferior crystal structure stability at the too high In3+/Mo6+ doping ratio.
First Principle DFT Calculations.
Collectively taking into account all of the experimental results from a wide array of instruments and methods, this study has found that there is a significant positive correlation between the compressive (minus) lattice strain and the CBE at least up to 10% In3+/Mo6+ dopant concentration. This positive correlation provides the physical driving force to transform (only) O2-evolving yellow BiVO4 into (both) H2/O2-evolving GBVOx by lifting the CBE, widening the band gap, and entailing the apparent color change. This finding based on a variety of experimental outcomes is also corroborated by the theoretical study using DFT calculations as follows.
The effect of In3+/Mo6+ dual doping was investigated by first principle DFT electronic structure calculation on scheelite type m-BiVO4 (space group: I2/a) and t-BiVO4 (space group: I41/amd). The detailed calculation procedures were based on those described in previous studies (1, 32). The lattice constants as well as atomic coordinates were simultaneously optimized, and the obtained lattice parameters for a conventional cell were a = 7.306 Å, b = 11.747 Å, c = 5.167 Å, and β = 135.003° for m-BiVO4 and a = b = 7.307 Å and c = 6.587 Å for t-BiVO4. To study the effect of In3+/Mo6+ substitution into Bi3+/V5+ sites, we used 96-atom BiVO4 host supercell of 2a × b × 2c and conventional cell of √2a × √2b × 2c for m-BiVO4 and t-BiVO4, respectively. We examined a single pair of In and Mo atoms’ substitution (i.e., the doping concentration x = 0.0625 in BiVO4). The calculated electronic density of states is shown in Fig. 4, where the origin of energy is set to be the valence band edge (VBE) of pristine m-BiVO4. The energy levels of m-BiVO4 and t-BiVO4 with and without doping are effectively compared by aligning the deep-lying O 2s orbital levels.
Fig. 4.

Density of states of the t-BiVO4 and m-BiVO4 system with and without In3+/Mo6+ dual doping. The energy levels in different structures are aligned comparing the deep-lying oxygen 2s orbital. The Fermi level of In3+/Mo6+ doped structure is near CBE, which is shown with vertical dotted lines. The VBE in t-structure is −0.32 eV lower than in m-BiVO4. In each crystal structure, doping causes band gap narrowing.
For m-BiVO4, the calculated band gap energy is 2.02 eV without doping and 1.84 eV with doping. This calculation means that the band gap is slightly narrowed on doping. The VBE of doped m-BiVO4 is at the same energy level as that of pristine m-BiVO4, and the CBE is 0.18 eV lower than pristine m-BiVO4. Both of these calculations are contradictory to the experimental findings of increased band gap energy and higher CBE induced by In3+/Mo6+ doping. In the case of t-BiVO4, the band gap in the equilibrium cell volume is 2.47 eV without doping and 2.31 eV with dual doping. Although the band gap of t-BiVO4 is larger (∼0.45 eV) than that of m-BiVO4, the CBE is slightly higher (∼0.13 eV) than its counterpart, because the average electronic potential is deeper (work function is higher) in t-BiVO4 compared with in m-BiVO4. Like m-BiVO4 mentioned above, the band gap of t-BiVO4 narrows upon doping, and the CBE of doped t-BiVO4 is slightly lower than that of pristine m-BiVO4, which is also contradictory to the experimental results.
This apparent inconsistency between calculation and experiments can be solved by considering the partial phase change of m-BiVO4 to t-BiVO4 on dual doping. Indeed, the best-performance water splitter, GBVO0.10, contains ∼60% of t-phase (Fig. 2B). Because the synthesized GBVOx crystal has the mixed structure of varying fraction m- and t-phases with respect to the doping concentration, each crystal structure would not have its equilibrium lattice constants. The conventional cell volume of t-BiVO4 (351.68 Å3) containing four BiVO4 units is quite larger than that of m-BiVO4 (313.54 Å3). The energy-level alignment and the total energy variation of m-BiVO4 and t-BiVO4 are illustrated with respect to the cell volume in Fig. 5. For doped m-BiVO4 with larger cell volume, the band gap is smaller than that of pristine m-BiVO4 with the equilibrium lattice constants. Thus, only the energy-level alignment of t-BiVO4 is compared with the CBE and VBE of pristine m-BiVO4, as shown in Fig. 5A. According to the comparison, compressed t-BiVO4 with In3+/Mo6+ substitution has larger band gap energy as well as higher CBE position than pristine m-BiVO4.
Fig. 5.

(A) Cell volume dependency of CBE and VBE positions in t-BiVO4 systems. The horizontal dashed lines indicate corresponding energy positions in pristine m-BiVO4. (B) Total energy vs. cell volume plot of m-BiVO4 and t-BiVO4. The points are calculation results, and the lines stand for the Birch fit to the equation of state.
This DFT calculation outcome supports the experimental finding of higher CBE. To be more specific, In3+/Mo6+ substitution causes partial phase transformation from pure m-BiVO4 to a mixture of m-BiVO4 and t-BiVO4, and the phase transformation results in unit cell volume growth and compressive lattice strain increase. As a result of the compressive lattice strain increase, GBVO0.10 (having the optimum mixed crystal structure of ∼60% t-BiVO4 and ∼40% m-BiVO4 to maximize the compressive lattice strain) can take advantage of uplifted conduction band edge to achieve OWS under visible light without any additives. The physical mechanism on how In3+/Mo6+ dual doping triggers the partial phase transformation from pure m-BiVO4 to a mixture of m-BiVO4 and t-BiVO4 is investigated in the following discussion.
To figure out the physical reason why t-phase expands as doping concentration grows, we estimated the total energy of each case as shown in Fig. 5B. Based on the total energy values, we could also estimate the formation energy of In3+/Mo6+ dual dopants within m-BiVO4 and t-BiVO4 according to the cell volume by calculating the total energy differences between pristine BiVO4 and doped BiVO4. There is no ambiguity to determine the chemical potentials, because the contributions of cation chemical potentials are compensated. The formation energy of In3+/Mo6+ dopants in m-BiVO4 is about 1.31 eV and independent of lattice strain, but that in t-BiVO4 strongly depends on cell volume variation. The formation energy of In3+/Mo6+ dopants in t-BiVO4 is 1.79 eV in its equilibrium cell volume (1,407 Å3), which is about 0.48 eV higher than that in m-BiVO4 at 1,253 Å3. However, as the cell volume goes down, the formation energy of In3+/Mo6+ dopants in t-BiVO4 decreases and eventually gets smaller than that in m-BiVO4 when the cell volume is smaller than ∼1,320 Å3. In addition, the total energy of doped t-BiVO4 gets smaller than that of undoped t-BiVO4 when the cell volume is smaller than 1,254 Å3. This tendency suggests that, as the cell volume becomes smaller and smaller, the formation of In3+/Mo6+ dopants is more promoted within t-BiVO4 rather than within m-BiVO4, and ultimately, the compressed t-crystal structure can be more stabilized with doping than without doping. This doping-induced phase change and the subsequent rise of the CBE represent a new concept to create a visible light-active OWS photocatalyst by the band structure engineering.
In summary, through In3+/Mo6+ dual doping into the host lattice of m-BiVO4, we have created a new visible light-responsive OWS photocatalyst. The resulting GBVOx splits water into H2 and O2 by one-step photoexcitation, because it has an upshifted CBE transforming (only) O2-evolving yellow BiVO4 into (both) H2/O2-evolving GBVOx, a one-photon OWS photocatalyst. The DFT calculation indicates that In3+/Mo6+ dual doping triggers partial phase transformation from pure m-BiVO4 to a mixture of m-BiVO4 and t-BiVO4, which sequentially leads to unit cell volume growth, compressive lattice strain increase, conduction band edge uplift, and band gap widening. This domino effect is also corroborated experimentally. The GBVOx is an active and stable one-photon OWS photocatalyst made of earth-abundant elements and works in pure water without any additives.
Materials and Methods
Sample Preparation.
Powder samples of GBVOx were prepared by solid-state reaction. Each stoichiometric amount of bismuth oxide (Aldrich; 99.999%), vanadium oxide (Aldrich; 98%), indium oxide (Alfa Aeser; 99.9%), and molybdenum oxide (Aldrich; 99.99%) was mixed well by grinding in an agate mortar and manually pelletized under 2,500 psi. All pellets were preheated at 873 K for 5 h, and then, they were vigorously reground and manually repelletized as mentioned earlier. These pellets were heated again at 1,073 K for 3 h. After this main heating, they were thoroughly reground to get powder samples.
Modification of GBVO0.10 with RuO2.
GBVO0.10 was modified by photodepositing 3 wt% RuO2 (the loading had been optimized). Specifically, the photodeposition was carried out at room temperature under atmospheric pressure in a closed Pyrex glass vessel containing nitrogen-purged suspension of GBVO0.10 powder and RuCl36H2O in 50 mL methanol and 50 mL distilled water under visible light irradiation for 8 h. The light source was an Hg-arc lamp (500 W; Oriel) equipped with a UV cutoff filter (λ ≥ 420 nm). After this photodeposition, RuO2-loaded GBVO0.10 was vacuum-filtered, washed with distilled water, and dried in an air oven at 70 °C for 12 h. Finally, the dried powder sample was heated at 623 K for 1 h.
Physicochemical Characterization.
The structural properties of GBVOx were investigated by using powder XRD (X’pert Pro; Phillips). Rietveld analysis was performed using X'pert Plus 3.0 software (PANalytical) to calculate the phase ratios of m-structure to t-structure in GBVOx. The UV-visible diffuse reflectance spectra were measured with a UV-visible spectrometer (UV-2401PC; Shimadzu Co.) equipped with integrated sphere method. The particle size and morphology were examined by field emission SEM (JEOL JSM-6330F). The HR-TEM images, high-angle annular dark field images, and corresponding elemental mapping images were obtained by using Cs-corrected high-resolution scanning transmission EM (JEM 2200FS; 200 kV; JEOL) at the National Research Center for Nanomaterial Technology at Pohang University of Science and Technology in Korea. The atomic ratios of bismuth to indium and vanadium to molybdenum in GBVOx were analyzed by using inductively coupled plasma.
Photocatalytic Activity Measurement Under Visible Light Irradiation.
The structural properties of GBVOx were investigated by using powder XRD. The photocatalytic activities under visible light were investigated by measuring H2 and O2 evolution at room temperature under atmospheric pressure in a closed Pyrex glass vessel (193.5, 187, and 189 mL) containing nitrogen-purged suspension of 0.3 g photocatalyst powder in 100 mL distilled water. The light source was an Hg-arc lamp (450 W; Oriel) equipped with a UV cutoff filter (λ ≥ 420 nm). The evolved amounts of H2 and O2 were analyzed by a gas chromatograph (HP5890) with a thermal conductivity detector and a molecular sieve 5-A column.
Supplementary Material
Acknowledgments
We thank N. H. Ahn (Pohang University of Science and Technology) for assistance with collecting X-ray diffraction data. W.J.J. thanks the Samsung Foundation of Culture for a Samsung Scholarship. This work was supported by the Brain Korea Plus Program of the Ministry of Education, Korean Centre for Artificial Photosynthesis Grant 2009-0093880, the Ministry of Trade, Industry and Energy of Republic of Korea Project 10050509, and the Ulsan National Institute of Science and Technology. This work was also supported, in part, by the US Army Research Laboratory and the US Army Research Office through Institute for Soldier Nanotechnologies Contract W911NF-13-D-0001.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1509674112/-/DCSupplemental.
References
- 1.Jo WJ, et al. Phosphate doping into monoclinic BiVO4 for enhanced photoelectrochemical water oxidation activity. Angew Chem Int Ed Engl. 2012;51(13):3147–3151. doi: 10.1002/anie.201108276. [DOI] [PubMed] [Google Scholar]
- 2.Grätzel M. Solar energy conversion by dye-sensitized photovoltaic cells. Inorg Chem. 2005;44(20):6841–6851. doi: 10.1021/ic0508371. [DOI] [PubMed] [Google Scholar]
- 3.Bard AJ, Fox MA. Artificial photosynthesis: Solar splitting of water to hydrogen and oxygen. Acc Chem Res. 1995;28(3):141–145. [Google Scholar]
- 4.Maeda K, et al. Photocatalyst releasing hydrogen from water. Nature. 2006;440(7082):295. doi: 10.1038/440295a. [DOI] [PubMed] [Google Scholar]
- 5.Maeda K, et al. GaN:ZnO solid solution as a photocatalyst for visible-light-driven overall water splitting. J Am Chem Soc. 2005;127(23):8286–8287. doi: 10.1021/ja0518777. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Liu L, Yu PY, Mao SS. Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science. 2011;331(6018):746–750. doi: 10.1126/science.1200448. [DOI] [PubMed] [Google Scholar]
- 7.Liu H, et al. Novel photocatalyst of V-based solid solutions for overall water splitting. J Mater Chem. 2011;21(41):16535–16543. [Google Scholar]
- 8.Wang D, et al. Photocatalytic water oxidation on BiVO4 with the electrocatalyst as an oxidation cocatalyst: Essential relations between electrocatalyst and photocatalyst. J Phys Chem C. 2012;116(8):5082–5089. [Google Scholar]
- 9.Kudo A, Omori K, Kato H. A novel aqueous process for preparation of crystal form-controlled and highly crystalline BiVO4 powder from layered vanadates at room temperature and its photocatalytic and photophysical properties. J Am Chem Soc. 1999;121(49):11459–11467. [Google Scholar]
- 10.Tokunaga S, Kato H, Kudo A. Selective preparation of monoclinic and tetragonal BiVO4 with scheelite structure and their photocatalytic properties. Chem Mater. 2001;13(12):4624–4628. [Google Scholar]
- 11.Hong SJ, Lee S, Jang JS, Lee JS. Heterojuction BiVO4/WO3 electrodes for enhanced photoactivity of water oxidation. Energy Environ Sci. 2011;4(5):1781–1787. [Google Scholar]
- 12.Yao W, Iwai H, Ye J. Effects of molybdenum substitution on the photocatalytic behavior of BiVO4. Dalton Trans. 2008;(11):1426–1430. doi: 10.1039/b713338c. [DOI] [PubMed] [Google Scholar]
- 13.Long M, Cai W, Kisch H. Visible light induced photoelectrochemical properties of n-BiVO4 and n-BiVO4/p-Co3O4. J Phys Chem C. 2008;112(2):548–554. [Google Scholar]
- 14.Jiang HQ, Endo H, Natori H, Nagai M, Kobayashi K. Fabrication and efficient photocatalytic degradation of methylene blue over CuO/BiVO4 composite under visible-light irradiation. Mater Res Bull. 2009;44(3):700–706. [Google Scholar]
- 15.Kim TW, Choi K-S. Nanoporous BiVO4 photoanodes with dual-layer oxygen evolution catalysts for solar water splitting. Science. 2014;343(6174):990–994. doi: 10.1126/science.1246913. [DOI] [PubMed] [Google Scholar]
- 16.Kohtani S, Tomohiro M, Tokumura K, Nakagaki R. Photooxidation reactions of polycyclic aromatic hydrocarbons over pure and Ag-loaded BiVO4 photocatalysts. Appl Catal B. 2005;58(3-4):265–272. [Google Scholar]
- 17.Kohtani S, et al. Adsorptive and photocatalytic properties of Ag-loaded BiVO4 on the degradation of 4-n-alkylphenols under visible light irradiation. Catal Commun. 2005;6(3):185–189. [Google Scholar]
- 18.Yao W, Ye J. Photophysical and photocatalytic properties of Ca(1-x)BixVxMo(1-x)O4 solid solutions. J Phys Chem B. 2006;110(23):11188–11195. doi: 10.1021/jp0608729. [DOI] [PubMed] [Google Scholar]
- 19.Luo W, et al. Solar hydrogen generation from seawater with a modified BiVO4 photoanode. Energy Environ Sci. 2011;4(10):4046–4051. [Google Scholar]
- 20.Parmar KPS, et al. Photocatalytic and photoelectrochemical water oxidation over metal-doped monoclinic BiVO(4) photoanodes. ChemSusChem. 2012;5(10):1926–1934. doi: 10.1002/cssc.201200254. [DOI] [PubMed] [Google Scholar]
- 21.Asahi R, Morikawa T, Ohwaki T, Aoki K, Taga Y. Visible-light photocatalysis in nitrogen-doped titanium oxides. Science. 2001;293(5528):269–271. doi: 10.1126/science.1061051. [DOI] [PubMed] [Google Scholar]
- 22.Park HS, et al. Factors in the metal doping of BiVO4 for improved photoelectrocatalytic activity as studied by scanning electrochemical microscopy and first-principles density-functional calculation. J Phys Chem C. 2011;115(36):17870–17879. [Google Scholar]
- 23.Wang Q, Liu H, Yuan J, Shangguan W. Synthesis and characterization of visible-light-responding Bi0.5La0.5VO4 solid solution for photocatalytic water splitting. Chin J Catal. 2009;30(6):565–569. [Google Scholar]
- 24.Sasaki Y, Nemoto H, Saito K, Kudo A. Solar water splitting using powdered photocatalysts driven by Z-schematic inter-particle electron transfer without an electron mediator. J Phys Chem C. 2009;113(40):17536–17542. [Google Scholar]
- 25.Seabold JA, Zhu K, Neale NR. Efficient solar photoelectrolysis by nanoporous Mo:BiVO4 through controlled electron transport. Phys Chem Phys. 2014;16(3):1121–1131. doi: 10.1039/c3cp54356k. [DOI] [PubMed] [Google Scholar]
- 26.Park Y, McDonald KJ, Choi K-S. Progress in bismuth vanadate photoanodes for use in solar water oxidation. Chem Soc Rev. 2013;42(6):2321–2337. doi: 10.1039/c2cs35260e. [DOI] [PubMed] [Google Scholar]
- 27.Zhong DK, Choi S, Gamelin DR. Near-complete suppression of surface recombination in solar photoelectrolysis by “Co-Pi” catalyst-modified W:BiVO4. J Am Chem Soc. 2011;133(45):18370–18377. doi: 10.1021/ja207348x. [DOI] [PubMed] [Google Scholar]
- 28.Abdi FF, Krol RVD. Nature and light dependence of bulk recombination in Co-Pi-catalyzed BiVO4 photoanodes. J Phys Chem C. 2012;116(17):9398–9404. [Google Scholar]
- 29.Abdi FF, Savenije TJ, May MM, Dam B, Krol RVD. The origin of slow carrier transport in BiVO4 thin film photoanodes: A time-resolved microwave conductivity. J Phys Lett. 2013;4(16):2752–2757. [Google Scholar]
- 30.Mills A, Lee S-K. Platinum group metals and their oxides in semiconductor photosensitization. Platin Met Rev. 2003;47(1):2–12. [Google Scholar]
- 31.Maeda K, Teramura K, Domen K. Effect of post-calcination on photocatalytic activity of (Ga1-xZnx)(N1-xOx) solid solution for overall water splitting under visible light. J Catal. 2008;254(2):198–204. [Google Scholar]
- 32.Yin WJ, Wei SH, Al-Jassim MM, Turner J, Yan Y. Doping properties of monoclinic BiVO4 studied by first-principles density-functional theory. Phys Rev B. 2011;83(15):1–11. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.