

Background: Ca2+ oscillations in mesothelial cells depend on Ca2+ influx.
Results: However, the “lanthanum insulation method” renders the oscillations independent of extracellular Ca2+.
Conclusion: Multiple pathways of Ca2+ shuttling are operating simultaneously during Ca2+ oscillations.
Significance: Experimental and mathematical approaches shed light on the mechanism of Ca2+ oscillations.
Keywords: calcium imaging, calcium intracellular release, calcium transport, calcium-binding protein, mitochondria, Ca2+ oscillations, calretinin, mitochondrial Ca2+ handling
Abstract
In some cell types, Ca2+ oscillations are strictly dependent on Ca2+ influx across the plasma membrane, whereas in others, oscillations also persist in the absence of Ca2+ influx. We observed that, in primary mesothelial cells, the plasmalemmal Ca2+ influx played a pivotal role. However, when the Ca2+ transport across the plasma membrane by the “lanthanum insulation method” was blocked prior to the induction of the serum-induced Ca2+ oscillations, mitochondrial Ca2+ transport was found to be able to substitute for the plasmalemmal Ca2+ exchange function, thus rendering the oscillations independent of extracellular Ca2+. However, in a physiological situation, the Ca2+-buffering capacity of mitochondria was found not to be essential for Ca2+ oscillations. Moreover, brief spontaneous Ca2+ changes were observed in the mitochondrial Ca2+ concentration without apparent changes in the cytosolic Ca2+ concentration, indicating the presence of a mitochondrial autonomous Ca2+ signaling mechanism. In the presence of calretinin, a Ca2+-buffering protein, the amplitude of cytosolic spikes during oscillations was decreased, and the amount of Ca2+ ions taken up by mitochondria was reduced. Thus, the increased calretinin expression observed in mesothelioma cells and in certain colon cancer might be correlated to the increased resistance of these tumor cells to proapoptotic/pronecrotic signals. We identified and characterized (experimentally and by modeling) three Ca2+ shuttling pathways in primary mesothelial cells during Ca2+ oscillations: Ca2+ shuttled between (i) the endoplasmic reticulum (ER) and mitochondria, (ii) the ER and the extracellular space, and (iii) the ER and cytoplasmic Ca2+ buffers.
Introduction
The calcium ion (Ca2+) is a universal intracellular messenger that controls a diverse range of cellular processes including cell proliferation, apoptosis, fertilization, neurotransmitter release, and heartbeat among many others (1). Ca2+ pumps in the plasma membrane (plasma membrane Ca2+-ATPase) and in endoplasmic reticulum (ER)2 membranes (SERCA) are responsible for the low cytosolic (ccyt) and nuclear free Ca2+ concentrations (cnucl) (50–100 nm) compared with the free Ca2+ concentrations in the extracellular space (1–2 mm) and the ER lumen (cER) (100–500 μm). At rest, the free Ca2+ concentration in the mitochondrial matrix (cmito) is close to the resting ccyt, but it rises to 20–30 μm during stimulation, e.g. in motor nerve terminals in Drosophila melanogaster (2). Cell activation in a wide range of cell types results in Ca2+ oscillations and in transient waves of increased ccyt (3–6). These oscillations (or waves) are not restricted to ccyt, but also cnucl (7), cER (8), and cmito show Ca2+ oscillations (9). The spatial extent of the oscillatory Ca2+ signal is also important. (i) In astrocytes, the area of Ca2+ oscillations is sometimes restricted to only one protrusion regulating the release of gliotransmitters; i.e. different oscillatory frequencies can coexist at the same time within the same cell (10). (ii) In Xenopus laevis oocytes, regenerative spiral waves of release of free Ca2+ spread through the entire cell (11). (iii) Intercellular Ca2+ waves spreading via gap junctions occur in rat liver epithelial cells upon mechanical stimulation (12).
In cells maintained in vitro, serum starvation followed by readministration leads to intracellular Ca2+ signals, most often in the form of oscillations (13, 14). The precise mechanism(s) leading to these oscillations is poorly understood because serum contains a large number of known and as yet unidentified growth factors and mitogenic compounds, all potentially participating in this oscillatory activity (15). In Swiss 3T3 cells, serum-induced Ca2+ changes are essential but not sufficient to induce NF-κB activation and subsequent DNA synthesis (16). In some cell types, Ca2+ oscillations even persist in the absence of Ca2+ influx across the plasma membrane (3, 4), whereas in others, Ca2+ oscillations strictly depend on Ca2+ influx (5, 8). Mitochondria influence cytosolic Ca2+ oscillations in at least two ways. First, mitochondria produce ATP, which is required for SERCA and plasma membrane Ca2+-ATPase function, that results in Ca2+ extrusion and thus lowering of ccyt. Second, during ccyt oscillations, cmito also manifests oscillations, indicative of a role of mitochondria in shaping and/or modulating ccyt oscillations (9). Ca2+ uptake into the mitochondria is determined by both the large negative voltage (−150 to −180 mV) across the inner membrane that results from the proton pumping by the respiratory chain and the Ca2+ concentration gradient between the cytoplasm and matrix (17). The mitochondrial calcium uniporter (MCU) is the key player responsible for the uptake of Ca2+ by mitochondria (18). The MCU has a rather low Ca2+ affinity and operates over a micromolar range of cytosolic Ca2+.
To address these questions, we performed lanthanum (La3+) insulation experiments where both the Ca2+ influx and efflux across the plasma membrane are blocked (19). We hypothesized that under these experimental conditions mitochondria serving as a Ca2+ store/source might substitute for this function normally exerted by the extracellular space. Using a genetically encoded Ca2+ indicator targeted to the mitochondria, we managed to verify this assumption in vitro. In addition, we investigated the effects of the following compounds on ccyt oscillations and mitochondrial Ca2+ handling: the proton uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), the mitochondrial Na+/Ca2+ blocker CGP-37157, the mitochondrial MCU blocker Ru-360, and finally the “Ca2+-buffering” protein calretinin. Based on the experimental findings, we built a mathematical model for Ca2+ oscillations taking into account the various processes implicated in these oscillations.
Materials and Methods
Reagents
Thapsigargin, LaCl3, and EGTA were purchased from Sigma-Aldrich. CGP-37157 and BAPTA-AM were obtained from Tocris Bioscience (Bristol, UK). Ru-360 was purchased from Calbiochem, and Rhodamine 123 was from Invitrogen. EGTA-AM and tetramethylrhodamine methyl ester were purchased from AAT Bioquest (Sunnyvale, CA). CGP-37157 was dissolved in pure ethanol as 100 mm stock solutions. Thapsigargin and Rhodamine 123 were dissolved as 100 mm stock solutions in DMSO. BAPTA-AM and EGTA-AM were dissolved as 30 mm stock solutions in DMSO. LaCl3 was dissolved in double distilled water. The final concentrations of the solvents were <0.1% in all experimental solutions. At these concentrations, the solvents did not modify the evoked Ca2+ signals in control experiments (data not shown).
Isolation of Primary Mouse Mesothelial Cells
Primary mouse mesothelial cells (prMC) were isolated from 4–6-month-old C57Bl/6J mice according to an established protocol (20) and as applied in our previous study (8). The pelleted cells enriched in mouse mesothelial cells were grown in DMEM/F-12 GlutaMAX medium supplemented with 15% FCS; 0.4 μg/ml hydrocortisone; 10 ng/ml epidermal growth factor; 1% insulin, transferrin, and selenium; 1 mm sodium pyruvate; 0.1 mm β-mercaptoethanol; 1% non-essential amino acids; 1% penicillin-streptomycin; and 2% Mycokill (PAA Laboratories, Pasching, Austria) (21). After a few days (>4 days in vitro), cultured cells showed the typical cobblestone-like morphology of mesothelial cells, and cell cultures maintained for ∼60 days in vitro were used for the measurements.
Plasmids and Lentiviral Infection
For the generation of cell lines stably expressing the Ca2+ indicator proteins GCaMP3 (Addgene plasmid 22692 (22)) and mito-CAR-GECO1 (Addgene plasmid 46022 (23)), the lentiviral expression vector pLVTHM (Addgene plasmid 12247 (24)) was used. The GFP cassette in pLVTHM was replaced with cDNAs coding for the respective Ca2+ indicator proteins. Briefly, pGCaMP3 was produced in SCS110 dam− bacteria and digested with AfeI and XbaI, and the fragment was inserted into the PmeI and SpeI sites of the backbone of pLVTHM to produce the final plasmid pLV-GCaMP3. The expression plasmid CMV-mito-CAR-GECO1 was used as template for the production of a DNA fragment coding for mito-CAR-GECO1. The required DNA fragment was synthesized by PCR using the following primers pairs: 5′-TAG CGT TTA AAC GGG CCC TC-3′ and 5′-GAG AAC TAG TTT ACT TCG CTG TCA TCA TTT GTA C-3′. The amplicon was digested with PmeI and SpeI and inserted into the unique sites of the pLVTHM vector to produce the final pLV-mito-CAR-GECO1 plasmid. Calretinin overexpression was achieved by the help of a lentiviral system. Briefly, the GFP cassette in pLVTHM was replaced with the human CALB2 cDNA coding for full-length calretinin using the previously described expression plasmid RSV-CALB2-neo (25) as template. The DNA fragment coding for full-length calretinin was synthesized by PCR using the primers PmeI-CALB2 (5′-AGT CGT TTA AAC ATG GCT GGC CCG CAG CAG CAG-3′) and SpeI-CALB2 (AGT CAC TAG TTT ACA TGG GGG GCT CGC TGC A-3′). The amplicon was digested with PmeI and SpeI and inserted into the unique sites of the pLVTHM vector to produce the final pLV-CALB2 plasmid. We also generated a lentivirus expressing calretinin (CR) tagged with the enhanced blue fluorescent protein (EBFP) allowing for the easy identification of cells overexpressing EBFP-CR. For this, the pLV-EBFP2-nuc plasmid (Addgene plasmid 36085) and CMV-CALB2-neo were used. The DNA fragment coding for full-length calretinin was synthesized by PCR using the primers XhoI-CALB2 (5′-GAG ACT CGA GTA GCT GGC CCG CAG CAG C-5′) XbaI-CALB2 (5′-GAG ATC TAG ATT ACA TGG GGG GCT CGC TGC A-3′). The amplicon was digested with XhoI and XbaI and inserted into the unique sites of the pLV-EBFP2-nuc vector to produce the final pLV-EBFP2-CR plasmid. As a control plasmid coding for EBFP only (pLV-EBFP2-X), the nuclear localization signal was removed in the plasmid pLV-EBFP2-nuc by deleting an XhoI fragment. All lentiviral plasmids were verified by restriction enzyme digestion and sequencing. Lentivirus was produced by the calcium phosphate transfection method using HEK293T cells and three plasmids: one of the expression plasmids (e.g. pLV-GCaMP3 or pLV-mito-CAR-GECO1), the envelope plasmid (pMD2G-VSVG, Addgene plasmid 12259), and the packaging plasmid (psPAX2, Addgene plasmid 12260). Virus-containing supernatants were collected after 48 and 72 h, filtered, aliquoted, and frozen at −80 °C (26). MCU expression was knocked down in prMC expressing GCaMP3 and mito-CAR-GECO1 using Mission lentiviral transduction particles (Sigma-Aldrich) TRCN0000267404 and TRCN0000265169. Mission transduction particles directed toward human parvalbumin (TRCN000056549) and non-infected cells served as controls. Infected cells were selected using 2 μg/ml puromycin dihydrochloride (Sigma-Aldrich) for 1 week. MCU transcript knockdown was verified by qRT-PCR analysis.
qRT-PCR
PrMC were seeded in 6-well plates, and RNA was extracted with 1 ml of PeqGold Trifast (PeqLab, Erlangen, Germany). cDNA synthesis (QuantiTect Reverse Transcription kit, Qiagen, Hombrechtikon, Switzerland) and qRT-PCR (Rotor-Gene SYBR Green PCR kit, Qiagen) were performed following the manufacturers' protocols. Primers were as follows: mUBC: forward, 5′-GGA CGC CAC CGT GAA ACA ACT C-3′; reverse, 5′-ACC TCC AGG GTG ATG GTC TTA CCA-3′; mMCU: forward, 5′-CTC ACC AGA TGG CGT TCG AGT CG-3′; reverse, 5′-GCG TCG CTG CAT CTT CAT GGC T-3′.
Calcium Imaging
prMC were isolated as described before (27) and grown on collagen-coated glass bottom 35-mm dishes (MatTek Corp., Ashland, MA). The buffer solution (Hepes-buffered saline) used for Ca2+ imaging experiments contained 120 mm NaCl; 5.4 mm KCl, 0.8 mm Mg2Cl, 20 mm Hepes, 1 mm CaCl2, and 10 mm glucose, pH 7.4 (adjusted by NaOH). In the low Ca2+ solution, CaCl2 was replaced with an equimolar concentration of NaCl. The drugs (thapsigargin, FCS, and EGTA) were added to the solutions and remained in the solution until the end of the experiments. In some experiments, cells were pretreated either with 250 μm CGP-37157 or with 10 μm Ru-360 for 30 min at 37 °C. Cells were loaded either with 30 μm BAPTA-AM or 30 μm EGTA-AM for 15 min at 37 °C. We used a DMI6000 inverted confocal microscope integrated to a Leica TCS-SP5 work station to examine fluorescence signals indirectly, reporting ccyt or cmito. The following excitation wavelengths were used to illuminate the fluorophores: 488 nm for GCaMP3 and 561 nm for mito-CAR-GECO1. Fluorescence emissions were recorded with a 20× objective and bandpass filters of 505–550 nm for GCaMP3 and 584–683 nm for mito-CAR-GECO1. Fluorescence images for ccyt or cmito measurements were collected every 3 s. Circle-shaped regions of interest (ROIs) were placed inside the cytoplasmic area of cells. The fluorescence values were calculated after background subtraction (fluorescence intensity of regions without cells). Fluorescence intensity values were normalized in each experiment to the averaged basal value preceding the treatment period. A bleaching correction was carried out when the baseline was not stable. LAS-AF (Leica) and Prism5 (GraphPad Software, Inc., San Diego, CA) software were used for data analysis.
ATP Measurements
PrMC were starved in serum-free DMEM supplemented with 1% penicillin-streptomycin for 24 h and distributed into 15 centrifuge tubes (50,000 cells/tube) in 50 μl of Hepes-buffered saline (+Ca2+). 1% FCS was added to 11 tubes, and 6 min later, 100 nm CCCP was added to five tubes. During the experiment, lysis buffer was added into each tube one after another with a delay of 1 min. ATP levels were determined using the ATP bioluminescence assay kit HS II according to the manufacturer's protocol (Roche Applied Science) with a microplate luminometer (PerkinElmer Life Sciences).
Mitochondrial Membrane Potential (ΔΨ) Measurements
Mito-CAR-GECO1-expressing prMC were seeded on glass bottom Petri dishes and incubated with 10 μm Rhodamine 123 for 20 min at room temperature. Cells were washed three times with Hepes-buffered saline (+Ca2+). During the recording using the confocal microscope, a 488-nm excitation wavelength was used to illuminate Rhodamine 123. Fluorescence emissions were recorded with a 20× objective and bandpass filters of 505–550 nm for Rhodamine 123. The distribution of Rhodamine 123 between the mitochondrial matrix and cytosol is proportional to the mitochondrial membrane potential. As the mitochondrial network is distributed within the entire cytoplasmic space, circle-shaped ROIs were randomly assigned to the cytoplasmic region for the fluorescence intensity measurements. The signal intensity is proportional to the amount of Rhodamine 123 dye incorporated by mitochondria in this ROI. For the normalization and thus the measurement of Rhodamine 123 released by mitochondria, an ROI within the nuclear region not containing mitochondria was selected, and the fluorescence intensity in this ROI was determined. The relative (rel.) ΔΨ was calculated according to the following equation.
 |
where Fmito and Fnucl are the fluorescence intensity of Rhodamine 123 in the mitochondrial and nuclear regions, respectively. The mitochondrial membrane potential was additionally measured with tetramethylrhodamine methyl ester. For these measurements, cells were preincubated with 50 nm tetramethylrhodamine methyl ester for 30 min.
Estimation of the Intracellular Calretinin Concentration by Western Blot Analysis
Protein samples were isolated from cultured prMC. Cells were grown in 25-cm2 flasks and harvested at near confluence. Total proteins were extracted with ice-cold radioimmune precipitation assay buffer. Serial dilutions of protein extracts (50, 5, 0.5, and 0.005 μg) from each cell culture sample as well as 40 ng of purified human recombinant calretinin were loaded onto SDS-polyacrylamide gels (12.5%). After separation, proteins were transferred onto nitrocellulose membranes (Bio-Rad) and incubated overnight at 4 °C with the calretinin-specific antibody CR7699/4 (Swant, Marly, Switzerland) at a dilution of 1:10,000. Rabbit secondary antibody linked to horseradish peroxidase (Sigma-Aldrich) was diluted at 1:10,000, and membranes were incubated for 4 h. For the detection, the chemiluminescent reagent Luminata Classico Forte (EMD Millipore Corp., Billerica, MA) was used. Chemiluminescent and normal illumination digital images were recorded on a system from Cell Biosciences (Santa Clara, CA). Area densities of calretinin bands were measured with ImageJ software. From the density curves, the cell protein concentration corresponding to 40 ng of calretinin was determined. This allowed determination of calretinin or more precisely that of the fusion protein EBFP-calretinin content in μg/mg of total protein. Based on previous estimation of a protein concentration of about 0.2 g/ml (28) in mammalian cells, the intracellular concentration of EBFP-calretinin was estimated.
Frequency Determination and Amplitude Scan
Computerized peak recognition for frequency and amplitude analyses was realized via the Microsoft Excel 2010 environment as described before (8); normalized recordings from >30 oscillating prMC were evaluated. The oscillation frequency as well as the average amplitude was determined for three time windows: 1–5, 5–9, and 9–13 min after serum administration.
Mathematical Simulation
To build the mathematical model, we considered four compartments: the extracellular space, cytoplasm, mitochondrial matrix, and ER lumen (Fig. 1). A fifth element placed within the cytoplasm in some simulations was the presence of a Ca2+ buffer. Membrane junctions between the ER and the plasma membrane ensured that the functional unit components (Ca2+ channels and pumps) are concentrated spatially in a very small space (29). Similarly close contacts were also assumed to exist between mitochondria and ER (30). One oscillatory unit represents an inositol trisphosphate receptor (InsP3R) cluster and its surrounding. We presumed that changes in ccyt, cER, and cmito of the entire cell were similar to that of individual units, i.e. spatially homogenous. In our view, this simplification is acceptable because the oscillations are slow and the cell size is small. In this case, the spatial diffusion of Ca2+ rapidly equilibrates the putative spatial differences and thus synchronizes the functions of individual functional units (31). In a cell with a 10-μm diameter, the diffusion is estimated to equilibrate spatial heterogeneity in ccyt in less than 0.1 s (32). However, because Ca2+ waves not only depend on Ca2+ diffusion but also on the action of Ca2+ pumps and channels, the Ca2+ wave is ∼10 times slower (33). Although our model is a minimal deterministic point model and cannot produce the stochastic and spatial phenomena of the Ca2+ oscillations, it is a useful tool to illuminate the observed characteristics of the mitochondrial Ca2+ handling. Our aim was to build the most simple model still able to produce the experimentally observed phenomena.
FIGURE 1.

Schematic model of cellular compartments and Ca2+ toolkit components implicated in Ca2+ oscillations in prMC. The plasma membrane contains components responsible for Ca2+ influx (JIN) and efflux (JEFF). The dominant intracellular Ca2+ release (JEREFF) and uptake (JSERCA) systems are localized in ER membranes. A small constant leak (JERLEAK) occurs independently of Ca2+ channels. The functions of the mitochondrial Ca2+ exchangers (Na+/Ca2+ exchanger and H+/Ca2+ exchanger) (JMEXC) and the mitochondrial calcium uniporter (JMCU) are responsible for the Ca2+ transport across the mitochondrial inner membrane; intracellular Ca2+ buffers (CaBP) such as calretinin acting as a transient cytosolic Ca2+ store/source modulate temporal aspects of ccyt and consequently affect Ca2+ fluxes across all membranes (plasma membrane, ER, and mitochondria (mito.)).
Ca2+ transports across the plasma membrane were defined as JIN and JEFF, and the transports across the ER membrane were termed JSERCA and JEREFF, respectively. JIN includes Ca2+ channels in the plasma membrane, e.g. voltage-gated Ca2+ channels, transient receptor potential channels, store-operated channels, P2X purinoreceptors, hyperpolarization-activated cyclic nucleotide-gated channels, etc. The JEFF represents the pumps involved in Ca2+ extrusion, plasma membrane Ca2+-ATPases and Na+/Ca2+ exchangers. The SERCA pumps transport Ca2+ from the cytoplasm to the ER, whereas the JEREFF represents the ER channels involved in emptying the ER, ryanodine receptor and InsP3R. The function of the mitochondrial exchangers (JMEXC) and the mitochondrial calcium uniporter (JMCU) are responsible for the Ca2+ transport across the mitochondrial inner membrane (see Fig. 1).
We denote by ccyt the Ca2+ concentration (in nm) in the cytosol and by cER that in the lumen of the ER. Mitochondrial matrix free concentration is denoted by cmito. The equations for the model are as follows.
 |
 |
 |
where JIN is the flux of Ca2+ ions entering the cell, JEFF is the Ca2+ flux pumped out of the cell, JSERCA denotes the Ca2+ flux pumped from the cytosol to ER, JEREFF is the flux of Ca2+ passing from the ER to the cytosol, JMCU denotes the function of MCU, JMEXC displays the function of mitochondrial Ca2+ exchangers (mitochondrial Na+/Ca2+ and H+/Ca2+ exchangers), and finally JERLEAK represents a small flux of Ca2+ diffusing from the ER to the cytosol (all values in nm/s).
The constant γ is the ratio between the changes in ccyt and cER caused by the same quantity of Ca2+ ions transported through the ER membrane. This value is derived from the difference in the effective volume of the ER lumen and the cytoplasm and from the different fraction of free and protein-bound Ca2+ in these compartments (34). The value of the γ parameter was estimated experimentally (8).
The quantity of Ca2+ pumped out of the cell through the plasma membrane increases as a function of the Ca2+ concentration in the cytosol. The Na+/Ca2+ exchangers have low Ca2+ affinity but high capacity for Ca2+ transport, whereas the plasma membrane Ca2+-ATPases have a high Ca2+ affinity but a low transport capacity. Although the individual components of extrusion systems are usually modeled by Hill equations (35), the overall flux can be simulated by a simple linear equation (36) based on the experimental results of Herrington et al. (37).
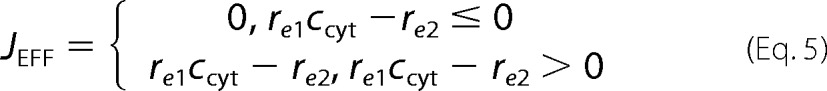 |
where re1 and re2 are two positive constants.
SERCAs pump the Ca2+ ions from the cytosol to the ER. The quantity of the transported Ca2+ ions depends on ccyt levels. We assume a linear relationship because the ER influx is also composed of different SERCA pumps with different Kd values (38). Nevertheless, our model can also work when JEFF and/or JSERCA is simulated with the conventional Hill equations.
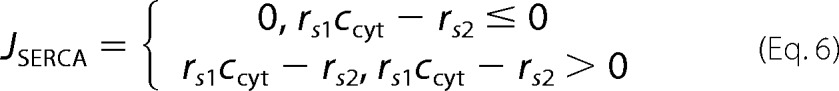 |
where rs1 and rs2 are two positive constants.
Ca2+ ions are released from the ER to the cytosol through InsP3R and ryanodine receptor. Because we found experimentally that ryanodine receptor does not play a role in serum-induced oscillations in mesothelial cells (8) similarly to other non-excitable cells (39), we focused on InsP3R. In our model, InsP3R is influenced both by ccyt and by cER but without an allosteric regulation between the two. InsP3R has Ca2+ binding sites not only on the cytoplasmic side but also on the luminal side (40). Experimental data show that an increase in inositol trisphosphate (InsP3) concentration causes a significant Ca2+ release from the ER in the absence of cytosolic Ca2+ (ccyt = 0) (41). Moreover, the effects of luminal Ca2+ do not affect the cytosolic binding sites (42, 43). Therefore we modeled InsP3R function as the sum of two individual contributions.
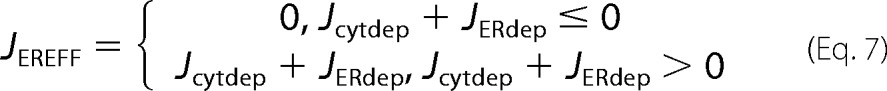 |
where
 |
and
with positive constants σ and ri1.
We introduced the dependence of InsP3R on the [InsP3], which has an influence both on Jcytdep and on JERdep. According to the experimental data from several studies (44–46), elevating cIP3 mainly changes the mean and the maximum (μ and ri,max) of the bell-shaped curve of ccyt dependence. Nevertheless, based on the experimental data presented (47, 48), elevating cIP3 also has an effect on the loading of the ER. Increased cIP3 reduces the amount of the stored Ca2+ ions. We simulated this effect by changing the ri2 parameter.
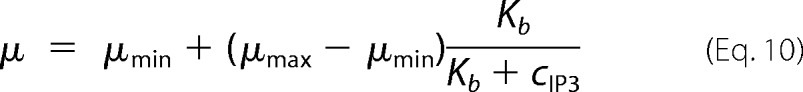 |
 |
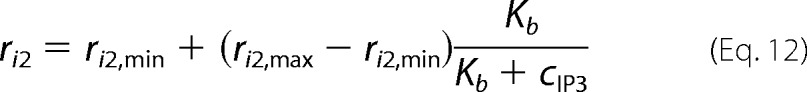 |
where Kb is the half-saturation constant of InsP3R for InsP3 and cIP3 represents the InsP3R sensitivity to the inositol trisphosphate molecule, which was taken equal to inositol trisphosphate concentration in μm. μmax, μmin, rim,min, rim,max, ri2,min, and ri2,max are positive constants. The parameter JERLEAK accounts for a Ca2+ flux from the ER to the cytoplasm independently of known Ca2+ channels, and this parameter is assumed to represent a small constant value (49).
The outer membrane of mitochondria is freely permeable for Ca2+ ions, but the inner mitochondrial membrane provides a barrier. The constant ρ is the ratio between the changes in ccyt and cER caused by the same quantity of Ca2+ ions transported through the mitochondrial inner membrane. This value is derived from the difference in the effective volume of the mitochondrial matrix and the cytoplasm and from the different fraction of free and protein-bound Ca2+ in these compartments.
There is a fast Ca2+ influx into the mitochondria matrix if ccyt reaches a certain value. This fast influx is attributable to the function of MCU. We used Hill equations with a very high Hill coefficient as was done in the work of Marhl et al. (50). For simplicity, we did not take into account the changes in mitochondrial transmembrane potential and in mitochondrial volume during the Ca2+ oscillations in line with Marhl et al. (50), but we have to consider it during a protonophore treatment. The passage of calcium ions through the MCU requires the large membrane potential difference generated by the action of the electron transport chain (51).
 |
where rΨ and vMCU,max are positive constants, Kd,MCU is the dissociation constant of MCU for Ca2+ ions, and H is the Hill coefficient. In our model, JMCU has a constant basal activity. That ensures that mitochondria can store a small amount of Ca2+ ions, which are released into the cytoplasm immediately after the collapse of the mitochondrial membrane potential. Higher ΔΨ means increased Ca2+ uptake but slower mitochondrial Ca2+ release.
To simulate the function of mitochondrial exchangers (Na+/Ca2+ and H+/Ca2+ exchangers), we consider that both will transport Ca2+ ions with a low velocity when there is a concentration gradient between the two sides of the mitochondrial inner membrane. For the simplicity, we neglected the changes in sodium and proton concentrations during the Ca2+ oscillations. Depending on the calcium concentration gradient, the exchangers can work in both directions.
 |
where rm1 and rm2 are positive constants.
The Ca2+ influx across the plasma membrane is composed of passive leakage and the agonist-activated fluxes: the capacitive (store-operated channel-dependent) and the non-capacitive (arachidonate- or diacylglycerol-regulated) Ca2+ influx (52). We simulated the changes in JIN starting from the beginning of the administration of serum (t1) using the following equations.
 |
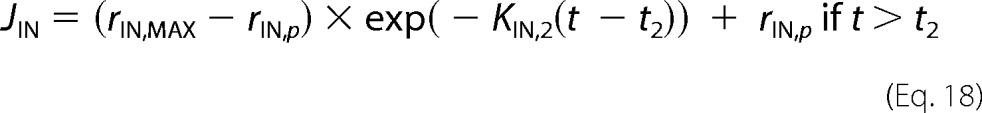 |
We simulated the changes in cIP3 from the beginning of the administration of serum (t1) with the following equations. The resting cIP3 was set to 15 nm (53).
 |
To simulate the effect of calretinin, we neglected its fast kinetics. Because this protein is considered as a fast Ca2+ buffer (54), calretinin reaches the Ca2+ steady state in a few milliseconds, which is much faster than our observed Ca2+ changes lasting for a few seconds. The fast kinetics of calretinin plays an important role at the mouth of voltage-gated Ca2+ channels in excitable cells (55) where fast and large changes in Ca2+ concentrations are expected.
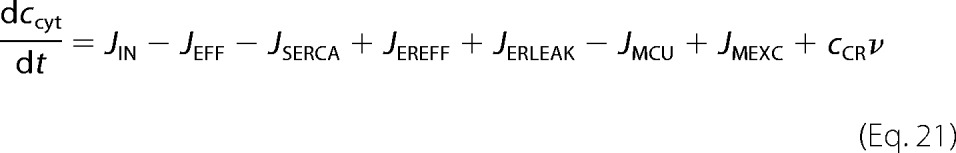 |
where cCR is the concentration of calretinin in the cytoplasm and ν is the average number of the Ca2+ binding sites of calretinin occupied by Ca2+. Calretinin has four high affinity Ca2+ binding sites and one low affinity binding site. The binding kinetics of the Ca2+ binding sites were simulated with Hill equations.
 |
where Kd1 is the dissociation constant for the high affinity Ca2+ binding sites, Kd2 is the dissociation constant for low affinity Ca2+ binding site, and h is the Hill coefficient for the high affinity binding sites. Among the high affinity Ca2+ binding sites, there is a positive cooperativity (h > 1). The values for the parameters came from the study of Faas et al. (54).
The values of each parameter are listed in Table 1. The initial values of parameters are derived either from our experiments in primary mesothelial cells or from fitting to experimental data previously reported in the above mentioned articles. The presented values are the result of the sequential fitting of the initial values to our in situ recordings. All computations of the model were implemented in the Microsoft Excel 2010 environment. The model system was discretized with a temporal resolution of 0.1 s (supplemental Excel document). There were no significant differences in the solution of the differential equations if we increased the temporal resolution (not shown). For visualization, Prism5 (GraphPad Software, Inc.) software was used.
TABLE 1.
Parameters used for the modeling
| Equation to determine | Parameter name | Value |
|---|---|---|
| γ | 450 | |
| ρ | 4 | |
| JEFF (Equation 5)a | re1 | 0.17/s |
| re2 | 18.8 nm/s | |
| JSERCA (Equation 6)a | rs1 | 0.27/s |
| rs2 | 22 nm/s | |
| JEREFF (Equations 8 and 9) | σ | 0.14142 nm |
| ri1 | 1300/s | |
| μ (Equation 10) | μmin | 2.4 nm |
| μmax | 2.18 nm | |
| Kb | 2 μm | |
| ri,max (Equation 11) | rim,min | 821.3 nm/s |
| rim,max | 24.3 nm/s | |
| Kb | 2 μm | |
| ri2 (Equation 12) | ri2,min | 6352 nm/s |
| ri2,max | 7042 nm/s | |
| Kb | 2 μm | |
| JERLEAK (Equation 13) | β | 2.5 nm/s |
| cER (initial) | 260 μm | |
| ccyt (initial) | 110 nm | |
| ΔΨ (initial) | 180 mV | |
| JMCU (Equation 14) | rΨ | 0.005555/mV |
| vMCU,MAX | 5 nm/s | |
| Kd,MCU | 208 nm | |
| H | 7.4 | |
| JMEXC (Equation 15) | rm1 | −0.01251/mV |
| rm2 | 2.295 | |
| ν (Equation 22) | Kd1 | 2.5 μm |
| Kd2 | 53 μm | |
| h | 2.4 | |
| JIN (Equations 16, 17, and 18) | t1 | 60 s |
| t2 | 72 s | |
| rIN,MAX | 5 nm/s | |
| KIN,1 | 0.01 s | |
| rIN,p | 1.05 nm/s | |
| KIN,2 | 2 s | |
| cIP3 (Equations 19 and 20) | cIP3,MAX | 1.8 μm |
| KIP3 | 0.1 μm |
a Alternatively, the JEFF and JSERCA can be simulated conventionally with a Hill equation with the following parameters: Vmax, 260 and 170 nm/s; Kd, 460 and 480 nm; and Hill coefficients, 3.5 and 2.4, respectively.
Results
Characterizing Ca2+ Fluctuations in Mitochondria of Primary Mouse Mesothelial Cells
In the absence of serum, prMC did not show Ca2+ oscillations as reported before (8). However, in a small fraction of cells (2–3%), isolated arrhythmic mitochondrial increases in cmito were present without detectable changes in ccyt (Fig. 2A). The addition of 1% FCS to the cell culture medium containing prMC that were grown in the absence of serum for 24 h resulted in a sudden rise of ccyt lasting, on average, for ∼40 s followed by Ca2+ oscillations (Fig. 2, B–D). The percentage of prMC responding to serum readministration with Ca2+ oscillations was in the order of 70%. Non-oscillatory cells showed only an initial single Ca2+ transient or a so-called peak-plateau response (53). A wide range of different oscillatory patterns in ccyt was present in a supposedly homogenous population of mesothelial cells. Most cells displayed long period (>10 min) baseline spiking oscillations with various frequencies of one spike per 3 min (Fig. 2B) up to 10 per min (Fig. 2D); also maximal spike amplitudes varied between individual cells. The baseline spiking oscillations represent discrete Ca2+ transients starting from a constant basal ccyt level (Fig. 2, B and C). Sinusoidal oscillation is a term for a continuous fluctuation in ccyt starting from a ccyt value that is higher than the resting (basal) ccyt (Fig. 2D). Most probably, sinusoidal oscillations are the result of high frequency overlapping baseline spiking oscillations (56). In prMC maintained in cell culture medium for longer periods (>10 passages), the percentage of the cells showing sinusoidal oscillations was increased as exemplified in Fig. 2D. However, the percentage of cells showing oscillatory activity was rather low at higher passages (∼20–40% at passages >10). The average frequency of the baseline spiking oscillations was found to be 15, 13, and 13 mHz in the following time segments: 1–5, 5–9, and 9–13 min after serum administration, respectively. The average amplitude of spikes (-fold increase in GCaMP3 fluorescence intensity) was found to be 2.66, 2.53, and 2.53 in the above mentioned time segments. By using two Ca2+ indicators targeted to either the mitochondrial matrix (mito-CAR-GECO1) or the cytoplasm (GCaMP3), we simultaneously monitored changes in cmito and ccyt, respectively. The initial serum-induced rise in ccyt was paralleled by a rapid rise in cmito that reached the peak value within 30 s after the addition of 1% FCS (Fig. 2, B–D); from then on, cmito decreased continuously until it reached its initial basal value, generally within the time span of 15 min monitored in most experiments. High frequency oscillations in ccyt resulted in continuous elevation in cmito (Fig. 2D). The rate of decay in cmito was also rather variable between cells, and oscillations in ccyt did not stop when cmito had reached its basal levels. In some cells during the decreasing phase in cmito, small fluctuations (short rises in cmito) coincided with the ccyt spikes but with a small delay (e.g. shown in Fig. 2B, inset). For the modeling, we took into account our previous results where basal and maximal ccyt values during Ca2+ spikes in prMC were found to be 100 and 200–300 nm, respectively (8). Similarly, the values for the resting cER were taken as 150–250 μm, and the values after serum readministration were taken as 100–150 μm (8). The pattern of cER changes is best described as a sawtooth wave (8). These data were incorporated to build the mathematical model where Ca2+ concentrations in all compartments (ccyt, cmito, and cER) were calculated and fitted to one ccyt recording (Fig. 2E). The model accurately recapitulated the experimental findings, in particular with respect to cmito, which had not been modeled in our previous study (8). The pattern in cmito is best described as a sudden rise after serum readministration followed by a rather smooth decay phase with small humps (increases in cmito) as the result of the oscillatory Ca2+ spikes.
FIGURE 2.

Ca2+ oscillations in ccyt, cER, and cmito in prMC. A, a representative recording shows spontaneous mitochondrial Ca2+ transients (red trace) in a resting cell without changes in ccyt (green trace). B–D, single cell fluorescence recordings derived from time lapse videos show the simultaneous changes in ccyt (green traces) and cmito (red traces) after serum readministration. Despite the different oscillatory frequencies (increasing from B to D) in ccyt, the traces for cmito were very similar in all three recordings: a fast initial rise followed by a quasiexponential decay with varying kinetics. The small inset in A shows (at an expanded time scale) that small fluctuations (short rises in cmito) coincide with ccyt spikes. The peak in cmito is slightly delayed in comparison with the peak in ccyt. E, an experimental recording (black dashed line) in prMC showing low frequency oscillations was selected for the fitting. The model shows the changes in ccyt (green trace), cmito (red trace), and cER (blue trace). The parameters and equations are presented in the supplemental Excel document. The changes in cmito consist of an initial rise after FCS administration followed by a slow return to basal levels. Each Ca2+ spike in ccyt results in a small hump in cmito reaching its relative maximum with a small delay compared with the maximum in ccyt. Original recordings for cER in prMC were previously presented (8); they show sawtooth-like waves at a semidepleted ER state during Ca2+ oscillations as shown in our model here (blue trace).
Modulation of Mitochondrial Ca2+ Transport Mechanisms Affects Ca2+ Oscillations
In the next series of experiments and modeling simulations, we investigated how altering mitochondrial function, mostly in relation to Ca2+ handling, affects ccyt oscillations. The simulation showed that inhibition of the mitochondrial Ca2+ release (JMEXC) or mitochondrial Ca2+ uptake (JMCU) during Ca2+ oscillations decreased the oscillation frequency (Fig. 3, A and B). The experimental verification of our predictions was hampered by the absence of pharmacological mitochondrially targeted compounds that immediately reach the mitochondrial inner membrane when added to the recording solution. Briefly, after serum readministration, the addition of CGP-37157 (50 μm), a nonspecific blocker of the mitochondrial Na+/Ca2+ exchanger, had no effect on the patterns of Ca2+ oscillations in either ccyt or cmito (data not shown). However, some cells pretreated with CGP-37157 for 30 min displayed stairlike increases in cmito (Fig. 3C), an effect predicted from our model (see Fig. 3A, right part (cmito)). Ruthenium compounds, e.g. ruthenium red and Ru-360, are potent and effective blockers of MCU in isolated mitochondria, but their usefulness for intact cells is limited by their poor membrane permeability and selectivity (57). Pretreatment of cells with 10 μm Ru-360 reduced the average oscillation frequency (approximately a 30% decrease during each time segment) and the initial mitochondrial Ca2+ uptake (Fig. 3D). However, it is currently still unclear whether in intact cells Ru-360 acts uniquely by the inhibition of the mitochondrial Ca2+ uptake or additionally by the inhibition of the extracellular Ca2+ influx. Therefore, we also used a molecular approach, i.e. down-regulation of MCU by shRNA to decrease the mitochondrial Ca2+ uptake. The down-regulation of MCU mRNA levels by 60–90% as determined by qRT-PCR resulted in a 40–60% decrease in the initial mitochondrial Ca2+ uptake (Fig. 3E). This led to an ∼20% reduction in the ccyt oscillation frequency calculated by frequency scan analysis (Fig. 3F) in line with the predictions from our model (Fig. 3B). In all time windows (bins of 3 s), the oscillation frequency was lower in prMC where MCU had been down-regulated. Thus, both approaches (Ru-360 and shMCU) underscore the importance of mitochondria in Ca2+ oscillations.
FIGURE 3.

Modulation of mitochondrial Ca2+ transport in prMC. A, in the simulation, blocking of the mitochondrial Na+/Ca2+ exchanger activity (JMEXC = 0) at 9 min predicts a staircase-like increase in cmito (red trace) following each Ca2+ spike, and the oscillation frequency is expected to decrease (black trace) compared with the unperturbed situation (green trace). B, in the simulation, MCU activity was stopped (JMCU = 0) at 9 min. This resulted in the absence of humps in cmito following the Ca2+ spikes and a decrease in oscillation frequency (compare green trace for ccyt (control conditions) with black trace) when JMCU = 0. C, a representative recording of cmito, obtained in prMC pretreated for 30 min with CGP-37157 (50 μm), which blocked mitochondrial Ca2+ release, is depicted. As predicted by the model (A), a staircase-like increase in cmito was observed after each cytosolic Ca2+ spike. D, reduction in mitochondrial Ca2+ uptake was observed in oscillatory prMC pretreated with 10 μm Ru-360; data are mean + S.D. from 10 cells each. Asterisks indicate significance at p < 0.05. E, reduction in mitochondrial Ca2+ uptake was observed in oscillatory prMC with down-regulated MCU; data are mean + S.D. from 20 cells each. Asterisks indicate significance at p < 0.05. F, frequency scan analyses of control (CTR) cells (green trace) and shMCU-treated (black trace) cells (n ≥ 50 oscillating cells for both conditions). In all time windows, the frequency was lower in cells where MCU had been down-regulated. Error bars represent S.D.
CCCP is an inhibitor of oxidative phosphorylation by acting as a protonophore; i.e. it allows H+ to cross the inner mitochondrial membrane, resulting in the collapse of ΔΨ. During Ca2+ oscillations, ΔΨ was slightly increased (more negative), but it collapsed immediately after CCCP (100 μm) treatment (Fig. 4A). The collapse of the membrane potential after addition of 10 or 100 μm CCCP was also confirmed by using the tetramethylrhodamine methyl ester indicator dye (data not shown). When applied during Ca2+ oscillations resulting from serum readministration, CCCP blocked Ca2+ oscillations at 100 μm but not at 10 μm (data not shown). An immediate drop in cmito was observed after CCCP treatment (Fig. 4B) followed by a continuous elevation in cmito in some cells (∼20%) but not in others (Fig. 4, A and C). In a few cases (∼5% of cells), administration of ATP (1 μm) partially reverted the CCCP-induced oscillation stop (Fig. 4C). However, addition of ATP to the recording solution in the absence of serum was equally able to evoke Ca2+ oscillations in some cells (data not shown). The reason for this effect is currently unknown; ATP might act on receptors on the surface of prMC but was also shown to cross the plasma membrane and to have an impact from the intracellular side (58). Application of CCCP before serum administration led to an immediate fall in the basal cmito, reaching a new plateau 1–2 min later; the fall in cmito was accompanied by a visible small rise in ccyt in ∼20% of cells, indicative of a release of mitochondrial Ca2+ to the cytosolic compartment. In addition, CCCP also decreased the basal level in ccyt (both at 100 and 10 μm), signifying that also the plasma membrane potential was affected. Serum readministration following CCCP (100 μm) treatment was still able to briefly elevate both ccyt and cmito, but the increase in cmito was smaller compared with cells not treated with CCCP; moreover, cmito returned quickly to the level reached after CCCP addition, i.e. not to basal levels before treatment (Fig. 4D). Serum readministration after treatment with the lower CCCP concentration (10 μm) evoked low amplitude Ca2+ oscillations, and the mitochondrial Ca2+ rise during a cytosolic Ca2+ spike was small, and cmito immediately returned to levels before serum administration but to lower levels than the basal cmito before CCCP administration (Fig. 4E). The model also correctly predicted that the collapse in ΔΨ (at t = 3 min) resulted in a lower cmito. Serum administration (modeled as increasing JIN and cIP3) led to an increase in ccyt and cmito followed by oscillations in ccyt and cmito (Fig. 4, compare F with the experimental recording shown in E). To provide more evidence for the presence of ΔΨ-independent mitochondrial Ca2+ uptake as shown in Fig. 4D, we induced Ca2+ release from the ER by thapsigargin after CCCP administration (Fig. 4G). We observed a rise not only as expected in ccyt but in parallel also in cmito, confirming the existence of a ΔΨ-independent mitochondrial Ca2+ uptake. Addition of 1% FCS also resulted in an increase in the intracellular ATP concentration that lasted during the entire period of Ca2+ oscillations. Shortly after the collapse of ΔΨ induced by CCCP, an immediate fall in ATP levels was observed (Fig. 4H). Overall, our findings indicate that the oscillation stop induced by the protonophore CCCP is not exclusively the result of the decreased mitochondrial Ca2+ uptake but also mediated via CCCP-induced changes in plasmalemmal Ca2+ influx and decreased ATP production.
FIGURE 4.

Effect of the proton uncoupler CCCP on Ca2+ oscillations. A, serum addition to prMC led to a rapid rise in cmito (red trace) followed by a gradual decay. Addition of CCCP (100 μm) resulted in a rapid collapse of the membrane potential as evidenced by measuring Rhodamine 123 fluorescence signals (green trace). As the result of CCCP application, cmito immediately returned to basal levels because the slightly elevated cmito level (compared with the ccyt level) could not be maintained when the driving force for mitochondrial Ca2+ uptake was eliminated. B–H, the role of the membrane potential and ATP treatment was investigated in greater detail, and one representative recording for each experimental condition is depicted. B, the addition of CCCP (100 μm) at t = 9 min blocked the serum-induced cytosolic Ca2+ oscillations. CCCP treatment led to a rapid fall in cmito; in some cells, a slow increase in cmito occurred afterward (B), whereas in others, cmito remained low (not shown). C, administration of ATP (1 μm) reestablished the CCCP-inhibited Ca2+ oscillations in some cells. D, addition of CCCP (100 μm) prior to serum readministration lowered the resting cmito (also reflected by the simultaneous small increase in ccyt). However, from then onward, ccyt dropped to levels lower then the basal ccyt before CCCP treatment. The serum readministration at t = 8 min evoked only a single Ca2+ transient. E, at a lower CCCP concentration (10 μm), addition of 1% FCS at t = 6 min resulted in elevations in ccyt and cmito followed by a few oscillations in ccyt. The small amount of Ca2+ ions taken up by mitochondria (red trace) during Ca2+ spikes (green trace) was released back to the cytosol almost immediately. F, in this simulation experiment, the mitochondrial membrane potential was switched off (ΔΨ = 0) at t = 3 min; this resulted in a slight increase in ccyt and a decrease in cmito to values lower than basal cmito as observed experimentally in D and E. Serum readministration evoked oscillatory activity in ccyt and cmito. G, after the CCCP-induced collapse in ΔΨ, ER Ca2+ release by thapsigargin (5 μm) resulted in mitochondrial Ca2+ uptake (red trace) independent of ΔΨ. A representative recording displays the simultaneous changes in cmito (red) and ccyt (green). H, during FCS-induced Ca2+ oscillations, ATP levels in prMC were increased but dropped quickly after CCCP administration. The panel shows the mean + S.D. of three independent experiments. Error bars represent S.D.
The Role of Ca2+ Influx on Ca2+ Oscillations and on Mitochondrial Ca2+ Handling
A decrease in extracellular [Ca2+] by the addition of 0.25 mm EGTA to the extracellular solution resulted in a reduction in the oscillation frequency (Fig. 5A). In this condition, i.e. when ccyt oscillations were not blocked completely, the amplitude of the Ca2+ signals was not affected, and the pattern of mitochondrial Ca2+ release/uptake was not affected (Fig. 5A). This could be accurately modeled in our simulation (Fig. 5B). When oscillations were induced by the addition of 1% FCS to the Ca2+-containing recording solutions ([Ca2+]o ≈ 1 mm), decreasing [Ca2+]o to <1 μm by the addition of 10 mm EGTA at t = 9 min resulted in an immediate stop of the oscillations, indicating the necessity of Ca2+ influx for the oscillations in ccyt (Fig. 5C). Removal of the extracellular Ca2+ had no visible effect on the decay curve of cmito, and basal levels were reached at the end of the observation period (15 min). When the serum readministration was carried out in the “Ca2+-free” condition, most prMC did not show any response in ccyt. In ∼5% of prMC, an initial small rise in ccyt was observed but without signs of Ca2+ oscillations in support of the hypothesis that extracellular Ca2+ is essential for the sustained oscillations (Fig. 5E). Interestingly, different results were obtained in the Ca2+-free condition when both Ca2+ influx and efflux across the plasma membrane were blocked by the addition of 1 mm La3+, the so-called lanthanum insulation (19, 59), prior to the serum readministration. After serum addition, an immediate rise in ccyt and cmito was detected; although ccyt decayed to basal levels within the next 2 min, cmito remained elevated and did not show the typical decay curve as seen e.g. in Fig. 5A, C, and E. Moreover, long lasting but slow oscillations in ccyt were observable (Fig. 5G), and at each cytosolic Ca2+ spike, a corresponding spike in cmito occurred. This indicates that during the La3+ insulation a considerable amount of Ca2+ ions released from the ER, leading to the transient increase in ccyt, is taken up by mitochondria as evidenced by the mitochondrial Ca2+ spikes (Fig. 5G). Thus, blocking the Ca2+ efflux across the plasma membrane leads to a shuttling of the Ca2+ ions between the ER and mitochondria, leading to these slow oscillations. Of note, the mitochondria remain in a rather Ca2+-loaded state because Ca2+ cannot be transported out of the cell. We estimate that the mitochondrial Ca2+ uptake and release velocities likely determine the frequency of Ca2+ oscillations. La3+-induced blocking of the Ca2+ transport across the plasma membrane at a time point when serum-induced Ca2+ oscillations were ongoing led to a complete block of the oscillations (Fig. 5, I and K). In some cases, La3+ treatment caused a final longer lasting Ca2+ spike (Fig. 5K), whereas in other prMC, La3+ completely blocked any further spikes (Fig. 5I). In all cases, the mathematical model could truthfully recapitulate the experimental findings by changing the parameters JIN and JEFF at different time points (Fig. 5, D, F, H, J, and L). Of note during La3+ insulations, the width (duration) of Ca2+ spikes was wider (longer) both in vitro and in silico. Moreover, the La3+-evoked oscillation block in the presence of extracellular Ca2+ ([Ca2+]o ≈ 1 mm) is, according to our model, mostly due to the decreased levels of Ca2+ ions present in the different cell compartments; i.e. the sum of ccyt + cER + cmito is smaller than the sum prior to agonist administration.
FIGURE 5.

Modulation of Ca2+ transport across the plasma membrane: representative experimental findings (left panels) and model simulations (right panels). A, a decrease in external [Ca2+] from 1 to ∼0.75 mm by administration of 0.25 mm EGTA results in a decreased oscillation frequency. B, the model was able to recapitulate the effect of reduction of Ca2+ influx on Ca2+ oscillations. The parameter rIN,p representing JIN was reduced from 0.85 to 0.6 nm/s at t = 9 min. C, removal of extracellular Ca2+ by the addition of EGTA (10 mm) at t = 9 min resulted in one final Ca2+ spike before cessation of oscillations. D, the model correctly predicted an immediate stop in Ca2+ oscillations when JIN was decreased to zero. E, in the absence of extracellular Ca2+ ([Ca2+]o < 1 μm), prMC did not show Ca2+ oscillations. In a few cells, a single Ca2+ transient was visible. F, model traces for ccyt (green) and cmito (red) in “zero Ca2+” (JIN = 0) confirmed the experimental findings shown in E. G, addition of Lanthanum chloride (La3+ insulation) prior to serum administration rendered Ca2+ oscillations independent of extracellular Ca2+ ions. Note that (i) cmito remained elevated during the entire period (no slow decay phase), (ii) the frequency of Ca2+ oscillations was lower than in control conditions (e.g. as shown in the initial period in A or C), and (iii) the half-width of Ca2+ transients representing the duration of a Ca2+ spike was increased. H, the model confirmed that mitochondria were able to substitute for the role of the extracellular Ca2+ reservoir during Ca2+ oscillations. In line with the experimental findings, the half-width of Ca2+ spikes was increased, and cmito remained elevated. I–K, La3+ insulation induced after serum administration (at t ≈ 8–10 min) blocked the Ca2+ oscillations either with (K) or without (I) a final large Ca2+ transient. J–L, the model was able to recapitulate both phenomena: it revealed that cTOTAL, i.e. the total amount of Ca2+ ions in the cell (cmito + ccyt + cER), determined the response to La3+ insulation.
Effect of the Intracellular Buffer Calretinin on Ca2+ Oscillations
Based on previous findings that human mesothelioma in vivo, mesothelioma cells in vitro, and reactive mesothelial cells express calretinin (60, 61), we hypothesized that prMC also might express this protein and that its presence might affect the Ca2+ oscillations. However, calretinin protein expression levels in prMC were found to be below the detection limit of our Western blot analysis (8), i.e. lower than ∼100 nm and thus unlikely to affect the Ca2+ oscillations as the result of the Ca2+-buffering capacity of calretinin. In support of this assumption, oscillation patterns (frequency, amplitude, and duration) in prMC from either wild type or calretinin knock-out (CR−/−) mice were indistinguishable (data not shown). However, to mimic the situation of calretinin-expressing reactive mesothelial cells and to investigate the putative role of calretinin in those cells, we overexpressed a fusion protein consisting of EBFP separated from full-length calretinin by a small linker peptide by infection of prMC with the appropriate lentivirus. We estimated in a semiquantitative way by Western blot analyses the expression levels of EBFP-calretinin. The expression level was found to be ∼75 pg of EBFP-calretinin/μg of total protein, leading to an estimated upper concentration of 250 μm calretinin. The EBFP tag on calretinin served as a marker for the distinction of the two populations with or without calretinin (Fig. 6A). The percentage of infected cells was usually higher than 90%. The fraction of prMC showing Ca2+ oscillations (∼10–20%) was considerably lower than in non-infected cells not expressing calretinin (∼60–70%) and moreover was restricted to cells showing faint blue fluorescence, i.e. low EBFP-calretinin expression levels. In the oscillating EBFP-calretinin-expressing prMC, the Ca2+ spike amplitudes were smaller, and the half-width of Ca2+ spikes (duration) was increased (Fig. 6B). The largest effect caused by EBFP-calretinin was the reduction of the amplitude of the first Ca2+ spike after serum readministration (Fig. 6C); on average it was half the size compared with the situation without calretinin. Likely as the consequence of the reduction in ccyt, the increase in cmito also was clearly diminished (Fig. 6D). The frequency of oscillation slightly decreased (∼10–20% reduction in each time segment). Our model simulations incorporating calretinin with the known Ca2+ binding characteristics (54) showed similar modifications: a decrease both in the amplitudes of ccyt spikes and in the amount of mitochondrial Ca2+ uptake (Fig. 6E). In our model, an increase in calretinin concentration resulted in an increase of the oscillation frequency, a prediction not supported by our experimental findings. One reason may be that calretinin, in addition to its buffering capacity, might act as a Ca2+ sensor in prMC. We had previously shown that calretinin is able to directly modify the activity of a Ca2+ channel (60), and direct targets for calretinin implicated in Ca2+ transportation might also be present in prMC. As a control to exclude that observed effects were mediated by the EBFP part of the fusion protein, prMC were infected with the lentivirus LV-EBFP2-X leading to the expression of EBFP only. No differences in the Ca2+ oscillations patterns were observed between cells expressing EBFP and non-infected control cells (data not shown). Based on the fact that Ca2+ oscillations in EBFP-calretinin-expressing cells were limited to those with faint fluorescence, we reasoned that the concentration in these cells was ∼10-fold (e.g. 25 μm) lower than the global concentration (250 μm) estimated from Western blot analyses. Thus, we tested whether the commonly used synthetic Ca2+ chelators BAPTA and EGTA, which have different properties (e.g. Kd, on-rate constant (kon), and diffusion coefficient (D)) than calretinin, were able to recapitulate the effects of calretinin. In prMC loaded with BAPTA-AM (30 μm), serum readministration evoked a slow and prolonged ccyt elevation paralleled by a minute increase in cmito (Fig. 6F). Most importantly, the initial rise in ccyt as also seen in EBFP-calretinin-expressing prMC (Fig. 6C) was completely abolished. In contrast, after EGTA-AM loading serum, readministration induced a short spike both in ccyt and cmito (Fig. 6E) followed by a rapid return to essentially baseline levels. No Ca2+ oscillations were observed in both cases. This further indicates that the properties of calretinin are clearly distinct from those of either BAPTA or EGTA.
FIGURE 6.

The effect of increased mobile cytosolic Ca2+-buffering capacity resulting from ectopic EBFP-calretinin expression on Ca2+ oscillations. A, fluorescence image of cultured prMC expressing the fusion protein EFBP-calretinin (blue). The faint green and red fluorescence signals represent the basal fluorescence intensities of GCaMP3 (cytosolic Ca2+ indicator) and mito-CAR-GECO1 (mitochondrial Ca2+ indicator), respectively. The faintly fluorescent non-blue cells are control cells not expressing EFBP-calretinin. The scale bar represents 250 μm. B, single cell fluorescence intensity recordings from time lapse videos show a representative Ca2+ response in ccyt and cmito of cells expressing EBFP-calretinin. Oscillations were only observed in cells with faint blue fluorescence, i.e. relatively low EBFP-calretinin expression levels. Note the relatively small signal in ccyt as compared with control cells (e.g. in Figs. 2C and 4B). C, Ca2+ oscillations were monitored in a mixed population of mito-CAR-GECO1/GCaMP3 prMC (green trace) and EBFP-calretinin/mito-CAR-GECO1/GCaMP3 prMC (black trace). Averages of ccyt values of 10 randomly selected oscillatory prMC each were plotted; data are mean + S.D. Serum-induced maximal amplitudes in ccyt were considerably lower in EBFP-calretinin-expressing prMC. D, a similar reduction was also seen for cmito in oscillatory prMC; data are mean + S.D. from eight cells each. E, in the simulation, the known Ca2+ binding properties of calretinin were incorporated. The addition of calretinin (0.5 μm) resulted in lower amplitudes of ccyt, and the mitochondrial uptake (reflected by cmito) was decreased in the presence of calretinin. F, BAPTA-AM preloading (30 μm for 15 min) resulted in a low amplitude plateau response in ccyt (green) and an even smaller one in cmito without Ca2+ oscillations. Also the initial serum-induced Ca2+ spike was completely abolished. G, in EGTA-AM preloaded (30 μm for 15 min) prMC, serum readministration evoked single Ca2+ spikes of short duration in ccyt (green) and cmito (red). Only a few cells (<1%) showed a second Ca2+ transient, but oscillations in ccyt were completely blocked by 30 μm EGTA. F and G, averages of ccyt and cmito values of 10 randomly selected prMC were plotted; data are mean + S.D. Asterisks indicate significance at p < 0.05. Error bars represent S.D. CTR, control.
Discussion
Characteristics of mitochondrial Ca2+ transport have not been examined in detail in most cell types. The main reason why we know relatively little about mitochondrial Ca2+ handling is because the molecular identity of the channels involved in mitochondrial transport have only recently been discovered (18, 62, 63), and specifically targeted, pH- and ΔΨ-insensitive Ca2+ indicators are only currently available (23). Nevertheless, there are few models for Ca2+ oscillations where the function of mitochondrial Ca2+ uptake has been taken into account (64).
Our experiments affirm previous data that mitochondria, even at the resting state, are able to transport and store Ca2+ ions (65). The fast release of the stored Ca2+ from the mitochondria due to the decrease/collapse of the membrane potential indicates that the strongly negative ΔΨ ensures a constant Ca2+ uptake into the mitochondria. This uptake is in a steady-state equilibrium with the constant Ca2+ efflux mediated by the mitochondrial exchangers (66), and the efflux is an electrogenic process (67). The electrochemical proton gradient across the inner mitochondrial is used to remove the excess Ca2+ ions (68). Our recordings show that this basal steady-state mitochondrial Ca2+ concentration can fluctuate, showing “spontaneous” mitochondrial Ca2+ spikes. Most probably this is mediated by an endogenous MCU activator that has not been identified at the molecular level yet. Ca2+ transients in ccyt were previously reported to evoke an increase in cmito, activating both cytoplasmic (19) and mitochondrial enzymes (2). Thus, Ca2+ transients observed selectively in cmito in some prMC (Fig. 2A) might allow for the autonomous activation of mitochondrial enzymes. The Ca2+ ions causing the mitochondrial spike are likely to originate from the cytosolic compartment; however, our results indicate that the amount of Ca2+ ions responsible for the increase in cmito was not sufficient to be detected as a decrease in ccyt. Alternatively, at basal conditions, the equilibrium level of ccyt might be regulated by a rather rapid constant exchange of Ca2+ ions among the cytosol, the extracellular space, and/or the ER compartment.
The Ca2+ oscillation models usually differ in how they simulate the functions of InsP3R, the channel that transports Ca2+ ions from the ER to the cytosol. The “ccyt/[InsP3]” models (for a review, see Ref. 69) postulate that the InsP3R has a binding site for InsP3, an activating binding site for Ca2+, and an inhibiting binding site for Ca2+. In these models, all binding sites are localized on the cytoplasmic side, and the function of InsP3 does not depend on cER. Binding of Ca2+ to the activating site and of InsP3 to the InsP3 binding site opens the channel, whereas Ca2+ binding to the inhibiting site closes the InP3R. Moreover, the binding of Ca2+ to the inhibiting site occurs rather slowly and with a lower affinity as compared with the activating site, subsequently resulting in oscillations in ccyt. In these models, the InsP3 concentration uniquely determines the oscillation frequency (70). In the “store loading” models (also called “ccyt/cER” models), the function of InsP3R depends not only on ccyt but also on cER. In these models, the Ca2+ influx across the plasma membrane plays a critical role in determining the oscillation frequency (8, 71, 72). At a constant [InsP3], the duration of the interspike period is determined by the velocity of cellular Ca2+ replenishment, which is manifested as a continuous ER loading together with a constant basal ccyt. The experimentally observable sawtooth wave oscillations in cER during the cytoplasmic baseline spiking oscillations are an important argument in favor of the store loading theory (8). However, the store loading-based models cannot cope with the fact that in some cells the Ca2+ oscillations do not depend on Ca2+ influx across the plasma membrane. Our experiments and modeling studies revealed that the incorporation of mitochondria as an additional Ca2+ source/store in the store loading-based models considerably augments the quality of the simulations. That is, the modeling predictions are more congruent with the experimental findings, which allows for a better mechanistic understanding. The mitochondrial Ca2+ transport enables the store loading-based models also to display Ca2+ oscillation in the absence of extracellular Ca2+.
The simulation of the La3+ insulation was previously endeavored by Sneyd et al. (73). Although their model does not contain mitochondria and moreover ccyt is continuously decreasing during the oscillations, their model reveals important aspects of the Ca2+ oscillations, namely their dependence on the total Ca2+ load of the cell. In their model, the cell has a high resting Ca2+; upon agonist stimulation, the activation of plasma membrane Ca2+-ATPases causes a net loss of Ca2+ from the cells even though the Ca2+ influx is augmented after stimulation (73). A similar phenomenon is also observed in our model; the total cellular Ca2+ content (ccyt + cER + cmito) determines the response to the La3+ insulation; blocking of the Ca2+ influx and efflux results in an oscillation stop that can either occur after a final Ca2+ spike or directly after La3+ addition, i.e. without a change in ccyt. In contrast to the previous model (73), basal ccyt levels during the interspike phase of the oscillations remain constant. This is in line with the experiments carried out by us and others (74).
Shuttling of Ca2+ ions between the ER and mitochondria was experimentally demonstrated in the study of Ishii et al. (9). They reported that in HeLa cells the cycles of ER/mitochondrion shuttling are repeated until cmito has reached the basal level prior to the stimulation. In our study with prMC, we observed Ca2+ oscillation even (i) when cmito had reached its basal levels or (ii) if cmito had been considerably lowered by CCCP administration. One has to keep in mind that CCCP also results in the collapse of the plasma membrane potential (75), which subsequently reduces the plasmalemmal Ca2+ influx (76). Thus, one reason for the CCCP-evoked stop in oscillations might be a disturbed Ca2+ influx. Moreover, the CCCP-mediated drop in ATP production likely leading to an impairment of the ER Ca2+ transport might also contribute to the oscillation arrest (77); i.e. the effects of protonophores are not exclusively attributed to the reduced mitochondrial Ca2+ uptake as was proposed in earlier studies (9). When CCCP was administered before serum, it caused a Ca2+ transient due to the mitochondrial release, which was followed by a period of lower resting ccyt. A lower ccyt is a sign of the reduced Ca2+ influx (resting plasmalemmal Ca2+ leakage). There was a similar decrease in resting ccyt when the extracellular free Ca2+ was chelated by EGTA (data not shown).
The Ca2+ influx across the plasma membrane is important to sustain the Ca2+ oscillations in prMC (78) but not in HeLa cells (9). The different dependence of these cell types on extracellular Ca2+ for the oscillations might be the result of differences in the contribution/importance of the various Ca2+ shuttling pathways between ER and mitochondria on the one hand and between ER and the extracellular space on the other. Our results indicate that plasmalemmal Ca2+ extrusion systems and mitochondrial Ca2+ uptake channels compete for the Ca2+ ions released from the ER. We hypothesize that in some cells, such as prMC and HEK cells (74), the shuttling between the extracellular space and the ER dominates over the shuttling between mitochondria and the ER. However, in HeLa cells and hepatocytes, the ER/mitochondrion shuttling prevails. This might explain why Ca2+ oscillations in some cells are strongly dependent on extracellular Ca2+ ions but not in others.
Another often neglected aspect about “Ca2+ shuttling” pathways is the contribution of cytosolic Ca2+ buffers present at rather high concentrations in the cytosol of some cell types. They are expected to modulate the Ca2+ shuttling among all compartments, extracellular space, ER, and mitochondria, as well as to transiently affect ccyt (Fig. 7). A strong interdependence between cytoplasmic Ca2+ buffers and mitochondria has been demonstrated before. The expression levels of parvalbumin, a Ca2+-buffering protein with slow binding kinetics, and the mitochondrial volume in fast twitch muscle cells and in parvalbumin-expressing neurons are inversely regulated (Ref. 79, and for more details, see Ref. 80). In our study, we observed that overexpression of calretinin modifies Ca2+ signals and associated oscillations. It reduces the amount of Ca2+ ions shuttling both between the ER and mitochondria and between the ER and the cytoplasm. Our model predicts that at calretinin concentrations >1 μm Ca2+ oscillations should be blocked in prMC. This is in apparent contradiction with the experimental results where oscillations still existed in EBFP-calretinin-expressing cells likely expressing levels higher than 1 μm (Fig. 6). However, in our modeling, the Ca2+ microdomain was not considered, and Ca2+ binding characteristics of calretinin (e.g. Kd and kon) might be different in the cytosol of prMC than the parameters determined in vitro (34). Furthermore, adaptation/compensation mechanisms might be induced in prMC overexpressing calretinin that would still allow for the generation of Ca2+ oscillations.
FIGURE 7.

Contribution of Ca2+ signaling toolkit components to serum-induced Ca2+ oscillations in prMC. A, in unperturbed (control) prMC in vitro, Ca2+ oscillations are primarily the result of the interplay between Ca2+ from the extracellular space and the ER with some minor contributions of mitochondria. The arrows indicate the shuttling of Ca2+ ions between the different compartments (the thicker the arrow, the more important is this pathway). In prMC, expression of calretinin is virtually absent, excluding an important role of this protein with respect to mobile Ca2+ buffering. B, if cells are subjected to La3+ insulation excluding the exchange of Ca2+ ions via the plasma membrane, the repetitive Ca2+ exchange between the ER and mitochondria allows for the generation of Ca2+ oscillations. C, the addition of the mobile Ca2+ buffer calretinin as observed in reactive mesothelial cells and mesothelioma cells affects Ca2+ oscillations. High expression levels (>1 μm in our model) completely block oscillations; lower levels (≈0.5 μm) reduce the amplitude of ccyt as well as of cmito during Ca2+ oscillations; i.e. calretinin competes with mitochondria, thus reducing the shuttling of Ca2+ ions between the ER and mitochondria.
Of relevance, calretinin reduced the mitochondrial Ca2+ uptake and Ca2+ accumulation. In human malignant mesothelioma, mostly of the epithelioid type, calretinin is overexpressed (81). This might cause changes, e.g. a delay or blocking of apoptotic/necrotic processes (78, 82). Thus, the increased calretinin expression in mesothelioma cells and moreover in certain colon cancer (83) and derived cell lines (84) might be correlated or causally linked to the increased resistance of these tumor cells to the apoptotic/necrotic signals either occurring in healthy physiological conditions or resulting from treatment with chemotherapy drugs such as oxaliplatin or 5-fluorouracil (85). In support, colon cancer cells resistant to aurora kinase inhibitors are characterized by higher calretinin expression levels (86). Moreover, down-regulation of calretinin by lentiviral infection induces apoptosis in mesothelioma cell lines in vitro via an intrinsic mitochondrion-mediated pathway (87). Also down-regulation of calretinin in colon cancer cells is associated with cell growth arrest and increased apoptosis (88).
Author Contributions
L. P. designed the study, performed the experiments with simulations, and wrote the paper. W. B. provided assistance, contributed to lentivirus production and cloning (CALB2), and performed qRT-PCR. B. S. secured funding, analyzed data, and wrote the paper.
Supplementary Material
Acknowledgments
We are grateful to Simone Eichenberger and Valérie Salicio for excellent technical assistance as well as to Michael Dougoud, Department of Mathematics, University of Fribourg, for verifying the mathematical equations. We thank Dr. D. Trono (École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) for providing pLVTHM (Addgene plasmid 12247), pMD2G-VSVG (Addgene plasmid 12259), and psPAX2 (Addgene plasmid 12260); Dr. L. Looger for GCaMP3 (Addgene plasmid 22692); and Dr. R. E. Campbell for mito-CAR-GECO1 (Addgene plasmid 46022). pLV-EBF2-nuc was a gift from Dr. P. Tsoulfas (Addgene plasmid 36085).
This work was partially supported by Swiss National Science Foundation Grants 130680 and 147697 (Sinergia) (to B. S.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains a supplemental Excel document showing an example of mathematical simulation.
- ER
- endoplasmic reticulum
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase
- ccyt
- cytosolic free Ca2+ concentration
- cnucl
- nuclear free Ca2+ concentration
- cER
- free Ca2+ concentration in the ER lumen
- cmito
- free Ca2+ concentration in the mitochondrial matrix
- MCU
- mitochondrial calcium uniporter
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- prMC
- primary mouse mesothelial cells
- EBFP
- enhanced blue fluorescent protein
- InsP3R
- inositol trisphosphate receptor
- InsP3
- inositol trisphosphate
- ΔΨ
- mitochondrial membrane potential
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- AM
- acetoxymethyl ester
- CR
- calretinin
- qRT-PCR
- quantitative RT-PCR
- ROI
- region of interest.
References
- 1.Berridge M. J., Bootman M. D., and Roderick H. L. (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 2.Ivannikov M. V., and Macleod G. T. (2013) Mitochondrial free Ca2+ levels and their effects on energy metabolism in Drosophila motor nerve terminals. Biophys. J. 104, 2353–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rooney T. A., Sass E. J., and Thomas A. P. (1989) Characterization of cytosolic calcium oscillations induced by phenylephrine and vasopressin in single fura-2-loaded hepatocytes. J. Biol. Chem. 264, 17131–17141 [PubMed] [Google Scholar]
- 4.Clapham D. E., Lechleiter J. D., and Girard S. (1993) Intracellular waves observed by confocal microscopy from Xenopus oocytes. Adv. Second Messenger Phosphoprotein Res. 28, 161–165 [PubMed] [Google Scholar]
- 5.Yule D. I., and Gallacher D. V. (1988) Oscillations of cytosolic calcium in single pancreatic acinar cells stimulated by acetylcholine. FEBS Lett. 239, 358–362 [DOI] [PubMed] [Google Scholar]
- 6.Martin S. C., and Shuttleworth T. J. (1994) Ca2+ influx drives agonist-activated [Ca2+]i oscillations in an exocrine cell. FEBS Lett. 352, 32–36 [DOI] [PubMed] [Google Scholar]
- 7.Estrada M., Uhlen P., and Ehrlich B. E. (2006) Ca2+ oscillations induced by testosterone enhance neurite outgrowth. J. Cell Sci. 119, 733–743 [DOI] [PubMed] [Google Scholar]
- 8.Pecze L., and Schwaller B. (2015) Characterization and modeling of Ca2+ oscillations in mouse primary mesothelial cells. Biochim. Biophys. Acta 1853, 632–645 [DOI] [PubMed] [Google Scholar]
- 9.Ishii K., Hirose K., and Iino M. (2006) Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep. 7, 390–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y. W., Tang X., Arizono M., Bannai H., Shih P. Y., Dembitskaya Y., Kazantsev V., Tanaka M., Itohara S., Mikoshiba K., and Semyanov A. (2014) Spatiotemporal calcium dynamics in single astrocytes and its modulation by neuronal activity. Cell Calcium 55, 119–129 [DOI] [PubMed] [Google Scholar]
- 11.Lechleiter J., Girard S., Peralta E., and Clapham D. (1991) Spiral calcium wave propagation and annihilation in Xenopus laevis oocytes. Science 252, 123–126 [DOI] [PubMed] [Google Scholar]
- 12.Frame M. K., and de Feijter A. W. (1997) Propagation of mechanically induced intercellular calcium waves via gap junctions and ATP receptors in rat liver epithelial cells. Exp. Cell Res. 230, 197–207 [DOI] [PubMed] [Google Scholar]
- 13.Foreman M. A., Smith J., and Publicover S. J. (2006) Characterisation of serum-induced intracellular Ca2+ oscillations in primary bone marrow stromal cells. J. Cell. Physiol. 206, 664–671 [DOI] [PubMed] [Google Scholar]
- 14.Wood A., Wing M. G., Benham C. D., and Compston D. A. (1993) Specific induction of intracellular calcium oscillations by complement membrane attack on oligodendroglia. J. Neurosci. 13, 3319–3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goustin A. S., Leof E. B., Shipley G. D., and Moses H. L. (1986) Growth factors and cancer. Cancer Res. 46, 1015–1029 [PubMed] [Google Scholar]
- 16.Sée V., Rajala N. K., Spiller D. G., and White M. R. (2004) Calcium-dependent regulation of the cell cycle via a novel MAPK-NF-κB pathway in Swiss 3T3 cells. J. Cell Biol. 166, 661–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rizzuto R., and Pozzan T. (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 86, 369–408 [DOI] [PubMed] [Google Scholar]
- 18.De Stefani D., Raffaello A., Teardo E., Szabò I., and Rizzuto R. (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Breemen C., Farinas B. R., Gerba P., and McNaughton E. D. (1972) Excitation-contraction coupling in rabbit aorta studied by the lanthanum method for measuring cellular calcium influx. Circ. Res. 30, 44–54 [DOI] [PubMed] [Google Scholar]
- 20.Bot J., Whitaker D., Vivian J., Lake R., Yao V., and McCauley R. (2003) Culturing mouse peritoneal mesothelial cells. Pathol. Res. Pract. 199, 341–344 [DOI] [PubMed] [Google Scholar]
- 21.Connell N. D., and Rheinwald J. G. (1983) Regulation of the cytoskeleton in mesothelial cells: reversible loss of keratin and increase in vimentin during rapid growth in culture. Cell 34, 245–253 [DOI] [PubMed] [Google Scholar]
- 22.Tian L., Hires S. A., Mao T., Huber D., Chiappe M. E., Chalasani S. H., Petreanu L., Akerboom J., McKinney S. A., Schreiter E. R., Bargmann C. I., Jayaraman V., Svoboda K., and Looger L. L. (2009) Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J., Liu L., Matsuda T., Zhao Y., Rebane A., Drobizhev M., Chang Y. F., Araki S., Arai Y., March K., Hughes T. E., Sagou K., Miyata T., Nagai T., Li W. H., and Campbell R. E. (2013) Improved orange and red Ca2+ indicators and photophysical considerations for optogenetic applications. ACS Chem. Neurosci. 4, 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiznerowicz M., and Trono D. (2003) Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 77, 8957–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D'Orlando C., Fellay B., Schwaller B., Salicio V., Bloc A., Gotzos V., and Celio M. R. (2001) Calretinin and calbindin D-28k delay the onset of cell death after excitotoxic stimulation in transfected P19 cells. Brain Res. 909, 145–158 [DOI] [PubMed] [Google Scholar]
- 26.Kutner R. H., Zhang X. Y., and Reiser J. (2009) Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 4, 495–505 [DOI] [PubMed] [Google Scholar]
- 27.Blum W., Pecze L., Felley-Bosco E., Worthmüller-Rodriguez J., Wu L., Vrugt B., de Perrot M., Schwaller B. (2015) Establishment of immortalized murine mesothelial cells and a novel mesothelioma cell line. In Vitro Cell. Dev. Biol. Anim. 51, 714–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milo R. (2013) What is the total number of protein molecules per cell volume? A call to rethink some published values. BioEssays 35, 1050–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manford A. G., Stefan C. J., Yuan H. L., Macgurn J. A., and Emr S. D. (2012) ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell 23, 1129–1140 [DOI] [PubMed] [Google Scholar]
- 30.Rizzuto R., Pinton P., Carrington W., Fay F. S., Fogarty K. E., Lifshitz L. M., Tuft R. A., and Pozzan T. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 [DOI] [PubMed] [Google Scholar]
- 31.Dupont G., Abou-Lovergne A., and Combettes L. (2008) Stochastic aspects of oscillatory Ca2+ dynamics in hepatocytes. Biophys. J. 95, 2193–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.al-Baldawi N. F., and Abercrombie R. F. (1995) Calcium diffusion coefficient in Myxicola axoplasm. Cell Calcium 17, 422–430 [DOI] [PubMed] [Google Scholar]
- 33.Young R. C., and Zhang P. (2001) The mechanism of propagation of intracellular calcium waves in cultured human uterine myocytes. Am. J. Obstet. Gynecol. 184, 1228–1234 [DOI] [PubMed] [Google Scholar]
- 34.Fridlyand L. E., Tamarina N., and Philipson L. H. (2003) Modeling of Ca2+ flux in pancreatic β-cells: role of the plasma membrane and intracellular stores. Am. J. Physiol. Endocrinol. Metab. 285, E138–E154 [DOI] [PubMed] [Google Scholar]
- 35.Caride A. J., Penheiter A. R., Filoteo A. G., Bajzer Z., Enyedi A., and Penniston J. T. (2001) The plasma membrane calcium pump displays memory of past calcium spikes. Differences between isoforms 2b and 4b. J. Biol. Chem. 276, 39797–39804 [DOI] [PubMed] [Google Scholar]
- 36.Fink C. C., Slepchenko B., Moraru I. I., Watras J., Schaff J. C., and Loew L. M. (2000) An image-based model of calcium waves in differentiated neuroblastoma cells. Biophys. J. 79, 163–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herrington J., Park Y. B., Babcock D. F., and Hille B. (1996) Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron 16, 219–228 [DOI] [PubMed] [Google Scholar]
- 38.Chandrasekera P. C., Kargacin M. E., Deans J. P., and Lytton J. (2009) Determination of apparent calcium affinity for endogenously expressed human sarco(endo)plasmic reticulum calcium-ATPase isoform SERCA3. Am. J. Physiol. Cell Physiol. 296, C1105–C1114 [DOI] [PubMed] [Google Scholar]
- 39.Turovsky E. A., Kaimachnikov N. P., Turovskaya M. V., Berezhnov A. V., Dynnik V. V., and Zinchenko V. P. (2012) Two mechanisms of calcium oscillations in adipocytes. Biochemistry (Moscow) 6, 26–34 [Google Scholar]
- 40.Sienaert I., De Smedt H., Parys J. B., Missiaen L., Vanlingen S., Sipma H., and Casteels R. (1996) Characterization of a cytosolic and a luminal Ca2+ binding site in the type I inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 271, 27005–27012 [DOI] [PubMed] [Google Scholar]
- 41.Horne J. H., and Meyer T. (1995) Luminal calcium regulates the inositol trisphosphate receptor of rat basophilic leukemia cells at a cytosolic site. Biochemistry 34, 12738–12746 [DOI] [PubMed] [Google Scholar]
- 42.Sienaert I., Missiaen L., De Smedt H., Parys J. B., Sipma H., and Casteels R. (1997) Molecular and functional evidence for multiple Ca2+-binding domains in the type 1 inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 272, 25899–25906 [DOI] [PubMed] [Google Scholar]
- 43.Missiaen L., De Smedt H., Parys J. B., and Casteels R. (1994) Co-activation of inositol trisphosphate-induced Ca2+ release by cytosolic Ca2+ is loading-dependent. J. Biol. Chem. 269, 7238–7242 [PubMed] [Google Scholar]
- 44.Mak D. O., McBride S., and Foskett J. K. (2001) Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 insp3 receptors. J. Gen. Physiol. 117, 435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaftan E. J., Ehrlich B. E., and Watras J. (1997) Inositol 1,4,5-trisphosphate (InsP3) and calcium interact to increase the dynamic range of InsP3 receptor-dependent calcium signaling. J. Gen. Physiol. 110, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joseph S. K., Rice H. L., and Williamson J. R. (1989) The effect of external calcium and pH on inositol trisphosphate-mediated calcium release from cerebellum microsomal fractions. Biochem. J. 258, 261–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanimura A., and Turner R. J. (1996) Inositol 1,4,5-trisphosphate-dependent oscillations of luminal [Ca2+] in permeabilized HSY cells. J. Biol. Chem. 271, 30904–30908 [DOI] [PubMed] [Google Scholar]
- 48.Zimmermann B. (2000) Control of InsP3-induced Ca2+ oscillations in permeabilized blowfly salivary gland cells: contribution of mitochondria. J. Physiol. 525, 707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Camello C., Lomax R., Petersen O. H., and Tepikin A. V. (2002) Calcium leak from intracellular stores—the enigma of calcium signalling. Cell Calcium 32, 355–361 [DOI] [PubMed] [Google Scholar]
- 50.Marhl M., Schuster S., and Brumen M. (1998) Mitochondria as an important factor in the maintenance of constant amplitudes of cytosolic calcium oscillations. Biophys. Chem. 2, 125–132 [PubMed] [Google Scholar]
- 51.Gunter T. E., and Pfeiffer D. R. (1990) Mechanisms by which mitochondria transport calcium. Am. J. Physiol. Cell Physiol. 258, C755–C786 [DOI] [PubMed] [Google Scholar]
- 52.Shuttleworth T. J., and Mignen O. (2003) Calcium entry and the control of calcium oscillations. Biochem. Soc. Trans. 31, 916–919 [DOI] [PubMed] [Google Scholar]
- 53.Tanimura A., Morita T., Nezu A., Shitara A., Hashimoto N., and Tojyo Y. (2009) Use of fluorescence resonance energy transfer-based biosensors for the quantitative analysis of inositol 1,4,5-trisphosphate dynamics in calcium oscillations. J. Biol. Chem. 284, 8910–8917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Faas G. C., Schwaller B., Vergara J. L., and Mody I. (2007) Resolving the fast kinetics of cooperative binding: Ca2+ buffering by calretinin. PLoS Biol. 5, e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Christel C. J., Schaer R., Wang S., Henzi T., Kreiner L., Grabs D., Schwaller B., and Lee A. (2012) Calretinin regulates Ca2+-dependent inactivation and facilitation of Cav2.1 Ca2+ channels through a direct interaction with the α12.1 subunit. J. Biol. Chem. 287, 39766–39775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berridge M. J. (1990) Calcium oscillations. J. Biol. Chem. 265, 9583–9586 [PubMed] [Google Scholar]
- 57.Hajnóczky G., Csordás G., Das S., Garcia-Perez C., Saotome M., Sinha Roy S., and Yi M. (2006) Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chaudry I. H. (1982) Does ATP cross the cell plasma membrane. Yale J. Biol. Med. 55, 1–10 [PMC free article] [PubMed] [Google Scholar]
- 59.Dupont G., Combettes L., Bird G. S., and Putney J. W. (2011) Calcium oscillations. Cold Spring Harb. Perspect. Biol. 3, a004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schwaller B. (2014) Calretinin: from a “simple” Ca2+ buffer to a multifunctional protein implicated in many biological processes. Front. Neuroanat. 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwaller B., Celio M. R., and Doglioni C. (2004) Identification of calretinin and the alternatively spliced form calretinin-22k in primary pleural mesotheliomas and in their metastases. Anticancer Res. 24, 4003–4009 [PubMed] [Google Scholar]
- 62.Perocchi F., Gohil V. M., Girgis H. S., Bao X. R., McCombs J. E., Palmer A. E., and Mootha V. K. (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waldeck-Weiermair M., Jean-Quartier C., Rost R., Khan M. J., Vishnu N., Bondarenko A. I., Imamura H., Malli R., and Graier W. F. (2011) Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 286, 28444–28455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marhl M., Schuster S., and Brumen M. (1998) Mitochondria as an important factor in the maintenance of constant amplitudes of cytosolic calcium oscillations. Biophys. Chem. 71, 125–132 [PubMed] [Google Scholar]
- 65.Haynes C. L., Buhler L. A., and Wightman R. M. (2006) Vesicular Ca2+-induced secretion promoted by intracellular pH-gradient disruption. Biophys. Chem. 123, 20–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Marchi U., Santo-Domingo J., Castelbou C., Sekler I., Wiederkehr A., and Demaurex N. (2014) NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 289, 20377–20385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Castaldo P., Cataldi M., Magi S., Lariccia V., Arcangeli S., and Amoroso S. (2009) Role of the mitochondrial sodium/calcium exchanger in neuronal physiology and in the pathogenesis of neurological diseases. Prog. Neurobiol. 87, 58–79 [DOI] [PubMed] [Google Scholar]
- 68.Kaasik A., Safiulina D., Zharkovsky A., and Veksler V. (2007) Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 292, C157–C163 [DOI] [PubMed] [Google Scholar]
- 69.Schuster S., Marhl M., and Höfer T. (2002) Modelling of simple and complex calcium oscillations. From single-cell responses to intercellular signalling. Eur. J. Biochem. 269, 1333–1355 [DOI] [PubMed] [Google Scholar]
- 70.De Young G. W., and Keizer J. (1992) A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc. Natl. Acad. Sci. U.S.A. 89, 9895–9899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Somogyi R., and Stucki J. W. (1991) Hormone-induced calcium oscillations in liver cells can be explained by a simple one pool model. J. Biol. Chem. 266, 11068–11077 [PubMed] [Google Scholar]
- 72.Dupont G., and Goldbeter A. (1993) One-pool model for Ca2+ oscillations involving Ca2+ and inositol 1,4,5-trisphosphate as co-agonists for Ca2+ release. Cell Calcium 14, 311–322 [DOI] [PubMed] [Google Scholar]
- 73.Sneyd J., Tsaneva-Atanasova K., Yule D. I., Thompson J. L., and Shuttleworth T. J. (2004) Control of calcium oscillations by membrane fluxes. Proc. Natl. Acad. Sci. U.S.A. 101, 1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bird G. S., and Putney J. W. Jr. (2005) Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells. J. Physiol. 562, 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lichtshtein D., Kaback H. R., and Blume A. J. (1979) Use of a lipophilic cation for determination of membrane potential in neuroblastoma-glioma hybrid cell suspensions. Proc. Natl. Acad. Sci. U.S.A. 76, 650–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Houamed K., Fu J., Roe M. W., and Philipson L. H. (2004) in Diabetes Mellitus: a Fundamental and Clinical Text (LeRoith D., Taylor S. I., and Olefsky J. M., eds) pp. 51–68, Lippincott, Williams, and Wilkins, Philadelphia [Google Scholar]
- 77.Walsh C., Barrow S., Voronina S., Chvanov M., Petersen O. H., and Tepikin A. (2009) Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta 1787, 1374–1382 [DOI] [PubMed] [Google Scholar]
- 78.Pecze L., Blum W., and Schwaller B. (2013) Mechanism of capsaicin receptor TRPV1-mediated toxicity in pain-sensing neurons focusing on the effects of Na+/Ca2+ fluxes and the Ca2+-binding protein calretinin. Biochim. Biophys. Acta 1833, 1680–1691 [DOI] [PubMed] [Google Scholar]
- 79.Ducreux S., Gregory P., and Schwaller B. (2012) Inverse regulation of the cytosolic Ca2+ buffer parvalbumin and mitochondrial volume in muscle cells via SIRT1/PGC-1α axis. PLoS One 7, e44837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schwaller B. (2012) The regulation of a cell's Ca2+ signaling toolkit: the Ca2+ homeostasome. Adv. Exp. Med. Biol. 740, 1–25 [DOI] [PubMed] [Google Scholar]
- 81.Doglioni C., Dei Tos A. P., Laurino L., Iuzzolino P., Chiarelli C., Celio M. R., and Viale G. (1996) Calretinin: a novel immunocytochemical marker for mesothelioma. Am. J. Surg. Pathol. 20, 1037–1046 [DOI] [PubMed] [Google Scholar]
- 82.Lukas W., and Jones K. A. (1994) Cortical neurons containing calretinin are selectively resistant to calcium overload and excitotoxicity in vitro. Neuroscience 61, 307–316 [DOI] [PubMed] [Google Scholar]
- 83.Gotzos V., Wintergerst E. S., Musy J. P., Spichtin H. P., and Genton C. Y. (1999) Selective distribution of calretinin in adenocarcinomas of the human colon and adjacent tissues. Am. J. Surg. Pathol. 23, 701–711 [DOI] [PubMed] [Google Scholar]
- 84.Gander J.-C., Bustos-Castillo M., Stüber D., Hunziker W., Celio M., and Schwaller B. (1996) The calcium-binding protein calretinin-22k, an alternative splicing product of the calretinin gene is expressed in several colon adenocarcinoma cell lines. Cell Calcium 20, 63–72 [DOI] [PubMed] [Google Scholar]
- 85.Boyer J., Allen W. L., McLean E. G., Wilson P. M., McCulla A., Moore S., Longley D. B., Caldas C., and Johnston P. G. (2006) Pharmacogenomic identification of novel determinants of response to chemotherapy in colon cancer. Cancer Res. 66, 2765–2777 [DOI] [PubMed] [Google Scholar]
- 86.Hrabakova R., Kollareddy M., Tyleckova J., Halada P., Hajduch M., Gadher S. J., and Kovarova H. (2013) Cancer cell resistance to aurora kinase inhibitors: identification of novel targets for cancer therapy. J. Proteome Res. 12, 455–469 [DOI] [PubMed] [Google Scholar]
- 87.Blum W., and Schwaller B. (2013) Calretinin is essential for mesothelioma cell growth/survival in vitro: a potential new target for malignant mesothelioma therapy? Int. J. Cancer 133, 2077–2088 [DOI] [PubMed] [Google Scholar]
- 88.Gander J. C., Gotzos V., Fellay B., and Schwaller B. (1996) Inhibition of the proliferative cycle and apoptotic events in WiDr cells after down-regulation of the calcium-binding protein calretinin using antisense oligodeoxynucleotides. Exp. Cell Res. 225, 399–410 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.