

Background: Polycomb proteins control transcription by regulating chromatin structure and dynamics.
Results: By developing and applying a novel Sm-ChIPi technique, we identified that one PRC1 binds multiple nucleosomes within cells, although two PRC2s can bind a single nucleosome.
Conclusion: PRC1 and PRC2 complexes employ distinct mechanisms to assemble on chromatin.
Significance: The cellular assembly stoichiometry provides insight into repressive polycomb chromatin structure.
Keywords: chromatin, chromatin immunoprecipitation (ChiP), epigenetics, polycomb, single-molecule biophysics, stoichiometry, PRC1, PRC2, Sm-ChIPi, histone chromosomal protein
Abstract
Epigenetic complexes play an essential role in regulating chromatin structure, but information about their assembly stoichiometry on chromatin within cells is poorly understood. The cellular assembly stoichiometry is critical for appreciating the initiation, propagation, and maintenance of epigenetic inheritance during normal development and in cancer. By combining genetic engineering, chromatin biochemistry, and single-molecule fluorescence imaging, we developed a novel and sensitive approach termed single-molecule chromatin immunoprecipitation imaging (Sm-ChIPi) to enable investigation of the cellular assembly stoichiometry of epigenetic complexes on chromatin. Sm-ChIPi was validated by using chromatin complexes with known stoichiometry. The stoichiometry of subunits within a polycomb complex and the assembly stoichiometry of polycomb complexes on chromatin have been extensively studied but reached divergent views. Moreover, the cellular assembly stoichiometry of polycomb complexes on chromatin remains unexplored. Using Sm-ChIPi, we demonstrated that within mouse embryonic stem cells, one polycomb repressive complex (PRC) 1 associates with multiple nucleosomes, whereas two PRC2s can bind to a single nucleosome. Furthermore, we obtained direct physical evidence that the nucleoplasmic PRC1 is monomeric, whereas PRC2 can dimerize in the nucleoplasm. We showed that ES cell differentiation induces selective alteration of the assembly stoichiometry of Cbx2 on chromatin but not other PRC1 components. We additionally showed that the PRC2-mediated trimethylation of H3K27 is not required for the assembly stoichiometry of PRC1 on chromatin. Thus, these findings uncover that PRC1 and PRC2 employ distinct mechanisms to assemble on chromatin, and the novel Sm-ChIPi technique could provide single-molecule insight into other epigenetic complexes.
Introduction
In the nucleus, genome organization is shaped by the nucleosome, the basic building unit of chromatin (1). The nucleosome is formed by wrapping ∼147 bp of DNA around a histone octamer consisting of two copies of H2A, H2B, H3, and H4 (2). The highly conserved basic N termini and, to a lesser extent, the globular domains of histones are extensively post-translationally modified by epigenetic regulatory complexes (3–5). Genomic compartments and chromatin-related activities are tightly correlated with histone modifications (1, 4, 6). These modifications either directly organize chromatin structure or recruit effectors that impact genome organization (1, 4, 6, 7). However, cellular molecular details about how epigenetic complexes assemble on and spread along chromatin are incompletely understood.
Polycomb group (PcG)2 proteins are a long-standing paradigm of studying the epigenetic inheritance of transcriptional states and are essential for the establishment and maintenance of transcriptional profiles during normal development and in cancer (8, 9). Two major PcG complexes, PRC1 and PRC2, exhibit distinct enzymatic activities (8). PRC2 is a methyltransferase that catalyzes di- and trimethylation of lysine 27 on H3 (H3K27me2/3) (8). The mammalian core PRC2 is composed of Ezh2, Suz12, Eed, and RbAp48. Ezh2 is the catalytic subunit (8), and Eed is involved in recognition of the H3K27me3 mark (10). PRC1 is a ubiquitin ligase that catalyzes ubiquitylation of lysine 119 on H2A (H2AK119Ub) (11). In mammals, six forms of PRC1 have been identified, each comprising one of six Pcgf subunits and the E3 ligase Ring1a/b (12). Further classification of PRC1 is determined by the mutually exclusive association of either Rybp or Yaf2 (variant PRC1s) or one of the Cbx proteins (canonical PRC1s) (12). Several mechanisms for the PcG-mediated gene silencing such as histone H3K27 trimethylation (13), histone H2A monoubiquitination (11), chromatin compaction (14), and organization of higher order chromatin structure have been proposed (15); however, it is not yet clear how PcG complexes assemble on chromatin within cells.
Although much is known about the interaction domains and the protein identities within PRC1 complexes, far less is known about their molecular architecture on chromatin within cells. Several studies have shown that the PRC1 subunits and their isolated domains self-associate in vitro (16–20). Clearly, these in vitro observations need to be verified within cells. In contrast with the individual PRC1 subunits, the reconstituted Drosophila PRC1 is a monomer having one copy of each subunit (14). Studies of the assembly stoichiometry of PRC1 on chromatin reached varying views on how PRC1 interacts with chromatin. The reconstituted Drosophila PRC1 packs nucleosomal arrays with a stoichiometry of one PRC1 per tetranucleosome (14). The reconstituted Drosophila Psc (homolog of Pcgfs) bridges nucleosomes with a stoichiometry of one Psc per mononucleosome (21). A recent crystal structure indicated that one PRC1 ubiquitylation module binds to each disk surface of a nucleosome (22). These variations could be due to the compositions of subunits used in the reconstitution reactions or the methods used in the experiments. Thus, it is important to resolve these disparities and to determine the cellular assembly stoichiometry of PRC1 complexes on chromatin.
Studies of the oligomerization status of PRC2 reached divergent opinions (23–27). The reconstituted PRC2 has been characterized as a monomer, dimer, or oligomer (23–25). By utilizing size exclusion chromatography, the endogenous PRC2 complex from both human and Drosophila was found to have a wide range of apparent molecular masses, ranging from 300 kDa to 1 mDa or higher (26, 27), whereas gel filtration of native complexes cannot exclude the possibility that PRC2 has extended structures or that non-PRC2 proteins are associated. The molecular stoichiometry of PRC2 within cells therefore remains elusive. Electron microscopy studies suggested that PRC2 is monomeric and may bind to a dinucleosome (25); however, whether the in vitro model recaptures the in vivo situation remains unknown.
A few approaches have been developed to quantify the stoichiometry of epigenetic modifications at histones of nucleosomes (28, 29) or in an entire proteome (30), but addressing the cellular assembly stoichiometry of epigenetic complexes at chromatin has so far been hampered by the absence of adequate techniques. Chromatin immunoprecipitation (ChIP) followed by high throughput sequencing (ChIP-Seq) maps global patterns of histone modifications and chromatin-binding proteins, but ChIP-Seq cannot directly reveal molecular stoichiometry. Sequential ChIP performed on native and purified nucleosomes can reveal the co-occurrence of epigenetic proteins on chromatin, but it is a formidable challenge to establish absolute stoichiometry. Sedimentation velocity analytical ultracentrifugation and gel filtration chromatography are often used to determine the apparent molecular sizes of native protein complexes; however, these techniques cannot exclude the influence of uncharacterized proteins and heterogeneous conformations. Single-molecule fluorescence microscopy is a powerful technique to quantify the absolute number of subunits of the macromolecular protein complex (31–33). The quantification is based on the photobleaching behaviors of fluorophores (32, 33) or the ratios of the fluorescent intensities of fluorophores to the reference fluorophores (31, 34, 35). Single-molecule techniques have been widely applied to chromatin biology and provide a wealth of information on nucleosome structure and dynamics (36–41). Here, we combined genetic engineering, chromatin biochemistry, and single-molecule fluorescence imaging to develop a novel and sensitive approach termed Sm-ChIPi to circumvent these limitations and to enable us to directly assess the cellular assembly stoichiometry. By using Sm-ChIPi, for the first time we present the cellular assembly stoichiometry of PcG complexes PRC1 and PRC2 on chromatin. We have found that PRC1 and PRC2 employ distinct mechanisms by which they assemble on chromatin, reflecting their distinct roles in establishing and maintaining repressive polycomb domains. These results contribute significantly to our quantitative understanding of the cellular architecture of PcG complexes, allowing us to suggest possible molecular mechanisms for the PcG-mediated epigenetic silencing. Sm-ChIPi is a direct and sensitive technique and could be applied to many other studies of epigenetic complex assembly on native chromatin.
Experimental Procedures
Cell Lines and Plasmids
The Cbx2−/− (42), Cbx7−/− (43), Ring1bfl/fl;Rosa26::CreERT2 (44), Bmi1−/−/Mel18−/− (Bmi1 and Mel18 double knock-out) (45), Eed−/− (44), Ezh2−/− (46), and PGK12.1 (47) mES cell lines were maintained in mES medium (DMEM (D5796; Sigma) supplemented with 15% FBS (SH30071.03; Hyclone), 2 mm glutamine (G7513; Life Technologies, Inc.), 100 units/ml penicillin/streptomycin (15140-122; Life Technologies, Inc.), 55 μm β-mercaptoethanol (21985-023; Life Technologies, Inc.), 103 units/ml leukemia inhibitor factor, and 0.1 mm non-essential amino acids (11140050; Life Technologies, Inc.)) at 37 °C in 5% CO2. Medium was changed every day unless otherwise indicated. To deplete Ring1b alleles, 4-hydroxytamoxifen (OHT; H7904; Sigma) was administered for 3 days under a concentration of 1.0 μm. HEK293T cells were maintained in DMEM supplemented with 10% FBS, 2 mm glutamine, and 100 units/ml penicillin/streptomycin at 37 °C in 5% CO2. H3.3−/−/H3.3-EGFP DT40 cells (48) were maintained in RPMI 1640 medium (11875093; Life Technologies, Inc.) supplemented with 5% FBS, 5% chicken serum (C5405–100ML; Sigma), 50 μm β-mercaptoethanol, and 100 units/ml penicillin/streptomycin at 37 °C in 5% CO2.
The plasmids pTRIPZ(M)-YFP-Cbx2 (49), pTRIPZ(M)-YFP-Cbx4 (49), pTRIPZ(M)-YFP-Cbx7 (49), pTRIPZ(M)-YFP-Cbx8 (49), pTRIPZ(M)-YFP-Ring1b (49), pTRIPZ(M)-YFP-Mel18 (49), and pEGFP-KAP1 (50) have been described previously. The sequences encoding Eed (Addgene) and Ezh2 (Addgene) were amplified by PCR and inserted downstream of the coding sequence of fluorescence protein in pTRIPZ(M) vector (49). The sequence encoding YFP was amplified by PCR and inserted into pGEX-6P-1 vector (GE Healthcare) to generate pGEX-6P-1-YFP (monomeric YFP) and pGEX-6P-1-YFP-YFP (dimeric YFP). The sequences encoding fusion proteins have been verified by DNA sequencing.
Establishing Transgenic mES Cell Lines
Establishing the transgenic mES cell lines was performed according to the procedure described previously (49). Briefly, pseudo-viruses were packaged in HEK293T cells by co-transfecting with 21 μg of pTRIPZ(M) containing the fusion gene, 21 μg of psPAX2, and 10.5 μg of pMD2.G. 60 h after transfection, medium was collected and used for transducing mES cells. Hexadimethrine bromide (Polybrene; H9268; Sigma) was added at a concentration of 8 μg/ml, and cells were seeded at ∼15% confluence on gelatin-coated plates. 2 days after infection, infected cells were selected by using 1.0–2.0 μg/ml puromycin (P8833; Sigma). The expression of transgenes was induced by doxycycline (Dox; D9891; Sigma).
Transfection
HEK293T cells at 85–90% confluence were transfected with pEGFP-KAP1 by calcium phosphate. After 24 h of transfection, the medium was replaced with fresh medium. 48 h later, cells were harvested for isolation of chromatin.
Preparation of YFP Proteins
The plasmids pGEX-6p-1-YFP and pGEX-6p-1-YFP-YFP were transformed into the BL21-competent cells, respectively. The protein expression was induced by isopropyl β-d-thiogalactopyranoside (AC121; Omega Bio-Tek, Norcross, GA) for 5 h at 37 °C. Cell pellets were collected, resuspended in PBS containing 0.1 mm phenylmethanesulfonyl fluoride (PMSF; 93482; Sigma) and protease inhibitor mixture (P8340; Sigma), and sonicated using Vibra-CellTM sonicator (VCX130; Newtown, CT). 1% Triton X-100 was added to the mixture. After centrifugation, prewashed GSH-Sepharose 4B beads (17-0756-01; GE Healthcare) were added to the supernatant. The mixture was incubated for 30 min at 4 °C. After washing four times with PBS containing 1.0% Triton X-100, the YFP proteins were eluted by 5 mm reduced glutathione (G4251; Sigma). The purity and identity of YFP proteins were assessed by SDS-PAGE.
Preparation of Nucleosomes from mES Cells
Approximately 5 × 108 cells were harvested by citrate saline solution (135 mm potassium chloride and 15 mm sodium citrate) for adherent cells or collected by centrifugation for DT40 cells, cross-linked with 2.0% paraformaldehyde for 10 min at 4 °C, and quenched with glycine. Cells were collected by centrifuging at 300 × g for 5 min at 4 °C, and the packed cell volume (PCV) was estimated. Pellets were resuspended in 2.5× PCV of buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 1.5 mm MgCl2, 340 mm sucrose, 10% glycerol, 50 μg/ml BSA, 1.0 mm Na3VO4, protease inhibitor mixture, and 0.1 mm PMSF). 2.5× PCV of buffer B (buffer A plus 0.2% Triton X-100) was added, and the mixture was incubated at 4 °C for 10 min. Pellets were collected by centrifuging at 1,300 × g for 5 min at 4 °C and resuspended with 6× PCV of buffer A. The mixture was loaded to the top layer of pre-chilled sucrose cushion (buffer A + 30% sucrose) and centrifuged at 1,300 × g for 12 min at 4 °C. Chromatin pellets were resuspended in buffer A containing 1.0 mm CaCl2 at the DNA concentration of 2.0 μg/ml. To generate mononucleosomes, chromatin was digested with 1.4 units/ml micrococcal nuclease (N5386; Sigma; the enzyme activity was defined as a Sigma unit) for 8 min at 37 °C. To produce polynucleosomes, chromatin was digested with 0.7 units/ml for 8 min at 37 °C. The reaction was stopped by 4.0 mm EGTA (pH 8.0). To purify mononucleosomes, 5–30% linear sucrose gradient was used. To purify polynucleosomes, 15–40% linear sucrose gradient was used. Linear sucrose gradients were prepared by dissolving sucrose in the buffer M (10 mm HEPES, pH 7.9, 50 μg/ml BSA, 10 mm KCl, 1.5 mm EDTA, 1.0 mm Na3VO4, 0.2 mm DTT and 0.5 mm PMSF). Approximately 300–400 μg of DNA in 0.5 ml were loaded on the top layer of the gradient, and samples were fractionated for 18–20 h at 200,000 × g using TH-641 Swinging Bucket Rotor and Sorvall WX ultracentrifuge (Thermo Fisher Scientific, Waltham, MA). 0.5 ml per fraction was collected. The DNA fragment size of each fraction was analyzed by agarose gel electrophoresis.
Preparation of Nucleosomes from Differentiated Cells
mES cells were induced to differentiate as described previously (51). Briefly, mES cell lines, Cbx2−/−/Y-Cbx2, Cbx7−/−/Y-Cbx7, Ring1bfl/fl/Y-Ring1b, and Bmi1−/−Mel18−/−/Y-Mel18, were cultured to reach 80–90% confluency. Approximately 6 × 106 cells were resuspended in 10 ml of DMEM supplemented with 10% FBS, 2 mm glutamine, and 100 units/ml penicillin/streptomycin, and plated in a 10-cm polystyrene stackable Petri dish (8609–0010; USA Scientific, Ocala, FL). Medium was changed every 48 h. On day 4, a final concentration of 500 μm retinoic acid (R2625; Sigma) was administered. On day 8, cells were changed with medium containing 2 μg/ml Dox or Dox with OHT for Ring1bfl/fl/Y-Ring1b. On day 10, chromatin was isolated, and nucleosomes were prepared as described above.
Preparation of Polynucleosomal Arrays and Their Interaction with PRC1
Tetranucleosome reconstitution was performed as described previously by salt dialysis (52, 53). Briefly, recombinant human histone octamer (H2A, H2B, H3.1, and H4), assembled from Escherichia coli-expressed individual histones as described (52), was added to DNA in approximately a 1:1 molar ratio in 10 mm Tris-HCl, pH 7.6, 2 m NaCl, 1 mm EDTA, 0.5 mg/ml BSA, 0.05% Nonidet P-40, and 5 mm β-mercaptoethanol. Salt dialysis was performed at 4 °C for ∼20 h from 2 m NaCl buffer to 50 mm NaCl and a final dialysis step for 1 h at 50 mm NaCl. Samples were incubated at 37 °C for 1 h before storage on ice up to 4 weeks. The extent of chromatinization was assessed by limited micrococcal nuclease digestion. The DNA template used was an 863-bp PCR fragment amplified from a plasmid construct3 containing two “601” nucleosome-positioning sequences (54) flanking five Gal4-binding sites and an adenoviral E4 promoter. The PCR product was amplified with one 5′-biotin-triethyleneglycol primer.
The 15-mer nucleosomal DNA template is a purified ∼3.1-kb biotinylated PCR product from the plasmid pUC18-G5cyc1G- (55). The 15-mer polynucleosome was reconstituted through a previously developed enzymatic assembly method (56, 57). Biotinylated DNA, human histone octamers (as described for tetranucleosome assembly), human histone chaperone NAP1, yeast nucleosome positioning factor yIsw1a, and an ATP regeneration system (final concentrations: 30 mm creatine phosphate, 3 mm ATP, 4.1 mm MgCl2, and 6.4 μg/ml creatine kinase) were incubated at 30 °C for 5 h in buffer containing 10 mm HEPES, pH 7.5, 10 mm KCl, 1.5 mm MgCl2, 500 μm EGTA, 10% glycerol, 2.5 mm β-glycerophosphate, 200 μm PMSF, and 1 mm DTT.
Ring1bfl/fl/Y-Ring1b mES cells were cultured in the presence of 0.5 μg/ml doxycycline and 1.0 μm OHT for 3 days. Nuclei were purified from 5 × 107 cells and lysed in 0.5 ml of buffer containing 20 mm Tris-HCl, pH 7.4, 0.5% Nonidet P-40, 350 mm NaCl, 0.25 mm EDTA, 10% glycerol, 0.1 mm Na3VO4, protein inhibitor mixture, and 0.1 mm PMSF. 120 μl of biotinylated nucleosomal arrays (18 nm) was incubated with 380 μl of nuclear extract at 4 °C overnight. The 0.5 ml of mixture was loaded into 15–40% sucrose gradient and fractionated as described above. 0.5 ml per fraction was collected and fixed with 0.2% of paraformaldehyde. DNAs were extracted and analyzed by agarose gel electrophoresis.
Construction and Passivation of Flow Chamber
Flow chambers were constructed as described previously with modifications (32). Two 0.75-mm holes across from each other were drilled in a quartz slide (12-550-15; Thermo Fisher Scientific). The slides and coverslips (48366-249; VWR, Radnor, PA) were sonicated with Milli-Q water for 30 min and incubated with methanol overnight. The coverslips were treated with 1.0 m KOH for 40 min, dried, and burned for 1–2 s using a propane torch. Then the coverslips were incubated with methanol supplemented with 1% aminosilane (N-2-aminoethyl-3-aminopropyltrimethoxysilane (A21541; Pfaltz & Bauer, Waterbury, CT)) and 5% acetic acid for 20 min in the dark at room temperature. After washing with methanol and water, the coverslips were dried with nitrogen gas and placed in a humidified box in the dark. To each coverslip, 70 μl of the passivated solution (10 mm sodium bicarbonate, pH 8.5,16 mg of mPEG-SVA (MPEG-SVA-5000; Laysan Bio, Arab, AL)), 0.3 mg of biotin PEG-SVA (256-586-9004; Laysan Bio) was added and incubated in a humidified box for 3–4 h in the dark. After washing with Milli-Q water, the coverslips were assembled on the quartz slide by sandwiching a piece of double-sided tape between the slide and the coverslip in the way that it creates an ∼6.0-mm channel where the inlet/outlet holes are located. The edges of the flow chambers were sealed with epoxy glue (14250; Devcon, Danvers, MA) and stored at −20 °C under nitrogen gas.
Imaging by Single-molecule Total Internal Reflection Fluorescence (TIRF) Microscopy
Samples intended for the Sm-ChIPi analysis were incubated with biotinylated antibodies, anti-GFP (ab6658; Abcam, Cambridge, UK), anti-histone H2B (60R-1215; Fitzgerald Industries, Acton, MA), and anti-histone H3 (5748; Cell Signaling Technology, Boston, MA), at 4 °C overnight. Flow chamber was loaded with 0.2 μg/ml NeutrAvidin (31000; Thermo Fisher Scientific) and washed with TE50 buffer. After cross-linking with 0.5% paraformaldehyde for 15 min at 4 °C, 100 μl of the samples were loaded into the flow chamber. After washing with TE50 buffer, images were acquired by using Zeiss Axio Observer D1 Manual Microscope (Zeiss, Germany) equipped with an Alpha Plan-Apochromatic 100×/1.46 NA Oil Objective (Zeiss, Germany) and an Evolve 512 × 512 EMCCD camera (Photometrics, Tucson, AZ). The fluorescent intensity of time traces was generated by ImageJ, and background intensity was subtracted from the area surrounding the spot of interest. The photobleaching steps were detected by Chang-Kennedy filtering (58). Histogram was constructed using data from three replicates with each measurement of over 100 individual spots analyzed. The functionalized blank slide surface showed ∼5 fluorescent spots per 1,000 μm2. The number of fluorescent spots was typically ∼300–600 spots per 1,000 μm2 via antibody immobilization by controlling the lysate or fraction dilution factor. At the same dilution factor and without antibody, the number of fluorescent spots was typically ∼10 fluorescent spots per 1,000 μm2, implying that ∼4% is from non-specifically adsorbed proteins to the surface.
Fluorescence Correlation Spectroscopy (FCS)
FCS measurements were performed at 37 °C on a Zeiss LSM780 using a C-Apochromat infinity color-corrected 1.2 NA 40× water objective. Cells were seeded on glass dishes the day before the experiment. Excitation of YFP was performed with the 488-nm line of a 20-milliwatt argon laser. For intracellular measurements, the desired recording position was chosen in the LSM image. Autocorrelation curves were derived from fluorescence fluctuation analysis using the ZEN2012 FCS module. Autocorrelation curves were fit to one-component models of free diffusion in three dimensions with triplet function of Equation 1 (59),
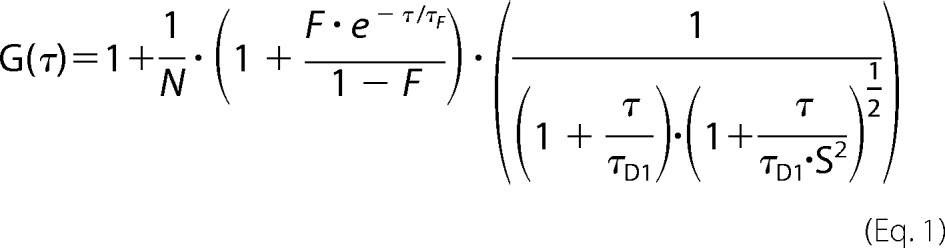 |
where τD1 and τF are the diffusion time and the triplet time, respectively; N and F are the number of molecules in the confocal volume and the triplet fraction, respectively; and S = wz/wxy, where wz and wxy represent the half height and radius of the confocal volume, respectively. The size of the confocal volume Veff was calibrated using a series dilution of rhodamine green dye in PBS. The concentration of YFP-Ring1b was determined by Equation 2,
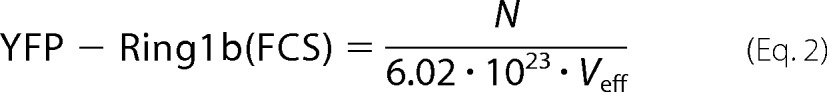 |
The concentration of endogenous Ring1b was determined by Equation 3,
 |
where 0.90 is the ratio of endogenous Ring1b to YFP-Ring1b, which was determined by Western blotting. The number of YFP-Ring1b molecules in single mES cell nucleus was calculated by Equation 4,
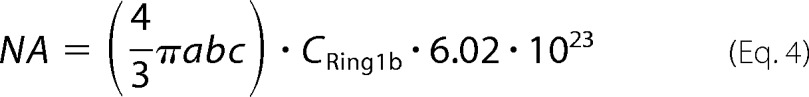 |
where a = b = 5 μm and c = 2.5 μm. CRing1b is the concentration of endogenous Ring1b protein.
ChIP
ChIP was performed as described previously (43, 51). Briefly, KO mES cells complemented with the YFP-PRC1 fusion gene were cross-linked with 1.2% formaldehyde (28908; Thermo Fisher Scientific) for 10 min at room temperature and quenched by 125 mm glycine. Cells were washed sequentially with LBI buffer (50 mm HEPES, pH 7.9, 140 mm NaCl, 1.0 mm EDTA, 10% glycerol, 0.5% Nonidet P-40, and 0.25% Triton X-100), LBII buffer (10 mm Tris-HCl, pH 8.0, 200 mm NaCl, and 15 mm EDTA), and LBIII buffer (10 mm Tris-HCl, 100 mm NaCl, 1.5 mm EDTA, 0.1% sodium deoxycholate, and 0.5% N-lauroylsarcosine). Chromatin was fragmented to the size of 200–500 bp by Vibra-CellTM sonicator (VCX130; Sonics, Newtown, CT). One-tenth of 10% Triton X-100 was added to the lysate. After pre-cleaning with protein G beads (101241; Life Technologies, Inc.), antibodies, anti-Cbx7 (sc-70232; Santa Cruz Biotechnology, Santa Cruz, CA), anti-Cbx2 (ab80044; Abcam, Cambridge, MA), anti-Mel18 (sc-10744; Santa Cruz Biotechnology), and anti-Ring1b (D139-1; MBL, Woburn, MA), were incubated with lysates, respectively. The immunoprecipitated DNAs were quantified using LightCycler 4800 SYBR Green I master mix (04707516001; Roche Applied Science) with AB Applied Biosystems. Triplicate PCRs were carried out for each sample. The efficiencies of ChIP were quantified relative to a standard curve prepared using input chromatin. The sequences of the primers used for quantitative PCR has been described previously (43, 51).
Immunoprecipitation (IP)
IP was performed as described previously (51). Briefly, nuclei were purified from 3 × 108 cells and lysed using buffer containing 20 mm Tris-HCl, pH 7.4, 0.1% Nonidet P-40, 350 mm NaCl, 0.25 mm EDTA, 20% glycerol, 0.1 mm Na3VO4, 0.1 mm PMSF, and protein inhibitor mixture. After pre-cleaning with protein G beads, the lysate was incubated with anti-GFP mAb-agarose beads (D153-8; MBL). The beads were washed using buffer containing 20 mm Tris-HCl, pH 8.0, 1% Nonidet P-40, 200 mm KCl, 0.2 mm EDTA, and 0.1 mm PMSF. The proteins were resolved using NuPAGE 4–12% BisTris gel (NP0321BOX; Life Technologies, Inc.) and were transferred to 0.45-μm Immobilon-FL polyvinylidene fluoride membrane (Millipore, Darmstadt, Germany). Specific proteins were probed with anti-Phc1 (6-1-3; Active Motif, Carlsbad, CA) and anti-Ring1b (D139-3; MBL), and detected with ECL Plus (GE Healthcare). Membranes were imaged using a ChemiDoc XRS system (Bio-Rad).
Immunofluorescence
Immunofluorescence was performed as described previously (51). Wild-type, Ezh2−/−, Eed−/−, Ezh2−/−/Y-Ezh2, and Eed−/−/Y-Eed mES cells were plated on coverslips and cultured for 24 h. Cells were fixed using 2.0% paraformaldehyde for 10 min. Cells were washed with PBS and incubated with 0.2% Triton X-100 for 10 min. After washing with basic blocking buffer (10 mm PBS, pH 7.2, 0.1% Triton X-100, and 0.05% Tween 20), cells were incubated with blocking buffer (basic blocking buffer plus 3% goat serum and 3% bovine serum albumin) for 1 h. Anti-H3K27me3 antibody (07-449; Millipore, Billerica, MA) diluted in blocking buffer was incubated with cells for 2 h at room temperature. After washing with basic blocking buffer, Alexa 488-labeled goat anti-rabbit antibody (A-11008; Life Technologies, Inc.) diluted in blocking buffer was incubated with cells for 1 h. Cells were rinsed with PBS and washed with basic blocking buffer. After incubating with 0.1 μg/ml Hoechst, cells were washed and mounted on slides with ProLong antifade reagents (P7481; Life Technologies, Inc.). The images were taken and processed as described previously (49).
Results
Development of a Novel Approach to Assess the Cellular Assembly Stoichiometry of PcG Complexes on Chromatin
To assess the cellular assembly stoichiometry of PcG complexes on chromatin, we developed the Sm-ChIPi approach (Fig. 1a). A YFP-PcG fusion gene was stably expressed in its corresponding KO mES cells, which allows incorporating the fusion protein into PcG complexes without the interference of the endogenous counterpart. Cells were cross-linked with paraformaldehyde to preserve complex association prior to cell lysis. After cleaning with a sucrose cushion, chromatin extraction was subject to micrococcal nuclease digestion. The PcG·nucleosome complexes were fractionated by sucrose gradient ultracentrifugation. The fractions intended to be analyzed were incubated with biotinylated antibodies. The resultant complexes were immobilized on a quartz slide that had been passivated and functionalized with NeutrAvidin. Alternatively, without sucrose gradient ultracentrifugation, the PcG·nucleosome complexes were directly immobilized on the surface by biotinylated anti-histone antibodies. Image stacks were acquired by using TIRF microscopy with laser excitation, which gives high sensitivity and low background. Such TIRF experiments detect surface-bound molecules as discrete spots. Individual spots represent single PcG·nucleosome complexes. The time traces of fluorescent intensity were generated from image stacks by using ImageJ. The photobleaching steps were detected by Chung-Kennedy filtering (58) and reflect the number of YFP tags within a spot and thus the number of protein subunits within a complex.
FIGURE 1.
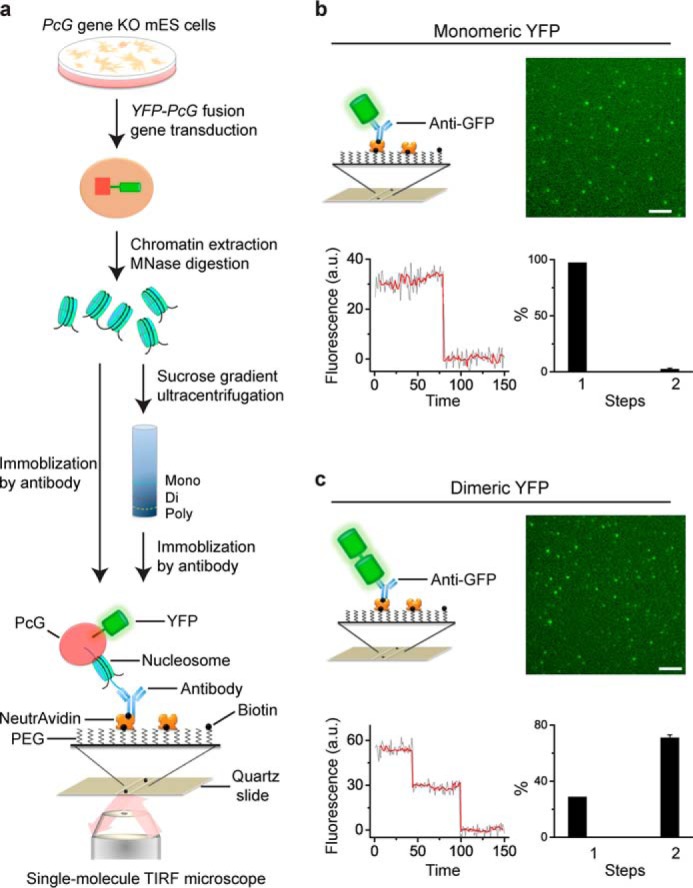
Development of Sm-ChIPi to assess the cellular assembly stoichiometry of PcG complexes on chromatin. a, schematic of the Sm-ChIPi approach. Nucleosomes generated from chromatin are directly immobilized on the surface or are separated by sucrose gradient ultracentrifugation and then immobilized on the surface. b and c, photobleaching behavior of monomeric (b) and dimeric (c) YFPs. The YFP proteins were immobilized by biotinylated anti-GFP antibody via interaction with NeutrAvidin. A sample single image of the acquired sequence is shown. A representative time course of fluorescence emission of the YFP protein is shown (black line). The photobleaching steps were detected by Chung-Kennedy filter (red line). Results are means ± S.D. a.u. denotes arbitrary unit. MNase is micrococcal nuclease. Scale bar, 5 μm.
To assess the detection efficiency of Sm-ChIPi approach, we generated monomeric and dimeric YFPs. The YFP proteins were immobilized on the surface via biotinylated anti-GFP antibody (Fig. 1, b and c). The discrete points were observed under TIRF microscopy, indicating individual YFP proteins on the surface. The functionalized coverslip greatly prevents nonspecific binding of YFPs to the surface (images not shown). Analysis of fluorescence trajectories of monomeric YFPs indicated that 97% of spots are one-step photobleaching, although 3% are two-step photobleaching, which accounts for random co-localization (Fig. 1b). For dimeric YFPs, 70% were two-step photobleaching (Fig. 1c), which is consistent with the previous report of a probability of p = 0.80 for an individual YFP protein to be fluorescent (60). The 3 and 70% values were used to predict the assembly stoichiometry of PcG complexes on chromatin, which was reported in the text unless otherwise indicated.
To validate the Sm-ChIP approach enabling us to accurately quantify stoichiometry of nucleosome complexes, we counted the number of H3.3-EGFP within a nucleosome (Fig. 2, a–e). Mononucleosomes were prepared from H3.3−/−/H3.3-EGFP DT40 cells where both H3F3A and H3F3B have been depleted (48). The H3.3-EGFP mononucleosomes were immobilized on the surface by biotinylated anti-H2B antibody. The functionalized surface efficiently prevents nonspecific binding of nucleosome complexes to the surface (images not shown). Analysis of fluorescence trajectories indicated that (75 ± 2)% of spots are two-step photobleaching. If the probability of an individual EGFP to be fluorescent was taken into account, 100% of EGFP-H3.3 nucleosome had a dimeric EGFP-H3.3.
FIGURE 2.

Validation of the Sm-ChIPi approach by using chromatin complexes with known stoichiometry. a–e, H3.3−/−/H3.3-EGFP DT40 cells contain dimer of H3.3-EGFP within a nucleosome. Agarose gel electrophoresis analysis of DNAs were extracted from nucleosomes prepared from H3.3−/−/H3.3-EGFP DT40 cells (a). Nucleosomes were immobilized on the surface by biotinylated anti-H2B antibody (b). A sample single image of the acquired sequence is shown (c). A representative two-step photobleaching of fluorescence trajectory (black line) detected by Chung-Kennedy filter (red line) is shown (d). The percentage of photobleaching steps of H3.3-EGFP within a nucleosome is shown (e). f–j, EGFP-KAP1 expressed in HEK293T cells oligomerizes on a nucleosome. Agarose gel electrophoresis analysis of DNAs extracted from nucleosomes was prepared from HEK293T/EGFP-KAP1 cells (f). Nucleosomes were immobilized on the surface by biotinylated anti-H2B antibody (g). The arrows imply that EGFP-KAP1 oligomerizes stepwise on a nucleosome. A representative single molecule image of the acquired sequence is shown (h). Samples of fluorescence trajectories (black line) and photobleaching steps detected by Chung-Kennedy filter (red line) are shown (i). The percentage of photobleaching steps of EGFP-KAP1 on a nucleosome. Results are means ± S.D. a.u. denotes arbitrary unit. Scale bar, 5 μm.
To evaluate whether the Sm-ChIPi approach can detect oligomers of protein on a nucleosome, we analyzed oligomerization of EGFP-KAP1 on a nucleosome (Fig. 2, f–j) because KAP1 forms trimer in solution (61). EGFP-KAP1 was transiently expressed in HEK293T cells. The presence of endogenous KAP1 protein prevents quantifying the exact stoichiometry of KAP1 on nucleosome, and overexpression allows assessing its oligomerization status. Mononucleosomes were prepared from EGFP-KAP1-transfected HEK293T cells and immobilized on the surface by biotinylated anti-H2B antibody. Analysis of fluorescence trajectories indicated that 4, 16, 25, 20, 17, and 18% of spots are one- to six-step photobleaching, respectively, suggesting that one nucleosome can associate with six KAP1 proteins. In summary, the Sm-ChIPi approach is a direct and sensitive technique to quantitatively assess assembly stoichiometry of epigenetic complex on chromatin.
Cellular Assembly Stoichiometry of YFP-PRC1 Proteins on a Mononucleosome
To assess the cellular assembly stoichiometry of PRC1 on chromatin, the YFP-PRC1 fusion genes, Y-Cbx2, Y-Cbx7, Y-Mel18, and Y-Ring1b, were introduced into KO mES cells, respectively. Cbx2, Cbx7, and Mel18 are the core subunits of canonical PRC1 complexes, and Ring1b is the core subunit of all PRC1 complexes (8). The expression of the fusion proteins were induced by 0.5 μg/ml Dox unless otherwise indicated. Ring1bfl/f;Rosa26::CreERT2 cells were incubated with 1.0 μm OHT for 3 days to deplete Ring1b locus (hereafter Ring1b−/−). The YFP-PRC1·nucleosome complexes were isolated from cells and fractionized by ultracentrifugation. Agarose gel electrophoresis was used to analyze the distribution of nucleosomal DNAs extracted from ultracentrifugation fractions (Fig. 3a). Fraction 18, the peak of mononucleosomes, was selected for the Sm-ChIPi analysis. The YFP-PRC1·mononucleosome complexes were immobilized by biotinylated anti-H3 antibody (Fig. 3b). The Sm-ChIPi analysis showed that 98.6, 97.2, 97.2, and 97.2% of individual fluorescent spots had one molecule of YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b, respectively (Fig. 3b), suggesting an assembly stoichiometry of PRC1 to mononucleosome is 1:1. To rule out issues of histone epitope accessibility, we immobilized the YFP-PRC1·mononucleosome complexes by biotinylated anti-H2B antibody as well (Fig. 3c). The immobilization by anti-H2B antibody gave the same results as the immobilization by anti-H3 antibody. We also immobilized the YFP-PRC1·mononucleosome complexes by biotinylated anti-GFP antibody (Fig. 3d). The immobilization produced the same results as the immobilization by antibodies against histones. To investigate whether the protein level affects the assembly stoichiometry, we prepared nucleosomes from Cbx2−/−/Y-Cbx2 mES cells in the presence of a variety of Dox concentrations. The assembly stoichiometry of YFP-Cbx2 to a mononucleosome was not affected by its protein level (Fig. 3e). Because sucrose gradient ultracentrifugation is based on the volume and mass of particles, it is possible that the assembly stoichiometry may be different among fractions. The Sm-ChIPi analysis showed that fractions 22 and 23 have the same assembly stoichiometry as fraction 18 (Fig. 3f).
FIGURE 3.

Cellular assembly stoichiometry of YFP-PRC1 proteins on a mononucleosome. a, agarose gel electrophoresis analysis of nucleosomal DNAs extracted from fractions of 5–30% sucrose gradient. Nucleosomes were prepared from Cbx2−/−/Y-Cbx2, Cbx7−/−/Y-Cbx7, Ring1b−/−/Y-Ring1b, and Mel18−/−/Y-Mel18 mES cells. A sample image of agarose gel is shown. Fraction 18 indicated by the arrow below the gel was used for single-molecule TIRF imaging. b–d, percentage of fluorescence photobleaching steps of YFP-Cbx2, YFP-Cbx7, YFP-Ring1b, and YFP-Mel18 on a mononucleosome from fraction 18. The YFP-PRC1·nucleosome complexes were immobilized on the surface by biotinylated antibodies directed against H3 (b), H2B (c), and GFP (d). Results are means ± S.D. e, percentage of fluorescence photobleaching steps of YFP-Cbx2 on a mononucleosome prepared from Cbx2−/−/Y-Cbx2 cells in the presence of Dox concentrations of 0 μg/ml (black bar), 0.5 μg/ml (red bar), or 2.0 μg/ml (green bar). The YFP-Cbx2·nucleosome complexes were immobilized on the surface by biotinylated anti-H3 antibody. Results are means ± S.D. f, percentage of fluorescence photobleaching steps of YFP-Cbx2 on a mononucleosome from fractions 22 and 23. The YFP-Cbx2·nucleosome complexes were immobilized on the surface by biotinylated anti-H3 antibody. Results are means ± S.D. g, single-molecule co-localization analysis. YFP-Cbx2 and mCherry-Cbx2 were stably co-expressed in Cbx2−/− mES cells (top). YFP-Cbx7 and mCherry-Cbx7 were stably co-expressed in Cbx7−/− mES cells (middle). YFP-Cbx2 and mCherry-Cbx7 were stably co-expressed in Cbx7−/− mES cells (bottom). The PRC1·nucleosome complexes from fraction 18 were immobilized by biotinylated anti-H3 antibody. YFP (left) and mCherry (center) were imaged. Overlay of the two images (right) shows 2–3% co-localization. Scale bar, 5 μm.
To seek out independent evidence for the assembly stoichiometry, we performed a single-molecule co-localization assay (Fig. 3g). Both YFP-Cbx2 and mCherry-Cbx2 fusion genes were co-expressed in the Cbx2−/− mES cells. The cross-linked mononucleosome fractions were prepared as above. The single-molecule co-localization analysis showed that 2.3% of YFP-Cbx2 and mCherry-Cbx2 overlap, which accounts for random co-localization. The same analysis showed 1.4% of YFP-Cbx7 and mCherry-Cbx7 co-localize. Additionally, both YFP-Cbx2 and mCherry-Cbx7 fusion proteins were co-expressed in Cbx7−/− mES cells. Analysis as above showed that 2.3% of YFP-Cbx2 and mCherry-Cbx7 co-localize. Thus, these data suggest that one PRC1 binds one mononucleosome.
To assess whether the YFP-PRC1 fusion proteins behave as their endogenous counterparts, we performed biochemical assays. Western blotting analysis indicated that the levels of YFP-Cbx7 and YFP-Ring1b are similar to that of their endogenous counterparts at 0.5 μg/ml Dox, whereas the background expression level of YFP-Cbx2 is similar to that of its endogenous counterpart (Fig. 4a). Co-IP indicated that YFP-Cbx2, YFP-Cbx7, and YFP-Mel18 precipitate endogenous Ring1b and Phc1, whereas YFP-Ring1b precipitates endogenous Phc1 (Fig. 4b). ChIP analysis indicated that YFP-Cbx2, YFP-Cbx7, YFP-Ring1b, and YFP-Mel18 are enriched at the promoters of known PRC1 target genes (Fig. 4c). Thus, these data indicate that the YFP fusion proteins test function as their endogenous counterparts.
FIGURE 4.

Fusion proteins tested recapture the functions of their endogenous counterparts. a, Western blot analysis of protein levels using antibodies directed against endogenous proteins. Ponceau S staining was used for the loading control. * indicates nonspecific bands. b, IP analysis of the interaction of endogenous Ring1b and Phc1 with YFP-PRC1 fusion proteins. Extracts were precipitated by anti-GFP antibody. The precipitates were analyzed by immunoblotting using antibodies directed against Ring1b and Phc1. The input contained 5% of the extract. WT denotes PGK12.1 mES cells. * indicates nonspecific bands. c, ChIP analysis of the binding YFP-PRC1 fusion proteins to endogenous target gene promoters. The fragmented chromatins isolated from Cbx2−/−/Y-Cbx2, Cbx7−/−/Y-Cbx7, Ring1b−/−/Y-Ring1b, and Mel18−/−/Y-Mel18 mES cells were precipitated using antibodies directed against Cbx2, Cbx7, Ring1b, and Mel18, respectively. Results are means ± S.D. d, quantification of the number and the concentration of Ring1b·PRC1 complexes in mES cells. The autocorrelation curves (black dot line) were fitted with the one component model of free diffusion in three dimensions with triplet function (red line). The table shows the ratio of endogenous to YFP-tagged Ring1b protein (En/Ex) detected by Western blotting (WB), the concentration of endogenous Ring1b and YFP-Ring1b fusion, and the number of endogenous Ring1b proteins.
To quantify the number and the concentration of PRC1 complexes in mES cells, we performed FCS experiments (Fig. 4d). The concentration and the numbers of Ring1b were estimated to be 0.12 μm and 18,000 molecules, respectively. The number of polycomb domains has been estimated to be about 16,000 (6). Thus, the number of PRC1 complexes roughly equals the number of polycomb domains.
Cellular Assembly Stoichiometry of YFP-PRC1 Proteins on a Polynucleosomal Array
Although fraction 23 of the sucrose gradients typically contains both mononucleosomes and dinucleosomes, 97.2% of individual fluorescent spots have one YFP-Cbx2 molecule, which suggests that at least a dinucleosome can associate with one PRC1 (Fig. 3, a and f). To further explore the assembly stoichiometry of PRC1 on polynucleosome, we generated a mixture of nucleosomes containing mono-, di-, and tri-nucleosomes (Fig. 5a). Fraction 19 used for the Sm-ChIPi analysis contained (48 ± 2)%, (31 ± 5)%, and (20 ± 6)% of mononucleosomes, dinucleosomes, and trinucleosomes, respectively. The mixture of nucleosomes was immobilized by biotinylated anti-H3 antibody (Fig. 5c). The Sm-ChIPi analysis indicated that 94.3, 94.3, 95.8, and 94.7% of individual fluorescent spots had one molecule of YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b, respectively (Fig. 5d). Thus, these data indicate that one PRC1 can bind a trinucleosome.
FIGURE 5.

Cellular assembly stoichiometry of YFP-PRC1 proteins on a polynucleosomal array. a and b, agarose gel electrophoresis analysis of nucleosomal DNAs extracted from fractions of 15–40% sucrose gradient. Nucleosomes were prepared from Cbx2−/−/Y-Cbx2, Cbx7−/−/Y-Cbx7, Ring1b−/−/Y-Ring1b, and Mel18−/−/Y-Mel18 mES cells. Representative images of agarose gel are shown. Fractions 19, 22, and 23 indicated by color-coded bars below the gels were used for single-molecule TIRF imaging. c, schematic depiction of the immobilization of YFP-PRC1·nucleosome complex on the surface by biotinylated anti-H3 antibody. d, percentage of fluorescence photobleaching steps of YFP-Cbx2, YFP-Cbx7, YFP-Ring1b, and YFP-Mel18 on a polynucleosomal array. The color-coded bars are described in a and b. The black bar indicates the percentage of photobleaching steps of YFP-PRC1 proteins on a mononucleosome, which is replicated from Fig. 3b. Results are means ± S.D. e, flow diagram describes the approach used for analyzing the assembly stoichiometry of YFP-Ring1b·PRC1 complex on a reconstituted tetranucleosomal array (f) and a 15-mer polynucleosomal array (g). f and g, agarose gel electrophoresis analysis of nucleosomal DNAs extracted from the fractions indicated above the gel (left). Representative single images of the acquired sequences are shown (middle). The percentage of fluorescence photobleaching steps for samples from fractions indicated is shown. Results are means ± S.D. Scale bar, 5 μm.
To further assess the assembly stoichiometry, we generated a mixture of nucleosomes containing nucleosomal arrays larger than trinucleosomes (Fig. 5b). Fraction 22 contained (16 ± 9)% and (10 ± 5)% of pentanucleosomes and hexanucleosomes, respectively. The Sm-ChIPi analysis of fraction 22 showed that 7.1, 8.6, 7.1, and 5.7% of individual fluorescent spots had two molecules of YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b, respectively (Fig. 5d), indicating that one PRC1 complex can associate with multiple nucleosomes. Fraction 23 contained (17 ± 6)% and (12 ± 5)% of hexanucleosomes and heptanucleosomes, respectively. The Sm-ChIPi analysis of fraction 23 showed that 20, 18.6, 22.9, and 18.6% of individual fluorescent spots had two molecules of YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b, respectively (Fig. 5d). Together, these data suggest that one PRC1 can associate with multiple nucleosomes.
To provide additional evidence of the PRC1 association with multiple nucleosomes, we reconstituted tetranucleosomal and 15-mer polynucleosomal arrays from recombinant histone octamers with biotin at one end (Fig. 5, e–g). The biotin-nucleosomal arrays were incubated with nuclear extract from Ring1b−/−/Y-Ring1b mES cells (Fig. 5e). The mixture was subjected to sucrose gradient ultracentrifugation, and fractions containing the nucleosomal arrays were analyzed. The YFP-Ring1b-PRC1·polynucleosome complexes were immobilized by biotin to the surface. Analysis of fluorescence trajectories indicated that 4.2 and 5.7% of individual fluorescent spots have two molecules of YFP-Ring1b on a tetranucleosome for fractions 22 and 23, respectively (Fig. 5f), and 30% of individual fluorescent spots have two molecules of YFP-Ring1b on a 15-mer polynucleosome. Notably, there were no three molecules of YFP-Ring1b on a 15-mer polynucleosome. Thus, these data suggest that one PRC1 can potentially associate with seven nucleosomes.
ES Cell Differentiation Selectively Alters the Assembly Stoichiometry of YFP-Cbx2 Protein on Chromatin
Features of chromatin are distinct between pluripotent and differentiated cells (62, 63). To assess whether the ES cell differentiation alters the PRC1-nucleosome stoichiometry, KO mES cells complemented with the YFP-PRC1 fusion gene were induced to differentiation by forming embryoid bodies. After a 10-day differentiation, the YFP-Ring1b-PRC1·polynucleosome complexes were prepared and immobilized (Fig. 6, a–c). The Sm-ChIPi analysis of the mononucleosome fraction indicated that a 1:1 assembly stoichiometry of PRC1 to mononucleosome (Fig. 6d), which is the same as undifferentiated mES cells. The Sm-ChIPi analysis of fraction 20 indicated that 17.1, 7.1, 6.6, and 8.6% of individual fluorescent spots have two molecules of YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b, respectively (Fig. 6d), indicating that a 2-fold larger fraction of the Cbx2-containing PRC1 complex associates with nucleosomes that have a second PRC1 complex bound, and that, in contrast with undifferentiated mES cells, a dinucleosome can associate with two molecules of Cbx2.
FIGURE 6.

ES cell differentiation selectively alters the assembly stoichiometry of YFP-Cbx2 protein on chromatin. a and b, agarose gel electrophoresis analysis of nucleosomal DNAs extracted from fractions of 15 to 40% sucrose gradient. Nucleosomes were prepared from Cbx2−/−/Y-Cbx2, Cbx7−/−/Y-Cbx7, Ring1b−/−/Y-Ring1b, and Mel18−/−/Y-Mel18 differentiated mES cells. Representative images of agarose gel are shown. Fractions 15, 20, 22, and 23 indicated by color-coded bars below the gels were used for single-molecule TIRF imaging. c, schematic depiction of the immobilization of YFP-PRC1·nucleosome complex on the surface by biotinylated anti-H3 antibody. d, percentage of fluorescence photobleaching steps of YFP-Cbx2, YFP-Cbx7, YFP-Ring1b, and YFP-Mel18 on a nucleosome prepared from the differentiated mES cells. The color-coded bars are described in a and b. Results are means ± S.D.
Fractions 22 and 23 contained nucleosomal array larger than a trinucleosome (Fig. 6b). The Sm-ChIPi analysis of fraction 22 indicated that 31.0, 12.9, 11.4, and 10.0% of individual fluorescent spots have two molecules of YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b, respectively (Fig. 6d). The Sm-ChIPi analysis of fraction 23 indicated that 52.8, 25.7, 27, and 31% of individual fluorescent spots have two molecules of YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b, respectively (Fig. 6d). Thus, these data indicated again that a 2-fold larger fraction of the Cbx2-containing PRC1 complexes associates with nucleosomes that have a second PRC1 complex bound. Altogether, these data suggest that ES cell differentiation selectively alters the assembly stoichiometry of YFP-Cbx2 protein on chromatin.
Assembly Stoichiometry Is Not Affected by the Depletion of PRC2 Subunit Eed
Previous studies have shown that the histone tails of the nucleosome are not required for compacting nucleosomal arrays by PRC1 (14). To test the effects of PRC2 on the native PRC1-nucleosome stoichiometry, we took advantage of the fact that Cbx4 and Cbx8 are not expressed in mES cells (64). YFP-Cbx4 and YFP-Cbx8 fusion genes were expressed in Eed KO mES cells, respectively. Nucleosomes were generated and immobilized by anti-H3 antibody (Fig. 7, a and c). The Sm-ChIPi analysis of the mononucleosome fraction 15 showed that 97.0 and 97.4% of individual fluorescent spots have one molecule of YFP-Cbx4 and YFP-Cbx8, respectively (Fig. 7d). The Sm-ChIPi analysis of the polynucleosome fraction 23 indicated that 26.9 and 26.3% of fluorescent spots have two molecules of YFP-Cbx4 and YFP-Cbx8, respectively (Fig. 7d). For a control, we established wild-type mES cells that stably express YFP-Cbx4 and YFP-Cbx8, respectively. Nucleosomes were prepared and immobilized by anti-H3 antibody as above (Fig. 7, b and c). The Sm-ChIPi analysis of the mononucleosome fraction 15 indicated that 97.1% of individual fluorescent spots have one molecule of both YFP-Cbx4 and YFP-Cbx8, respectively (Fig. 7e). The Sm-ChIPi analysis of the polynucleosome fraction 23 showed that 25.4 and 27.8% of individual fluorescent spots have two molecules of YFP-Cbx4 and YFP-Cbx8, respectively (Fig. 7e). Thus, these data suggest that the PRC2 Eed protein does not affect the cellular assembly stoichiometry of YFP-Cbx4 and YFP-Cbx8 on chromatin.
FIGURE 7.

Cellular assembly stoichiometry is not affected by the depletion of PRC2 subunit Eed. a and b, agarose gel electrophoresis analysis of nucleosomal DNAs extracted from fractions of 15 to 40% sucrose gradient. Nucleosomes were prepared from Eed−/−/Y-Cbx4 and Eed−/−/Y-Cbx8 (left) and Eed+/+/Y-Cbx4 and Eed+/+/Y-Cbx8 (right). Representative images of agarose gel are shown. Fractions 15 and 23 indicated by color-coded bars below the gels were used for single-molecule TIRF imaging. c, schematic depiction of the immobilization of YFP-PRC1·nucleosome complex on the surface by biotinylated anti-H3 antibody. d and e, percentage of fluorescence photobleaching steps of YFP-Cbx4 and YFP-Cbx8 on a nucleosome isolated from Eed−/−/Y-Cbx4 and Eed−/−/Y-Cbx8 (d) and from Eed+/+/Y-Cbx4 and Eed+/+/Y-Cbx8 (e) is shown. The color-coded bars are described in a and b. Results are means ± S.D.
Nucleoplasmic PRC1 Is Monomeric
To assess the stoichiometry of individual subunits of PRC1 within the nucleoplasm of cells, we employed a recently developed single-molecule immunoprecipitation approach (32). Nucleoplasmic fractions were extracted from mES cell lines and cross-linked with paraformaldehyde. YFP-PRC1 was immobilized on the surface by biotinylated anti-GFP antibody (Fig. 8a). Single-molecule image stacks were acquired using TIRF microscopy. Analysis of the numbers of YFP-PRC1 fusion proteins showed that 98.9, 97.9, 99.0, and 97.7% of individual fluorescent spots are one molecule of YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b (Fig. 8b), respectively, indicating a stoichiometry of 1:1:1:1 molecule for YFP-Cbx2, YFP-Cbx7, YFP-Mel18, and YFP-Ring1b and one copy of each subunit of PRC1.
FIGURE 8.

Nucleoplasmic PRC1 is monomeric. a, schematic depiction of the immobilization of YFP-PRC1 proteins on the surface by biotinylated anti-GFP antibody. The YFP-PRC1 complexes extracted from Cbx2−/−/Y-Cbx2, Cbx7−/−/Y-Cbx7, Ring1b−/−/Y-Ring1b, and Mel18−/−/Y-Mel18 mES cells were pulled down by biotinylated anti-GFP antibody via interaction with NeutrAvidin. b, percentage of fluorescence photobleaching steps of YFP-Cbx2, YFP-Cbx7, YFP-Ring1b, and YFP-Mel18 is shown. Results are means ± S.D.
PRC2 Is a Mixture of Monomer and Dimer and Binds to Nucleosome in a 1:1 or 2:1 Stoichiometry
To investigate whether PRC2 self-interacts within cells, YFP-PRC2 fusion genes, Y-Eed and Y-Ezh2, were stably expressed in Eed−/− and Ezh2−/− mES cells, respectively. Introduction of Y-Eed and Y-Ezh2 fusions into their respective KO mES cells restored H3K27me3 levels as demonstrated by immunofluorescence (Fig. 9a), suggesting that the two fusion proteins function as their endogenous counterparts. The residual H3K27me3 in Ezh2−/− mES cells may be generated by Ezh1. YFP-PRC2 from the nucleoplasm was immobilized by anti-GFP antibody (Fig. 9b). Single-molecule immunoprecipitation analysis showed that 18.6 and 15.7% of individual fluorescent spots have two molecules of YFP-Eed and YFP-Ezh2, indicating a mixture of monomeric and dimeric PRC2.
FIGURE 9.

PRC2 is a mixture of monomer and dimer and binds to mononucleosome in a 1:1 or 2:1 stoichiometry. a, immunostaining of H3K27me3 in Ezh2+/+, Eed+/+, Ezh2−/−, Eed−/−, Ezh2−/−/Y-Ezh2, and Eed−/−/Y-Eed mES cells by using antibody directed against H3K27me3 (green). DNAs were stained with Hoechst (blue). Overlay images are shown. Scale bar is 5 μm. b, nucleoplasmic YFP-Eed and YFP-Ezh2 are a mixture of monomers and dimers. The YFP-PRC2 complexes extracted from Ezh2−/−/Y-Ezh2 and Eed−/−/Y-Eed mES cells were pulled down by biotinylated anti-GFP antibody via interaction with NeutrAvidin (left). The percentage of fluorescence photobleaching steps of YFP-Eed and YFP-Ezh2 is shown as black bar (right). For a comparison, the red bar for the monomeric YFP is replicated from Fig. 1b. Results are means ± S.D. c, PRC2 binds to mononucleosome in a 1:1 or 2:1 stoichiometry. The YFP-PRC2·mononucleosome complexes from Ezh2−/−/Y-Ezh2 and Eed−/−/Y-Eed mES cells were immobilized by biotinylated antibodies directed against H3 (left). The percentage of fluorescence photobleaching steps of YFP-Eed and YFP-Ezh2 on a mononucleosome is shown as black bar (right). Results are means ± S.D. For comparison, the red bar for the monomeric YFP is replicated from (Fig. 1b). d and e, percentage of fluorescence photobleaching steps of YFP-Eed on a mononucleosome. The YFP-Eed·PRC2·mononucleosome complexes were immobilized by biotinylated antibodies directed against H2B (d) and GFP (e). For a comparison, the red bar for the monomeric YFP is replicated from Fig. 1b. Results are means ± S.D.
To assess the assembly stoichiometry of PRC2 on chromatin, the YFP-PRC2·mononucleosome complexes were prepared and immobilized by biotinylated anti-H3 antibody (Fig. 9c). The Sm-ChIPi analysis showed that 19.5 and 19.2% of fluorescent spots have two molecules of Y-Eed and Y-Ezh2, indicating that two PRC2 complexes can bind to a nucleosome. The Y-Eed·mononucleosome complexes were also immobilized by biotinylated anti-H2B (Fig. 9d) or anti-GFP antibodies (Fig. 9e). The Sm-ChIPi analysis gave similar results among these antibodies. Together, these data indicate that PRC2 binds to nucleosome in a 1:1 or 2:1 stoichiometry.
Discussion
In this study, we devised a novel approach to assess the cellular assembly stoichiometry of epigenetic complexes on chromatin and provided evidence that the PcG complexes PRC1 and PRC2 employ distinct mechanisms by which they assemble on chromatin. The cellular assembly stoichiometry reflects the mechanism by which the PcG complexes initiate, establish, and maintain repressive polycomb domains.
Molecular counting based on single-molecule fluorescence microscopy is a powerful approach to quantitatively assess the number of molecules within a macromolecular protein complex. By analyzing fluorescence photobleaching steps, Ulbrich and Isacoff (33) counted subunit composition of membrane-bound proteins expressed in Xenopus laevis oocytes. By developing and applying single-molecule fluorescence two-color coincidence detection, Balasubramanian and co-workers (31) characterized subunit composition within a reconstituted telomerase complex. By combining immunoprecipitation and single-molecule imaging, Ha and co-workers (32) developed single-molecule pull down (SiMPull) to probe how many proteins and which kinds are present in individual cellular protein complexes. Here, by integrating genetic engineering, chromatin immunoprecipitation, and single-molecule imaging, we developed Sm-ChIPi to quantify cellular assembly stoichiometry of epigenetic complex on chromatin. Both SiMPull and Sm-ChIPi are based on immunoprecipitation; however, Sm-ChIPi has been specifically developed and optimized for nucleosome complexes.
Biological Significance of the Cellular Assembly Stoichiometry of PRC1 on Chromatin
Although a nucleosome has 2-fold symmetry of histone organization, histone tails have been shown to be asymmetrically modified (28). The H3K27me3 mark has been suggested to be a dock site for the canonical PRC1 via interaction with the Cbx proteins (65). By a biochemical principle, we should detect a mixture of nucleosomes bound with one or two Cbx·PRC1 complexes. However, our Sm-ChIPi analysis indicated that one PRC1 can potentially associate with seven nucleosomes, suggesting that the PRC1 complex has multiple binding sites for nucleosomes or that the nuclear environment directs the PRC1 complex assembly on multiple nucleosomes. These data also implicate that the binding of a PRC1 to one disk surface of a nucleosome prevents association of the second nucleosomal disk surface with an additional PRC1. In mammalian cells, only a few large polycomb domains cover multiple neighboring genes, and the vast majority of polycomb domains cover individual promoter regions (6). Considering that the polycomb peaks of ChIP-Seq at promoters is less than 10 kb on average (6), we predict that only a small number of PRC1s reside at the promoter of each gene. To assess the stoichiometric relationship between PRC1 and polycomb domains, we measured the number of PRC1s in mES cells by FCS and found that the number of Ring1bs in mES cells roughly equals the number of polycomb domains. These data imply that only a small number of PRC1 complexes are decorated on chromatin to repress one gene.
The reconstituted Drosophila PRC1 packs nucleosomal arrays where histone tails have been depleted (14). The in vitro observations are consistent with the cellular assembly stoichiometry of PRC1 on chromatin where the depletion of the PRC2 Eed has no effect on the PRC1 assembly stoichiometry. The mechanism by which PRC1 mediates compaction of chromatin may be distinct from HP1 and L3MBTL1 proteins because both require histone lysine methylation in vitro (66, 67).
Previous studies have shown that the features of chromatin are distinct between pluripotent and differentiated cells and reflect the importance in establishing and maintaining lineage-specific gene transcription profile (62, 63). Our observations that the assembly stoichiometry of Cbx2 on chromatin is distinct between mES and differentiated cells suggest that the Cbx proteins diversify their functions during cell differentiation. Recent studies have shown that Cbx2 possesses unique characteristics during development and cell cycle progression (49, 68). In a mouse zygote, Cbx2 targets PRC1 to constitutive heterochromatin in a parent-of-origin-dependent manner (68). In mES cells, Cbx2 targets PRC1 to mitotic chromosomes in a PRC2-independent manner and binds stably to mitotic chromosomes without dissociation (49). Cbx2 is the active subunit of mammalian PRC1 for both inhibition of remodeling and compaction of chromatin in vitro via a stretch of charged amino acids (69). The charged domain has been proposed to interact with a nucleosome and to create more interactions with other nucleosomes. We propose that ES cell differentiation induces more Cbx2 proteins to be loaded onto chromatin, which may facilitate further chromatin compacting to establish and maintain stable epigenetic silencing. Further studies are needed to explore the mechanisms and functional roles of the unique Cbx2 protein.
Our single-molecule immunoprecipitation analysis indicated that the nucleoplasmic PRC1 proteins do not self-interact within cells under their expression levels similar to endogenous counterparts. However, several studies of the individual PRC1 subunits showed that they can self-associate in vitro (16–19), suggesting that the complex formation may prevent the self-association of individual subunits or that the in vitro observations do not reflect the physiological conditions. Recent studies showed that both Drosophila Ph and mammalian homolog Phc2 form oligomers (70, 71), and the oligomerization can be prevented by O-GlcNAcylation (71). Such oligomerization of Ph/Phc2 may play an architectural role in the long range organization of large polycomb domains that cover multiple neighboring genes, such as the Hox gene clusters and the inactive X chromosome in female cells. However, because the vast majority of polycomb domains are relatively small and usually overlap with promoter regions, the Ph/Phc2 oligomerization may not be required for genes with small polycomb domains. This hypothesis is consistent with the depletion of Ph/Phc2 mainly affecting expression of genes with larger polycomb domains (70, 71). Clearly, it will be important to test how the oligomerization of Ph/Phc regulates long range organization of chromatin structure in vivo.
Role of a Dimeric PRC2 in the Formation of a Repressive Polycomb Domain
Here, we provide direct evidence that nucleoplasmic PRC2 is a mixture of monomers and dimers. Previous studies of the reconstituted PRC2 reached divergent views about states of PRC2 oligomerization (23–25). The reconstituted PRC2 with five core subunits has been identified as a dimer (23). The reconstituted PRC2 with four subunits has been shown to be monomeric by electron microscopy (25). The reconstituted PRC2 with three subunits has been found to a mixture of monomers, dimers, trimers, and higher order oligomers (24). These variations could be due to the numbers of PRC2 subunits used in the reconstituted assay or the methods used to characterize the oligomerization states. Gel filtration fractionation of nuclear extracts from both mammals and Drosophila suggested that the apparent molecular weight of PRC2 is consistent with a mixture of monomers, dimers, and oligomers (26, 27). However, the gel filtration could not exclude non-PRC2 proteins or an extended structure of PRC2. Our observations by using ultra-sensitive single-molecule immunoprecipitation resolved these disparities.
We found that PRC2 binds to a nucleosome in a 1:1 or 2:1 stoichiometry in vivo. We suggest that a monomeric PRC2 might play a role in the initial establishment stage of polycomb domain formation and that the subsequent assembly and spreading of PRC2 proteins along the chromatin may require dimeric PRC2. In this model, the initial recruitment of PRC2 to specific loci by noncoding RNAs or sequence-specific DNA binding factors would promote trimethylation of H3K27 on an adjacent nucleosome (46, 72–75). This would lead to binding a dimeric PRC2 to the nucleosome via Eed interaction with H3K27me3 modification, which then facilitates methylation of an adjacent nucleosome or within a nucleosome and repeats the cycle (Fig. 10). This model is analogous to the formation of heterochromatin by the SIR proteins (76). This model can explain the previous discoveries that PRC2 favors di- and oligonucleosome substrates over mononucleosomes (77, 78). In a dimeric PRC2, one Eed binding to a nucleosome would position the second PRC2 to methylate the histone tail of H3 within the second nucleosome.
FIGURE 10.
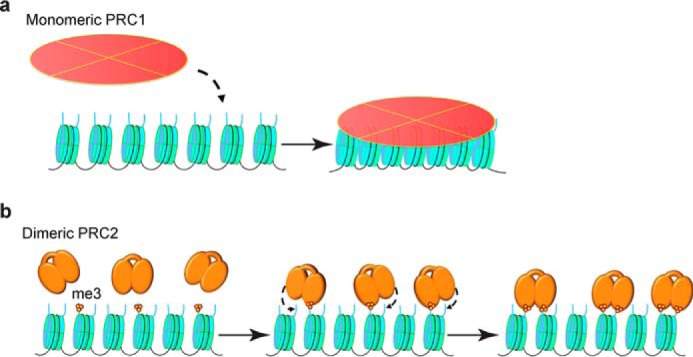
Models for the PRC1- and PRC2-mediated chromatin fibers. a, PRC1 can potentially pack seven nucleosomes within cells. b, dimeric PRC2 facilitates trimethylation of H3K27 within a nucleosome or an adjacent nucleosome.
In summary, we developed a novel approach of ChIP-coupled single-molecule fluorescence imaging to assess the cellular assembly stoichiometry of epigenetic complexes on chromatin. Sm-ChIPi could provide insights to other epigenetic complexes. The cellular assembly stoichiometry of the PcG complexes PRC1 and PRC2 on chromatin presented here provides us with the first assembly stoichiometry of these two complexes on chromatin within cells and offers invaluable in vivo data to understand previous in vitro biochemical data. The in vivo data of the PcG interaction with chromatin leads to novel insights and testable hypotheses that should inspire further studies of both PRC1 and PRC2 in the establishment and maintenance of repressive polycomb domains.
Author Contributions
X. R. conceived and designed the study, supervised the experiments, and wrote the paper. R. T. constructed plasmids, established transgenic mES cell lines, performed ChIP assays, and produced data of single-molecule imaging. C. Y. Z. performed Western blotting, immunoprecipitation, immunofluorescence, and fluorescence correlation spectroscopy. H. N. D. constructed plasmids and performed transfection. M. M. B. and A. M. J. provided reconstituted nucleosomal arrays. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Haruhiko Koseki for providing the Cbx2−/−, Ring1bfl/fl;Rosa26::CreERT2, Bmi1−/−/Mel18−/−, and Eed−/− mES cell lines; Dr. Julian Sale for providing H3.3−/−/H3.3-EGFP DT40 cell line, and Dr. Stuart Orkin and Dr. Xiaohua Shen for providing Ezh2−/− mES cell line. We thank the members of the Ren laboratory for constructive criticism.
This work was supported, in whole or in part, by National Institutes of Health Pathway to Independence Award K99/R00 GM094291 (to A. J.). This work was also supported by grants from the University of Colorado Denver (to X. R.), the CU-Denver Office Research Service (to X. R.), and American Cancer Society Grant IRG 57-001-53 subaward (to X. R.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.
M. Balas and A. Johnson, unpublished data.
- PcG
- polycomb group
- ChIP-Seq
- chromatin immunoprecipitation followed by high-throughput sequencing
- Dox
- doxycycline
- IP
- immunoprecipitation
- mES
- mouse embryonic stem
- OHT
- 4-hydroxytamoxifen
- PRC
- polycomb repressive complex
- Sm-ChIPi
- single-molecule chromatin immunoprecipitation imaging
- TIRF
- total internal reflection fluorescence
- EGFP
- enhanced GFP
- PCV
- packed cell volume
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- FCS
- fluorescence correlation spectroscopy.
References
- 1.Misteli T. (2007) Beyond the sequence: cellular organization of genome function. Cell 128, 787–800 [DOI] [PubMed] [Google Scholar]
- 2.Luger K., Dechassa M. L., and Tremethick D. J. (2012) New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 13, 436–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oliver S. S., and Denu J. M. (2011) Dynamic interplay between histone H3 modifications and protein interpreters: emerging evidence for a “Histone Language”. Chembiochem. 12, 299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zentner G. E., and Henikoff S. (2013) Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259–266 [DOI] [PubMed] [Google Scholar]
- 5.Musselman C. A., Lalonde M. E., Côté J., and Kutateladze T. G. (2012) Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bickmore W. A., and van Steensel B. (2013) Genome architecture: domain organization of interphase chromosomes. Cell 152, 1270–1284 [DOI] [PubMed] [Google Scholar]
- 7.Bowman G. D., and Poirier M. G. (2015) Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 115, 2274–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon J. A., and Kingston R. E. (2013) Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 49, 808–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan K. M., Fang D., Gan H., Hashizume R., Yu C., Schroeder M., Gupta N., Mueller S., James C. D., Jenkins R., Sarkaria J., and Zhang Z. (2013) The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 27, 985–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Margueron R., Justin N., Ohno K., Sharpe M. L., Son J., Drury W. J. 3rd, Voigt P., Martin S. R., Taylor W. R., De Marco V., Pirrotta V., Reinberg D., and Gamblin S. J. (2009) Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H., Wang L., Erdjument-Bromage H., Vidal M., Tempst P., Jones R. S., and Zhang Y. (2004) Role of histone H2A ubiquitination in polycomb silencing. Nature 431, 873–878 [DOI] [PubMed] [Google Scholar]
- 12.Gao Z., Zhang J., Bonasio R., Strino F., Sawai A., Parisi F., Kluger Y., and Reinberg D. (2012) PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45, 344–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., and Zhang Y. (2002) Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 14.Francis N. J., Kingston R. E., and Woodcock C. L. (2004) Chromatin compaction by a polycomb group protein complex. Science 306, 1574–1577 [DOI] [PubMed] [Google Scholar]
- 15.Eskeland R., Leeb M., Grimes G. R., Kress C., Boyle S., Sproul D., Gilbert N., Fan Y., Skoultchi A. I., Wutz A., and Bickmore W. A. (2010) Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 38, 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cowell I. G., and Austin C. A. (1997) Self-association of chromo domain peptides. Biochim. Biophys. Acta 1337, 198–206 [DOI] [PubMed] [Google Scholar]
- 17.Fujisaki S., Ninomiya Y., Ishihara H., Miyazaki M., Kanno R., Asahara T., and Kanno M. (2003) Dimerization of the Polycomb-group protein Mel-18 is regulated by PKC phosphorylation. Biochem. Biophys. Res. Commun. 300, 135–140 [DOI] [PubMed] [Google Scholar]
- 18.Lo S. M., and Francis N. J. (2010) Inhibition of chromatin remodeling by polycomb group protein posterior sex combs is mechanistically distinct from nucleosome binding. Biochemistry 49, 9438–9448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czypionka A., de los Paños O. R., Mateu M. G., Barrera F. N., Hurtado-Gómez E., Gómez J., Vidal M., and Neira J. L. (2007) The isolated C-terminal domain of Ring1B is a dimer made of stable, well-structured monomers. Biochemistry 46, 12764–12776 [DOI] [PubMed] [Google Scholar]
- 20.Kim C. A., Gingery M., Pilpa R. M., and Bowie J. U. (2002) The SAM domain of polyhomeotic forms a helical polymer. Nat. Struct. Biol. 9, 453–457 [DOI] [PubMed] [Google Scholar]
- 21.Lo S. M., McElroy K. A., and Francis N. J. (2012) Chromatin modification by PSC occurs at one PSC per nucleosome and does not require the acidic patch of histone H2A. PLoS ONE 7, e47162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGinty R. K., Henrici R. C., and Tan S. (2014) Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davidovich C., Goodrich K. J., Gooding A. R., and Cech T. R. (2014) A dimeric state for PRC2. Nucleic Acids Res. 42, 9236–9248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu L., Murat P., Matak-Vinkovic D., Murrell A., and Balasubramanian S. (2013) Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry 52, 9519–9527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciferri C., Lander G. C., Maiolica A., Herzog F., Aebersold R., and Nogales E. (2012) Molecular architecture of human polycomb repressive complex 2. Elife 1, e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao R., and Zhang Y. (2004) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15, 57–67 [DOI] [PubMed] [Google Scholar]
- 27.Tie F., Prasad-Sinha J., Birve A., Rasmuson-Lestander A., and Harte P. J. (2003) A 1-megadalton ESC/E(Z) complex from Drosophila that contains polycomblike and RPD3. Mol. Cell. Biol. 23, 3352–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voigt P., LeRoy G., Drury W. J. 3rd, Zee B. M., Son J., Beck D. B., Young N. L., Garcia B. A., and Reinberg D. (2012) Asymmetrically modified nucleosomes. Cell 151, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy P. J., Cipriany B. R., Wallin C. B., Ju C. Y., Szeto K., Hagarman J. A., Benitez J. J., Craighead H. G., and Soloway P. D. (2013) Single-molecule analysis of combinatorial epigenomic states in normal and tumor cells. Proc. Natl. Acad. Sci. U.S.A. 110, 7772–7777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baeza J., Dowell J. A., Smallegan M. J., Fan J., Amador-Noguez D., Khan Z., and Denu J. M. (2014) Stoichiometry of site-specific lysine acetylation in an entire proteome. J. Biol. Chem. 289, 21326–21338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alves D., Li H., Codrington R., Orte A., Ren X., Klenerman D., and Balasubramanian S. (2008) Single-molecule analysis of human telomerase monomer. Nat. Chem. Biol. 4, 287–289 [DOI] [PubMed] [Google Scholar]
- 32.Jain A., Liu R., Ramani B., Arauz E., Ishitsuka Y., Ragunathan K., Park J., Chen J., Xiang Y. K., and Ha T. (2011) Probing cellular protein complexes using single-molecule pull-down. Nature 473, 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulbrich M. H., and Isacoff E. Y. (2007) Subunit counting in membrane-bound proteins. Nat. Methods 4, 319–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren X., Gavory G., Li H., Ying L., Klenerman D., and Balasubramanian S. (2003) Identification of a new RNA.RNA interaction site for human telomerase RNA (hTR): structural implications for hTR accumulation and a dyskeratosis congenita point mutation. Nucleic Acids Res. 31, 6509–6515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren X., Li H., Clarke R. W., Alves D. A., Ying L., Klenerman D., and Balasubramanian S. (2006) Analysis of human telomerase activity and function by two color single molecule coincidence fluorescence spectroscopy. J. Am. Chem. Soc. 128, 4992–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ngo T. T., Zhang Q., Zhou R., Yodh J. G., and Ha T. (2015) Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell 160, 1135–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee J. Y., Lee J., Yue H., and Lee T. H. (2015) Dynamics of nucleosome assembly and effects of DNA methylation. J. Biol. Chem. 290, 4291–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deindl S., Hwang W. L., Hota S. K., Blosser T. R., Prasad P., Bartholomew B., and Zhuang X. (2013) ISWI remodelers slide nucleosomes with coordinated multi-base-pair entry steps and single-base-pair exit steps. Cell 152, 442–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li M., Hada A., Sen P., Olufemi L., Hall M. A., Smith B. Y., Forth S., McKnight J. N., Patel A., Bowman G. D., Bartholomew B., and Wang M. D. (2015) Dynamic regulation of transcription factors by nucleosome remodeling. Elife 4, e06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simon M., North J. A., Shimko J. C., Forties R. A., Ferdinand M. B., Manohar M., Zhang M., Fishel R., Ottesen J. J., and Poirier M. G. (2011) Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci. U.S.A. 108, 12711–12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo Y., North J. A., Rose S. D., and Poirier M. G. (2014) Nucleosomes accelerate transcription factor dissociation. Nucleic Acids Res. 42, 3017–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katoh-Fukui Y., Tsuchiya R., Shiroishi T., Nakahara Y., Hashimoto N., Noguchi K., and Higashinakagawa T. (1998) Male-to-female sex reversal in M33 mutant mice. Nature 393, 688–692 [DOI] [PubMed] [Google Scholar]
- 43.Cheng B., Ren X., and Kerppola T. K. (2014) KAP1 represses differentiation-inducible genes in embryonic stem cells through cooperative binding with PRC1 and derepresses pluripotency-associated genes. Mol. Cell. Biol. 34, 2075–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Endoh M., Endo T. A., Endoh T., Fujimura Y., Ohara O., Toyoda T., Otte A. P., Okano M., Brockdorff N., Vidal M., and Koseki H. (2008) Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development 135, 1513–1524 [DOI] [PubMed] [Google Scholar]
- 45.Elderkin S., Maertens G. N., Endoh M., Mallery D. L., Morrice N., Koseki H., Peters G., Brockdorff N., and Hiom K. (2007) A phosphorylated form of Mel-18 targets the Ring1B histone H2A ubiquitin ligase to chromatin. Mol. Cell 28, 107–120 [DOI] [PubMed] [Google Scholar]
- 46.Shen X., Kim W., Fujiwara Y., Simon M. D., Liu Y., Mysliwiec M. R., Yuan G. C., Lee Y., and Orkin S. H. (2009) Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell 139, 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penny G. D., Kay G. F., Sheardown S. A., Rastan S., and Brockdorff N. (1996) Requirement for Xist in X chromosome inactivation. Nature 379, 131–137 [DOI] [PubMed] [Google Scholar]
- 48.Frey A., Listovsky T., Guilbaud G., Sarkies P., and Sale J. E. (2014) Histone H3.3 is required to maintain replication fork progression after UV damage. Curr. Biol. 24, 2195–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhen C. Y., Duc H. N., Kokotovic M., Phiel C. J., and Ren X. (2014) Cbx2 stably associates with mitotic chromosomes via a PRC2- or PRC1-independent mechanism and is needed for recruiting PRC1 complex to mitotic chromosomes. Mol. Biol. Cell 25, 3726–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ziv Y., Bielopolski D., Galanty Y., Lukas C., Taya Y., Schultz D. C., Lukas J., Bekker-Jensen S., Bartek J., and Shiloh Y. (2006) Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 8, 870–876 [DOI] [PubMed] [Google Scholar]
- 51.Ren X., and Kerppola T. K. (2011) REST interacts with Cbx proteins and regulates polycomb repressive complex 1 occupancy at RE1 elements. Mol. Cell. Biol. 31, 2100–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luger K., Rechsteiner T. J., and Richmond T. J. (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304, 3–19 [DOI] [PubMed] [Google Scholar]
- 53.Johnson A., Li G., Sikorski T. W., Buratowski S., Woodcock C. L., and Moazed D. (2009) Reconstitution of heterochromatin-dependent transcriptional gene silencing. Mol. Cell 35, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li G., and Widom J. (2004) Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 11, 763–769 [DOI] [PubMed] [Google Scholar]
- 55.Johnson A., Wu R., Peetz M., Gygi S. P., and Moazed D. (2013) Heterochromatic gene silencing by activator interference and a transcription elongation barrier. J. Biol. Chem. 288, 28771–28782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vary J. C., Fazzio T. G., and Tsukiyama T. (2004) Assembly of yeast chromatin using ISWI complexes. Method Enzymol. 375, 88–102 [DOI] [PubMed] [Google Scholar]
- 57.An W., and Roeder R. G. (2004) Reconstitution and transcriptional analysis of chromatin in vitro. Methods Enzymol. 377, 460–474 [DOI] [PubMed] [Google Scholar]
- 58.Chung S. H., and Kennedy R. A. (1991) Forward-backward non-linear filtering technique for extracting small biological signals from noise. J. Neurosci. Methods 40, 71–86 [DOI] [PubMed] [Google Scholar]
- 59.Schwille P., Meyer-Almes F. J., and Rigler R. (1997) Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys. J. 72, 1878–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coffman V. C., and Wu J. Q. (2012) Counting protein molecules using quantitative fluorescence microscopy. Trends Biochem. Sci. 37, 499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peng H., Feldman I., and Rauscher F. J. 3rd. (2002) Hetero-oligomerization among the TIF family of RBCC/TRIM domain-containing nuclear cofactors: a potential mechanism for regulating the switch between coactivation and corepression. J. Mol. Biol. 320, 629–644 [DOI] [PubMed] [Google Scholar]
- 62.Meshorer E., and Misteli T. (2006) Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 7, 540–546 [DOI] [PubMed] [Google Scholar]
- 63.Fisher C. L., and Fisher A. G. (2011) Chromatin states in pluripotent, differentiated, and reprogrammed cells. Curr. Opin. Genet. Dev. 21, 140–146 [DOI] [PubMed] [Google Scholar]
- 64.Morey L., Pascual G., Cozzuto L., Roma G., Wutz A., Benitah S. A., and Di Croce L. (2012) Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell 10, 47–62 [DOI] [PubMed] [Google Scholar]
- 65.Min J., Zhang Y., and Xu R. M. (2003) Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 17, 1823–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Canzio D., Chang E. Y., Shankar S., Kuchenbecker K. M., Simon M. D., Madhani H. D., Narlikar G. J., and Al-Sady B. (2011) Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell 41, 67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trojer P., Li G., Sims R. J. 3rd, Vaquero A., Kalakonda N., Boccuni P., Lee D., Erdjument-Bromage H., Tempst P., Nimer S. D., Wang Y. H., and Reinberg D. (2007) L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 129, 915–928 [DOI] [PubMed] [Google Scholar]
- 68.Tardat M., Albert M., Kunzmann R., Liu Z., Kaustov L., Thierry R., Duan S., Brykczynska U., Arrowsmith C. H., and Peters A. H. (2015) Cbx2 targets PRC1 to constitutive heterochromatin in mouse zygotes in a parent-of-origin-dependent manner. Mol. Cell 58, 157–171 [DOI] [PubMed] [Google Scholar]
- 69.Grau D. J., Chapman B. A., Garlick J. D., Borowsky M., Francis N. J., and Kingston R. E. (2011) Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev. 25, 2210–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Isono K., Endo T. A., Ku M., Yamada D., Suzuki R., Sharif J., Ishikura T., Toyoda T., Bernstein B. E., and Koseki H. (2013) SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell 26, 565–577 [DOI] [PubMed] [Google Scholar]
- 71.Gambetta M. C., and Müller J. (2014) O-GlcNAcylation prevents aggregation of the Polycomb group repressor polyhomeotic. Dev. Cell 31, 629–639 [DOI] [PubMed] [Google Scholar]
- 72.Davidovich C., Wang X., Cifuentes-Rojas C., Goodrich K. J., Gooding A. R., Lee J. T., and Cech T. R. (2015) Toward a consensus on the binding specificity and promiscuity of PRC2 for RNA. Mol. Cell 57, 552–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rinn J. L., Kertesz M., Wang J. K., Squazzo S. L., Xu X., Brugmann S. A., Goodnough L. H., Helms J. A., Farnham P. J., Segal E., and Chang H. Y. (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peng J. C., Valouev A., Swigut T., Zhang J., Zhao Y., Sidow A., and Wysocka J. (2009) Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 139, 1290–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Landeira D., Sauer S., Poot R., Dvorkina M., Mazzarella L., Jørgensen H. F., Pereira C. F., Leleu M., Piccolo F. M., Spivakov M., Brookes E., Pombo A., Fisher C., Skarnes W. C., Snoek T., et al. (2010) Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat. Cell Biol. 12, 618–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Swygert S. G., Manning B. J., Senapati S., Kaur P., Lindsay S., Demeler B., and Peterson C. L. (2014) Solution-state conformation and stoichiometry of yeast Sir3 heterochromatin fibres. Nat. Commun. 5, 4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuzmichev A., Jenuwein T., Tempst P., and Reinberg D. (2004) Different Ezh2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol. Cell 14, 183–193 [DOI] [PubMed] [Google Scholar]
- 78.Martin C., Cao R., and Zhang Y. (2006) Substrate preferences of the EZH2 histone methyltransferase complex. J. Biol. Chem. 281, 8365–8370 [DOI] [PubMed] [Google Scholar]