

Background: Advanced glycation end products (AGEs) are involved in the development and progression of diabetes-associated osteoporosis.
Results: High dose AGEs induced cell apoptosis. Low dose AGEs stimulated cell proliferation and effected cell function by increasing autophagy via the RAGE/Raf/MEK/ERK pathway.
Conclusion: AGE-induced autophagy correlated with the proliferation and function of osteoblast.
Significance: Autophagy is a potential therapeutic molecular target for diabetic osteoporosis.
Keywords: autophagy, cell proliferation, extracellular-signal-regulated kinase (ERK), osteoblast, receptor for advanced glycation end products (RAGE), advanced glycation end products (AGEs)
Abstract
The interaction between advanced glycation end products (AGEs) and receptor of AGEs (RAGE) is associated with the development and progression of diabetes-associated osteoporosis, but the mechanisms involved are still poorly understood. In this study, we found that AGE-modified bovine serum albumin (AGE-BSA) induced a biphasic effect on the viability of hFOB1.19 cells; cell proliferation was stimulated after exposure to low dose AGE-BSA, but cell apoptosis was stimulated after exposure to high dose AGE-BSA. The low dose AGE-BSA facilitates proliferation of hFOB1.19 cells by concomitantly promoting autophagy, RAGE production, and the Raf/MEK/ERK signaling pathway activation. Furthermore, we investigated the effects of AGE-BSA on the function of hFOB1.19 cells. Interestingly, the results suggest that the short term effects of low dose AGE-BSA increase osteogenic function and decrease osteoclastogenic function, which are likely mediated by autophagy and the RAGE/Raf/MEK/ERK signal pathway. In contrast, with increased treatment time, the opposite effects were observed. Collectively, AGE-BSA had a biphasic effect on the viability of hFOB1.19 cells in vitro, which was determined by the concentration of AGE-BSA and treatment time. A low concentration of AGE-BSA activated the Raf/MEK/ERK signal pathway through the interaction with RAGE, induced autophagy, and regulated the proliferation and function of hFOB1.19 cells.
Introduction
Both osteoporosis and diabetes are common in the elderly. Numerous studies have demonstrated a relationship between osteoporosis and diabetes. Recent studies showed that the incidence of hip fracture and spinal fracture in patients with type 1 or type 2 diabetes increased significantly compared with healthy subjects (1). However, the increased incidence of osteoporosis-associated fractures in diabetic patients cannot be explained by a decrease in bone mineral density in these patients; different from type 1 diabetes, bone mineral density in patients with type 2 diabetes often increases (2, 3). Moreover, in patients with type 2 diabetes, the risk of fracture in patients with a long disease history is significantly higher than in newly diagnosed patients (4). Therefore, the increased incidence of fracture in diabetic patients cannot be ascribed to bone structure damage only but is also associated with the long term pathogenic effects of diabetes.
Advanced glycation end products (AGEs)3 are the end products of glucose and other reducing sugars, proteins, lipids, and nucleic acids via a non-enzymatic glycosylation reaction (5). During aging, renal failure, inflammation, particularly diabetes, and the formation and deposit of AGEs accelerate. The deposit of AGEs can be aggravated by disease progression. AGEs can bind to multiple membrane receptor proteins. The main receptor of AGEs is receptor of advanced glycation end products (RAGE). The interaction between AGEs and RAGE is involved in the pathophysiological processes of many diseases (6). In diabetes, an abnormal increase in AGEs and RAGE in bone tissues may be associated with the development and progression of diabetes-associated osteoporosis (7). However, there is a dispute concerning the effects of AGEs on osteoblasts. McCarthy et al. (8) found that the short term effect of AGEs may promote the proliferation of osteoblasts, and the long term effect of AGEs may inhibit the proliferation of osteoblasts. However, other studies did not observe this phenomenon. These studies suggested that AGEs significantly inhibit the proliferation and induce apoptosis of osteoblasts, and neither long term nor short term treatment with AGEs promoted the proliferation of osteoblasts (9–11).
Autophagy is the primary metabolic process by which eukaryotic cells degrade and recover damaged macromolecules and organelles (12, 13). During this process, substances in the cytoplasm are phagocytosed by autophagosomes, which are spherical structures with double layer membranes, and transported to lysosomes for degradation. After binding to late endosomes or lysosomes, autophagosomes and their contents are degraded. The degradation products can be re-used in the syntheses of macromolecules and in energetic metabolism (12). In all cells, low level autophagy ensures the recycling of longevity proteins and organelles (14). The level of autophagy can be up-regulated in stressful conditions (15). However, excessive autophagy is harmful to cells and leads to damage or massive death of cells (16, 17). Recent studies have proven that autophagy is closely associated with the functions of osteoblasts. Autophagy deficiency can cause increased oxidative stress levels in osteoblasts, secretion of receptor activator for nuclear factor-κ B ligand (RANKL), and decreased mineralization (18). Autophagy is also helpful in maintaining the proliferation and function of osteoblasts in high glucose levels (19). Studies on cardiovascular diseases and cancer have confirmed that an increase in AGEs as well as RAGE can activate autophagy-associated signal pathways and induce autophagy (20–22). However, there are no reports on whether AGEs in osteoblasts can regulate autophagy.
The primary aim of this study is to determine the effects of AGEs on the proliferation and function of osteoblasts, assess whether autophagy plays a key role in these processes, and the likely mechanisms involved.
Experimental Procedures
Cell Culture and Materials
The human fetal osteoblastic cell line hFOB 1.19, kindly provided by Dr. M. Subramaniam (23), was maintained in a 1:1 mixture of Ham's F-12 medium/Dulbecco's modified Eagle's medium without phenol red (Gibco) and supplemented with 10% fetal bovine serum (FBS) (HyClone) and 0.3 g/liter G418 (Sigma) in a humidified 5% CO2 atmosphere at 33.5 °C, and the medium was changed every other day. The cells were subcultured using trypsin/EDTA to replace the cells and begin the experiment. The hFOB 1.19 cells were plated at 104 cells/cm2 for 24 h before treatment.
Bovine serum albumin (BSA), PD98059, the 3-(4,5-dimethyl-thiazol-2-yl)- 2,5-diphenyl-tetrazolium bromide (MTT), and Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-CH2F (Z-DEVD-fmk) were obtained from Sigma. The EGFP-LC3 plasmid was kindly provided by Addgene. The RAGE-shRNA lentiviral and ctrl-shRNA lentiviral were purchased from Genechem (China). Primary antibodies for LC3, phospho-c-Raf (p-c-Raf), total ERK1/2 (t-ERK1/2), p-ERK1/2, p-MEK1/2, and t-MEK were purchased from Cell Signaling Technology, and antibodies for beclin-1, p62/SQSTM1, OPG, OCN, RANKL, and RAGE were purchased from Abcam.
Preparation of AGE Protein
AGE-BSA was prepared as described previously. BSA was added into 10 mmol/liter phosphate-buffered saline (PBS) (pH 7.4, concentration of 5 g/liter) and incubated with 50 mmol/liter d-glucose in 5% CO2/95% air at 37 °C for 12 weeks. Unincorporated glucose was removed by dialysis overnight against PBS. AGE-BSA-specific fluorescence determinations were performed by measuring emission at 440 nm on excitation at 370 nm using a fluorescence spectrophotometer (Hitachi, Japan). The fluorescence intensity of AGE-BSA was 50 times higher than BSA. AGE-BSA content was estimated by fluorescence intensity at a protein concentration of 1 mg/ml. AGE-BSA was stored at −70 °C until use.
Cell Viability and Proliferation Analysis
Cell viability was measured using MTT. Briefly, the cells were seeded onto 96-well plates (6000 cells/well) for 24 h, and the medium was then replaced with 10% serum medium. After treatment, culture media were changed for serum-free culture media. MTT dissolved in PBS was added to each well and then incubated for 4 h. After this interval, the serum-free culture medium containing MTT was discarded, and dimethyl sulfoxide (DMSO) was added to each well dissolving the precipitate. The optical densities were measured at 490 nm spectral wavelength using a microplate reader (Spectra Thermo, Switzerland). Cell proliferation was estimated using a BrdU kit (Roche Applied Science, China) following the protocol of the manufacturer. Cell viability and proliferation results were expressed as percentages. The absorbance measured from untreated cells was taken to be 100%.
Cell Apoptosis Analysis
Apoptosis was quantified by cell death detection ELISAPLUS (Roche Applied Science, China) according to the manufacturer's protocol. Briefly, the cytoplasmic histone/DNA fragments from cells were extracted and bound to the immobilized anti-histone antibody. Subsequently, the peroxidase-conjugated anti-DNA antibody was added for the detection of immobilized histone/DNA fragments. After addition of substrate for peroxidase, the optical densities were measured at 405 nm spectral wavelength using a microplate reader (Spectra Thermo, Switzerland). Cell death results were expressed as percentages. The absorbance measured from untreated cells was taken to be 100%.
Apoptosis was also determined by detecting phosphatidylserine exposure on cell plasma membrane with the fluorescent dye annexin V-FITC/PI apoptosis detection kit according to the manufacturer's protocols (KeyGEN, China). In brief, cells were harvested and washed in ice-cold PBS twice, resuspended in 500 μl of binding buffer, and incubated with 5 μl of annexin V-FITC and 5 μl of PI solution for 15 min at room temperature in the dark, and then immediately analyzed by bivariate flow cytometry using a FACS SCAN flow cytometer equipped with Modfit LT 3.0 (BD Biosciences). Approximately 5 × 105 cells were analyzed in each of the samples.
Western Blotting
After treatment, the cells were extracted with lysis buffer (150 mm NaCl, 1% Nonidet P-40, 0.1% SDS, 2 μg/ml aprotinin, 1 mm PMSF) for 30 min at 4 °C. The supernatants were centrifuged at 12,000 × g for 15 min at 4 °C. The supernatant containing total protein was harvested. Aliquots containing 50 μg of proteins were separated by SDS-12% PAGE and transferred to PVDF membranes at 60 or 40 V for 2 h at low temperature. The membranes were soaked in blocking buffer (5% skimmed milk) for 2 h. Subsequently, proteins were detected using primary antibodies at 1:500 or 1:1000 dilution overnight at 4 °C and then visualized using anti-goat or anti-rabbit IgG conjugated with peroxidase (HRP) at 1:6000 or 1:8000 dilution for 2 h at room temperature. The EC3 imaging system (UVP Inc. Upland, CA) was used to catch up the specific bands, and the optical density of each band was measured using an ImageJ software (National Institutes of Health, Bethesda). The rate between interesting proteins and GAPDH of the same sample was calculated as relative content and expressed graphically.
Transmission Electron Microscopy
The cells from each group were digested after 24 h of culture, followed by centrifugation, and the floating cells were collected. The cells were washed twice with cold PBS and fixed in 5% glutaraldehyde. Subsequently, the cells were conventionally dehydrated, embedded, sectioned, and stained, and the formation of autophagosomes was observed using transmission electron microscopy. The number of intracellular autophagosomes in every 10 fields was counted.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde at room temperature for 15 min. After washing with PBS, cells were permeabilized with 0.2% Triton X-100 for 5 min. After washing with PBS, sections were incubated in a blocking buffer containing 5% BSA for 30 min at room temperature, followed by incubation with anti-LC3 (1:200) antibody overnight at 4 °C. Secondary antibodies labeled with fluorescein (1:500, Abcam) were applied for 120 min. After incubating with 0.1% DAPI for 5 min and another washing step with PBS, coverslips were transferred onto glass slides. Images were captured on a wide field fluorescent microscope (Olympus, Japan).
EGFP-LC3 Expression Analysis
The cells from each group were seeded into 24 wells (25,000 cells/well) and incubated for 12 h. The cells were transiently transfected with the pEGFP-LC3B plasmid with the use of Lipofectamine 2000 (Invitrogen). Briefly, the cells were transfected with 4.0 μg of vector DNA and 10 μl of Lipofectamine 2000 in 2 ml of Opti-MEM medium. At 6 h after transfection, the medium was replaced with normal DMEM/F-12 medium with 10% FBS for 24 h. Images were captured on a wide field fluorescent microscope (Olympus, Japan). The number of EGFP-LC3 dots was determined by manual counting in five fields, and nuclear number was evaluated by counting DAPI-stained nuclei in the same fields using the same magnification. The number of EGFP-LC3 puncta/cell was evaluated as the total number of dots divided by the number of nuclei in each microscopic field.
RNA Interference
For the gene knockdown experiments, we obtained lentiviral shRNA purchased from Shanghai GenePharma. The shRNA was designed against RAGE (sense, 5′-CCG GTG AGA CAG GGC TCT TCA CAC TCT CGA GAG TGT GAA GAG CCC TGT CTC ATT TTT G-3′; antisense, 5′-AAT TCA AAA ATG AGA CAG GGC TCT TCA CAC TCT CGA GAG TGT GAA GAG CCC TGT CTC A-3′). A nontargeting stem-loop DNA vector was also generated for use as a negative control. RAGE-shRNA lentivirus or negative control lentivirus was added into target hFOB 1.19 cells at multiplicity of infection (40) with ENi.S and 5 μg/ml Polybrene to obtain stably transfected RAGE-shRNA and ctrl-shRNA cells.
Real Time RT-PCR
Total RNA was extracted from the hFOB cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions and quantified spectrophotometrically at 260 nm with acceptable 260:280 ratios between 1.8 and 2.0. The RNA quality was also checked by 1% agarose gel electrophoresis, stained with 1 μg/ml ethidium bromide. Residual genomic DNA was removed by incubating RNA with RNase-free DNase (Promega). Total RNA was reverse-transcribed using a reverse transcription kit (TaKaRa, China) according to the manufacturer's protocols.
Real time PCR was performed on ABI Prism 7900HT Fast System (Applied Biosystems) using SYBR Premix Ex TaqTM II (TaKaRa, China). Amplifications were carried out in a total volume of 20 μl and cycled 40 times after initial denaturation (95 °C for 30 s) with the following parameters: 95 °C for 5 s and 60 °C for 30 s. Primers sequences were listed in Table 1, and β-actin was used as an internal control. The relative mRNA expression was quantified through a comparison of the cycle threshold (Ct) values. The experimental data were processed using the 2−ΔΔCt method as follows: ΔΔCt = (Ct target − Ct internal control) experiment group − (Ct target − Ct internal control) normal control group. Each experimental group was repeated three times.
TABLE 1.
Primer sequences used in real time PCR experiments
The following abbreviations are used: F, forward; R, reverse.
| Gene | Primer sequence 5′–3′ | Accession no. | Product size |
|---|---|---|---|
| bp | |||
| RANKL | F: CAACATATCGTTGGATCACAGCA | GenBankTM NM_033012 | 161 |
| R: GACAGACTCACTTTATGGGAACC | |||
| OPG | F: GCGCTCGTGTTTCTGGACA | GenBankTM NM_002546 | 226 |
| R: AGTATAGACACTCGTCACTGGTG | |||
| OCN | F: CACTCCTCGCCCTATTGGC | GenBankTM NM_199173 | 112 |
| R: CCCTCCTGCTTGGACACAAAG | |||
| ALP | F: AACATCAGGGACATTGACGTG | GenBankTM NM_000478 | 159 |
| R: GTATCTCGGTTTGAAGCTCTTCC | |||
| β-Actin | F: GACAGGATGCAGAAGGAGATTACT | GenBankTM NM_001101 | 142 |
| R: TGATCCACATCTGCTGGAAGGT |
ALP Detection
The cells from each group were washed three times with PBS and lysed with 0.1% Triton X-100 using three cycles of freezing and thawing to verify that the cells were completely lysed. The cell lysates were centrifuged at 15,000 × g for 5 min at 4 °C, and the supernatant was collected. 50 μl of cell lysate was transferred to a 96-well plate, and 150 ml of p-nitrophenyl phosphate (Sigma, Irvine, UK) was added to each well as substrate for 20 min at 37 °C. p-Nitrophenol was quantified based on the spectrophotometric absorbance at 405 nm. ALP activity was normalized to the total protein concentration for each sample using a BCA protein assay (Pierce).
Statistical Analyses
The experiments were repeated three times. The quantitative data are presented as means ± S.E. The Statistical Package for Social Science (SPSS) 17.0 software was used for analysis. Comparisons among multiple groups were performed using one-way analysis of variance. Pairwise comparisons were performed using the t test. p < 0.05 indicated that the observed difference was significant.
Results
AGE-BSA Had a Biphasic Effect on the Viability of hFOB1.19 Osteoblasts and the Effect Was Associated with Autophagy
To determine the influence of AGE-BSA on the viability of osteoblasts, we treated human hFOB1.19 osteoblasts with AGE-BSA or BSA at different concentrations (50–250 mg/liter) for different time periods (0, 24, 48, and 72 h) and then assessed the viability of treated cells using MTT analysis. Interestingly, we found that AGE-BSA had a biphasic effect on the viability of hFOB1.19 cells; treatment with a low concentration of AGE-BSA (150 mg/liter) for 24 and 48 h significantly increased cell viability, particularly at 24 h. This effect decreased after 72 h of treatment. Treatment with high concentrations of AGE-BSA (above 200 mg/liter) for 24, 48, and 72 h significantly inhibited cell viability (Fig. 1A). The control BSA at various concentrations did not have a significant effect on cell viability (Fig. 1B).
FIGURE 1.

Effect of AGE-BSA on cell viability and proliferation in hFOB 1.19 cells, and autophagy is involved in the effect of AGE-BSA. A, cells were treated with AGE-BSA at various concentrations for 0, 24, 48, or 72 h, and cell viability was estimated using MTT assay. Values represent mean ± S.E. of at least three independent experiments. *, p < 0.05 versus control; **, p < 0.01 versus control. B, cells were treated with BSA at various concentrations for 0, 24, 48, or 72 h, and cell viability was estimated using MTT assay. Values represent mean ± S.E. of at least three independent experiments. C and D, cells were treated with or without 3-MA (5 mm) incubated with AGE-BSA (150 μg/ml) for 0, 24, 48, or 72 h. C, cell viability was estimated using MTT assay; D, cell proliferation was estimated using a BrdU kit. Values represent mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control; #, p < 0.05 versus AGE-BSA treatment; ##, p < 0.01 versus AGE-BSA treatment. E, cells were treated with or without 3-MA (5 mm) incubated with AGE-BSA (150 μg/ml) for 24, 48, or 72 h; cell death was estimated using a cell death detection ELISAPlus. Values represent mean ± S.E. of at least three independent experiments.
To assess whether increased viability of hFOB1.19 osteoblasts is associated with enhanced proliferation of cells, we assessed the proliferation of treated cells using a BrdU kit, and the results were similar to the MTT assay (Fig. 1D). Furthermore, to determine whether the decreased effect of AGE-BSA (150 mg/liter) for long term on osteoblast proliferation is related to cell death, cell death detection ELISAPLUS was carried out. No significant cell death was observed at any time point examined, even after the AGE-BSA (150 mg/liter) treatment for 72 h (Fig. 1E). In addition, to further investigate the effect of autophagy on enhanced proliferation, 3-methyladenine (3-MA), a classic inhibitor of autophagy, was used to inhibit cell autophagy. The result showed that 3-MA significantly inhibited the effect of AGE-BSA (150 mg/liter) in promoting the viability and proliferation of hFOB1.19 cells (Fig. 1, C and D), suggesting the effect of AGE-BSA (150 mg/liter) on promoting proliferation might be related to cell autophagy.
To determine whether the inhibitory effect of high concentrations of AGE-BSA (above 200 mg/liter) on osteoblast viability is related to cell apoptosis, a cell death detection ELISAPLUS and an annexin V-FITC/PI apoptosis detection kit were performed. The DNA fragmentation ratio of AGE-BSA (200 mg/liter)-treated groups was predominantly elevated, compared with the control group, as examined by cell death detection ELISAPlus (Fig. 2B). Similar to this observation, the percentage of apoptotic cells detected by the annexin V-FITC/PI apoptosis detection kit increased remarkably in AGE-BSA (200 mg/liter)-treated hFOB1.19 cells (Fig. 2C). To investigate the underlying mechanisms involved in AGE-BSA-induced apoptosis of hFOB1.19 cells, we incubated cells with the caspase inhibitor Z-DEVD-fmk (25 μm) for 1 h prior to treatment with AGE-BSA (200 mg/liter). As illustrated, Z-DEVD-fmk pre-treatment significantly inhibited the effect of AGE-BSA (200 mg/liter) on decreasing viability (Fig. 2A) and inducing apoptosis of hFOB1.19 cells (Fig. 2, B and C). In contrast, 3-MA markedly enhanced the AGE-BSA (200 mg/liter)-induced inhibition of cell viability (Fig. 2A) and promoted apoptotic cell death (Fig. 2, B and C). These findings demonstrate that AGE-BSA-induced apoptotic cell death in a caspase-dependent manner was significantly exacerbated by the inhibition of autophagy.
FIGURE 2.

Autophagy as a protective response against high concentrations of AGE-BSA-induced apoptosis in hFOB 1.19 cells. A–C, cells preincubated with or without Z-DEVD-fmk (25 μm) for 1 h were treated with or without 3-MA (5 mm) incubated with AGE-BSA (200 μg/ml) for 24 h. A, cell viability was estimated using MTT assay. B, cell death was estimated using a cell death detection ELISAPlus. Values represent mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment. C, cell apoptosis detection using annexin V-FITC/PI kit. Viable cells (annexin V−/PI−), early apoptotic cells (annexin V+/PI−), late apoptotic cells (annexin V−/PI+), and necrotic cells (annexin V+/PI+) are located in the bottom left, bottom right, top right, and top left quadrants, respectively. The numbers in each quadrant represent the percentage of cells.
AGE-BSA-induced Autophagy of hFOB1.19 Cells
LC3II and beclin1 are known biomarkers in the formation of autophagosomes (24). Their expression is positively correlated with autophagy level. In addition, p62/SQSTMI is a biomarker of the degradation of autolysosomes. Impaired autophagy is often accompanied by the accumulation of p62/SQSTMI, and its expression is negatively correlated with autophagy level. To determine whether a low concentration of AGE-BSA can affect the autophagy level of hFOB1.19 cells, we examined the changes in expression of autophagy-associated proteins, including LC3I/LC3II, beclin1, and p62/SQSTMI. We found that AGE-BSA at the concentration of 150 mg/liter induced cell autophagy, and the effect was time-dependent. The effect was most significant when the treatment time was 24 h (Fig. 3, A and D). With increased concentration of AGE-BSA, autophagy levels increased accordingly, which suggested that the effect was also concentration-dependent (Fig. 3, B and E). 3-MA significantly inhibited the autophagy induced by 150 mg/liter AGE-BSA (Fig. 3, C and F).
FIGURE 3.

Effect of AGE-BSA on the expression of autophagy related protein beclin-1, LC3, and p62 in hFOB 1.19 cells. A, Western blotting showing beclin-1, LC3, and p62 protein levels in hFOB 1.19 cells treated with AGE-BSA (150 μg/ml) for 0, 0.5, 1, 3, 6, 12, 24, or 48 h. B, Western blotting showing beclin-1, LC3, and p62 protein levels in hFOB 1.19 cells treated with AGE-BSA at various concentrations for 24 h. C, Western blotting showing beclin-1, LC3, and p62 protein levels in hFOB 1.19 cells treated with or without 3-MA (5 mm) incubated with AGE-BSA (150 μg/ml) or BSA (150 μg/ml) for 24 h. D–F, quantitative analysis of LC3, beclin-1, and p62 protein expression. Each bar represents mean ± S.E. of at least thee independent experiments. *, p < 0.05 versus control; **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment.
We confirmed the inductive effects of AGE-BSA on cell autophagy by counting the number of autolysosomes using a transmission electron microscope (TEM) and fluorescence microscope. TEM is the recognized gold standard for monitoring autophagy (24). The results indicated that after 24 h of treatment with 150 mg/liter AGE-BSA, the number of autolysosomes in hFOB1.19 cells increased significantly compared with controls, and the addition of 3-MA significantly lowered the number of autolysosomes (Fig. 4, A and C). LC3 is mainly located on the surface of pre-autophagosomes and autophagosomes and is known as a general biomarker of autophagy (24). We used a fluorescence microscope to observe the punctate aggregation (autolysosome) of internal LC3 and external EGFP-LC3. The results were consistent with those obtained using TEM. The punctate aggregation of both proteins increased significantly after 24 h of treatment with 150 mg/liter AGE-BSA compared with controls, and when 3-MA was added, the punctate aggregation decreased significantly compared with the AGE-BSA group without 3-MA (Fig. 4, B, D, and E). These results suggested that a low concentration of AGE-BSA up-regulated the autophagy level of hFOB1.19 cells.
FIGURE 4.

AGE-BSA (150 μg/ml)-induced autophagy in hFOB 1.19 cells, and the autophagy was blocked by 3-MA. A, transmission electron microscopy showing autophagosomes (bold arrows) in hFOB 1.19 cells treated with or without 3-MA (5 mm) incubated with AGE-BSA (150 μg/ml) for 24 h. C, quantitation of autophagosomes. Each bar represents the mean ± S.E. of three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment. B, fluorescence microscopy showing endogenous LC3 (green fluorescence), exogenous EGFP-LC3 (green fluorescence), and nucleus (blue fluorescence) in hFOB 1.19 cells treated with or without 3-MA (5 mm) incubated with AGE-BSA (150 μg/ml) for 24 h. D, quantitation of LC3 puncta. Each bar represents the mean ± S.E. of three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment. E, quantitation of EGFP-LC3 puncta. Each bar represents the mean ± S.E. of three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment.
Autophagy of hFOB1.19 Cells Induced by AGE-BSA Was Mediated via the RAGE/Raf/MEK/ERK Signal Pathway
As one of the major receptors of AGEs, RAGE participates in multiple signal transduction pathways (25). To investigate the functions of RAGE in cell autophagy induced by AGE-BSA, we first determined the expression of RAGE protein in hFOB1.19 cells treated with different concentrations (50, 100, and 150 mg/liter) of AGE-BSA for 24 h. We found that when the concentration of AGE-BSA increased, the expression level of RAGE increased accordingly (Fig. 5, A and B), which suggested that AGE-BSA up-regulated the expression of RAGE. We then transfected hFOB1.19 cells with RAGE-shRNA, and this decreased the level of RAGE successfully (Fig. 5, C and D). Up-regulation of RAGE can activate the Raf/MEK/ERK signal pathway (25), and this pathway is closely associated with autophagy (26). It is thought that this pathway may be involved in the autophagy induced by AGE-BSA. Western blotting was used to determine the expression of proteins in the Raf/MEK/ERK signal pathway associated with autophagy (Fig. 5E). The results showed that treatment with 150 mg/liter AGE-BSA for 24 h activated the Raf/MEK/ERK signal pathway and increased the autophagy level. Furthermore, when the pathway was inhibited by PD98059 and RAGE-shRNA, the activity of both the pathway and autophagy level decreased significantly (Fig. 5, E–G). This further proved the function of the RAGE/Raf/MEK/ERK signal pathway in autophagy of hFOB1.19 cells induced by AGE-BSA.
FIGURE 5.

RAGE/Raf/MEK/ERK pathway is involved in AGE-BSA induced autophagy. A, Western blotting showing RAGE protein levels in hFOB 1.19 cells treated with AGE-BSA at various concentrations for 24 h. B, quantitative analysis of RAGE protein expression. Each bar represents mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control. C, Western blotting showing RAGE protein levels in hFOB 1.19 cells infected lentiviral with control shRNA or RAGE shRNA for 24 h. D, quantitative analysis of RAGE protein expression. Each bar represents mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus control shRNA treatment. E, Western blotting showing Raf/MEK/ERK pathway relative protein (c-Raf, MEK1/2, or ERK1/2) levels and autophagy relative protein (Beclin-1, LC3, or p62) levels in hFOB 1.19 cells treated with or without PD98059 (50 μm), control shRNA, or RAGE shRNA incubated with or without AGE-BSA (150 μg/ml) for 24 h. F and G, quantitative analysis of c-Raf, MEK1/2, ERK1/2, LC3, beclin-1, or p62 protein expression. Each bar represents mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment.
Pro-proliferative Function of AGE-BSA Was Inhibited by RAGE-shRNA and PD98059
To determine the function of the RAGE/Raf/MEK/ERK signal pathway in cell proliferation induced by AGE-BSA, we first investigated the cell viability using MTT analysis following treatment with RAGE-shRNA or PD98059. We found that compared with the AGE-BSA group, the viability of cells in the RAGE-shRNA group and the PD98059 group decreased significantly (Fig. 6A). To further confirm this, BrdU incorporation assay was performed, and the results also showed that RAGE-shRNA and PD98059 significantly inhibited AGE-BSA-induced hFOB1.19 cell proliferation (Fig. 6B). These results further confirmed the role of the RAGE/Raf/MEK/ERK signal pathway in the pro-proliferative effect of AGE-BSA on hFOB1.19 cells.
FIGURE 6.
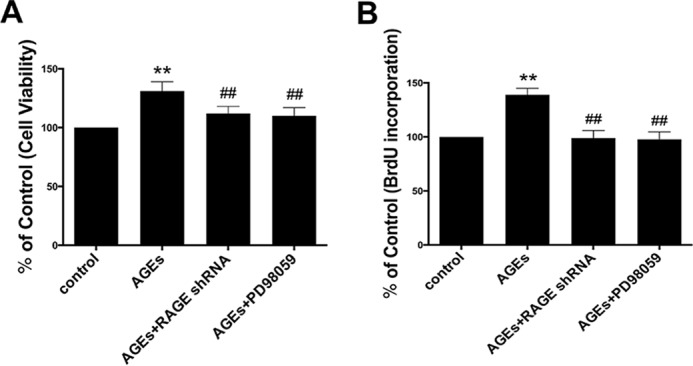
RAGE shRNA and PD98059 attenuated the effect of AGE-BSA on cell proliferation in hFOB 1.19 cells. A and B, cells were treated with or without PD98059 (50 μm) or RAGE shRNA incubated with AGE-BSA (150 μg/ml) for 24 h. A, cell viability was estimated using MTT assay; B, cell proliferation was estimated using a BrdU kit. Values represent mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment.
Regulatory Function of AGE-BSA on Osteogenesis and Osteoclastogenesis in Vitro May Be Mediated by the RAGE/Raf/MEK/ERK Signal Pathway and Autophagy
To further investigate the effects of AGE-BSA on the bone remodeling function of hFOB1.19 cells, we evaluated the mRNA and protein levels of osteoprotegerin (OPG) and RANKL, which are biomarkers of osteoclastogenesis (27), and ALP and osteocalcin (OCN), which are biomarkers of osteogenesis. Real time PCR showed that compared with the controls, the mRNA level of RANKL was down-regulated by treatment with AGE-BSA (150 mg/liter) for 24 h, and the mRNA levels of alkaline phosphatase (ALP), OCN and OPG were up-regulated. After 48 h of treatment, the levels of the above mRNAs did not change significantly. However, the effect of 72 h of treatment was opposite that of 24 h of treatment. Compared with the controls, 72 h of treatment with AGE-BSA significantly up-regulated the mRNA level of RANKL and down-regulated the mRNA levels of ALP, OCN, and OPG. 3-MA, RAGE-shRNA, and PD98059 inhibited the effects of AGE-BSA and maintained the levels of the above biomarkers at baseline levels (Fig. 7, A–D). The protein levels of RANKL, OCN, and OPG were determined by Western blotting. The activity of ALP was measured using an ALP kit. The results of these tests were consistent with those obtained by real time PCR (Fig. 7, E–I). It is thought that AGE-BSA affected the bone remodeling functions of hFOB1.19 cells, and the effect was associated with the RAGE/Raf/MEK/ERK signal pathway and autophagy.
FIGURE 7.
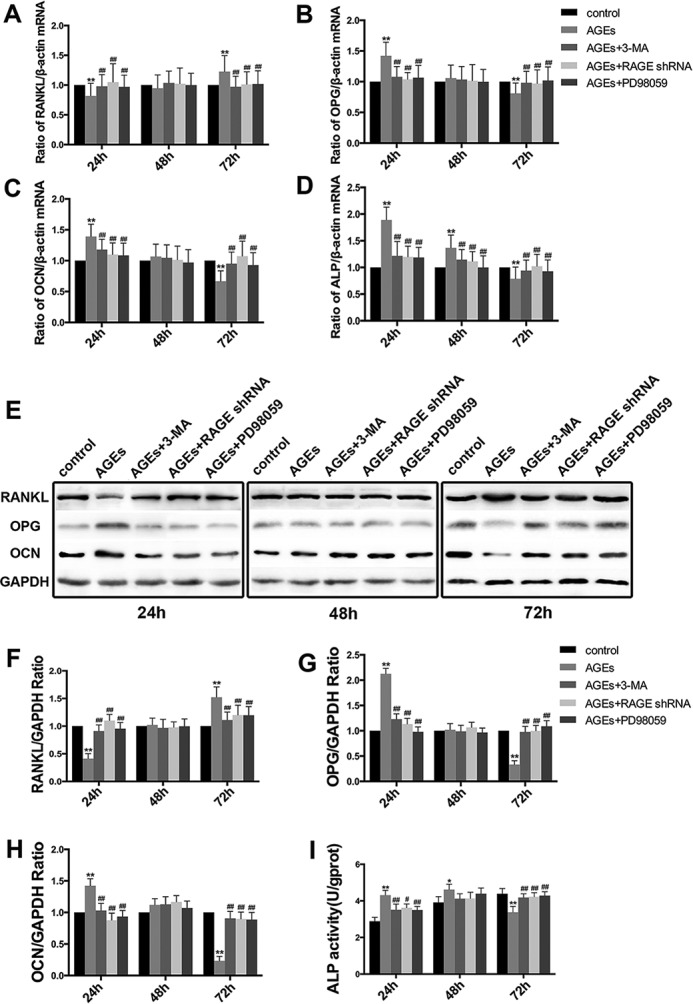
3-MA, RAGE shRNA and PD98059 attenuated the effect of AGE-BSA on the expression of a complex set of bone formation parameters (ALP and OCN) and bone resorption parameters (OPG and RANKL) in hFOB 1.19 cells. A–D, real time RT-PCR showing RANKL (A), OPG (B), OCN (C), or ALP (D) mRNA levels in hFOB 1.19 cells treated with or without 3-MA(5 mm), PD98059 (50 μm), or RAGE shRNA incubated with AGE-BSA (150 μg/ml) for 24, 48, 72 h. Values represent mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment. E, Western blotting showing RANKL, OPG, or OCN protein levels in hFOB 1.19 cells treated with or without 3-MA (5 mm), PD98059 (50 μm), or RAGE shRNA incubated with AGE-BSA (150 μg/ml) for 24, 48, and 72 h. F–H, quantitative analysis of RANKL, OPG, or OCN protein expression. Each bar represents mean ± S.E. of at least three independent experiments. **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment. I, ALP activity kit showing ALP activity levels in hFOB 1.19 cells treated with or without 3-MA (5 mm), PD98059 (50 μm), or RAGE shRNA incubated with AGE-BSA (150 μg/ml) for 24, 48, and 72 h. Values represent mean ± S.E. of at least three independent experiments. *, p < 0.05 versus control; **, p < 0.01 versus control; ##, p < 0.01 versus AGE-BSA treatment.
Discussion
Many studies suggest that AGEs are involved in the development and progression of osteoporosis (7, 28–30). The binding of AGEs to organic bone matrix may also increase the fragility of bones (30, 31). However, there is controversy regarding the effects of AGEs on cell proliferation in vitro. Some studies suggested that AGEs accelerate the maturation of dendritic cells and promote the proliferation of T cells (32). Studies have also found that a low concentration of AGEs can promote the proliferation of vascular smooth muscle cells; however, AGEs at a similar concentration inhibited the proliferation of vascular endothelial cells (33). In this study, we rely on the hFOB1.19 cell line as a cellular model not only because these cells are osteoblasts homologous to human osteoblasts, but also because they have strong proliferation ability (34). To mimic in vivo, we used AGE-BSA, which is described to have a high affinity to RAGE (35). As glycated BSA is modified by a variety of AGE structures and its complexity resembles in vivo generated forms, a physiologically plausible model is approximated. We found that AGE-BSA had a biphasic effect on the viability of hFOB1.19 cells; AGE-BSA (150 mg/liter) induced an early and transient pro-proliferative effect on hFOB1.19 cells, but AGE-BSA (200 mg/liter) continuously inhibited cell viability and induced cell apoptosis. Therefore, we suggest that the pro-proliferative effect of AGE-BSA on osteoblasts not only depends on time or concentration alone but on the synergistic effects of both factors. This partially explains the controversial results regarding the effects of AGE-BSA on osteoblast proliferation.
The protective role of autophagy under various stress conditions is well documented (36). A recent study showed that autophagy had a pro-survival role under high glucose and oxidative stress conditions in osteoblastic MC3T3-E1 cells (19). Also related to the findings, a more recent study has shown that the increased autophagy elicited by simvastatin protected MC3T3-E1 osteoblastic cells from H2O2-mediated cell death (37). Moreover, defective autophagy has been reported to contribute to aging, insulin resistance, and cancer (38, 39). In this study, by inhibiting autophagy using 3-MA, we showed the deleterious consequences of autophagy suppression on cell survival in hFOB1.19 cells cultured with AGE-BSA (200 mg/liter). Further studies will investigate the protective mechanisms of autophagy against AGE-BSA.
Numerous studies have demonstrated the close relationship between autophagy and cell proliferation. A recent study found that autophagy induced the proliferation of bone marrow-derived mesenchymal stem cells under hypoxic conditions (40). Another study found that AGE-BSA accelerated the proliferation of rat vascular smooth muscle cells, and this effect was associated with autophagy (41). However, there are no studies on the role of autophagy on the effects of AGE-BSA on osteoblast proliferation. In this study, we found that 3-MA effectively inhibited the increased cell viability and proliferation induced by the low concentration of AGE-BSA, suggesting a possible pro-proliferative role of autophagy in hFOB1.19 cells when exposed to AGE-BSA (150 mg/liter). Furthermore, to validate this hypothesis by monitoring autophagy using several experimental procedures, we found these results were able to support the important function of autophagy in the effects of AGE-BSA on hFOB1.19 cells proliferation.
As the most important receptor of AGEs, RAGE is extensively expressed in a variety of human tissues and is associated with many diseases, including diabetic complications, neuropathy, and inflammatory disease (42). In this study, we found that the AGE-BSA-activated Raf/MEK/ERK signal pathway in hFOB1.19 cells was clearly inhibited in RAGE-shRNA-expressing hFOB1.19 cells. Also related to the findings of this study, more studies had shown that AGE-BSA up-regulated the expression of RAGE, activated the ERK1/2 signal pathway, and accelerated the proliferation, migration, and invasion of cells (43, 44). These results suggest that RAGE plays a key role in activation of the Raf/MEK/ERK signal pathway of cells induced by AGE-BSA.
The Raf/MEK/ERK signal pathway, which is the main component of the MAPK pathway, plays a key role in regulating cell survival, cell cycle, and differentiation (45, 46). Recent studies have shown that the Raf/MEK/ERK signal pathway participates in the regulation of autophagy by regulating the expression of LC3B and p62, which are the key proteins associated with autophagy (26). Studies have also suggested that RAGE has a role in regulating autophagy and facilitates cell survival by up-regulating cell autophagy (21, 47). The effect of AGE/RAGE on promoting autophagy may be mediated by activation of the ERK pathway (41). Kang et al. (48) suggested that the activation of ERK by RAGE can lead to the subsequent stimulation of death-associated protein kinase, which in turn phosphorylates and activates beclin1 to promote autophagy. In this study, addition of PD98059 or RAGE-shRNA inhibited the autophagy and proliferation of hFOB1.19 cells induced by AGE-BSA (150 mg/liter), suggesting that RAGE and the Raf/MEK/ERK signal pathway have important functions in mediating the autophagy and proliferation of hFOB1.19 cells induced by AGE-BSA.
In light of the complex role of osteoblasts in bone remodeling, this study also examined the impact of AGEs on the expression of osteogenic (ALP and OCN) and osteoclastogenic (RANKL and OPG) markers. Interestingly, we found that the short term effects of AGE-BSA (150 mg/liter) increased osteogenic function and decreased osteoclastogenic function, which are likely mediated by autophagy and the RAGE/Raf/MEK/ERK signal pathway. With increased treatment time, the opposite effects were observed. The effects of AGE-BSA on inhibiting osteogenesis and promoting osteoclastogenic function have been reported in previous studies (11, 49). However, the role of AGE-BSA in promoting osteogenesis has not been reported. The role of autophagy in protein secretion and trafficking is a relatively recently recognized function of the autophagic machinery. Autophagy affects the secretion of proteins, not only via the conventional method (endoplasmic reticulum-Golgi pathway) but also via a non-conventional method (autophagy-dependent pathway) (50). For the first time, we demonstrated that autophagy participated in the regulation of functional proteins in hFOB1.19 cells culturing AGE-BSA. The dual effects of AGE-BSA were dependent on the duration of treatment. Further studies will elucidate how autophagy influences the production and secretion of these proteins.
Autophagy is regarded as a double-edged sword. Autophagy is an important factor in maintaining cellular function and viability. However, activation of autophagy is unnecessarily beneficial for cells. Excessive autophagy leads to cell injury and even apoptosis (16, 17). In our study, the activation of autophagy had short term favorable effects on hFOB1.19 cells treated with a low concentration AGE-BSA as it promoted cellular proliferation, increased osteogenic function, and inhibited osteoclastogenic function. In contrast, long term treatment had the opposite effects. Therefore, the increase in autophagy alone did not effectively improve the functions of hFOB1.19 cells treated with AGE-BSA. These results suggest that autophagy should be controlled to an appropriate degree depending on the different stages of diabetic osteoporosis.
In summary, AGE-BSA had a biphasic effect on the viability of hFOB1.19 cells in vitro, which was determined by the concentration of AGEs and treatment time. A low concentration of AGE-BSA activated the Raf/MEK/ERK signal pathway through the interaction with RAGE, induced autophagy of hFOB1.19 cells, and regulated the proliferation and function of hFOB1.19 cells. Elucidation of the mechanisms of AGEs is valuable for identifying novel targets for the prevention and therapy of diabetic osteoporosis.
Author Contributions
All authors conceived the study. H. Z. M. and W. L. Z. wrote the study. M. W. Y. coordinated the study. F. L. provided technical assistance and contributed to the preparation of the figures. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Dr. M. Subramaniam for kindly providing the hFOB 1.19 cell lines and Li X for help with LC3 staining.
This work was supported by National Natural Science Foundation of China Grants 81471094 and 81170808. The authors declare that they have no conflicts of interest with the contents of this article.
- AGE
- advanced glycation end product
- RAGE
- receptor of AGE
- AGE-BSA
- AGE-modified bovine serum albumin
- RANKL
- receptor activator for nuclear factor-κ B ligand
- 3-MA
- 3-methyladenine
- OPG
- osteoprotegerin
- OCN
- osteocalcin
- ALP
- alkaline phosphatase
- TEM
- transmission electron microscopy
- MTT
- 3-(4,5-dimethyl-thiazol-2-yl)- 2,5-diphenyl-tetrazolium bromide
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- PI
- propidium iodide.
References
- 1.Vestergaard P. (2007) Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes–a meta-analysis. Osteoporos. Int. 18, 427–444 [DOI] [PubMed] [Google Scholar]
- 2.Pietschmann P., Patsch J. M., and Schernthaner G. (2010) Diabetes and bone. Horm. Metab. Res. 42, 763–768 [DOI] [PubMed] [Google Scholar]
- 3.Oei L., Rivadeneira F., Zillikens M. C., and Oei E. H. (2015) Diabetes, diabetic complications, and fracture risk. Curr. Osteoporos. Rep. 13, 106–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Liefde I. I., van der Klift M., de Laet C. E., van Daele P. L., Hofman A., and Pols H. A. (2005) Bone mineral density and fracture risk in type-2 diabetes mellitus: the Rotterdam Study. Osteoporos. Int. 16, 1713–1720 [DOI] [PubMed] [Google Scholar]
- 5.Goh S. Y., and Cooper M. E. (2008) Clinical review: the role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 93, 1143–1152 [DOI] [PubMed] [Google Scholar]
- 6.Barbezier N., Tessier F. J., and Chango A. (2014) Receptor of advanced glycation endproducts RAGE/AGER: an integrative view for clinical applications. Ann. Biol. Clin. 72, 669–680 [DOI] [PubMed] [Google Scholar]
- 7.Yamagishi S., Nakamura K., and Inoue H. (2005) Possible participation of advanced glycation end products in the pathogenesis of osteoporosis in diabetic patients. Med. Hypotheses 65, 1013–1015 [DOI] [PubMed] [Google Scholar]
- 8.McCarthy A. D., Etcheverry S. B., Bruzzone L., and Cortizo A. M. (1997) Effects of advanced glycation end products on the proliferation and differentiation of osteoblast-like cells. Mol. Cell. Biochem. 170, 43–51 [DOI] [PubMed] [Google Scholar]
- 9.Alikhani M., Alikhani Z., Boyd C., MacLellan C. M., Raptis M., Liu R., Pischon N., Trackman P. C., Gerstenfeld L., and Graves D. T. (2007) Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone 40, 345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mercer N., Ahmed H., Etcheverry S. B., Vasta G. R., and Cortizo A. M. (2007) Regulation of advanced glycation end product (AGE) receptors and apoptosis by AGEs in osteoblast-like cells. Mol. Cell. Biochem. 306, 87–94 [DOI] [PubMed] [Google Scholar]
- 11.Franke S., Rüster C., Pester J., Hofmann G., Oelzner P., and Wolf G. (2011) Advanced glycation end products affect growth and function of osteoblasts. Clin. Exp. Rheumatol. 29, 650–660 [PubMed] [Google Scholar]
- 12.Levine B., and Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizushima N., Levine B., Cuervo A. M., and Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L., Vicencio J. M., Kepp O., Tasdemir E., Maiuri M. C., and Kroemer G. (2008) To die or not to die: that is the autophagic question. Curr. Mol. Med. 8, 78–91 [DOI] [PubMed] [Google Scholar]
- 15.Debnath J., Baehrecke E. H., and Kroemer G. (2005) Does autophagy contribute to cell death? Autophagy 1, 66–74 [DOI] [PubMed] [Google Scholar]
- 16.Alva A. S., Gultekin S. H., and Baehrecke E. H. (2004) Autophagy in human tumors: cell survival or death? Cell Death Differ. 11, 1046–1048 [DOI] [PubMed] [Google Scholar]
- 17.Platini F., Pérez-Tomás R., Ambrosio S., and Tessitore L. (2010) Understanding autophagy in cell death control. Curr. Pharm. Des. 16, 101–113 [DOI] [PubMed] [Google Scholar]
- 18.Nollet M., Santucci-Darmanin S., Breuil V., Al-Sahlanee R., Cros C., Topi M., Momier D., Samson M., Pagnotta S., Cailleteau L., Battaglia S., Farlay D., Dacquin R., Barois N., Jurdic P., et al. (2014) Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 10, 1965–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartolomé A., López-Herradón A., Portal-Núñez S., García-Aguilar A., Esbrit P., Benito M., and Guillén C. (2013) Autophagy impairment aggravates the inhibitory effects of high glucose on osteoblast viability and function. Biochem. J. 455, 329–337 [DOI] [PubMed] [Google Scholar]
- 20.Kang R., Tang D., Lotze M. T., and Zeh H. J. 3rd. (2011) RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy 7, 442–444 [DOI] [PubMed] [Google Scholar]
- 21.Xie Y., You S. J., Zhang Y. L., Han Q., Cao Y. J., Xu X. S., Yang Y. P., Li J., and Liu C. F. (2011) Protective role of autophagy in AGE-induced early injury of human vascular endothelial cells. Mol. Med. Rep. 4, 459–464 [DOI] [PubMed] [Google Scholar]
- 22.Hou X., Hu Z., Xu H., Xu J., Zhang S., Zhong Y., He X., and Wang N. (2014) Advanced glycation endproducts trigger autophagy in cardiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc. Diabetol. 13, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramaniam M., Jalal S. M., Rickard D. J., Harris S. A., Bolander M. E., and Spelsberg T. C. (2002) Further characterization of human fetal osteoblastic hFOB 1.19 and hFOB/ERα cells: bone formation in vivo and karyotype analysis using multicolor fluorescent in situ hybridization. J. Cell. Biochem. 87, 9–15 [DOI] [PubMed] [Google Scholar]
- 24.Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., Ahn H. J., Ait-Mohamed O., Ait-Si-Ali S., Akematsu T., Akira S., et al. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie J., Méndez J. D., Méndez-Valenzuela V., and Aguilar-Hernández M. M. (2013) Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 25, 2185–2197 [DOI] [PubMed] [Google Scholar]
- 26.Kim J. H., Hong S. K., Wu P. K., Richards A. L., Jackson W. T., and Park J. I. (2014) Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp. Cell Res. 327, 340–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofbauer L. C., and Heufelder A. E. (2001) Role of receptor activator of nuclear factor-κB ligand and osteoprotegerin in bone cell biology. J. Mol. Med. 79, 243–253 [DOI] [PubMed] [Google Scholar]
- 28.Odetti P., Rossi S., Monacelli F., Poggi A., Cirnigliaro M., Federici M., and Federici A. (2005) Advanced glycation end products and bone loss during aging. Ann. N.Y. Acad. Sci. 1043, 710–717 [DOI] [PubMed] [Google Scholar]
- 29.Hein G. E. (2006) Glycation end products in osteoporosis–is there a pathophysiologic importance? Clin. Chim. Acta 371, 32–36 [DOI] [PubMed] [Google Scholar]
- 30.Saito M., and Marumo K. (2010) Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos. Int. 21, 195–214 [DOI] [PubMed] [Google Scholar]
- 31.Saito M., Kida Y., Kato S., and Marumo K. (2014) Diabetes, collagen, and bone quality. Curr. Osteoporos. Rep. 12, 181–188 [DOI] [PubMed] [Google Scholar]
- 32.Ge J., Jia Q., Liang C., Luo Y., Huang D., Sun A., Wang K., Zou Y., and Chen H. (2005) Advanced glycosylation end products might promote atherosclerosis through inducing the immune maturation of dendritic cells. Arterioscler. Thromb. Vasc. Biol. 25, 2157–2163 [DOI] [PubMed] [Google Scholar]
- 33.Satoh H., Togo M., Hara M., Miyata T., Han K., Maekawa H., Ohno M., Hashimoto Y., Kurokawa K., and Watanabe T. (1997) Advanced glycation endproducts stimulate mitogen-activated protein kinase and proliferation in rabbit vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 239, 111–115 [DOI] [PubMed] [Google Scholar]
- 34.Harris S. A., Enger R. J., Riggs B. L., and Spelsberg T. C. (1995) Development and characterization of a conditionally immortalized human fetal osteoblastic cell line. J. Bone Miner. Res. 10, 178–186 [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto S., Yoshida T., Murata H., Harada S., Fujita N., Nakamura S., Yamamoto Y., Watanabe T., Yonekura H., Yamamoto H., Ohkubo T., and Kobayashi Y. (2008) Solution structure of the variable-type domain of the receptor for advanced glycation end products: new insight into AGE-RAGE interaction. Biochemistry 47, 12299–12311 [DOI] [PubMed] [Google Scholar]
- 36.Kroemer G., Mariño G., and Levine B. (2010) Autophagy and the integrated stress response. Mol. Cell 40, 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai E. H., Hong C. Y., Kok S. H., Hou K. L., Chao L. H., Lin L. D., Chen M. H., Wu P. H., and Lin S. K. (2012) Simvastatin alleviates the progression of periapical lesions by modulating autophagy and apoptosis in osteoblasts. J. Endod. 38, 757–763 [DOI] [PubMed] [Google Scholar]
- 38.Yang L., Li P., Fu S., Calay E. S., and Hotamisligil G. S. (2010) Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rubinsztein D. C., Mariño G., and Kroemer G. (2011) Autophagy and aging. Cell 146, 682–695 [DOI] [PubMed] [Google Scholar]
- 40.Li L., Li L., Zhang Z., and Jiang Z. (2015) Hypoxia promotes bone marrow-derived mesenchymal stem cell proliferation through apelin/APJ/autophagy pathway. Acta Biochim. Biophys. Sin. 47, 362–367 [DOI] [PubMed] [Google Scholar]
- 41.Hu P., Lai D., Lu P., Gao J., and He H. (2012) ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int. J. Mol. Med. 29, 613–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chavakis T., Bierhaus A., and Nawroth P. P. (2004) RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect. 6, 1219–1225 [DOI] [PubMed] [Google Scholar]
- 43.Lander H. M., Tauras J. M., Ogiste J. S., Hori O., Moss R. A., and Schmidt A. M. (1997) Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 272, 17810–17814 [DOI] [PubMed] [Google Scholar]
- 44.Sharaf H., Matou-Nasri S., Wang Q., Rabhan Z., Al-Eidi H., Al Abdulrahman A., and Ahmed N. (2015) Advanced glycation endproducts increase proliferation, migration and invasion of the breast cancer cell line MDA-MB-231. Biochim. Biophys. Acta 1852, 429–441 [DOI] [PubMed] [Google Scholar]
- 45.McCubrey J. A., Steelman L. S., Chappell W. H., Abrams S. L., Wong E. W., Chang F., Lehmann B., Terrian D. M., Milella M., Tafuri A., Stivala F., Libra M., Basecke J., Evangelisti C., Martelli A. M., and Franklin R. A. (2007) Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 1773, 1263–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cagnol S., and Chambard J. C. (2010) ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 277, 2–21 [DOI] [PubMed] [Google Scholar]
- 47.Kang R., Tang D., Schapiro N. E., Livesey K. M., Farkas A., Loughran P., Bierhaus A., Lotze M. T., and Zeh H. J. (2010) The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 17, 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang R., Tang D., Loze M. T., and Zeh H. J. (2011) Apoptosis to autophagy switch triggered by the MHC class III-encoded receptor for advanced glycation endproducts (RAGE). Autophagy 7, 91–93 [DOI] [PubMed] [Google Scholar]
- 49.Franke S., Siggelkow H., Wolf G., and Hein G. (2007) Advanced glycation endproducts influence the mRNA expression of RAGE, RANKL and various osteoblastic genes in human osteoblasts. Arch. Physiol. Biochem. 113, 154–161 [DOI] [PubMed] [Google Scholar]
- 50.Deretic V., Jiang S., and Dupont N. (2012) Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 22, 397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]