

Background: Retinoblastoma protein (RB) is frequently targeted for proteasomal degradation by oncoproteins.
Results: NRBE3 promotes RB degradation as an E3 and is transcriptionally activated by E2F1.
Conclusion: NRBE3 is an E3 ubiquitin ligase for RB and regulates the cell cycle.
Significance: This study identified a novel E3 ubiquitin ligase for RB that might be a potential oncoprotein in human cancers.
Keywords: cell cycle; E2F transcription factor; E3 ubiquitin ligase; protein degradation; retinoblastoma protein (pRb, RB)
Abstract
Retinoblastoma protein (RB) plays critical roles in tumor suppression and is degraded through the proteasomal pathway. However, E3 ubiquitin ligases responsible for proteasome-mediated degradation of RB are largely unknown. Here we characterize a novel RB E3 ubiquitin ligase (NRBE3) that binds RB and promotes RB degradation. NRBE3 contains an LXCXE motif and bound RB in vitro. NRBE3 interacted with RB in cells when proteasome activity was inhibited. NRBE3 promoted RB ubiquitination and degradation via the ubiquitin-proteasome pathway. Importantly, purified NRBE3 ubiquitinated recombinant RB in vitro, and a U-box was identified as essential for its E3 activity. Surprisingly, NRBE3 was transcriptionally activated by E2F1/DP1. Consequently, NRBE3 affected the cell cycle by promoting G1/S transition. Moreover, NRBE3 was up-regulated in breast cancer tissues. Taken together, we identified NRBE3 as a novel ubiquitin E3 ligase for RB that might play a role as a potential oncoprotein in human cancers.
Introduction
The retinoblastoma susceptibility gene (Rb)4 is the first tumor suppressor gene to be cloned (1). Rb was originally identified by its absence in hereditary cases of retinoblastoma (2, 3). Rb loss is also found in a variety of cancers such as small cell lung carcinoma (∼90%) (4) and osteosarcoma (∼50%) (1, 5, 6). In addition, homozygous mutation of Rb is embryonic lethal with defective development of neurons, liver, and erythrocytes (7, 8). Mice carrying a single Rb mutant allele are prone to develop tumors of the pituitary and thyroid glands (7, 9, 10). RB protein is a major negative regulator of multiple cellular processes including cell cycle, differentiation, and apoptosis (11–13). RB exerts its tumor suppressor function in the hypophosphorylated form through arresting cells at G1 of the cell cycle by interacting and suppressing the activity of the transcription factors E2Fs (13–15). During G1 of the cell cycle, mitogens promote activation of cyclin-dependent kinase (CDK)-cyclin complexes that phosphorylate RB. Phosphorylation of RB releases E2Fs to activate its downstream genes, which are essential for G1/S transition of cell cycle, and eventually drives cell proliferation (12, 16–19). Given its central role in regulating cell cycle and proliferation, inactivation of RB is one of the most fundamental events in cancer.
The functions of RB are impaired in a variety of cancers by different mechanisms. For example, cyclin D is up-regulated in cancers, which inactivates RB through phosphorylation by increased cyclin D/CDK4/CDK6 activity (20). LXCXE-containing proteins such as viral oncoproteins including E1A, SV40 large T antigen, and human papilloma virus early protein 7 (HPV E7) and cellular proteins RBP-1 and RBP-2 inactivate RB by binding RB and disrupting the interaction of RB with E2F1 (21–25). It is of importance that multiple viral oncoproteins transform cells by inducing proteasome-mediated degradation of RB during tumorigenesis including the high risk HPV E7 (26), human cytomegalovirus (CMV) pp71 protein (27), Epstein-Barr virus nuclear antigen EBNA3C (28), hepatitis C virus N55B (29), and human T-lymphotropic virus type 1 Tax oncoprotein (30). In addition, two cellular oncoproteins, MDM2 and gankyrin, also promote proteasomal degradation of RB (31–33).
Proteasome-mediated protein degradation plays essential roles in the biological functions of cells and in tumorigenesis. Ubiquitin-dependent proteasomal degradation involves polyubiquitination of substrate catalyzed by an enzymatic cascade including the activation of ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, and ubiquitin ligase E3 before substrate protein is degraded in the proteasome (34). By binding specifically to the substrates and facilitating the correct transfer of ubiquitin from E2 to the substrates (35, 36), ubiquitin E3 ligases for tumor suppressors play crucial roles during tumorigenesis. For example, the E3 ligases for p53 including MDM2, Pirh2, COP1, and TRIM24 have been found to play oncogenic roles (37–44). Given the important role of RB in tumor suppression, the above mentioned viral and cellular oncoproteins transform cells by promoting RB degradation. However, E3 ubiquitin ligases responsible for proteasome-mediated degradation of RB are largely unknown.
Transformation of rodent fibroblasts is a frequently used experimental approach to study the biological functions of oncoproteins. KIAA0649 transforms mouse NIH3T3 fibroblast cells as we described previously (45). However, the biological processes involved in cell transformation by KIAA0649 are poorly understood. Bioinformatics analysis suggested that KIAA0649 harbored an RB-binding motif, LXCXE. In this study, we identify KIAA0649 as a novel RB E3 ubiquitin ligase (NRBE3) that binds RB, promotes RB degradation, and is transcriptionally activated by E2F1.
Experimental Procedures
Reagents and Plasmids
Plasmids encoding GST-RB fusion protein and its deletion mutants GST-RBLP (aa 379–928), GST-RB (A+B) (aa 379–792), and GST-RBC (aa 792–928); FLAG-NRBE3 and deletion mutants; pCI-neo-FLAG-HPV E7; pCMV-HA-Ub; pCMV-HA-K48R-Ub; and pGL3-NRBE3-Luc were cloned in our laboratory. These constructed plasmids were verified by DNA sequencing. MG132 was purchased from Calbiochem. Cycloheximide was purchased from Sigma. Lipofectamine 2000TM was from Invitrogen. Polyclonal anti-NRBE3 antibody was developed in our laboratory (46). Monoclonal antibody against RB was purchased from Pharmingen (G3-245) and Santa Cruz Biotechnology (C-15). Antibodies against β-actin and GFP were purchased from Santa Cruz Biotechnology. Antibody against FLAG (M2) was obtained from Sigma. Peroxidase-conjugated goat anti-mouse, goat anti-rabbit secondary antibodies, FITC-conjugated goat anti-mouse/rabbit, and TRITC-conjugated goat anti-rabbit/mouse IgGs were obtained from Zhongshan Co. (China).
Cell Culture and Transfection
HeLa, MCF-7, U2OS, HCT116, and H1299 cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS). Cells were incubated in a humidified atmosphere with 5% CO2 at 37 °C. Transfection was performed with Lipofectamine 2000 according to the manufacturer's instruction. The sequences of siRNAs were as follows: NRBE3 siRNA-1, 5′-CGCUUCUCCAGUGGUUGCU-3′; NRBE3 siRNA-2, 5′-AACUUGUACCUGGAUCAGGUG-3′; NRBE3 siRNA-3, 5′-AAUUCCGCCAAGUCACUCUUG-3′; and control siRNA, 5′-CGUACGCGGAAUACUUCGA-3′ (47).
Western Blot Analyses
Whole cell lysates were prepared in EBC250 buffer (32). Proteins from cell lysate were separated by SDS-PAGE and transferred onto PVDF membrane (Amersham Biosciences). Blots were hybridized with appropriate antibodies after being blocked with 5% milk in PBS/T (0.5% Tween 20 in phosphate-buffered saline). After extensive washing with PBS/T, blots were incubated with HRP-conjugated secondary antibodies. Immunocomplexes were detected with the ECL kit (GE Healthcare) before exposure to x-ray film.
GST Pulldown Assay
GST pulldown assays were performed as described previously(48). In brief, GST and GST-RB fusion proteins were expressed in Escherichia coli and immobilized on glutathione-Sepharose beads. FLAG-tagged NRBE3 proteins were transcribed/translated with TnT® lysate according to the instructions of the manufacturer (Promega) and incubated with GST or GST fusion proteins immobilized on glutathione-Sepharose beads. The GST fusion protein-bound FLAG-NRBE3 proteins were evaluated by Western blotting with anti-FLAG antibody. Amounts of input GST or GST fusion proteins were confirmed as equal by staining the protein gel with Coomassie Brilliant Blue R-250.
Immunoprecipitation
Cell lysates were prepared in buffer A (25 mm Tris-Cl, pH 7.5, 100 mm KCl, 1 mm dithioerythritol, 2 mm EDTA, 0.5 mm phenylmethylsulfonyl fluoride, 0.1% Nonidet P-40). Cell lysates used for in vivo ubiquitination assays were prepared in lysis buffer A (33). Cell lysates were used directly for immunoprecipitation. Antibody was coupled with a 50% suspension of protein A-Sepharose beads (Amersham Biosciences) in IPP500 (500 mm NaCl, 10 mm Tris-Cl, pH 8.0, 0.1% Nonidet P-40). Coupled beads were incubated with cellular extracts for 2 h at 4 °C. After washes, precipitated proteins were evaluated by Western blotting.
Immunofluorescence
Immunofluorescence was performed as described previously(48). In brief, cells were plated on coverslips in 6-well plates. Cells were washed with PBS and fixed with methanol/acetone (1:1) at −20 °C for 20 min. Cells were blocked with 10% goat serum and incubated with appropriate antibodies in 3% goat serum at 4 °C overnight. After washes with PBS, cells were incubated with TRITC-conjugated goat anti-mouse/rabbit IgG and FITC-conjugated goat anti-rabbit/mouse IgG. The immunofluorescence signals were recorded by confocal laser-scanning microscopy (Leica TCS-ST2).
In Vitro Ubiquitination Assays
FLAG-NRBE3-His and FLAG-NRBE3(Δaa225–240)-His were produced in insect Sf9 cells using Bac-to-Bac® Baculovirus Expression System (Invitrogen). These proteins were purified using nickel-nitrilotriacetic acid beads (Qiagen). The reactions were carried out at 30 °C for 1 h in a 40 μl of reaction buffer (50 mm HEPES, pH8.0, 0.5 mm DTT) containing 4 μl of 10× Energy solution (Boston Biochem catalog number K-960), 2 μg of ubiquitin (Boston Biochem catalog number K-960), 50 ng of recombinant human full-length RB (Active Motif Co. catalog number 31128), 50 ng of purified FLAG-NRBE3 or 50 ng of purified FLAG-NRBE3(Δaa225–240), 10 μg of Conjugation Fraction A (containing purified predominantly E1 and E2 enzymes, Boston Biochem catalog number K-960), and 1 μg of ubiquitin aldehyde (Boston Biochem catalog number U-201). The reactions were terminated, and the proteins were subjected to immunoblotting using specific monoclonal RB antibody.
Luciferase Assays
pGL3-NRBE3 promoter-luciferase reporter plasmid (pGL3-NRBE3-Luc) was co-transfected into 293 cells with E2F1 alone and/or DP1. The Renilla luciferase control reporter vector (Promega) was used in each transfection for normalizing transfection efficiency. After 24 h of transfection, the cells were harvested using Passive Lysis Buffer (Promega), and luciferase activity was assayed using the Dual-Luciferase® Reporter Assay System (Promega) with a Berthold luminometer (Berthold, Wildbad, Germany) according to the manufacturers' instructions. Data are presented as relative luciferase activity compared with the pGL3-Basic control, which is normalized to 1.0. Experiments were repeated at least three times in triplicates.
Flow Cytometry Cell Cycle Analysis
Exponentially growing cells were trypsinized and collected by centrifugation. After washes with PBS, cells were resuspended in 70% ice-cold ethanol and kept at 4 °C overnight. Cells were rehydrated in PBS at a density of 1 × 106 cells/ml. Following RNase digestion, cells were stained with 50 μg/ml propidium iodide. Flow cytometry analysis was performed using red (propidium iodide) emission (at 630 nm). The data from 104 cells were collected and analyzed using CellQuest software (BD Biosciences).
Tissue Collection and Sample Preparation
Breast cancer tissues and adjacent non-cancerous breast tissues were collected from 16 breast cancer patients who underwent tumor resection at the Third Clinical Medical College of Peking University. None of these patients received any antineoplastic therapy prior to surgery. After resection, specimens were rinsed thoroughly in ice-cold normal saline and snap frozen in liquid nitrogen. Access to human tissues complied with the guidelines of the Ethics Committee of Peking University. For each sample, tissue was homogenized in 3 volumes of lysis buffer (50 mm Tris-Cl, pH7.5, 2 mm EDTA, 150 mm NaCl, 0.5 mm DTT) on ice using a Polytron homogenizer. The samples were centrifuged for 30 min at 10,000 × g to remove particulate materials. Protein concentrations were determined by the Bradford method (Bio-Rad) and subjected to Western blot analysis.
Results
NRBE3 Interacted with RB in Vitro and in Vivo
Bioinformatics analysis showed that NRBE3 contained an RB-binding sequence, LXCXE (Fig. 1A). To investigate whether NRBE3 binds RB, GST pulldown was performed with in vitro transcribed/translated FLAG-NRBE3 and E. coli-expressed GST-RB fusion proteins. FLAG-NRBE3 interacted with the large pocket of RB, whereas no interaction between NRBE3 and RB (A+B) or RB C-pocket was observed (Fig. 1B). The RB-binding domain of NRBE3 was further narrowed down by GST pulldown experiments using in vitro translated FLAG-NRBE3 deletion mutants (Fig. 1C, upper left) and E. coli-expressed GST-RBLP fusion protein. Both the N terminus containing residues 1–681 and the C terminus containing residues 805–1209 of NRBE3 bound the large pocket of RB (Fig. 1C, upper right). Because residues 1–681 of NRBE3 contained an LXCXE motif, we wanted to know whether the LXCXE motif was responsible for the interaction with RB. We constructed LXCXE motif-mutated plasmid FLAG-NRBE3aa1–681-RXRXH and performed GST pulldown with in vitro transcribed/translated FLAG-NRBE3aa1–681-RXRXH and E. coli-expressed GST-RB fusion proteins. The results showed that FLAG-NRBE3aa1–681-RXRXH lost the capability of binding RB (Fig. 1C, lower panel), demonstrating that the LXCXE motif was required for the N terminus of NRBE3 to bind RB.
FIGURE 1.

NRBE3 interacted with RB in vitro and in vivo. A, schematic structure of NRBE3. The alignment of the LXCXE sequence in NRBE3 was plotted with that in HPV16 E7, AD5 E1A, SV40 large T antigen, cyclin D1, and gankyrin by ClustalW. B, GST pulldown was performed with E. coli-expressed GST or GST-RBLP, GST-RB (A+B), or GST-RBC fusion protein and in vitro transcribed/translated FLAG-NRBE3 protein. GST-RB-binding FLAG-NRBE3 proteins were evaluated by Western blotting with anti-FLAG antibody M2 (upper panel). Ten percent of the FLAG-NRBE3 protein was loaded as input control. GST or GST-RB deletion mutant proteins used in the GST pulldown experiment were stained with Coomassie Brilliant Blue R-250 as a loading control (lower panel). C, GST pulldown was performed with E. coli-expressed GST or GST-RBLP fusion proteins and in vitro transcribed/translated FLAG-NRBE3 and its deletion mutant proteins. GST-RBLP-binding FLAG-NRBE3 proteins were evaluated by Western blotting with anti-FLAG antibody M2 (right). Ten percent of FLAG-NRBE3 protein was loaded as input control. Schematic structures of FLAG-tagged NRBE3 deletion mutants are shown on the left. D, U2OS cells were transfected with FLAG-NRBE3, and immunoprecipitation was performed with anti-RB antibody (left) or anti-FLAG antibody (right). Immunoprecipitated RB and FLAG-NRBE3 proteins were evaluated by Western blotting with anti-RB or anti-FLAG antibody. Mouse IgG was used as a nonspecific antibody control. Five percent of cellular extracts for immunoprecipitation was loaded as input control. U2OS cells were transfected with FLAG-NRBE3, and cells were treated with the proteasome inhibitor MG132 for 4 h before harvest (lower). E, U2OS cells were transfected with FLAG-NRBE3 or FLAG, and immunoprecipitation were performed as described in D. F, HeLa cells were transfected with FLAG-NRBE3 expression plasmid. Cells were fixed 24 h post-transfection, and double immunofluorescence staining was performed with monoclonal anti-FLAG antibody M2 and polyclonal anti-NRBE3 antibody. Immunocomplexes were probed with TRITC-conjugated goat anti-mouse IgG and FITC-conjugated goat anti-rabbit IgG. Scale bars represent 16 μm. G, HeLa cells were fixed, and immunofluorescence staining was performed with polyclonal anti-NRBE3 antibody. TRITC-conjugated goat anti-rabbit IgG was used as the secondary antibody. Scale bars represent 16 μm. H, double immunofluorescence staining was performed with mouse monoclonal anti-RB and rabbit polyclonal anti-NRBE3 antibodies in HeLa cells. TRITC-conjugated goat anti-rabbit IgG and FITC-conjugated goat anti-mouse IgG were used as secondary antibodies. Scale bars represent 25 μm. I, HeLa cells were treated with MG132 before double immunofluorescence and immunofluorescence staining was performed as described in H. Nuclei were stained with DAPI. Scale bars represent 25 μm. The image was taken under confocal microscopy. Rr refers to the Pearson correlation coefficients. mIgG, mouse IgG; IP, immunoprecipitation; IB, immunoblot, DMSO, dimethyl sulfoxide; Hypo, hypophosphorylated; Hyper, hyperphosphorylated.
To verify whether NRBE3 interacts with RB in cells, immunoprecipitation was performed. However, RB was not found in the immunocomplexes of either endogenous NRBE3 or ectopically expressed FLAG-NRBE3 under normal culture conditions (Fig. 1D, upper panel). We therefore transfected cells with FLAG-NRBE3 and treated cells with a proteasome inhibitor, MG132, before harvest of cells and performed immunoprecipitation. It was plausible that NRBE3 and RB interacted with each other in cells when the proteasome was inhibited, suggesting that NRBE3 might promote RB degradation through proteasome. Importantly, the majority of RB co-precipitated with FLAG-NRBE3 was hypophosphorylated RB, whereas both hypo- and hyperphosphorylated RB were observed in the RB-specific immunoprecipitate, demonstrating that the active RB interacted with NRBE3 in cells (Fig. 1D, lower panel). To confirm this result, we transfected cells with FLAG-NRBE3 or FLAG vector plasmids, and immunoprecipitation was performed with anti-FLAG or anti-RB antibodies after cells were treated with MG132 or dimethyl sulfoxide. FLAG-NRBE3 associated with RB only when cells were treated with MG132 (Fig. 1E), suggesting that NRBE3 might promote RB degradation in the proteasome.
Next, we wanted to know in which cellular compartment NRBE3 interacted with RB. We first determined the localization of endogenous NRBE3 and ectopically expressed FLAG-NRBE3. FLAG-NRBE3 showed the same sparkle pattern in the nucleus as endogenous NRBE3 (Fig. 1, F and G). To determine whether NRBE3 co-localizes with RB, indirect immunofluorescence staining of endogenous NRBE3 and RB was performed, and the overlapping signals of NRBE3 and RB were quantified by Pearson correlation coefficient using a standard technique (49). Results showed that endogenous NRBE3 co-localized partially with RB in the nucleus under normal culture conditions (Pearson correlation coefficient = 0.763–0.5) (Fig. 1H), and MG132 treatment strongly enhanced the co-localization of NRBE3 with RB in the nucleus (Pearson correlation coefficient = 0.864), further confirming that NRBE3 associated with RB when proteasome is inhibited.
NRBE3 Resulted in an Active Proteolysis of RB Protein
Immunoprecipitation experiments showed that NRBE3 interacted with RB only when proteasome was inhibited; thus we speculated that NRBE3 might affect RB function through the proteasomal pathway. We first determined whether NRBE3 affects RB protein levels. As shown in Fig. 2A, ectopic expression of FLAG-NRBE3 resulted in decreased levels of endogenous RB protein in a dose-dependent manner. The NRBE3-caused decrease of RB protein was further confirmed by indirect immunofluorescence staining, which showed that the endogenous RB protein level in FLAG-NRBE3 transfected cells was lower than that in the untransfected cells (Fig. 2B). In contrast, knockdown of endogenous NRBE3 caused increased RB protein levels (Fig. 2C).
FIGURE 2.
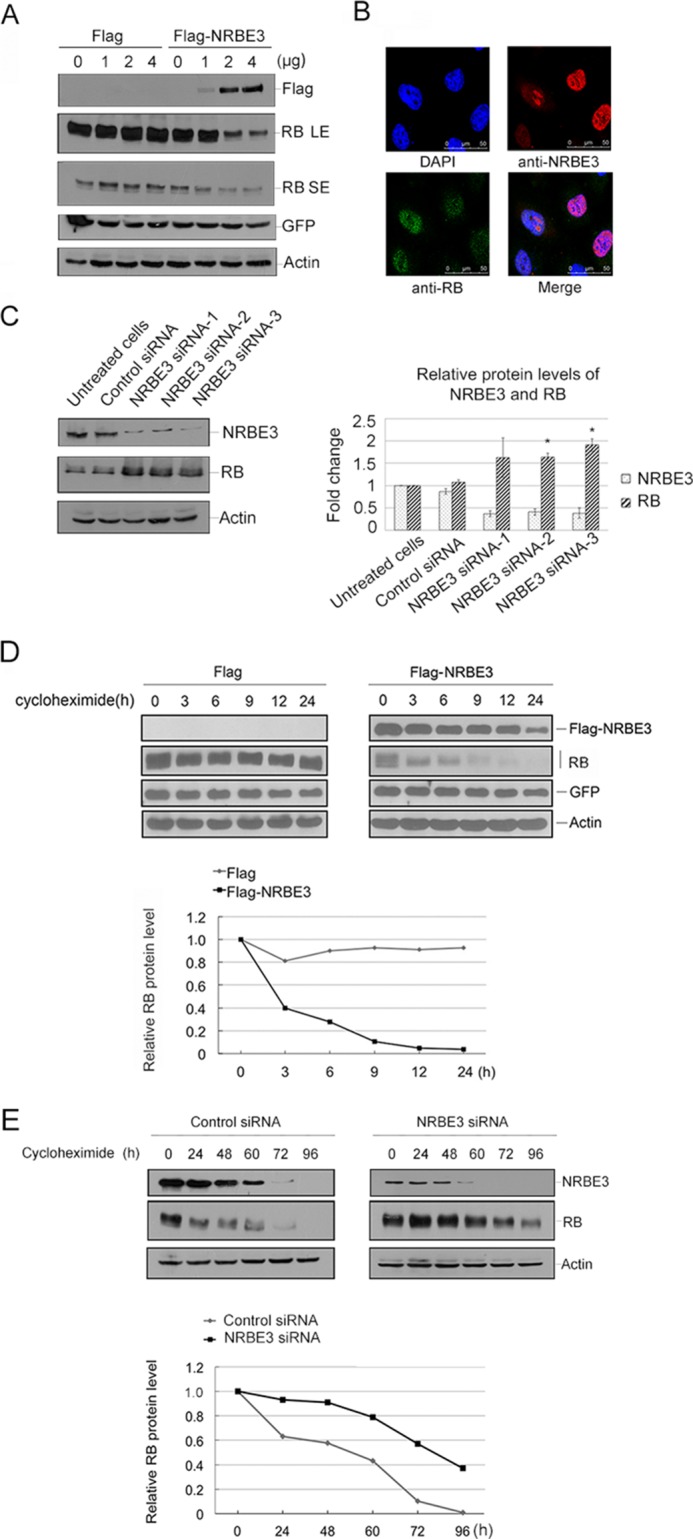
Expression of NRBE3 resulted in an active proteolysis of RB. A, U2OS cells were transfected with increasing amounts of FLAG or FLAG-NRBE3 plasmid and the same dose of GFP plasmid. Western blotting was performed with proteins from cell lysates. The upper part of the blot was probed with anti-FLAG, and the lower part was probed with anti-RB antibody. GFP was evaluated as a transfection efficiency control, and β-actin was evaluated as a loading control. LE, long exposure bands; SE, short exposure bands. B, U2OS cells were transfected with FLAG-NRBE3 expression plasmid. Cells were fixed 24 h post-transfection, and double immunofluorescence staining was performed with monoclonal anti-RB antibody and polyclonal anti-NRBE3 antibody. Immunocomplexes were probed with TRITC-conjugated goat anti-rabbit IgG and FITC-conjugated goat anti-mouse IgG. Nuclei were stained with DAPI. The image was taken under confocal microscopy. Scale bars represent 50 μm. C, HCT116 cells were transfected with NRBE3-specific siRNAs or a control siRNA, respectively. Western blotting was performed with proteins from cell lysates. The upper part of the blot was probed with anti-NRBE3 antibody, and the lower part was probed with anti-RB antibody. β-Actin was evaluated as a loading control. -Fold induction of the relative protein levels of RB is summarized from three independent experiments. Error bars represent S.E. *, p < 0.05 versus untreated cells (right panel). D, U2OS cells were transfected with either FLAG-NRBE3 or FLAG vector plasmid. Cells were treated with 10 μg/ml cycloheximide at 16 h post-transfection. Cells were harvested at the indicated time points, and cell lysates were prepared. Proteins from cell lysates were subjected to Western blotting with anti-FLAG and anti-RB antibodies as described in A (upper panel). GFP and β-actin were evaluated as transfection efficiency and loading controls, respectively. Relative RB levels were plotted with the integrated optical density of the RB bands on the Western blot (lower panel). E, U2OS cells were transfected with NRBE3-specific siRNAs or a control siRNA, respectively. Cells were treated with 10 μg/ml cycloheximide at 48 h post-transfection. Cells were harvested at the indicated time points, and cell lysates were prepared for Western blotting as described in A (upper panel). Relative RB levels were plotted with the integrated optical density of the RB bands on the Western blot (lower panel).
To test whether NRBE3-induced RB reduction is due to a decrease of protein stability, the half-life of RB protein was determined in the presence or absence of FLAG-NRBE3. FLAG-NRBE3 was transfected into U2OS cells, and RB levels were evaluated by Western blotting at different time points after de novo protein synthesis was blocked by cycloheximide. The half-life of RB was reduced from more than 24 h to less than 3 h when FLAG-NRBE3 was ectopically expressed (Fig. 2D). As expected, the half-life of RB protein was prolonged to more than 72 h in U2OS cells when NRBE3 was knocked down (Fig. 2E). These results demonstrate that NRBE3 promoted a rapid protein degradation of RB.
NRBE3 Promoted RB Protein Degradation through Proteasome-Ubiquitin Pathway
To verify whether NRBE3-induced RB degradation is proteasome-dependent, H1299 cells were transfected with FLAG-NRBE3, and cells were treated with MG132 before harvest. The results of Western blotting showed that FLAG-NRBE3 promoted RB degradation, and this FLAG-NRBE3-induced RB degradation was blocked by MG132 treatment, demonstrating that NRBE3 promoted RB degradation through the proteasomal pathway (Fig. 3A). Importantly, NRBE3 specifically promoted hypophosphorylated RB rather than its hyperphosphorylated form.
FIGURE 3.

NRBE3 promoted RB degradation through proteasome-ubiquitination pathway. A, RB and GFP plasmids were co-transfected either with FLAG-NRBE3 or FLAG vector plasmid into H1299 cells. Cells were treated either with 10 μm MG132 or dimethyl sulfoxide (DMSO) for 4 h before harvest. Equal amounts of protein of whole cell lysates were subjected to Western blotting for the indicated proteins. GFP was evaluated as a transfection efficiency control, and β-actin was evaluated as a loading control. B, H1299 cells were transfected with FLAG-NRBE3 or FLAG vector plasmid in the presence of HA-tagged wild-type ubiquitin plasmid (wt-Ub), HA-tagged K48R ubiquitin mutant plasmid (K48R-Ub), or empty vector (Mock). Equal amounts of protein from whole cell lysates were subjected to immunoblotting for evaluation of expression of FLAG-NRBE3 and RB as indicated. β-Actin was evaluated as a loading control. LE, long exposure bands; SE, short exposure bands (left panel). -Fold induction of the relative protein levels of RB is summarized from three independent experiments. Error bars represent S.E. (right panel). C, H1299 cells were co-transfected with RB, HA-Ub, and FLAG-NRBE3, FLAG-HPV16 E7, or FLAG vector plasmids, respectively. Cells were treated with 10 μm MG132 for 4 h before harvest. RB protein was immunoprecipitated from cell lysates with anti-RB antibody or mouse IgG. Proteins from the precipitates were subjected to Western blotting with anti-HA antibody (upper panel). Expression of RB, FLAG-NRBE3, FLAG-HPV16 E7, or HA-Ub was evaluated by immunoblotting with anti-RB, anti-FLAG anti-E7, or anti-HA antibody on cell lysates as indicated (lower panel). D, HCT116 cells were co-transfected with HA-Ub and NRBE3-specific siRNA or control siRNA, respectively. Cells were treated with 10 μm MG132 for 4 h before harvest. RB protein was immunoprecipitated from cell lysates with anti-RB antibody or mouse IgG. Proteins from the precipitates were subjected to Western blotting with anti-HA antibody (upper panel). NRBE3 and RB in cell lysates were evaluated by immunoblotting with anti-NRBE3 and anti-RB (lower panel). mIgG, mouse IgG; IP, immunoprecipitation; IB, immunoblot, hypo, hypophosphorylated; hyper, hyperphosphorylated.
To uncover the mechanism by which NRBE3 promotes RB proteasomal degradation, we examined the effect of wild-type ubiquitin (HA-Ub) or a mutated ubiquitin (HA-K48R-Ub) on NRBE3-mediated RB degradation in that ubiquitin-K48R might be deficient to mediate ubiquitin-dependent protein degradation. FLAG-NRBE3 expression plasmid was co-transfected into H1299 cells with empty vector, HA-Ub, or HA-K48R-Ub plasmid. FLAG-NRBE3-mediated RB degradation was dramatically enhanced by co-expression of wild-type HA-Ub, whereas mutated HA-K48R-Ub did not show any effect on NRBE3-mediated RB degradation (Fig. 3B). These results indicated that NRBE3 might promote RB degradation through the ubiquitin-proteasomal pathway.
To further confirm whether NRBE3 promotes RB polyubiquitination, an in vivo ubiquitination experiment was performed. H1299 cells were co-transfected with RB, HA-Ub, and FLAG-NRBE3 plasmids. Immunoprecipitation was performed with anti-RB antibody or mouse IgG, and the ubiquitination of RB was evaluated by Western blotting with anti-HA antibody. As shown in Fig. 3C, HPV16 E7 promoted RB ubiquitination as described previously (50), and FLAG-NRBE3 promoted RB ubiquitination much more strongly than did HPV16 E7. In contrast, knockdown of NRBE3 reduced polyubiquitination of RB (Fig. 3D).
A U-box Was Required for NRBE3-induced RB Degradation
The Pfam database based on hidden Markov models was searched for a ubiquitin E3 ligase domain in NRBE3. Two potential U-box-like domains were found between residues 135 and 147 of NRBE3 with an E-value of 3.6 and between residues 225 and 240 with an E-value of 0.16 (Fig. 4A). According to the E-value, it was more likely that residues 225–240 of NRBE3 were the conserved U-box. To determine which fragment of NRBE3 functions as a real U-box, two NRBE3 deletion mutants, FLAG-NRBE3(Δaa135–147) and FLAG-NRBE3(Δaa225–240), were constructed (Fig. 4B), and their capabilities of promoting RB degradation were evaluated. As shown in Fig. 4C, FLAG-NRBE3(Δaa225–240) lost the capability of inducing RB degradation, whereas FLAG-NRBE3(Δaa135–147) retained the ability of promoting RB degradation in H1299 cells. This experiment was also performed in U2OS cells, and a similar result was obtained (Fig. 4D). To further verify whether residues 225–240 are required for NRBE3 to destabilize RB protein, the half-life of RB protein was determined in the presence of FLAG, FLAG-NRBE3, or FLAG-NRBE3(Δaa225–240). Consistent with previous results, the half-life of RB was reduced to 3 h by FLAG-NRBE3, whereas FLAG-NRBE3(Δaa225–240) lost the capability of shortening the half-life of RB. As expected, FLAG-NRBE3(Δaa225–240) failed to promote RB ubiquitination even in the presence of HA-Ub (Fig. 4F) in cells. These data demonstrate that residues 225–240 functioned as a U-box in NRBE3 and suggested that NRBE3 might possess a ubiquitin E3 ligase function to promote RB degradation.
FIGURE 4.

The NRBE3-mediated RB degradation depended on its U-box. A, bioinformatics analyses of NRBE3 by the Pfam database. Two U-box domains were predicted in aa 135–147 and 225–240 of NRBE3. The alignment of U-box sequences in NRBE3 is shown in the table. HMM indicates that this analysis was based on hidden Markov models. B, schematic structure of the two potential U-boxes in NRBE3. NRBE3 deletion mutant plasmids with potential U-boxes deleted, NRBE3(Δaa135–147) and NRBE3(Δaa225–240), were constructed as shown. C, H1299 cells were transfected with FLAG-NRBE3, NRBE3(Δaa135–147), or NRBE3(Δaa225–240), respectively. Proteins from cell lysates were subjected to Western blotting. The upper part of the blot was probed with anti-FLAG, and the lower part was probed with anti-RB antibody. β-Actin was evaluated as a loading control. D, U2OS cells were transfected with FLAG-NRBE3, NRBE3(Δaa135–147), or NRBE3(Δaa225–240), respectively, and proteins from cell lysates were subjected to Western blotting as described in C. E, U2OS cells were transfected with FLAG-NRBE3, FLAG-NRBE3(Δaa225–240), or FLAG vector plasmid. Cells were treated with 10 μg/ml cycloheximide at 16 h post-transfection. Cells were harvested at the indicated time points, and cell lysates were prepared. Proteins from cell lysates were subjected to Western blotting with anti-FLAG and anti-RB antibodies as described in A (upper panel). β-Actin was evaluated as both transfection efficiency and loading controls. Relative RB levels were plotted with the integrated optical density of the RB bands on the Western blot (lower panel). F, H1299 cells were co-transfected with RB, HA-Ub, and FLAG vector, FLAG-NRBE3(Δaa225–240), or FLAG-NRBE3 plasmids, respectively. Cells were treated with MG132 (10 μm) for 4 h before harvest. Immunoprecipitation was performed with anti-RB antibody and cell extract. Proteins from the precipitates were subjected to Western blotting with anti-HA antibody. Expression of RB, FLAG-NRBE3, FLAG-NRBE3(Δaa225–240), or HA-Ub was evaluated by immunoblotting with anti-RB, anti-FLAG, or anti-HA antibody on cell lysates as indicated (lower panel). WCE, whole cell extract; IB, immunoblotting; IP, immunoprecipitation; SEQ, sequence.
NRBE3 Was a Bona Fide Ubiquitin E3 Ligase for RB
To determine whether NRBE3E3 acts as an E3 ligase for RB, we expressed and purified FLAG-NRBE3-His protein from Sf9 insect cells and performed in vitro ubiquitination with recombinant RB. As shown in Fig. 5A, purified FLAG-NRBE3-His polyubiquitinated RB, whereas RB was not ubiquitinated when E1, E2, or ubiquitin was missing. Sf9 cell lysate was used as a positive control for the ubiquitination experiment. Importantly, Sf9 purified FLAG-NRBE3(Δaa225–240)-His failed to ubiquitinate RB in vitro (Fig. 5B). Purified proteins used in the in vitro ubiquitin assay were verified by silver staining to show equal loading (Fig. 5B). Taken together, we demonstrated that NRBE3 was a bona fide E3 ligase for RB and that the U-box played an essential role in its E3 ligase function.
FIGURE 5.
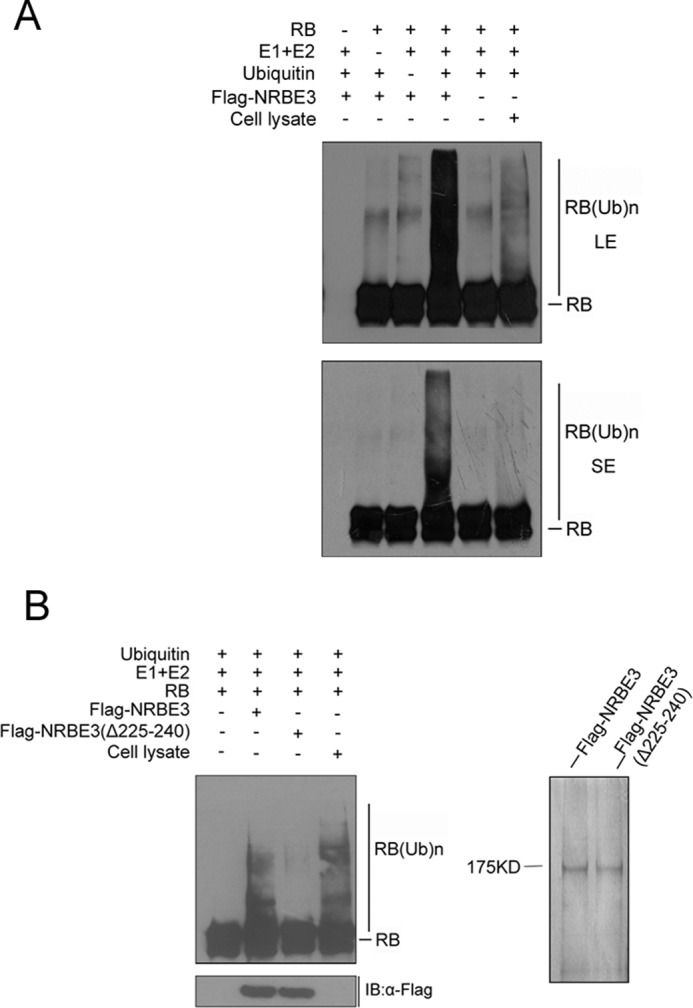
NRBE3 was a bona fide ubiquitin E3 ligase for RB. A, purified recombinant full-length RB was incubated with Sf9 purified FLAG-NRBE3-His together with E1, mixed E2s, and ubiquitin in an in vitro ubiquitin buffer at 30 °C for 1 h. The reactants were collected and separated by SDS-PAGE, transferred onto PVDF membrane, and immunoblotted using RB monoclonal antibody. The reactants missing RB, ubiquitin, or E1 and E2s were used as negative controls. Sf9 cell lysates were used to provide E3 ligases as a positive control (last lane). LE, long exposure bands; SE, short exposure bands. B, purified recombinant full-length RB was incubated with Sf9 purified FLAG-NRBE3-His or FLAG-NRBE3(Δaa225–240)-His together with E1, mixed E2s, and ubiquitin in an in vitro ubiquitination buffer at 30 °C for 1 h. The reactants were subjected to immunoblotting using anti-RB antibody. Purified FLAG-NRBE3-His and FLAG-NRBE3(Δaa225–240)-His proteins used in the in vitro ubiquitination assay were evaluated by immunoblotting using anti-FLAG monoclonal antibody and silver-stained as loading controls (left lower and right panels). IB, immunoblotting.
NRBE3 Was a Downstream Gene of E2F1
Because NRBE3 transforms NIH3T3 cells, we wanted to know how NRBE3 transcription is regulated. Bioinformatics analysis using PROMO at the ALGGEN server (51) showed that E2F1 and p53 might be potential main transcriptional factors on the NRBE3 promoter (Fig. 6A). NRBE3 promoter-luciferase reporter plasmid pGL3-NRBE3-Luc was constructed and co-transfected into 293 cells with E2F1 alone and/or DP1. E2F1 activated NRBE3 promoter reporter in a dose-dependent manner at low doses including 20 and 40 ng, but no further activation of reporter was observed by 60 ng of E2F1 (Fig. 6B). However, E2F1 showed a perfect dose-dependent activation of NRBE3 promoter reporter when it was co-expressed with DP1. These results demonstrate that E2F1 needs DP1 for its transactivation activity as described previously (52). In contrast, NRBE3 promoter reporter was not regulated by p53 (Fig. 6C). To further confirm the transactivation of E2F1 on NRBE3 transcription, E2F1 was transfected into U2OS cells with or without DP1, and the mRNA level of NRBE3 was determined by real time PCR. The mRNA levels of NRBE3 increased when E2F1 was ectopically expressed alone or together with DP1 (Fig. 6D). As expected, NRBE3 protein levels were also elevated by ectopic expression of E2F1 or E2F1/DP1 (Fig. 6E). These results demonstrated that NRBE3 was a downstream gene of E2F1.
FIGURE 6.

Transcription of NRBE3 was regulated by E2F1. A, DNA sequence of NRBE3 promoter. The transcription initiation site is indicated with an arrow. The binding sites for potential transcription factors are underlined, and the transcription factors are given below the sequence. B, pGL3-luciferase reporter plasmid (Basic) or NRBE3 promoter-luciferase reporter plasmid (NRBE3-Luc) was co-transfected with E2F1 and/or DP1 plasmids. Cells were harvested, and cell lysates were prepared 24 h post-transfection. Luciferase activity was measured with the Dual-Luciferase Reporter Assay System using a Berthold luminometer. -Fold induction of the luciferase activity was summarized from three independent experiments in triplicates. Error bars represent S.E. *, p < 0.05; **, p < 0.01 versus NRBE3-Luc. C, pGL3-Basic or NRBE3-Luc was co-transfected with p53 plasmids. Luciferase activities were measured as described in B. D, U2OS cells were transfected with E2F1 and/or DP1 plasmids. Cells were harvested 24 h post-transfection, and total RNA was extracted. Real time PCR was performed with NRBE3-specific primers, and GAPDH was amplified as an internal control. Shown is the relative mRNA level of NRBE3. Error bars represent S.E. *, p < 0.05 versus empty control. E, U2OS cells were transfected with E2F1 and/or DP1 plasmids. Proteins from cell lysates were subjected to Western blotting probed with anti-NRBE3, anti-E2F1, or anti-DP1 antibody. β-Actin was evaluated as a loading control. The experiment was repeated three times, and Western blot band intensity was scanned and plotted (lower panel). Error bars represent S.E. *, p < 0.05; **, p < 0.01 versus empty control.
Knockdown of NRBE3 Arrested Cell Cycle at G1
One of the best characterized activities of RB protein is inhibition of E2F1-mediated transcription with resultant effects on cell cycle regulation. To study the biological significance of NRBE3-induced RB degradation, we first evaluated the expression of E2F1 downstream genes cyclin E and cyclin A, which in turn activate RB phosphorylation and promote G1/S transition (12). FLAG-NRBE3 resulted in increases of both cyclin E and cyclin A, which was concomitant with degradation of RB protein in a dose-dependent fashion (Fig. 7A). In contrast, knockdown of NRBE3 caused decreases of both cyclin E and cyclin A (Fig. 7B). Thereafter, the cell cycle was analyzed when NRBE3 was silenced in U2OS cells. Knockdown of NRBE3 in U2OS cells arrested the cell cycle at G1 (Fig. 7C). To confirm this observation, we performed the same experiment in the MCF-7 cell line, which expresses wild-type RB like U2OS. As shown in Fig. 7D, knockdown of NRBE3 arrested the cell cycle at G1 in MCF-7 cells. The above described experiments were also conducted in an RB-null cell line, SAOS-2. In the absence of RB, either ectopic expression of FLAG-NRBE3 or knockdown of NRBE3 failed to affect the cell cycle (Fig. 7, E and F). These results demonstrated that NRBE3 promoted G1/S transition of the cell cycle at least partially by promoting RB degradation.
FIGURE 7.

NRBE3 promoted G1/S transition. A, U2OS cells were transfected with increasing doses of FLAG-NRBE3, and cells were harvested 24 h post-transfection. Expression of FLAG-NRBE3, RB, cyclin E, and cyclin A in the cells was evaluated by Western blotting performed with proteins from cell lysates. GFP was evaluated as a transfection efficiency control, and β-actin was used as a loading control. B, HCT116 cells were transfected with specific NRBE3 siRNA or control siRNA, and cells were harvested 72 h post-transfection. Expression of NRBE3, RB, cyclin E, and cyclin A in the cell lysates was evaluated by Western blotting as described in A. C, U2OS cells were transfected with siRNA targeting NRBE3 or a control siRNA. The cell cycle was analyzed by flow cytometry 72 h post-transfection. A representative result is shown on the left. The average percentage of cells in the cell cycle phases summarized from three independent experiments is shown on the right. D, MCF-7 cells were transfected with NRBE3 siRNA or control siRNA. The cell cycle was analyzed by flow cytometry 72 h post-transfection. The average percentage of cells in the cell cycle phases summarized from three independent experiments is shown on the right. E, SAOS-2 cells were transfected with either FLAG or FLAG-NRBE3 plasmids. The cell cycle was analyzed by flow cytometry 24 h post-transfection. A representative result is shown on the left. The average percentage of cells in the cell cycle phases summarized from three independent experiments is shown on the right. F, SAOS-2 cells were transfected with siRNA targeting NRBE3 or a control siRNA. The cell cycle was analyzed by flow cytometry 72 h post-transfection. A representative result is shown on the left. The average percentage of cells in the cell cycle phases summarized from three independent experiments is shown on the right.
NRBE3 Is Up-regulated in Human Breast Cancer Tissues
To address whether NRBE3 is related to human cancer, we examined NRBE3 expression in human breast cancer tissues. Proteins extracted from human breast cancer tissues and paired adjacent non-cancerous breast tissues were subjected to Western blotting to evaluate NRBE3 expression. In 16 cases of breast cancer, no NRBE3 protein was detected in the adjacent non-cancerous breast tissues, whereas NRBE3 was detected in the cancer tissues in 14 of 16 breast cancer patients (87.5%) (Fig. 8).
FIGURE 8.
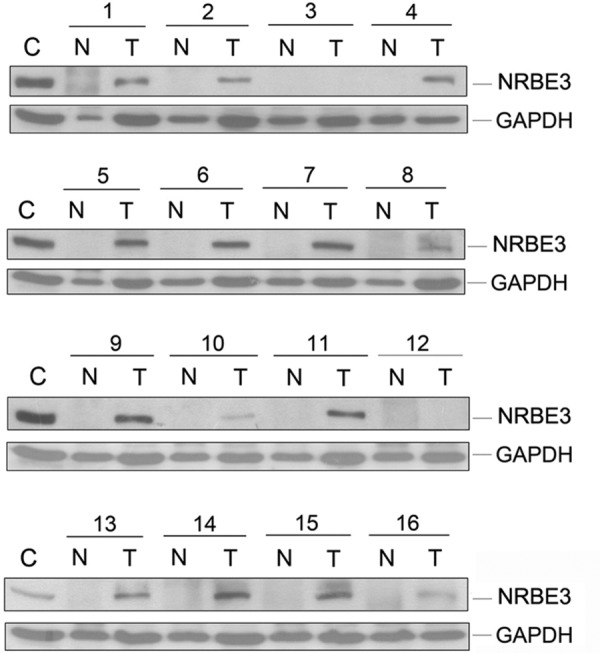
NRBE3 was up-regulated in breast carcinoma tissues. Proteins extracted from 16 cases of human breast cancer tissues and paired adjacent non-cancerous breast tissues were subjected to Western blotting with anti-NRBE3 antibody. GAPDH was used as a loading control. C represents proteins from MCF-7 cell lysates. T, cancer tissues; N, normal tissues.
Discussion
Because NRBE3 contained an LXCXE motif, we set out to investigate the interaction between NRBE3 and RB. We first demonstrated that NRBE3 interacted with RB in vitro by GST pulldown experiment. As expected, NRBE3, specifically N-terminal residues 1–681 that contained an LXCXE motif, interacted with the large pocket of RB protein. This was further confirmed by the mutant NRBE3aa1–681-RXRXH, which lost the capability of binding RB when LXCXE were mutated. However, we surprisingly found that the C terminus of NRBE3 containing residues 805–1209 interacted with RB as well. Given that the RB-binding proteins E1A and HPV E7 also contain other unknown RB binding modules besides their LXCXE motifs (53, 54), we conducted amino acid sequence alignment with residues 805–1209 in NRBE3 against the RB-interacting modules in E1A and HPV E7. However, no known conserved motif was found. The RB-binding modules in the C terminus of NRBE3 need further study.
It was of importance that NRBE3 selectively bound to and promoted degradation of the hypophosphorylated RB rather than hyperphosphorylated RB. Given that RB releases E2F1 when it is phosphorylated by cyclinD/CDK4/CDK6, hypophosphorylated RB plays an active role by interacting with and inhibiting the transcriptional activity of E2F1 (15, 16, 55), and hypophosphorylated RB is the natural target for some oncoproteins such as E1A, HPV E7, SV40 large T antigen, and MDM2. They preferably bind to and promote degradation of the active RB, thereby allowing E2F to be constitutively activated, resulting in uncontrolled cell cycle progression (26, 56–58). Here, we identified a novel cellular protein NRBE3 that acted as an oncoprotein in tumorigenesis by targeting active RB for degradation.
In the present study, we showed that NRBE3 interacted with RB in cells when proteasome activity was inhibited, and NRBE3 promoted RB turnover by a proteasomal pathway. In the proteasome, proteins are degraded by either ubiquitin-dependent or ubiquitin-independent pathways (59). Because ubiquitin-dependent protein degradation is mainly mediated through lysine 48 of ubiquitin (60), the ubiquitin mutant K48R blocks the conjugation of ubiquitin chain to the substrate and thus is used to distinguish the ubiquitin-dependent pathway from ubiquitin-independent pathway (61). We showed that wild-type ubiquitin dramatically enhanced NRBE3-mediated RB degradation, whereas the K48R ubiquitin was not able to do so, suggesting that NRBE3 promoted RB degradation through the ubiquitin-dependent pathway. In addition, an in vivo ubiquitination experiment demonstrated that NRBE3 promoted RB ubiquitination in cells. Taken together, we demonstrated that NRBE3 promoted RB ubiquitination and degradation through the proteasome.
Because typical E3 ligases contain conserved domains such as HECT (homologous to E6-AP C terminus) domain, RING (the really interesting new gene) finger domain (62, 63) and U-box (64–67), we searched for conserved E3 ligase domains in NRBE3 by sequence alignment. We found that two potential U-boxes existed, i.e. residues 135–147 and residues 225–240 in NRBE3. Our results demonstrated that the NRBE3 fragment containing residues 225–240 was required for promoting RB ubiquitination and degradation.
Ubiquitin E3 ligases for tumor suppressors play important roles in tumorigenesis. As for tumor suppressor RB, only two E3 ligases, MDM2 and anaphase-promoting complex, have been found to interact with RB; however, only MDM2 mediates RB protein degradation (33, 68). Thus, MDM2 is the only known E3 ligase for RB. It has been found that HPV E7 promotes RB ubiquitination and degradation (26). In addition, HPV16 E7 associates with the Cul2 ubiquitin ligase and the Cul2-E7 complex ubiquitinates RB in Caski cells. However, there is no in vitro evidence showing that the Cul2 complex is an E3 ligase for RB (69), and it has been demonstrated that MDM2 is not involved in the E7-induced proteolysis of RB (70). These findings suggest that some other cellular E3 ligase(s) may exist. In the present study, we demonstrated that NRBE3 acted as an E3 ubiquitin ligase by in vivo ubiquitination experiments. It was plausible that purified FLAG-NRBE3 from insect cell Sf9 ubiquitinated recombinant RB in vitro, demonstrating that NRBE3 was a bona fide E3 ligase for RB. Furthermore, we demonstrated that residues 225–240 in NRBE3 functioned as the U-box for its E3 ligase activity. Thus far, we identified NRBE3 as the second E3 ubiquitin ligase for RB. Whether NRBE3 mediates HPV E7-induced RB degradation and is associated with HPV E7-induced cervical tumorigenesis is currently under study.
Because NRBE3 expression was up-regulated in some types of human cancer tissues,5 we next set out to uncover the transcriptional regulation of NRBE3. We showed that NRBE3 transcription was activated by E2F1/DP1 instead of p53. So NRBE3 formed positive regulation feedback with RB/E2F1 where NRBE3 inactivated RB by promoting degradation of the hypophosphorylated RB and released E2F1, which in turn activated NRBE3 expression. Consequently, NRBE3 promoted G1/S transition. It will be significant to verify whether the existence of this positive regulation feedback magnifies the tumorigenic effect of other RB degradation oncoproteins.
It was reported that RB deficiency promotes proliferation of breast cancer cells and tumor growth in nude mouse xenografts and has highly significant effects on the therapeutic response (71). In breast cancer, inactivation of RB is believed to occur via multiple mechanisms to facilitate tumorigenesis. Loss of heterozygosity at the Rb locus has been defined in 20–30% of breast cancer, and histological loss of RB protein has been documented with varying frequency (15, 72). Cyclin D1 is up-regulated in ∼50% of breast cancers (73), and the CDK inhibitor p16Ink4a is down-regulated in some breast cancer cases (74–76). Because we already demonstrated that NRBE3 targeted active RB for degradation and a G1 arrest in the cell cycle was observed in breast cancer cells after knockdown of NRBE3, we were driven to ask whether NRBE3 is related to human breast cancers. We examined the expression of NRBE3 in 16 cases of human breast carcinoma tissues by Western blot. NRBE3 was detected in 14 of 16 (87.5%) cases of breast cancer, whereas no NRBE3 was detected in the paired adjacent non-cancerous breast tissues. Does NRBE3 play a role in promoting breast cancer progress? In cyclin D1-up-regulated or p16Ink4a-down-regulated breast cancer cases, cyclin-CDK complex-induced aberrant phosphorylation/inactivation of RB leads to deregulation of E2F1 (77–79), which is the transcriptional activator for NRBE3. Once the expression of NRBE3 is activated, it in turn targets RB for degradation and forms a positive regulation loop with RB/E2F1. It is worthwhile to examine whether activation and accumulation of NRBE3 are related to breast cancer progression. Although Western blotting is not the best measurement of the histological expression of NRBE3 in human tumor tissues, this result nonetheless suggests a great difference in NRBE3 expression between breast cancer tissues and adjacent non-cancerous breast tissues. Immunohistochemical staining with NRBE3 in breast cancer cases ranked in various clinical stages and following statistic analysis is underway in our laboratory. It will be imperative to determine whether NRBE3 is related with the prognosis of breast cancer.
Author Contributions
X. D. and Y. K. designed and coordinated the study. X. D. wrote the paper. Yi. W. performed and analyzed the experiments shown in Figs. 1 (D, E, H, and I), 2 (A, B, and C), 3 (C and D), 4, 5, 6, and 7 (A and B) and participated in drafting and revising the manuscript. Z. Z. performed and analyzed the experiments shown in Figs. 1 (A, B, C, F, and G), 2D, 3A, 7 (C, D, E, and F), and 8. J. Z. performed and analyzed the experiments shown in Figs. 2E and 3B. Yo. W. and J. L. participated in the construction of some plasmids and expression and purification of GST fusion proteins. R. K. and Y. Z. provided technical assistance and contributed to the preparation of the figures. H. D. revised the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Yin and Dr. Luo for helpful discussions of our work, Dr. Michael A McNutt for editing the English in this manuscript, Dr. Qihua He for assistance with confocal microscopy, and Dr. Hounan Wu for flow cytometry.
This work was supported by National Natural Science Foundation of China Grants 30571038 and 81372222, the Innovation team of the Ministry of Education (Grant IRT13001), and Beijing Municipal Natural Science Foundation Grant 7122032. The authors declare that they have no conflict of interest with the contents of this article.
Y. Wang, Z. Zheng, J. Zhang, Y. Wang, R. Kong, J. Liu, Y. Zhang, H. Deng, X. Du, and Y. Ke, unpublished data.
- Rb
- retinoblastoma susceptibility gene
- RB
- retinoblastoma protein
- CDK
- cyclin-dependent kinase
- HPV E7
- human papilloma virus early protein 7
- NRBE3
- novel RB E3 ubiquitin ligase
- aa
- amino acids
- TRITC
- tetramethylrhodamine isothiocyanate
- Ub
- ubiquitin.
References
- 1.Friend S. H., Bernards R., Rogelj S., Weinberg R. A., Rapaport J. M., Albert D. M., and Dryja T. P. (1986) A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323, 643–646 [DOI] [PubMed] [Google Scholar]
- 2.Knudson A. G., Jr. (1971) Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U.S.A. 68, 820–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavenee W. K., Hansen M. F., Nordenskjold M., Kock E., Maumenee I., Squire J. A., Phillips R. A., and Gallie B. L. (1985) Genetic origin of mutations predisposing to retinoblastoma. Science 228, 501–503 [DOI] [PubMed] [Google Scholar]
- 4.Kaye F. J. (2002) RB and cyclin dependent kinase pathways: defining a distinction between RB and p16 loss in lung cancer. Oncogene 21, 6908–6914 [DOI] [PubMed] [Google Scholar]
- 5.Deshpande A., and Hinds P. W. (2006) The retinoblastoma protein in osteoblast differentiation and osteosarcoma. Curr. Mol. Med. 6, 809–817 [DOI] [PubMed] [Google Scholar]
- 6.Viatour P., and Sage J. (2011) Newly identified aspects of tumor suppression by RB. Dis. Model. Mech. 4, 581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacks T., Fazeli A., Schmitt E. M., Bronson R. T., Goodell M. A., and Weinberg R. A. (1992) Effects of an Rb mutation in the mouse. Nature 359, 295–300 [DOI] [PubMed] [Google Scholar]
- 8.Clarke A. R., Maandag E. R., van Roon M., van der Lugt N. M., van der Valk M., Hooper M. L., Berns A., and te Riele H. (1992) Requirement for a functional Rb-1 gene in murine development. Nature 359, 328–330 [DOI] [PubMed] [Google Scholar]
- 9.Hu N., Gutsmann A., Herbert D. C., Bradley A., Lee W. H., and Lee E. Y. (1994) Heterozygous Rb-1 Δ20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene 9, 1021–1027 [PubMed] [Google Scholar]
- 10.Wikenheiser-Brokamp K. A. (2006) Retinoblastoma family proteins: insights gained through genetic manipulation of mice. Cell. Mol. Life Sci. 63, 767–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 12.Cobrinik D. (2005) Pocket proteins and cell cycle control. Oncogene 24, 2796–2809 [DOI] [PubMed] [Google Scholar]
- 13.Dick F. A., and Rubin S. M. (2013) Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinberg R. A. (1995) The retinoblastoma protein and cell cycle control. Cell 81, 323–330 [DOI] [PubMed] [Google Scholar]
- 15.Knudsen E. S., and Knudsen K. E. (2008) Tailoring to RB: tumour suppressor status and therapeutic response. Nat. Rev. Cancer 8, 714–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flemington E. K., Speck S. H., and Kaelin W. G. Jr. (1993) E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc. Natl. Acad. Sci. U.S.A. 90, 6914–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helin K., Harlow E., and Fattaey A. (1993) Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol. Cell. Biol. 13, 6501–6508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan W., Cox S., Hoess R. H., and Grafstrom R. H. (2001) A cyclin D1/cyclin-dependent kinase 4 binding site within the C domain of the retinoblastoma protein. Cancer Res. 61, 2885–2891 [PubMed] [Google Scholar]
- 19.Henley S. A., and Dick F. A. (2012) The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diehl J. A. (2002) Cycling to cancer with cyclin D1. Cancer Biol. Ther. 1, 226–231 [DOI] [PubMed] [Google Scholar]
- 21.Jones R. E., Wegrzyn R. J., Patrick D. R., Balishin N. L., Vuocolo G. A., Riemen M. W., Defeo-Jones D., Garsky V. M., Heimbrook D. C., and Oliff A. (1990) Identification of HPV-16 E7 peptides that are potent antagonists of E7 binding to the retinoblastoma suppressor protein. J. Biol. Chem. 265, 12782–12785 [PubMed] [Google Scholar]
- 22.Defeo-Jones D., Huang P. S., Jones R. E., Haskell K. M., Vuocolo G. A., Hanobik M. G., Huber H. E., and Oliff A. (1991) Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature 352, 251–254 [DOI] [PubMed] [Google Scholar]
- 23.Lee J. O., Russo A. A., and Pavletich N. P. (1998) Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 391, 859–865 [DOI] [PubMed] [Google Scholar]
- 24.Dick F. A., Sailhamer E., and Dyson N. J. (2000) Mutagenesis of the pRB pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol. Cell. Biol. 20, 3715–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai A., Lee J. M., Yang W. M., DeCaprio J. A., Kaelin W. G. Jr., Seto E., and Branton P. E. (1999) RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Mol. Cell. Biol. 19, 6632–6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyer S. N., Wazer D. E., and Band V. (1996) E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 56, 4620–4624 [PubMed] [Google Scholar]
- 27.Kalejta R. F., Bechtel J. T., and Shenk T. (2003) Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23, 1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight J. S., Sharma N., and Robertson E. S. (2005) Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 102, 18562–18566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munakata T., Liang Y., Kim S., McGivern D. R., Huibregtse J., Nomoto A., and Lemon S. M. (2007) Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 3, 1335–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kehn K., Fuente Cde L., Strouss K., Berro R., Jiang H., Brady J., Mahieux R., Pumfery A., Bottazzi M. E., and Kashanchi F. (2005) The HTLV-I Tax oncoprotein targets the retinoblastoma protein for proteasomal degradation. Oncogene 24, 525–540 [DOI] [PubMed] [Google Scholar]
- 31.Higashitsuji H., Itoh K., Nagao T., Dawson S., Nonoguchi K., Kido T., Mayer R. J., Arii S., and Fujita J. (2000) Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat. Med. 6, 96–99 [DOI] [PubMed] [Google Scholar]
- 32.Sdek P., Ying H., Chang D. L., Qiu W., Zheng H., Touitou R., Allday M. J., and Xiao Z. X. (2005) MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 20, 699–708 [DOI] [PubMed] [Google Scholar]
- 33.Uchida C., Miwa S., Kitagawa K., Hattori T., Isobe T., Otani S., Oda T., Sugimura H., Kamijo T., Ookawa K., Yasuda H., and Kitagawa M. (2005) Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 24, 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hershko A., and Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 35.Pickart C. M. (2004) Back to the future with ubiquitin. Cell 116, 181–190 [DOI] [PubMed] [Google Scholar]
- 36.Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 37.Honda R., Tanaka H., and Yasuda H. (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 420, 25–27 [DOI] [PubMed] [Google Scholar]
- 38.Dornan D., Wertz I., Shimizu H., Arnott D., Frantz G. D., Dowd P., O'Rourke K., Koeppen H., and Dixit V. M. (2004) The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429, 86–92 [DOI] [PubMed] [Google Scholar]
- 39.Leng R. P., Lin Y., Ma W., Wu H., Lemmers B., Chung S., Parant J. M., Lozano G., Hakem R., and Benchimol S. (2003) Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112, 779–791 [DOI] [PubMed] [Google Scholar]
- 40.Jain A. K., and Barton M. C. (2009) Regulation of p53: TRIM24 enters the RING. Cell Cycle 8, 3668–3674 [DOI] [PubMed] [Google Scholar]
- 41.Jung Y. S., Qian Y., and Chen X. (2012) Pirh2 RING-finger E3 ubiquitin ligase: its role in tumorigenesis and cancer therapy. FEBS Lett. 586, 1397–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai W. W., Wang Z., Yiu T. T., Akdemir K. C., Xia W., Winter S., Tsai C. Y., Shi X., Schwarzer D., Plunkett W., Aronow B., Gozani O., Fischle W., Hung M. C., Patel D. J., and Barton M. C. (2010) TRIM24 links a non-canonical histone signature to breast cancer. Nature 468, 927–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marine J. C. (2012) Spotlight on the role of COP1 in tumorigenesis. Nat. Rev. Cancer 12, 455–464 [DOI] [PubMed] [Google Scholar]
- 44.Brown D. R., Thomas C. A., and Deb S. P. (1998) The human oncoprotein MDM2 arrests the cell cycle: elimination of its cell-cycle-inhibitory function induces tumorigenesis. EMBO J. 17, 2513–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L., Zhao J., Lü W., Li Y., Du X., Ning T., Lu G., and Ke Y. (2005) KIAA0649, a 1A6/DRIM-interacting protein with the oncogenic potential. Biochem. Biophys. Res. Commun. 334, 884–890 [DOI] [PubMed] [Google Scholar]
- 46.Zheng Z. F., Han W., He Q. H., Ke Y., and Du X. J. (2009) Preparation of the polyclonal antibody against KIAA0649 and its cellular localization. Beijing Da Xue Xue Bao 41, 613–619 [PubMed] [Google Scholar]
- 47.Wu J., Zhang Y., Wang Y., Kong R., Hu L., Schuele R., Du X., and Ke Y. (2012) Transcriptional repressor NIR functions in the ribosome RNA processing of both 40S and 60S subunits. PLoS One 7, e31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu L., Wang J., Liu Y., Zhang Y., Zhang L., Kong R., Zheng Z., Du X., and Ke Y. (2011) A small ribosomal subunit (SSU) processome component, the human U3 protein 14A (hUTP14A) binds p53 and promotes p53 degradation. J. Biol. Chem. 286, 3119–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.French A. P., Mills S., Swarup R., Bennett M. J., and Pridmore T. P. (2008) Colocalization of fluorescent markers in confocal microscope images of plant cells. Nat. Protoc. 3, 619–628 [DOI] [PubMed] [Google Scholar]
- 50.Wang J., Sampath A., Raychaudhuri P., and Bagchi S. (2001) Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene 20, 4740–4749 [DOI] [PubMed] [Google Scholar]
- 51.Farré D., Roset R., Huerta M., Adsuara J. E., Roselló L., Albà M. M., and Messeguer X. (2003) Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Helin K., Wu C. L., Fattaey A. R., Lees J. A., Dynlacht B. D., Ngwu C., and Harlow E. (1993) Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev. 7, 1850–1861 [DOI] [PubMed] [Google Scholar]
- 53.Dyson N., Guida P., McCall C., and Harlow E. (1992) Adenovirus E1A makes two distinct contacts with the retinoblastoma protein. J. Virol. 66, 4606–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chemes L. B., Sánchez I. E., Smal C., and de Prat-Gay G. (2010) Targeting mechanism of the retinoblastoma tumor suppressor by a prototypical viral oncoprotein. Structural modularity, intrinsic disorder and phosphorylation of human papillomavirus E7. FEBS J. 277, 973–988 [DOI] [PubMed] [Google Scholar]
- 55.Adams P. D., Li X., Sellers W. R., Baker K. B., Leng X., Harper J. W., Taya Y., and Kaelin W. G. Jr. (1999) Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol. Cell. Biol. 19, 1068–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whyte P., Buchkovich K. J., Horowitz J. M., Friend S. H., Raybuck M., Weinberg R. A., and Harlow E. (1988) Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334, 124–129 [DOI] [PubMed] [Google Scholar]
- 57.Ludlow J. W., DeCaprio J. A., Huang C. M., Lee W. H., Paucha E., and Livingston D. M. (1989) SV40 large T antigen binds preferentially to an underphosphorylated member of the retinoblastoma susceptibility gene product family. Cell 56, 57–65 [DOI] [PubMed] [Google Scholar]
- 58.Sdek P., Ying H., Zheng H., Margulis A., Tang X., Tian K., and Xiao Z. X. (2004) The central acidic domain of MDM2 is critical in inhibition of retinoblastoma-mediated suppression of E2F and cell growth. J. Biol. Chem. 279, 53317–53322 [DOI] [PubMed] [Google Scholar]
- 59.Adams J. (2004) The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer 4, 349–360 [DOI] [PubMed] [Google Scholar]
- 60.Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 [DOI] [PubMed] [Google Scholar]
- 61.Ward C. L., Omura S., and Kopito R. R. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 62.Deshaies R. J., and Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 63.Rotin D., and Kumar S. (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409 [DOI] [PubMed] [Google Scholar]
- 64.Patterson C. (2002) A new gun in town: the U box is a ubiquitin ligase domain. Sci. STKE 2002, pe4. [DOI] [PubMed] [Google Scholar]
- 65.Koegl M., Hoppe T., Schlenker S., Ulrich H. D., Mayer T. U., and Jentsch S. (1999) A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644 [DOI] [PubMed] [Google Scholar]
- 66.Hatakeyama S., Yada M., Matsumoto M., Ishida N., and Nakayama K. I. (2001) U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276, 33111–33120 [DOI] [PubMed] [Google Scholar]
- 67.Nordquist K. A., Dimitrova Y. N., Brzovic P. S., Ridenour W. B., Munro K. A., Soss S. E., Caprioli R. M., Klevit R. E., and Chazin W. J. (2010) Structural and functional characterization of the monomeric U-box domain from E4B. Biochemistry 49, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Binné U. K., Classon M. K., Dick F. A., Wei W., Rape M., Kaelin W. G. Jr., Näär A. M., and Dyson N. J. (2007) Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat. Cell Biol. 9, 225–232 [DOI] [PubMed] [Google Scholar]
- 69.Huh K., Zhou X., Hayakawa H., Cho J. Y., Libermann T. A., Jin J., Harper J. W., and Munger K. (2007) Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 81, 9737–9747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oh K. J., Kalinina A., and Bagchi S. (2010) Destabilization of Rb by human papillomavirus E7 is cell cycle dependent: E2-25K is involved in the proteolysis. Virology 396, 118–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bosco E. E., Wang Y., Xu H., Zilfou J. T., Knudsen K. E., Aronow B. J., Lowe S. W., and Knudsen E. S. (2007) The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J. Clin. Investig. 117, 218–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bosco E. E., and Knudsen E. S. (2007) RB in breast cancer: at the crossroads of tumorigenesis and treatment. Cell Cycle 6, 667–671 [DOI] [PubMed] [Google Scholar]
- 73.Arnold A., and Papanikolaou A. (2005) Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol. 23, 4215–4224 [DOI] [PubMed] [Google Scholar]
- 74.Ertel A., Dean J. L., Rui H., Liu C., Witkiewicz A. K., Knudsen K. E., and Knudsen E. S. (2010) RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 9, 4153–4163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dublin E. A., Patel N. K., Gillett C. E., Smith P., Peters G., and Barnes D. M. (1998) Retinoblastoma and p16 proteins in mammary carcinoma: their relationship to cyclin D1 and histopathological parameters. Int. J. Cancer 79, 71–75 [DOI] [PubMed] [Google Scholar]
- 76.Hui R., Macmillan R. D., Kenny F. S., Musgrove E. A., Blamey R. W., Nicholson R. I., Robertson J. F., and Sutherland R. L. (2000) INK4a gene expression and methylation in primary breast cancer: overexpression of p16INK4a messenger RNA is a marker of poor prognosis. Clin. Cancer Res. 6, 2777–2787 [PubMed] [Google Scholar]
- 77.Casimiro M. C., Crosariol M., Loro E., Li Z., and Pestell R. G. (2012) Cyclins and cell cycle control in cancer and disease. Genes Cancer 3, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Musgrove E. A., Caldon C. E., Barraclough J., Stone A., and Sutherland R. L. (2011) Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 11, 558–572 [DOI] [PubMed] [Google Scholar]
- 79.Musgrove E. A., Lee C. S., Buckley M. F., and Sutherland R. L. (1994) Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc. Natl. Acad. Sci. U.S.A. 91, 8022–8026 [DOI] [PMC free article] [PubMed] [Google Scholar]