

Abstract
Adipose stromal cells (ASC) secrete various trophic factors that assist in the protection of neurons in a variety of neuronal death models. In this study, we tested the effects of human ASC conditional medium (ASC-CM) in human amyotrophic lateral sclerosis (ALS) transgenic mouse model expressing mutant superoxide dismutase (SOD1G93A). Treating symptomatic SOD1G93A mice with ASC-CM significantly increased post-onset survival time and lifespan. Moreover, SOD1G93A mice given ASC-CM treatment showed high motor neuron counts, less activation of microglia and astrocytes at an early symptomatic stage in the spinal cords under immunohistochemical analysis. SOD1G93A mice treated with ASC-CM for 7 days showed reduced levels of phosphorylated p38 (pp38) in the spinal cord, a mitogen-activated protein kinase that is involved in both inflammation and neuronal death. Additionally, the levels of α-II spectrin in spinal cords were also inhibited in SOD1G93A mice treated with ASC-CM for 3 days. Interestingly, nerve growth factor (NGF), a neurotrophic factor found in ASC-CM, played a significant role in the protection of neurodegeneration inSOD1G93A mouse. These results indicate that ASC-CM has the potential to develop into a novel and effective therapeutic treatment for ALS.
Amyotrophic lateral sclerosis (ALS), a progressive neurodegenerative disease, is defined by progressive loss of motor neurons, resulting in paralysis and ultimately death. The process is generally takes 2 to 5 years after diagnosis1. The degeneration of motor neurons in the spinal cord results in muscle atrophy and paralysis2. There are about two ALS incidences per 100,000 persons in the United States annually3. The majority cases are sporadic in origin and 5–10% are familial (FALS)4. Not only are both cases highly similar in clinical course, pathophysiology, and outcome, the mechanisms underlying disease progression are also posited to be the same5. 15–20% FALS are linked to mutant Zn/Cu superoxide dismutase6,7 (SOD1), which led to the creation of SOD1G93A mouse model. SOD1G93A mouse is currently the most extensively studied animal model of ALS8,9 that exhibit the major hallmarks of ALS, which include motor neuron pathology and progressive paralysis8,9. Researchers using this model has discovered an array of processes from ALS onset to end, including glutamate excitotoxicity, glial cell activation, oxidative damage, neuroinflammation, aberrant protein folding, mitochondrial dysfunction and axonal transport4,10.
Oxidative stress and motor neuron excitotoxic death have been linked to neuroinflammatory responses, for example, elevations of pro-inflammatory cytokines in the CNS11,12, astrocyte13 and microglia activation14. These pathogenic hallmarks are thought to play key roles in motor neuron death and ALS progression.
It has been reported that p38 mitogen activated protein kinase (MAPK) are involved in the motor neuron death15,16,17. Phosphorylated p38 MAPK was increased in the spinal cords of SOD1G93A mice15,16,18 and human ALS patients19. Moreover, the inhibition of p38 MAPK pathway extended motor neuron survival and also decreased the activation of microglial in ALS mouse spinal cord17, this suggests that the pathogenic aspect of p38 MAPK pathway play a significant role in ALS motor neuron cell death and neuroinflammation.
Activity of the Ca2+-activated protease, calpain, has also been observed to be increased in the SOD1G93A mouse20,21 due to increased levels of calcium in the cytosol caused by excitotoxicity22,23. Activated calpain cleaves cytoskeletal proteins, such as α-II spectrin and results in the accumulation of α-II spectrin and formation of inclusions located in motor neurons20. Inhibition of calpain via expression of its endogenous inhibitor, calpastatin, slowed degeneration of SOD1G93A motor neurons23.
Since multiple pathways for neuronal death are involved in the development and maintenance of this disease, an effective multi-target approach may be required to treat ALS10. Additionally, ALS treatment can only be given after disease onset, because there are no specific predictors, clinical diagnosis, or biomarkers4. Lots of pharmacologic therapies used in both ALS clinical trials and stringent testing have been unsuccessful24. These failures at pre-clinical or clinical stage may attribute to the treatment’s inability to target more than one neuronal death pathway, resulting in an incomplete or ineffective blocking of motor neuron cell loss and ALS disease progression.
Adipose-derived stem cell conditioned media (ASC-CM), a biologically-derived reagent containing a multitude of neuroprotective and neurotrophic factors, such as brain-derived neurotropic factor (BDNF), nerve growth factor (NGF), vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), and insulin-like growth factor-1 (IGF-1)25,26,27,28 was selected as ASC-CM has been previously shown to be neuroprotective by using both animal and cell culture models of neurodegeneration27,29,30. Our previous studies show that ASC-CM protected against hypoxia-ischemia-induced damage by blocking the activation of p38 MAPK27. NGF is a secreted growth factor in the central and peripheral nervous system. It is important in survival, growth and maintenance of specific types of neurons. The neurodegenerative disorders were due to the lack of hormones or growth factors31. NGF level was decreased in ALS dorsal spinal cord32. In this study, we evaluated the effectiveness of ASC-CM and investigated the role of NGF, a neurotrophic factor identified in the ASC-CM, in ASC-CM treatment for SOD1G93A mouse.
Materials and Methods
Animals
SOD1G93A heterozygous mice that overexpressing human SOD1-G93A mutation and wild type mice (WT) (all with B6SJL genetic background) were purchased form the Jackson Laboratory (Bar Harbor, ME, USA) and bred in the Animal Center of Indiana University School of Medicine. All animal procedures were performed in accordance with the protocols approved and authorized by the Institutional Animal Care and Use Committee at Indiana University School of Medicine. SOD1G93A mice were identified by PCR performance with DNA derived from tail tissue using a protocol provided by The Jackson Laboratory33.
Animal behavioral Assessment
As described in a previous paper33, behavioral assessment was performed beginning at 90 days of age. SOD1G93A mice were randomly assigned to 5 different treatments of “ASC-CM”, “ASC-CM-NGF Ab”, “NGF Ab”, “NGF” or “vehicle”. SOD1G93A mice were tested twice a week on a Rotarod apparatus at a speed of 15 rpm (ENV-575M; Med Associates, Inc., St. Albans, VT, USA) and up to 3 trials per day. Mice that were unable to remain on the Rotarod for 10 minutes were determined as disease onset, this performance was further tested the following day under same parameters to further verify disease onset. Animal that could not right itself in 20 seconds when gently rolled on its side indicated the end stage (surrogate death time point). Mice were checked every morning for mortality and morbidity, and every afternoon for the righting reaction.
Poteomic analysis of the presence of neurotrophic factors in human ACS-CM
Human ASC secrete over 400 proteins into the medium during culture34. We detected interesting factors in ASC-CM using an antibody array (RayBio Human Growth Factor Array I, RayBiotech, GA)
Quantification of NGF by ELISA
NGF level in ASC-CM was measured using Human NGF ELISA kits (Abcam, Cambridge, MA, USA), an enzyme-linked immunosorbent assay (ELISA) kit, according to the manufacturer’s instructions35.
Animal Treatment
Human adipose-derived stem cell conditioned media (ASC-CM) was made as previously described29,36 and injected intraperitoneally (i.p.) at a volume of 200 μl once daily. To neutralize the activity of NGF, a polyclonal anti-NGF antibody (Abcam, Cambridge, MA, USA) was introduced and tested for its specificity. No cross-reactivity has been detected after using it against VEGF, GDNF, IGF1 and BDNF.1 μg/μl of the antibody was incubated with ASC-CM at 5 μg/ml overnight at 4 °C28,37. NGF antibody neutralized ASC-CM treated mice group (ASC-CM-NGF Ab) received 200 ul ASC-CM-NGF antibody once daily. Additionally, as controls, the NGF Ab group daily received 5 μg/ml NGF antibody in 200 μl Basal Media Eagle (BME), the NGF group daily had 200 pg/ml human NGF (R&D systems, Minneapolis, MN, USA) in 200 μl BME, and the vehicle group mice were injected with 200 μl BME. One daily dose of ASC-CM, NGF antibody neutralized ASC-CM, NGF antibody, NGF or BME was given until a humane death endpoint for survival studies, and 3 or 7 days after onset for biochemical and immunohistochemical studies.
Western Blot
Western blot analysis was performed with tissue lysates as previously described33,38 after 3 or 7 days of ASC-CM or BME treatment. Radio-immunoprecipitation assay (RIPA) buffer and protease inhibitor (Roche Diagnostics Corp., Indianapolis, IN, USA) was used to homogenize spinal cord samples. 15 μg/well of protein was loaded onto a 4–12% Bis-Tris gel, electrophoresed, then transferred to nitrocellulose membrane (Hybond N; Amersham Biosciences, Piscataway, NJ, USA). Blots were probed with rabbit polyclonal primary antibody against pp38 (1:1000; Millipore, Temecula, CA, USA), mouse monoclonal antibody against glial fibrillary acidic protein (1:1000, GFAP; Millipore), rabbit polyclonal antibody against ionized calcium binding adaptor molecule 1 (1:1000, Iba-1; Wako Chemicals USA, Inc., Richmond, VA, USA), and goat polyclonal antibody against α-II spectrin (1:200, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) followed by horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Finally, visualized through utilizing enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ, USA), band intensities were quantitated by densitometric analysis (ImageJ, http://rsbweb.nih.gov/ij/).
Immunohistochemistry
Mice were anesthetized and perfusion-fixed with 4% paraformaldehyde (PFA) after treatments with ASC-CM or BME. PFA-fixed and paraffin embedded spinal cords were sectioned (15 μm) serially via the lumber enlargement and immunostained with anti-microtubule-associated protein 2 antibody (1:1000, MAP2; Millipore), then followed by a biotinylated anti-mouse IgG antibody (Sigma-Aldrich Corp.). Sections were visualized under the microscope after applying 3,3′-diaminobenzidine (DAB) substrate solution. A 40x objective light microscope (Nikon, Japan) was used to count large motor neurons in the ventral horn of the lumbar spinal cord39,40. 6–8 sections of each lumbar were counted. Data was reported as number of motor neurons per section as previously described33,41.
Statistical Analysis
One-way analysis of variance (ANOVA) was used for statistical analyses and comparisons for the differences between groups. All data are expressed as mean ± standard error of the mean (SEM). Differences between two means were considered significant when p was equal or less than 0.05.
Results
Neurotrophic factors in ASC-CM
We tested interesting neurotrophic factors in ASC-CM by using a Human Growth Factor Antibody Array (containing 41 growth factor antibodies). Image showed the presence of NGF, VEGF, GDNF, IGF-1 in human ASC-CM. BDNF was detected in human ASC-CM by using enzyme-linked immunosorbent assay (ELISA) kit (Chemicon, Temecula, CA, USA)27.
ASC-CM significantly reduced disease progression and increased life span of SOD1G93A mice, while NGF antibody attenuated this effect
SOD1G93A mice were treated with ASC-CM from disease onset to a humane death endpoint. The duration from disease onset to death was measured. Data was reported as number of days of post-onset survival (Fig. 1A). The SOD1G93A mice given ASC-CM had increased post-onset survival times (31.6 ± 4.1 days, n = 5; ***p < 0.005) as compared to vehicle-treated mice (16.1 ± 1.6 days, n = 7). Interestingly, NGF antibody neutralized ASC-CM didn’t affect post-onset survival times (13.4 ± 2.7 days, n = 5) as compared to vehicle-treated mice. ASC-CM treated mice also had significantly longer lifespans (138.8 ± 3.1 days, n = 5, Fig. 1B) versus the vehicle counterparts (124.4 ± 1.5 days, n = 7) and as expected, NGF antibody attenuated this effect (121.7 ± 1.2, n = 5). As controls, we treated NGF Ab group mice and NGF group mice with 5 μg/ml NGF antibody and 200 pg/ml NGF, respectively and did not observe any effects of the NGF antibody and NGF on post-onset survival (14.6 ± 3.8 days, 18.0 ± 3.2 days vs. 16.1 ± 1.6 days) and lifespans (125.3 ± 5.8 days, 120.1 ± 3.9 days vs. 124.4 ± 1.5 days).
Figure 1. Treatment with ASC-CM extends post-onset survival and lifespan of SOD1G93A mice.
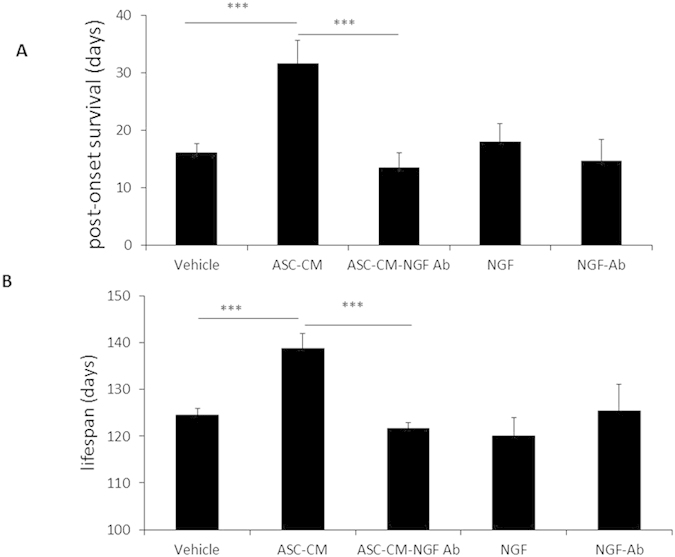
Symptomatic SOD1G93A mice were given daily ASC-CM, NGF depleted ASC-CM treatment or vehicle until endpoint. Duration of disease is reported as days of post-onset survival and measured by days between disease onset and humane death endpoint. Lifespan is the number of days from birth to the humane death endpoint. Symptomatic mice treated with ASC-CM daily (n = 5) had a significant increased survival time as compared to vehicle treated SOD1G93A mice (n = 7). The averages of post-onset survival time were significantly increased in mice given ASC-CM compared to vehicle. ASC-CM-treated mice with disease onset (n = 5) had a significant lifespan extension when compared to vehicle treated SOD1G93A mice (n = 7), but NGF antibody neutralized ASC-CM, NGF antibody, or NGF did not affect post-onset survival times (n = 5) (A). Increased post-onset survival time of ASC-CM treated mice was responsible for the significantly extended lifespan relative to vehicle mice. However, NGF antibody attenuated this effect in ALS mice. As controls, NGF antibody or NGF did not affect lifespans of mice (B). Statistical analysis was performed by using one-way ANOVA. ***p < 0.005 vs. vehicle.
ASC-CM increased the number of motor neurons in the lumbar spinal cord
A positive correlation was observed between extended survival time of ASC-CM treated SOD1G93A mice and the prevention of motor neuron loss. Changes in motor neuron number at 7 days following onset were analyzed in cross-sections of the lumbar spinal cords of SOD1G93A mice (Fig. 2A). BME-treated control mice showed a marked (***p < 0.005) loss of motor neurons in the ventral horn of the lumbar spinal cord versus WT control (7.8 ± 0.84 and 21.9 ± 0.79, respectively; Fig. 2B), but this loss was inhibited by ASC-CM treatment (15.9 ± 2.0, p < 0.01 vs. vehicle). Although there was still significant motor neuron loss (p < 0.05) compared to age-matched and wild type mice, this loss was not as severe in the vehicle group (Fig. 2B).
Figure 2. ASC-CM 7-day treatment of SOD1G93A mice with disease showed increase number of motor neurons in the lumbar spinal.
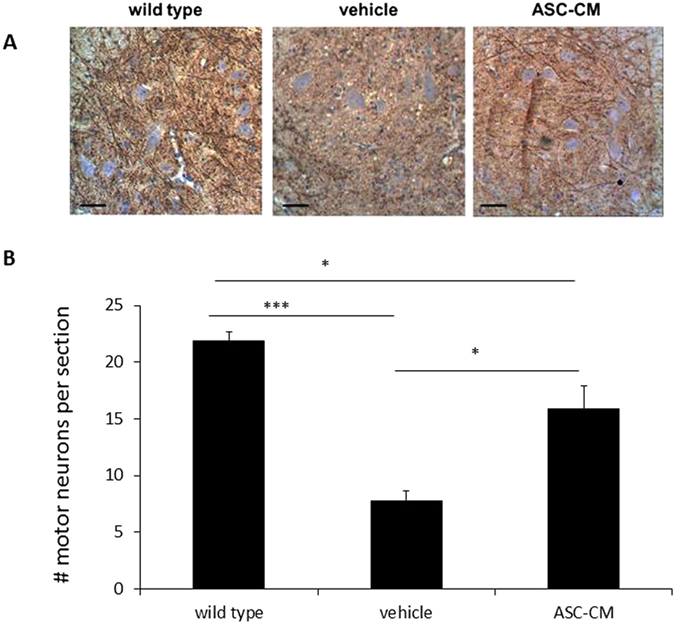
SOD1G93A mice were given ASC-CM or vehicle for 7 days after disease onset. Motor neurons in the spinal cord area were identified by immunoreactivity to MAP2 antibody. Representative images showed an increased number of MAP2-positive neurons in lumbar spinal cords of SOD1G93A mice treated with ASC-CM compared to vehicle. Scale bars: 100 μm. (A). MAP2 immunoreactive images covering the entire cross-sectional area from6–8 lumbar spinal regions for each mouse were quantitated. With ASC-CM treatment, a higher number of motor neurons was observed in SOD1G93A mice with disease onset (n = 5) versus vehicle (n = 5). Wild type control data were generated from 4 mice (B). Experimental group data are presented as averages (±SEM) and statistical analyses were performed using one-way ANOVA. *p < 0.05; ***p < 0.005.
Administration of ASC-CM had no effect on mouse SOD1wild type and mutant human SOD1G93A expression in SOD1G93A mice spinal cords
Studies have shown that the transgene copy number (copies of the transgene with the G → A at position 93, human mutant form of SOD1) affects survival of the SOD1G93A mouse9. In order to determine if ASC-CM has an effect on SOD1 gene expression, immunoblot analyses were performed on spinal cord homogenates after 7 days of ASC-CM treatment (Fig. 3A). Band density measurements showed that levels of both wild type endogenous mouse SOD1 (mSOD1WT) and mutated human SOD1 (hSOD1G93A) exhibited no differences with or without 7-day ASC-CM treatment (Fig. 3B). “Wild type/WT” mice express wild type endogenous mSOD1WT but does not carry the mutated hSOD1G93A transgene; “vehicle” and “ASC-CM” mice are the experimentally-treated SOD1G93A transgenic mice that express wild type endogenous mouse SOD1 (mSOD1WT) in addition to the hSOD1G93A transgene. Actin levels remain unchanged and assayed as loading controls for endogenous mSOD1WT. There were differences in endogenous mSOD1WT levels, which then served as an internal control for mutated hSOD1G93A expression.
Figure 3. Treatment with ASC-CM did not affect expression of endogenous mouse wild type SOD1 and mutated human SOD1 transgene in spinal cord.

Wild type control (WT) and SOD1G93A mice (vehicle and ASC-CM) spinal cords were prepared as detailed in materials and methods. Levels of actin were unchanged and used as an internal loading control for endogenous mouse wild type SOD1 (mSOD1WT). There was no difference in mSOD1WT levels, which was the loading control for mutated human SOD1 (hSOD1G93A). After 7 days of ASC-CM treatment, there was no change in expression of either endogenous mSOD1WT or hSOD1G93A mice in symptomatic SOD1G93A mice. n = 3 per group.
Daily ASC-CM treatment reduced p38 MAP kinase phosphorylation in SOD1G93A mice spinal cords
Phosphorylated p38 (pp38) levels in spinal cords were examined at 3 days (Fig. 4A) and 7 days (Fig. 4B) following disease onset with or without ASC-CM administration during that time period. In agreement with other reports using the SOD1G93A mouse17,19, p38 phosphorylation was elevated in SOD1G93A mice after disease onset at both 3 days (0.72 ± 0.008, p < 0.005 vs. wild type and ASC-CM; Fig. 4A) and 7 days post-onset (0.41 ± 0.05, p < 0.01 vs. wild type; Fig.4B). pp38 levels in spinal cords were significantly decreased with ASC-CM treatment for 3 days (0.36 ± 0.01, p < 0.005 vs. vehicle and wild type; Fig. 4A) and for 7 days (0.18 ± 0.07, p < 0.05 vs. vehicle; Fig.4B) as compared to the vehicle groups. Levels of pp38 in ASC-CM treated animals were higher than WT controls at 3 days (0.14 + 0.02, p < 0.005 vs. ASC-CM group; Fig.4A).
Figure 4. pp38 reduction was observed after 3 or 7 days of ASC-CM treatment in SOD1G93A mice spinal cords after disease onset.

SOD1G93A mice with onset received ASC-CM or vehicle for 3 days or 7 days, while spinal cords were removed for Western blot. (A): (a) Representative immuoblots and (b) quantitation of band density showed reduced pp38 expression in SOD1G93A mice spinal cords after 3 days ASC-CM treatment versus BME treatment. (B): (a) Representative immunoblots and (b) densitometric quantitation demonstrated reduced pp38 expression in 7 days ASC-CM-treated mice compared to BME-treated mice. Unchanged total p38 levels were used as internal loading controls. Values are shown as mean density ± SEM and statistical analyses were performed using one-way ANOVA. *p < 0.05; **p < 0.01; ***p < 0.005; n = 3 per group.
GFAP expression was reduced in the spinal cords of symptomatic SOD1G93A mice treated with ASC-CM
It has been demonstrated that motor neuron loss occurs partly through activated inflammatory cells in the surrounding area42. Because treatment with ASC-CM showed significant neuroprotection of motor neurons possibly via anti-inflammatory mechanisms thus far, glial activation after treatment with ASC-CM was examined to further explore this possibility. Expression of GFAP was measured in the spinal cords of SOD1G93A mice after 3 days of treatment with ASC-CM or vehicle. Representative immunoblots demonstrated that there is significantly reduced GFAP expression with ASC-CM treatment (2.71 ± 0.08) versus vehicle (4.12 ± 0.07; ***p < 0.005; Fig. 5). The GFAP levels observed in the ASC-CM treated group were comparable to that of the wild type controls (2.20 ± 0.08).
Figure 5. 3-day treatment with ASC-CM showed reduced GFAP expression in the spinal cords of SOD1G93A mice after disease onset.
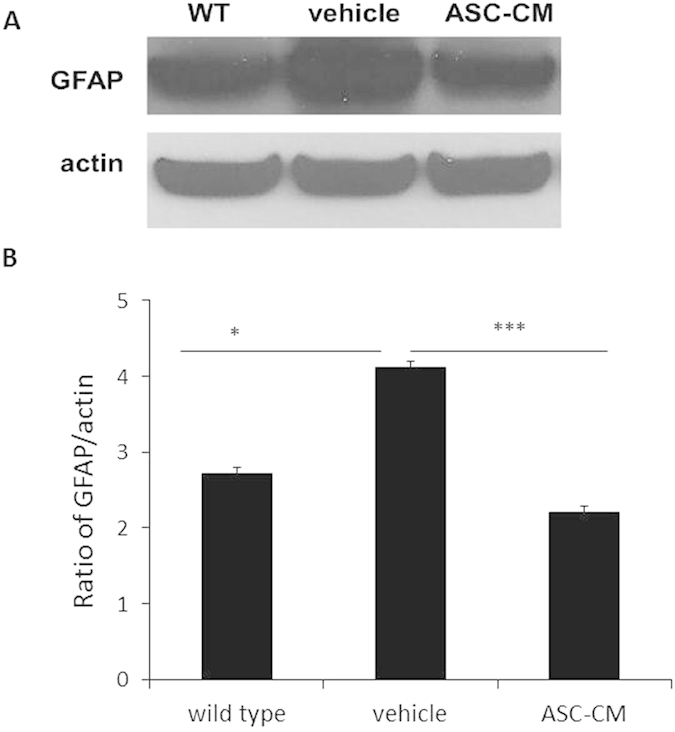
Figure SOD1G93A mice with disease onset were given ASC-CM or vehicle for 3 days. Spinal cords were removed for immunoblot. Actin expression levels remained constant in all groups and were used as internal loading controls. (A) Representative immunoblots and (B) band quantitation demonstrated a decrease in spinal cord GFAP expression after 3-day ASC-CM treatment as compared to vehicle. Experiments were completed in triplicate with n = 3 per group. *p < 0.05; ***p < 0.005.
Administration of ASC-CM to SOD1G93A mice with disease onset decreased CD11b expression in spinal cords
In order to further investigate glial activation after ASC-CM treatment, spinal cords were prepared for Western blot and examined for expression of CD11b, a glial marker found to be induced by neuroinflammation14. CD11b expression was observed at 3 days (Fig. 6A) and 7 days (Fig. 6B) after ASC-CM or vehicle treatment. 3 days of ASC-CM treatment demonstrated no differences in CD11b expression in all groups (group averages ± SEM: 0.073 ± 0.018 for wild type; 0.068 ± 0.012 for vehicle; 0.066 ± 0.018 for ASC-CM, Fig. 6A). However, 7 days of ASC-CM treatment demonstrated a significant decrease in CD11b expression in the spinal cord of ASC-CM-treated animals (0.109 ± 0.017, Fig. 6B)when compared to vehicle mice (0.251 ± 0.015 for vehicle, ***p < 0.005 versus wild type and ASC-CM), whose CD11b expression level was at a similar level to wild type mice (0.100 ± 0.005) after 7 days.
Figure 6. CD11b expression in spinal cords of symptomatic SOD1G93A mice after 3 days or 7 days of ASC-CM treatment.

SOD1G93A mice were given 3 days or 7 days of ASC-CM treatment or vehicle upon disease onset. Spinal cords were processed Western blot as described in Experimental Procedures. Unchanged actin levels in all groups were used as internal loading controls. (A) (a) The representative immunoblots and (b) band densities that were measured showed no change in spinal cord CD11b expression after 7 days of ASC-CM treatment. (B) (a) The representative immunoblots and (b) band quantitation demonstrated a decrease in spinal cord CD11b expression after 7 days of ASC-CM treatment as compared to vehicle. n = 3, ***p < 0.005.
ASC-CM treatment of symptomatic SOD1G93A mice resulted in decreased expression of α-II spectrin in spinal cords
ASC-CM’s effect on the calpain system in SOD1G93A mice was determined by measuring expression of α-II spectrin, a known cleavage target of calpain43. After 3 days following disease onset, activity of the Ca2+-activated protease, calpain, was increased in SOD1G93A mice spinal cords given vehicle(0.81 ± 0.002, n = 4, ***p < 0.005) as compared to wild type controls (0.10 ± 0.006, n = 3), consistent to a previous report20. Cleaved α-II spectrin was significantly decreased in SOD1G93A mice receiving ASC-CM treatment as compared to mice given vehicle (0.15 ± 0.026, n = 3, ***p < 0.005, Fig. 7). Levels of actin remained unchanged for all groups and were used as internal loading controls.
Figure 7. ASC-CM treatment for 3 days decreased α-II spectrin cleavage in symptomatic SOD1G93A mice.
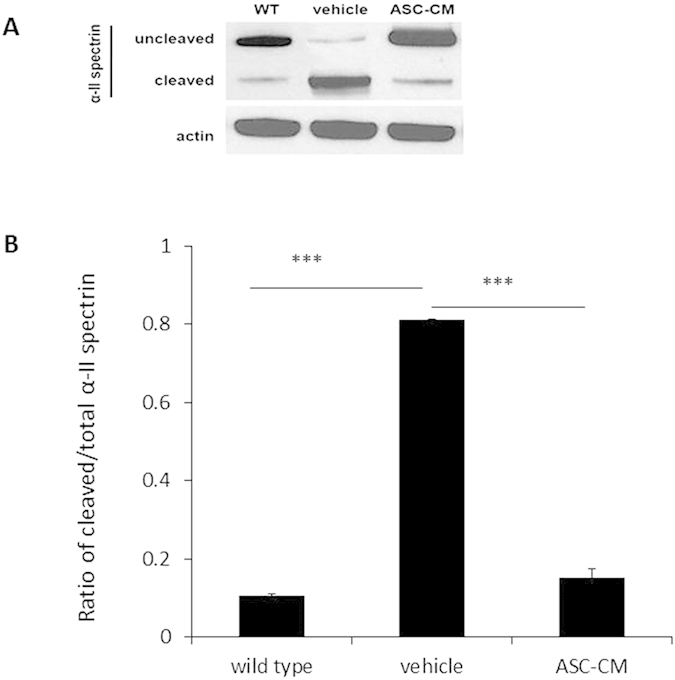
SOD1G93A mice were given ASC-CM or vehicle for 3 days. Western blot analysis was done to measure α-II spectrin. Unchanged actin levels expression were used as internal loading controls. (A) Representative immunoblots and (B) quantitation of band densities revealed a decrease in cleaved α-II spectrin expression at 3 days post-onset when ASC-CM treatment was given. Wild type, n = 5; vehicle, n = 3; ASC-CM, n = 4. ***p < 0.005.
Discussion
This study was designed to confirm the effects of a biologically-derived reagent on an ALS animal model that is clinically relevant such that treatment was applied after disease onset was established in the widely used SOD1G93A mouse model8. The data collected from this study demonstrate the effectiveness of ASC-CM treatment in prolonging post-onset survival and extending the lifespan of symptomatic SOD1G93A mouse. These results are in agreement with previous experiments from our laboratory, showing that the treatment with ASC-CM is beneficial and protective of neurons in vivo during neuronal injury or challenge, such as in the hypoxic-ischemic rat neonatal brain27. The findings from this study and the previous in vivo studies suggest that ASC-CM has the potential as a therapeutic for neuronal degeneration.
Although the etiology of sporadic ALS is currently unknown, through the use of animal models and correlation with neuropathological findings from human ALS patients, several key mechanisms of neuronal death have been proposed to disrupt normal motor neuron function and lead to their degeneration. Current studies implicate glutamate-induced excitotoxicity, mitochondrial dysfunction, oxidative stress, neuroinflammation, aberrant axonal transport systems, endoplasmic reticulum (ER) stress, inhibition of proteosomal function, compromised blood-brain barrier, and abnormal protein aggregation44. There is much debate as to which processes contribute towards disease etiology and which events are secondary to the established disease state. This presents ALS drug development with difficulties as neuropathology-specific targeting of treatment may be ineffective.
Despite the mechanistic uncertainties underlying events leading to ALS neuropathologic changes, treatments like ASC-CM may prove to be beneficial as they are able to block or modify several neuronal death pathways due to the heterogeneity in their composition. ASC-CM has been found to contain a multitude of neurotrophic factors that could be beneficial for the diseased motor neuron27. It was shown that neurotrophic factors may play a critical role in protecting or delaying motor neuronal death in ALS mice45. In this study, neurotrophic factors in ASC-CM could access affected neurons in CNS by either directly crossing the blood brain barrier46,47 or going through the retrograde transport mechanism45,46. Additionally, since motor neuronal death in ALS mice showed a distal–proximal gradient, it is also possible some neurotrophic factors in ASC-CM increased lifespan of ALS mice by directly protecting the neuromuscular junctions in peripheral45. Furthermore, in order to overcome the short half-life of polypeptides in ASC-CM peripherally, we i.p. injected ASC-CM daily until the end of experiments48,49. Here, after post-onset treatment with ASC-CM peripherally, symptomatic SOD1G93A mice have significant prolongation of post-onset survival times, which translated into an overall extension of lifespan. However, since many neurotrophic factors existing in ASC-CM may exert neuroprotective effects together as a neuroprotective mixture body27, deletion of single factor may fully remove its neuroprotective effects. In this study, we test this hypothesis by targeting the NGF contained in ASC-CM50. As predicted, NGF neutralized ASC-CM did not elongate the post-onset survival times and lifespan of SOD1G93A mice.
Additionally, the lengthening of post-onset lifespan following ASC-CM administration was correlated with markedly increased numbers of motor neuron survival in the spinal cord lumbar area. Further examinations at the cellular level determined that the increase in motor neuron survival after ASC-CM treatment was accompanied by decreased expression of glial activation markers and phosphorylated p38 MAP kinase, which are important components of the neuroinflammation pathway51 contributing to neuronal death in the spinal cords of SOD1G93A mice17. Other data from this study shows lower levels of cleaved α-II spectrin, which is a known substrate of both calpain and caspase-352,53. Decreased cleavage of α-II spectrin in the spinal cords of ASC-CM-treated SOD1G93A mice may indicate that calpain has protective effects against glutamate-induced neuronal apoptosis20,21,23.
Growing evidence has shown that NGF induces neuronal survival with low-affinity binding to the p75 neurotrophin receptor (p75NTR)54. Additionally, it was shown that NGF-induced neuroprotection against OGD insult by inhibiting OGD-induced p38 activation55 and against UV neurotoxicity by inhibiting calpain activity56. Furthermore, increased production of NGF in central nervous system (CNS) during diseases is able to suppress inflammation by switching the immune response to an anti-inflammatory57. Therefore, it is quite possible that NGF induced neuroprotection in ASC-CM-treated SOD1G93A mice via neuroprotection, inhibition of p38 and calpain, as well as anti-inflammation. It would be interesting to investigate by using appropriate models how NGF protects motor neurons in ASC-CM-treated SOD1G93A mice and whether glial activation or inflammatory processes play important roles in this neuroprotective process.
Taken together, this study demonstrated that ASC-CM provides significant neuroprotection in the SOD1G93A mouse model of ALS and extendsSOD1G93A mice life span. NGF in ASC-CM plays a significant role in this neuroprotection. This protective ability may associate with the preservation of motor neurons and inflammatory pathway inhibition in the spinal cord. This study establishes the therapeutic potential of this agent for treating ALS. Additionally, ASC-CM also holds great promise as potential therapies for many other neurodegenerative diseases due to the validated beneficial effects in many experimental models.
Additional Information
How to cite this article: Fontanilla, C. V. et al. Adipose-derived Stem Cell Conditioned Media Extends Survival time of a mouse model of Amyotrophic Lateral Sclerosis. Sci. Rep. 5, 16953; doi: 10.1038/srep16953 (2015).
Footnotes
Author Contributions C.F. prepared figures. H.G. prepared figures and wrote the main manuscript text. Q.L. prepared figures. T.Z. prepared figure 1. C.Z., B.J., K.M. and R.P. provided the study materials. M.F. designed experiments and performed administrative support. Y.D. designed experiments, performed financial support and did final approval of manuscript. All authors reviewed the manuscript.
References
- Appel S. H., Zhao W., Beers D. R. & Henkel J. S. The microglial-motoneuron dialogue in ALS. Acta Myol 30, 4–8 (2011). [PMC free article] [PubMed] [Google Scholar]
- Tandan R. & Bradley W. G. Amyotrophic lateral sclerosis: Part 1. Clinical features, pathology, and ethical issues in management. Ann Neurol 18, 271–280, doi: 10.1002/ana.410180302 (1985). [DOI] [PubMed] [Google Scholar]
- Sejvar J. J., Holman R. C., Bresee J. S., Kochanek K. D. & Schonberger L. B. Amyotrophic lateral sclerosis mortality in the United States, 1979–2001. Neuroepidemiology 25, 144–152, doi: 10.1159/000086679 (2005). [DOI] [PubMed] [Google Scholar]
- Rowland L. P. & Shneider N. A. Amyotrophic lateral sclerosis. N Engl J Med 344, 1688–1700, doi: 10.1056/NEJM200105313442207 (2001). [DOI] [PubMed] [Google Scholar]
- Bruijn L. I., Miller T. M. & Cleveland D. W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27, 723–749, doi: 10.1146/annurev.neuro.27.070203.144244 (2004). [DOI] [PubMed] [Google Scholar]
- Rosen D. R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62, doi: 10.1038/362059a0 (1993). [DOI] [PubMed] [Google Scholar]
- Siddique T. et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med 324, 1381–1384, doi: 10.1056/NEJM199105163242001 (1991). [DOI] [PubMed] [Google Scholar]
- Gurney M. E. et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994). [DOI] [PubMed] [Google Scholar]
- Alexander G. M. et al. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res Mol Brain Res 130, 7–15, doi: 10.1016/j.molbrainres.2004.07.002 (2004). [DOI] [PubMed] [Google Scholar]
- Rothstein J. D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 65 Suppl 1, S3–9, doi: 10.1002/ana.21543 (2009). [DOI] [PubMed] [Google Scholar]
- Almer G. et al. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol 49, 176–185 (2001). [PubMed] [Google Scholar]
- Wu D. C., Re D. B., Nagai M., Ischiropoulos H. & Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci USA 103, 12132–12137, doi: 10.1073/pnas.0603670103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K. et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11, 251–253, doi: 10.1038/nn2047 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S. et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392, doi: 10.1126/science.1123511 (2006). [DOI] [PubMed] [Google Scholar]
- Bendotti C. et al. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J Neuropathol Exp Neurol 63, 113–119 (2004). [DOI] [PubMed] [Google Scholar]
- Wengenack T. M. et al. Activation of programmed cell death markers in ventral horn motor neurons during early presymptomatic stages of amyotrophic lateral sclerosis in a transgenic mouse model. Brain Res 1027, 73–86, doi: 10.1016/j.brainres.2004.08.054 (2004). [DOI] [PubMed] [Google Scholar]
- Dewil M., dela Cruz V. F., Van Den Bosch L. & Robberecht W. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1(G93A)-induced motor neuron death. Neurobiol Dis 26, 332–341, doi: 10.1016/j.nbd.2006.12.023 (2007). [DOI] [PubMed] [Google Scholar]
- Tortarolo M. et al. Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol Cell Neurosci 23, 180–192 (2003). [DOI] [PubMed] [Google Scholar]
- Hu J. H., Chernoff K., Pelech S. & Krieger C. Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J Neurochem 85, 422–431 (2003). [DOI] [PubMed] [Google Scholar]
- Stifanese R. et al. Adaptive modifications in the calpain/calpastatin system in brain cells after persistent alteration in Ca2+ homeostasis. J Biol Chem 285, 631–643, doi: 10.1074/jbc.M109.031674 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootz H., Hansson I., Korhonen L. & Lindholm D. XIAP decreases caspase-12 cleavage and calpain activity in spinal cord of ALS transgenic mice. Exp Cell Res 312, 1890–1898, doi: 10.1016/j.yexcr.2006.02.021 (2006). [DOI] [PubMed] [Google Scholar]
- Roy J., Minotti S., Dong L., Figlewicz D. A. & Durham H. D. Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J Neurosci 18, 9673–9684 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradewell M. L. & Durham H. D. Calpastatin reduces toxicity of SOD1G93A in a culture model of amyotrophic lateral sclerosis. Neuroreport 21, 976–979, doi: 10.1097/WNR.0b013e32833ddd45 (2010). [DOI] [PubMed] [Google Scholar]
- Scott S. et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler 9, 4–15, doi: 10.1080/17482960701856300 (2008). [DOI] [PubMed] [Google Scholar]
- Kim I. G. et al. Effect of an adipose-derived stem cell and nerve growth factor-incorporated hydrogel on recovery of erectile function in a rat model of cavernous nerve injury. Tissue Eng Part A 19, 14–23, doi: 10.1089/ten.TEA.2011.0654 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagami H. et al. Novel autologous cell therapy in ischemic limb disease through growth factor secretion by cultured adipose tissue-derived stromal cells. Arterioscler Thromb Vasc Biol 25, 2542–2547, doi: 10.1161/01.ATV.0000190701.92007.6d (2005). [DOI] [PubMed] [Google Scholar]
- Wei X. et al. IFATS collection: The conditioned media of adipose stromal cells protect against hypoxia-ischemia-induced brain damage in neonatal rats. Stem Cells 27, 478–488, doi: 10.1634/stemcells.2008-0333 (2009). [DOI] [PubMed] [Google Scholar]
- Zhao L. et al. Adipose stromal cells-conditional medium protected glutamate-induced CGNs neuronal death by BDNF. Neurosci Lett 452, 238–240, doi: 10.1016/j.neulet.2009.01.025 (2009). [DOI] [PubMed] [Google Scholar]
- Gu H. et al. Adipose stromal cells-conditioned medium blocks 6-hydroxydopamine-induced neurotoxicity and reactive oxygen species. Neurosci Lett 544, 15–19, doi: 10.1016/j.neulet.2013.02.057 (2013). [DOI] [PubMed] [Google Scholar]
- Jeon D. et al. Neuroprotective effect of a cell-free extract derived from human adipose stem cells in experimental stroke models. Neurobiol Dis 54, 414–420, doi: 10.1016/j.nbd.2013.01.015 (2013). [DOI] [PubMed] [Google Scholar]
- Appel S. H. A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and Alzheimer disease. Ann Neurol 10, 499–505, doi: 10.1002/ana.410100602 (1981). [DOI] [PubMed] [Google Scholar]
- Anand P. et al. Regional changes of ciliary neurotrophic factor and nerve growth factor levels in post mortem spinal cord and cerebral cortex from patients with motor disease. Nat Med 1, 168–172 (1995). [DOI] [PubMed] [Google Scholar]
- Fontanilla C. V. et al. Caffeic acid phenethyl ester extends survival of a mouse model of amyotrophic lateral sclerosis. Neuroscience 205, 185–193, doi: 10.1016/j.neuroscience.2011.12.025 (2012). [DOI] [PubMed] [Google Scholar]
- Kim J. et al. Comparative analysis of the secretory proteome of human adipose stromal vascular fraction cells during adipogenesis. Proteomics 10, 394–405, doi: 10.1002/pmic.200900218 (2010). [DOI] [PubMed] [Google Scholar]
- Shimko M. J. et al. Nerve growth factor reduces amiloride-sensitive Na+ transport in human airway epithelial cells. Physiol Rep 2, doi: 10.14814/phy2.12073 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. et al. Adipose stem cells-conditioned medium blocks 6-hydroxydopamine-induced neurotoxicity via the IGF-1/PI3K/AKT pathway. Neurosci Lett 581, 98–102, doi: 10.1016/j.neulet.2014.08.033 (2014). [DOI] [PubMed] [Google Scholar]
- Du S. et al. TIMP1 in conditioned media of human adipose stromal cells protects neurons against oxygen-glucose deprivation injury. Neurosci Lett 584, 56–59, doi: 10.1016/j.neulet.2014.09.045 (2015). [DOI] [PubMed] [Google Scholar]
- Wei X. et al. Caffeic acid phenethyl ester prevents neonatal hypoxic-ischaemic brain injury. Brain 127, 2629–2635, doi: 10.1093/brain/awh316 (2004). [DOI] [PubMed] [Google Scholar]
- Grossman S. D., Rosenberg L. J. & Wrathall J. R. Temporal-spatial pattern of acute neuronal and glial loss after spinal cord contusion. Exp Neurol 168, 273–282, doi: 10.1006/exnr.2001.7628 (2001). [DOI] [PubMed] [Google Scholar]
- Sato M. et al. Cyclosporin A reduces delayed motor neuron death after spinal cord ischemia in rabbits. Ann Thorac Surg 75, 1294–1299 (2003). [DOI] [PubMed] [Google Scholar]
- Dodge J. C. et al. Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol Ther 16, 1056–1064, doi: 10.1038/mt.2008.60 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S., Vande Velde C. & Cleveland D. W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59, doi: 10.1016/j.neuron.2006.09.018 (2006). [DOI] [PubMed] [Google Scholar]
- Strong M. J. Neurofilament metabolism in sporadic amyotrophic lateral sclerosis. J Neurol Sci 169, 170–177 (1999). [DOI] [PubMed] [Google Scholar]
- Ilieva H., Polymenidou M. & Cleveland D. W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187, 761–772, doi: 10.1083/jcb.200908164 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould T. W. & Oppenheim R. W. Motor neuron trophic factors: therapeutic use in ALS? Brain Res Rev 67, 1–39, doi: 10.1016/j.brainresrev.2010.10.003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C., Nennesmo I., Fadeel B. & Henter J. I. Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Ann Neurol 56, 564–567, doi: 10.1002/ana.20223 (2004). [DOI] [PubMed] [Google Scholar]
- Pitzer C. et al. Granulocyte-colony stimulating factor improves outcome in a mouse model of amyotrophic lateral sclerosis. Brain 131, 3335–3347, doi: 10.1093/brain/awn243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom A. L. et al. Retrograde axonal transport and motor neuron disease. J Neurochem 106, 495–505, doi: 10.1111/j.1471-4159.2008.05393.x (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. Y. et al. Intramuscular injection of AAV-GDNF results in sustained expression of transgenic GDNF, and its delivery to spinal motoneurons by retrograde transport. Neurosci Res 45, 33–40 (2003). [DOI] [PubMed] [Google Scholar]
- Tan B. et al. AMP-activated kinase mediates adipose stem cell-stimulated neuritogenesis of PC12 cells. Neuroscience 181, 40–47, doi: 10.1016/j.neuroscience.2011.02.038 (2011). [DOI] [PubMed] [Google Scholar]
- Cuenda A. & Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 1773, 1358–1375, doi: 10.1016/j.bbamcr.2007.03.010 (2007). [DOI] [PubMed] [Google Scholar]
- Siman R., Noszek J. C. & Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci 9, 1579–1590 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. K. et al. Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem 273, 22490–22497 (1998). [DOI] [PubMed] [Google Scholar]
- Chen L. W., Yung K. K., Chan Y. S., Shum D. K. & Bolam J. P. The proNGF-p75NTR-sortilin signalling complex as new target for the therapeutic treatment of Parkinson’s disease. CNS Neurol Disord Drug Targets 7, 512–523 (2008). [DOI] [PubMed] [Google Scholar]
- Tabakman R., Jiang H., Schaefer E., Levine R. A. & Lazarovici P. Nerve growth factor pretreatment attenuates oxygen and glucose deprivation-induced c-Jun amino-terminal kinase 1 and stress-activated kinases p38alpha and p38beta activation and confers neuroprotection in the pheochromocytoma PC12 Model. J Mol Neurosci 22, 237–250 (2004). [DOI] [PubMed] [Google Scholar]
- McCollum A. T. & Estus S. NGF acts via p75 low-affinity neurotrophin receptor and calpain inhibition to reduce UV neurotoxicity. J Neurosci Res 77, 552–564, doi: 10.1002/jnr.20184 (2004). [DOI] [PubMed] [Google Scholar]
- Villoslada P. & Genain C. P. Role of nerve growth factor and other trophic factors in brain inflammation. Prog Brain Res 146, 403–414, doi: 10.1016/S0079-6123(03)46025-1 (2004). [DOI] [PubMed] [Google Scholar]