

Background: Sec6 has been reported to inhibit SNARE complex assembly, in contrast to other tethering complexes.
Results: Sec6 does not inhibit SNARE assembly. Rather, it binds assembled SNARE complexes. Disrupting this interaction results in growth defects in yeast.
Conclusion: This interaction is important for a functional exocytic pathway.
Significance: The exocyst is likely to have mechanisms of SNARE regulation comparable with other multisubunit tethering complexes.
Keywords: exocytosis, intracellular trafficking, intrinsically disordered protein, MS, protein cross-linking, protein-protein interaction, SNARE proteins, exocyst, tethering complex
Abstract
In eukaryotic cells, membrane-bound vesicles carry cargo between intracellular compartments, to and from the cell surface, and into the extracellular environment. Many conserved families of proteins are required for properly localized vesicle fusion, including the multisubunit tethering complexes and the SNARE complexes. These protein complexes work together to promote proper vesicle fusion in intracellular trafficking pathways. However, the mechanism by which the exocyst, the exocytosis-specific multisubunit tethering complex, interacts with the exocytic SNAREs to mediate vesicle targeting and fusion is currently unknown. We have demonstrated previously that the Saccharomyces cerevisiae exocyst subunit Sec6 directly bound the plasma membrane SNARE protein Sec9 in vitro and that Sec6 inhibited the assembly of the binary Sso1-Sec9 SNARE complex. Therefore, we hypothesized that the interaction between Sec6 and Sec9 prevented the assembly of premature SNARE complexes at sites of exocytosis. To map the determinants of this interaction, we used cross-linking and mass spectrometry analyses to identify residues required for binding. Mutation of residues identified by this approach resulted in a growth defect when introduced into yeast. Contrary to our previous hypothesis, we discovered that Sec6 does not change the rate of SNARE assembly but, rather, binds both the binary Sec9-Sso1 and ternary Sec9-Sso1-Snc2 SNARE complexes. Together, these results suggest a new model in which Sec6 promotes SNARE complex assembly, similar to the role proposed for other tether subunit-SNARE interactions.
Introduction
Intracellular trafficking is an essential, conserved, and highly regulated process in eukaryotic cells, characterized by the movement of membrane-bound vesicles between distinct organelles and cellular compartments. Vesicles bud from donor compartments, are trafficked to the proper target membrane, and subsequently fuse. These trafficking processes must be tightly regulated to ensure spatial and temporal delivery of vesicles to their cellular destination. Many conserved protein families are essential for trafficking, including the SNARE proteins that provide the impetus for membrane fusion (1) and the multisubunit tethering complexes (MTCs)2 proposed to tether vesicles to sites of fusion (2, 3). However, the details of these processes are still poorly understood at the mechanistic level.
The SNARE complex is the core of the membrane fusion apparatus. Proteins in this family are identified by their “SNARE motif” (4), a characteristic coiled-coil heptad repeat of hydrophobic residues generally followed by a transmembrane domain. In yeast, the plasma membrane target (t-) SNAREs Sso1/2 (syntaxin family in mammals) and Sec9 (SNAP-25 family in mammals) form a binary complex that binds the vesicle (v-) SNAREs Snc1/2 (VAMP/synaptobrevin family in mammals) to drive membrane fusion (5–9). Prior to complex formation, both Sec9 and Snc2 are intrinsically disordered proteins (IDPs) and fold into their helical SNARE conformations during assembly of the SNARE complex (10–12). This assembled SNARE complex is stable and requires the ATPase activity of the NSF and α-SNAP complex (Sec18 and Sec17 in yeast, respectively) for disassembly. Because of the energetic favorability of assembling the SNARE complex and the stability of the assembled complex, regulation of vesicle fusion necessitates the regulation of SNARE complex assembly (13, 14).
Several lines of evidence indicate that specific control of SNARE complex assembly is required to prevent the formation of premature, mislocalized, or nonspecific SNARE complexes that may result in the incorrect delivery of important cargo. First, yeast SNAREs can form fusion-competent complexes promiscuously in vitro. Vesicles containing the exocytic Sso1-Sec9 t-SNARE complex can fuse with vesicles containing v-SNAREs other than Snc1/2 (15), and vesicles loaded with the vacuolar t-SNARE complex (Vam3, Vti1, and Vam7) can fuse with vesicles containing non-vacuolar v-SNAREs, including the exocytic/endocytic Snc2 (16). This promiscuity extends to mammalian cells; SNAREs from various intracellular compartments can form stable non-cognate SNARE complexes, suggesting that SNARE complex formation is not the sole determinant of specificity (17–19). Secondly, the localization of Sec9 and Sso1 is not restricted to the yeast bud tips and mother-daughter necks where secretion is occurring. Instead, they are distributed along the plasma membrane (6). One level of regulation comes from Sso1. Like many syntaxins, Sso1 contains an autoinhibitory domain that can prevent premature binary Sso1-Sec9 complex assembly (20, 21). However, this autoinhibition is not absolute because SNARE complex assembly can proceed in vitro without the addition of putative “opening” factors, albeit at non-physiological rates (11, 22). Similarly, the mammalian exocytic SNAREs can form SNARE complexes that are not competent for fusion at the Golgi when not inhibited prior to trafficking to the cell surface (23). Therefore, other levels of activation and/or inhibition are necessary to prevent inappropriate complex formation and subsequent vesicle fusion.
In contrast to the SNAREs, several protein families are localized at sites of secretion and, therefore, well placed to provide additional control of exocytic SNARE complex assembly. One such example is the exocyst complex, the MTC that is thought to recognize and tether secretory vesicles to the plasma membrane prior to SNARE complex assembly (3 and references therein). Consistent with a putative upstream tethering role, three of the eight exocyst subunits interact with lipids and small Rab and Rho family GTPases on the opposing membranes, although tethering has not yet been directly demonstrated (24–32). The exocyst appears to function prior to SNARE assembly and vesicle fusion; temperature-sensitive mutants of individual exocyst subunits result in vesicle accumulation in yeast (33, 34). In addition, the exocyst has been implicated in other essential membrane trafficking processes such as autophagy, ciliogenesis, and pathogen invasion, likely because of either a “hijacking” or relocalization of the putative tethering function of the exocyst (35–37). Although there is no high-resolution structure of the entire complex, structures of domains of the individual exocyst subunits Sec6 (38), Sec15 (39), and Exo70 and Exo84 (40–42) reveal that they are composed of evolutionarily conserved helical bundles. The remaining subunits are predicted to have similar structures (43). This structural characterization places the exocyst into the conserved CATCHR (complexes associated with tethering containing helical rods) family of tethering complexes (2).
All MTCs (conserved oligomeric Golgi complex (COG), Golgi-associated retrograde protein complex (GARP), Dsl1 complex, homotypic vacuole fusion protein complex (HOPS), C core vacuole/endosome tethering complex (CORVET), and transport particle protein complex (TRAPP)), of which the CATCHR family is a subset, interact directly with their cognate SNARE proteins (44–54). These interactions, where characterized, promote the assembly or proofreading of their cognate SNARE complexes. The HOPS complex binds to and protects properly assembled complexes from disassembly while having a reduced affinity for improperly formed complexes (55, 56). In mammalian cells, knockdown of individual COG subunits leads to an increase in uncomplexed SNAREs and a decrease in overall SNARE expression (45). However, for many MTCs, only the binding interactions with the SNAREs have been identified. The functional implications of most of these interactions have not been studied at the molecular level.
Consistent with other MTC-SNARE interactions, we have shown previously that the yeast exocyst subunit Sec6 interacts with the C-terminal SNAP-25 domain of the plasma membrane t-SNARE Sec9 (Sec9CT, residues 414–651) (46). However, Sec6 appeared to inhibit, rather than promote or stabilize, in vitro formation of the Sso1-Sec9 binary SNARE complex (46). Therefore, we proposed that the Sec6-Sec9 interaction prevented premature assembly of the Sso1-Sec9 SNARE complex and that assembly of the exocyst complex could release Sec9 to form fusogenic SNARE complexes. To test these hypotheses, we sought to disrupt the Sec6-Sec9 interaction through mutagenesis without disrupting other protein-protein interactions with their critical binding partners. These mutants would then be useful reagents for testing the importance of this interaction in vivo.
Because Sec9 is an IDP, the process of identifying the critical residues for the Sec6-Sec9 interaction was not straightforward. Previous attempts to identify a minimal region of Sec9 necessary for the interaction with Sec6, including limited proteolysis and truncation experiments, were inconclusive (46). Identification of the minimal binding region of Sec6 was hindered by the insolubility of the N-terminal domain of Sec6. However, the C-terminal half of the protein has been shown to be insufficient for binding (46). We were also limited by the flexible nature of Sec9. The binding sites of IDPs like Sec9 are difficult to identify because the protein can compensate for loss of one or more binding residues, and the native state of the IDP-ordered partner interaction often involves more than one locus with varying degrees of preference (57–60).
Here we used a chemical cross-linking and tandem mass spectrometry approach to define the determinants of the Sec6-Sec9 interaction. Our analysis revealed a patch of charged and hydrophobic residues in the linker region of Sec9 that we predicted would mediate the interaction with Sec6. Mutation of these residues resulted in synthetic growth defects in combination with exocyst subunit temperature-sensitive mutations. Intriguingly, we also observed novel interactions between Sec6 and both the binary Sec9-Sso1 and ternary Sec9-Sso1-Snc2 SNARE complexes. These interactions were disrupted by the Sec9 mutation. The identification of these novel Sec6-SNARE complex interactions necessitated a revision of our previous model. We show that Sec6 does not inhibit SNARE complex assembly to the extent observed previously. These data suggest that Sec6 shares a SNARE regulatory role with other tethering complex subunits, including HOPS and COG.
Experimental Procedures
Protein Expression and Purification
Mutations in recombinant proteins were generated by overlap extension PCR, cloned into the T7 expression vectors pET15b (Sec6 constructs) or pETDuet-1 (Sec9 and Sso1 constructs), and confirmed by sequencing. Full-length yeast Sec6(1–805), Sec9(414–651), and the cytosolic domain of Sso1(1–265) proteins were overexpressed in BL21(DE3) or BL21(DE3) RIL Escherichia coli cells and purified as described previously (11, 46). Protein concentrations were determined by a quantitative ninhydrin assay (61). The C-terminal domain of Sec9 is homologous to the mammalian homolog SNAP-25 and has been shown previously to be functional in yeast (6).
Cross-linking and Protein Digestion
Sec6 and Sec9 were combined 1:1 (7 μm each) in a solution of 10 mm potassium phosphate buffer (pH 7.4), 140 mm KCl, and 4% glycerol and incubated for 2 h at room temperature. EDC (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride, Thermo Scientific) was then added at 1000× molar excess in 5 μl of 10 mm potassium phosphate buffer (pH 7.4). Variations on this reaction were also performed to improve detection sensitivity: with the addition of either 500× molar excess NHS (Thermo Scientific) or 500× molar excess sulfo-NHS (Thermo Scientific) to increase the stability of the partially cross-linked complex; and at low concentrations of reactants (1 micromolar) to detect only early-forming crosslinks. The reaction proceeded for 15, 30, 60, and 120 min before quenching with Laemmli sample loading buffer (Bio-Rad) and boiling for 10 min. One additional experiment was run at 30 s, 1 min, 5 min, and 10 min to again detect only early-forming crosslinks. Approximately 15 μg of protein from each reaction was loaded and run on a 4–20% Mini-Protean TGX precast SDS gel (Bio-Rad). Bands corresponding to the monomeric Sec6-Sec9 cross-linked complex were excised and destained twice with 200 μl of 25 mm ammonium bicarbonate in 50% acetonitrile, reduced in 10 μl of 50 mm DTT for 10 min at 60 °C, and then alkylated in 10 μl of 100 mm iodoacetamide for 1 h in the dark at room temperature. Proteins were digested overnight at 30 °C with 100 ng of trypsin (Promega). Following digestion, peptides were transferred into a clean tube and then extracted further twice with 50% acetonitrile containing 5% (v/v) formic acid. The combined extracts were dried in a SpeedVac and brought to 20 μl with 0.1% (v/v) formic acid for LC-MS/MS.
LC-MS/MS
Peptide digests were injected (2 μl) and loaded at 4 μl/min (5% acetonitrile containing 0.1% formic acid) onto a custom-packed trap column (100-μm inner diameter fused silica with Kasil frit) consisting of 2.0 cm of 200 Å, 5 μm Magic C18AQ particles (Bruker-Michrom) configured to a custom-packed analytical column (75-μm inner diameter fused silica) packed with 25 cm 100 Å, 5 μm Magic C18AQ particles (Bruker-Michrom) to a gravity-pulled tip. Peptides were separated at 300 nl/min using a Proxeon Easy-nLC (Thermo Scientific) system using a linear gradient from 100% A (5% acetonitrile with 0.1% (v/v) formic acid) to 35% B (acetonitrile with 0.1% formic acid) in 90 min and eluted directly into an LTQ Orbitrap Velos hybrid mass spectrometer (Thermo Scientific) (62). Data were acquired using a data-dependent acquisition routine of acquiring one mass spectrum from m/z 350–2000 in the Orbitrap (resolution 60,000), followed by tandem mass spectrometry scans of the 10 most abundant precursor ions found in the mass spectrum. Alternate runs collected either collision-induced dissociation or higher-energy collisional dissociation spectra acquired in the Orbitrap mass analyzer. Data acquisition utilized charge state rejection of singly, doubly, and triply charged ions, and dynamic exclusion was utilized to minimize data redundancy and maximize peptide identification (62).
Data Analysis
A concatenated peptide database was generated by xComb (University of Washington) software (63). The database considered all inter- and intramolecular Sec6 to Sec9 EDC-linked tryptic peptides up to two missed cleavages. The raw data were converted to peak lists and searched using the Sequest search engine contained in Proteome Discoverer (version 1.3, Thermo Scientific). Briefly, no enzyme specificity was considered. Parent ion tolerances were set to 15 ppm, fragment ion tolerances were set to 0.05 Da, methionine oxidation was considered as a variable modification, and carbamidomethylation of cysteine was considered as a fixed modification. The peptide results were then filtered by removing hits with an XCorr × Sp product of less than 146. Label-free quantitation using peptide-extracted ion chromatograms was done using ProteoIQ (version 2.3.08, NuSep) quantitative analysis software using the replicate method (64). Cross-linked peptides with a precursor ion intensity of less than 1.0 × 105 from the mass spectrum in the 120-min reaction time samples were filtered and removed from the dataset. For the generation of the cross-linking (Fig. 1), all experimental designs were combined and examined together to determine the effect of the cross-linker on the Sec6-Sec9 complex.
FIGURE 1.

Sec6 and Sec9 cross-link specifically, and different classes of cross-links highlight residues important for the binding interaction. All cross-links identified in each of the experiments described under “Experimental Procedures” were mapped onto the C-terminal structure of Sec6 (amino acids 411–805) (38) and linear segments representing the N-terminal domain of Sec6 and the SNARE domains of Sec9. Gray boxes indicate helical secondary structure (the known SNARE helices on Sec9 and the predicted helical regions of the Sec6 (amino acids 1–410) (SOPM algorithm, Ref. 77)). A, all possible cross-linkable residues (lysine, blue; glutamic acid/aspartic acid, red) are mapped onto the proteins. B, cross-linked residues are colored on the basis of the classes of cross-links identified in Fig. 2A (early-forming, yellow, late-forming, blue, combination residues that participate in both classes, green). Only the late-forming cross-links are shown. On the basis of the hypothesized model presented in Fig. 2B, residues likely to be involved in the binding interaction are circled in red. The sec9-142 mutation is located between the two blue residues (Lys-533 and Glu-537) in the red circle.
Effect of Sec9 Mutations in Vivo
The full-length sec9-142 (1–651) mutant gene was cloned into the yeast integration vector pRS306, linearized with EcoRI, and transformed into both BY4741 and BY4742 (Ref. 65), Open Biosystems) using a yeast high-efficiency transformation protocol (66). The wild-type SEC9 and residuals from the pRS306 plasmid were selected against using 5-fluoro-orotic acid (67), and the mutations were confirmed by sequencing. Double mutant strains containing sec9-142 and exocytic temperature-sensitive alleles were generated by mating the above sec9-142 strains to the exocytic deletion strains stated (33), sporulation, and confirmation by temperature sensitivity and sequencing of the sec9 locus. Growth defects were detected by a serial dilution assay, where log phase cultures were diluted to 1.0 OD600 units/ml. This culture was diluted 10-fold over six samples, and dilutions were spotted onto plates containing YPD media and incubated for 72 h at the indicated temperature.
SNARE Complex Assembly Monitored by Native Gel Mobility Shift Assay
Purified recombinant Sso1 (1–265) was mixed with the indicated proteins at 10 μm final concentration of each protein in a 15-μl final reaction volume of 10 mm sodium phosphate (NaPhos) (pH 7.4), 200 mm sodium chloride (NaCl), and 1 mm DTT and incubated at 18 °C for various times (0–72 h). 3 μl of 6× native gel loading dye (0.6% w/v bromcresol green, 30% v/v glycerol, 25 mm histidine, and 30 mm MOPS) was added to each reaction in the cold room, and 5 μl was loaded on a 200-ml slab of native PAGE gel (pH 6.6) (6.0% acrylamide, 25 mm histidine, 30 mm MOPS, and 2.5% w/w glycerol polymerized with 2.0 ml 10% w/v APS (ammonium persulfate) and 200 μl of TEMED (N,N,N',N'-tetramethylethylenediamine) that was pre-equilibrated for 1 h at 4 °C. The gel was run for 4 h at 100 V at 4 °C in native gel buffer (pH 6.6) (25 mm histidine and 30 mm MOPS) (68), and protein bands were visualized by Coomassie Blue staining (69). The percent free Sso1 was quantified by densitometry (Photoshop, CS5). The density of each band was divided by the Sso1 band from the zero time point. The resulting curve was fit using the derived second-order rate equation (11) using GraphPad Prism, with Y at t = 0 constrained to 1.0 and Y at t = ∞ constrained to >0. All four replicates were fit to a global rate constant, k, and presented as mean ± S.E. of the fit as reported by GraphPad Prism.
SNARE Complex Assembly Monitored by Gel Filtration
All proteins were incubated together in 200 μl of final volume at 10 μm final concentration in 30 mm NaPhos (pH 7.4), 200 mm NaCl, and 1 mm DTT for the indicated time (0–72 h) and injected on a Superdex 200 10/30 column (GE LifeSciences) pre-equilibrated in the same buffer. 200-μl fractions were collected from 0.41–0.71 column volumes and run on a 10% SDS-PAGE gel. Proteins were visualized by fluorescent Krypton staining (Pierce) and imaged on a Typhoon fluorescence stage with a 532-nm laser (GE LifeSciences). Sso1 band intensities of the three fractions corresponding to the top of the A280 free Sso1 peak (0.61–0.63 column volumes) were quantified by densitometry (Photoshop CS5), and each fraction was plotted as a curve across the time course. Because the free Sso1 peak is approximately Gaussian, the resulting curves were fit using the derived second-order rate equation (11), as described above, in GraphPad Prism, with each curve fit to a global rate constant with the surrounding fractions (three in total) to control for loading error. A representative fit of two replicate experiments is shown, and the k value is presented as mean ± S.E. of the fit as presented by GraphPad Prism.
Sec6:Sec9 and Sec6:SNARE Binding
GST-Sec9 and GST-Sec9-142 were purified by expression of each construct as described previously (46). GST-tagged binary and ternary SNARE complexes were purified by mixing E. coli lysates containing each of the individual components (Sec9CT, GST-Sso1CTb (residues 179–265), and Snc2 (residues 1–92)), expressed as described previously (11), with a limiting concentration of GST-Sso1CTb. All GST-protein-containing lysates were incubated for 1 h at 4 °C with glutathione-agarose resin, washed in wash buffer without glutathione (50 mm Tris (pH 8.0), 150 mm NaCl, 10% glycerol, 0.5% IPEGAL, and 3 mm DTT), and eluted in wash buffer + 40 mm reduced glutathione. All constructs except GST-Sec9-142 were diluted 2-fold in 10 mm HEPES (pH 7.4), loaded onto a MonoQ 10/10 column (GE LifeSciences) pre-equilibrated in 10 mm HEPES (pH 7.4) and 100 mm NaCl, and eluted with a gradient from 100 mm to 1 m NaCl. Fractions containing the SNAREs were concentrated and frozen in 10 mm NaPhos (pH 7.4) and 140 mm NaCl. GST-Sec9-142 was diluted 30× in 10 mm HEPES (pH 7.0) and 150 mm NaCl, concentrated to 1 ml in an Amicon 10-kDa spin concentrator (Millipore), and frozen in 10 mm NaPhos (pH 7.4). The final concentration of the protein was determined by a quantitative ninhydrin assay (61).
To test for binding to Sec6, 25 nm GST-protein (or GST alone) was incubated with the indicated concentration of Sec6 in binding buffer (30 mm NaPhos (pH 7.4), 200 mm NaCl, 5% glycerol, 0.5% IPEGAL, and 1 mm DTT) in a final volume of 100 μl for 1 h at 4 °C. 90 μl of the incubated proteins was added to 2 μl of magnetic glutathione resin slurry (Thermo Fischer, 25% slurry) and incubated for an additional 1 h at 4 °C. The supernatant was removed, and the resin was resuspended in 1× Laemmli loading buffer before being heated at 95 °C for 5 min. The beads were not washed prior to boiling to maximize the bound protein signal. 10 μl of each sample was loaded on a 12% SDS-PAGE gel, and proteins were visualized by Krypton staining (Pierce) and imaging on a Typhoon fluorescence stage (GE LifeSciences). The extent of binding was measured by calculating the molar Sec6:GST-protein ratio (densitometry of Sec6 divided by the densitometry of the GST-protein, normalized for protein size) and dividing by the Sec6:GST molar ratio to normalize by background binding. Each bar represents three replicates ± S.E., and statistical significance was calculated using an ordinary one-way analysis of variance test with multiple comparisons corrected for by Holm-Sidak multiple comparisons test (GraphPad Prism).
Results
Sec6 Specifically Cross-links to the IDP Sec9, Which Identifies Potential Binding Residues
To examine the function of the Sec6-Sec9 interaction in vivo, we wanted to identify the residues necessary for the binding interaction and generate a loss-of-binding mutant. We chose to use the chemical cross-linker EDC and mass spectrometry analyses to map this interaction to circumvent difficulties caused by the flexible nature of the IDP Sec9. Chemical cross-linkers create covalent bonds between side chains (usually Lys-Lys or Asp/Glu-Lys) separated by specific distances; in the case of EDC, the reaction of EDC with Asp/Glu and Lys residues that are in close proximity results in a peptide bond between the free carboxyl and free amine groups. Purified Sec6 and Sec9 were mixed together and cross-linked by EDC (with or without NHS or sulfo-NHS), and a portion of the total reaction was quenched at various time points (1–120 min depending on the experiment). The reaction from each time point was run on SDS-PAGE, and the band corresponding to the molecular weight of the 1:1 Sec6-Sec9 complex was excised and subjected to mass spectrometry analysis (see “Experimental Procedures”). The identified cross-linked peptides mapped across the entire length of Sec9 (36 residues of 63 total Glu/Lys/Asp residues) and Sec6 (68 residues of 200 total Glu/Lys/Asp residues; all Glu/Lys/Asp residues in the Sec6 C-terminal domain are located on the surface (38)), with 152 unique cross-links between them. This large number of cross-links was expected because of the flexible nature of the IDP Sec9. However, only ∼50% of the available Asp/Glu/Lys residues on Sec9 and only ∼35% of the Asp/Glu/Lys residues on Sec6 were identified as participating in a cross-link (Fig. 1A), indicating that the cross-linking reaction was not random.
Because of the large number of cross-links identified, we sought to limit our analysis to only those likely involved in the binding interaction. Because this experiment was carried out as a time course, we categorized each cross-link on the basis of its presence/absence and intensity at different time points (Fig. 2A). These analyses identified two classes of cross-links: those that had constant intensities over time, and those whose intensities changed over time. The cross-links that do not change over time are ones that form early in the reaction, and those that are more variable form later as cross-links that are secondary to the “early-forming” cross-links (Fig. 2B). We hypothesized that residues participating in salt bridges in the binding interface will initially be protected from EDC; therefore, the early-forming cross-links lie outside of the core of the protected binding site. After formation of the early cross-links, the now limited flexibility of the IDP Sec9 and low micromolar affinity of the Sec6-Sec9 interaction (∼0.5 μm (46)) would result in disruption of some of the salt bridges, allowing those residues to participate in “late-forming” cross-links. Some residues are capable of participating in both early and late-forming cross-links, likely because of the bulk nature of this assay and the variable accessibility of those residues due to the flexibility of Sec9.
FIGURE 2.

Cross-linking over a time course results in two classes of cross-linked peptides. A, at each time point in the cross-linking reaction (0–120 min depending on the experiment), a portion of the reaction was quenched, and the cross-linked peptides were identified and quantified by mass spectrometry. The resulting cross-links were grouped into two classes: early-forming cross-links (top panel), which form early in the reaction and do not change intensity over time, and late-forming cross-links (bottom panel), which increase in intensity over time. B, model for how an intrinsically disordered protein may interact with a well folded binding partner over time in the presence of EDC. 1, Sec9 binds to Sec6, and these side chain interactions (red circle) protect these residues from reaction with EDC. 2, early-forming cross-links (yellow) form as chemically capable side chains come into contact. 3, equilibrium dissociation of the local interaction occurs. Early-forming cross-links (yellow) restrict the proteins to remain in close proximity despite the absence of the original binding interaction (red). 4, late forming cross-links (blue) form as chemically capable side chains come into contact through random motion of the IDP protein. These new cross-links can occur in the binding site (bottom) or outside of the binding site (top).
On the basis of these hypotheses, we focused on residues that participate in late-forming cross-links, specifically those that are located nearby early-forming cross-links (Fig. 1B). After filtering the dataset accordingly, we identified a five amino acid stretch on Sec9 that satisfied the above conditions. The first (Lys-533) and last (Glu-537) residues cross-link to adjacent regions on the Sec6 C-terminal domain (Sec9 Lys-533 to Sec6 Glu-447 and Sec9 Glu-537 to Sec6 Lys-516), and these cross-links are near early-forming cross-links (Lys-531 and Lys-532 on Sec9). The intervening residues contain a mixture of hydrophobic and electrostatic side chains that are incapable of forming cross-links (Leu-534, Met-535, and Arg-536). These residues lie in the middle of the 80- to 90-residue linker region of Sec9, between the two regions that will fold into the SNARE motif helices upon SNARE assembly. Therefore, mutations in these residues will not disrupt the stability of the assembled SNARE complex. To generate a mutant sec9 allele for studying the importance of these residues in the Sec6-Sec9 interaction, the entire stretch was mutated to oppositely charged residues, generating the sec9-142 allele (Lys-533 to Glu, Leu-534 to Glu, Met-535 to Glu, Arg-536 to Glu, and Glu-537 to Arg).
The sec9-142 Allele Causes Growth Defects in Sensitized Backgrounds but Does Not Disrupt Sec6-Sec9 Direct Binding
We first tested whether this allele could function as the sole copy of SEC9 under the control of its endogenous promoter in yeast. We predicted that the interaction between Sec6 and Sec9 is necessary and that disruption of the interaction would result in a growth defect because of disruption of the secretory pathway. To test this, we integrated the sec9-142 allele into the SEC9 genomic locus, replacing the wild-type SEC9. We performed a serial dilution assay to compare the growth of wild-type and sec9-142 yeast at various temperatures (WT versus sec9-142, Fig. 3A). In this assay, there were no observable differences between the two strains, suggesting that this mutant protein is sufficient for growth under otherwise wild-type conditions.
FIGURE 3.

The sec9-142 mutant causes synthetic growth defects in combination with other components of the late secretory pathway, but does not disrupt Sec6-Sec9 direct binding. A, the sec9-142 mutation was integrated into the genomic SEC9 locus and tested either alone or in combination with other secretory pathway temperature-sensitive mutants at various temperatures. The sec1-1/sec9-142, sec3-2/sec9-142, sec8-6/sec9-142, and sec15-2/sec9-142 combinations showed synthetic growth defects. B, the indicated concentration of Sec6 was incubated with 25 nm GST-protein and immobilized on glutathione magnetic resin, and then the fold change over GST-alone binding was quantified. The differences between all pairwise interactions were determined by a one-way analysis of variance statistical test on non-matched parametric data with Holm-Sidak correction for multiple comparisons in GraphPad Prism. n = 3, graphed as mean ± S.E. *, p ≤ 0.05; **, p ≤ 0.01; ****, p ≤ 0.001.
Although the sec9-142 strain grew similarly to the wild type, it may have a synthetic/additive effect with other mutants when the Sec9-142 protein causes a mild disruption in the exocytic pathway. To test for such synthetic effects, we replaced the wild-type SEC9 gene with the sec9-142 allele in strains containing temperature-sensitive mutations that have exocytic defects at non-permissive temperatures: an exocytic SNARE regulator (sec1-1), and subunits of the exocyst complex (sec3-2, sec5-24, sec6-4, sec8-6, sec10-2 and sec15-2) (33). Each double-mutant haploid strain was then tested for growth at various temperatures (23–37 °C). In these sensitized backgrounds, sec9-142 showed synthetic growth defects in combination with sec1-1, sec3-2, sec8-6, and sec15-2. These results revealed a modest defect for the sec9-142 mutant in exocytosis and cell growth.
We hypothesized that the synthetic growth defect was due to a decreased affinity of Sec6 for Sec9, and therefore tested whether these mutations were sufficient to disrupt Sec6-Sec9 direct binding. A low concentration of GST-Sec9 or GST-Sec9-142 (25 nm) was incubated with three different concentrations of Sec6 (1, 0.1, and 0.01 μm), and the extent of binding was quantified as a -fold change over binding to GST alone. We found that Sec6 binds to both GST-Sec9 and GST-Sec9-142 significantly over the background (Fig. 3B) but detected no significant differences in binding between the two Sec9 constructs. This is likely due to the capability of an IDP to bind to an ordered partner with multiple low-affinity binding sites and our disruption of only one of those sites (58–60).
Sec6 Does Not Affect the Rate of SNARE Assembly in Vitro
Although we could detect no significant loss of the Sec6-Sec9 interaction, the function of Sec6 as a SNARE assembly inhibitor may be affected in the presence of the Sec9-142 protein, resulting in the observed synthetic growth defect. To further investigate this, we tested the effect of the Sec9-142 mutation on SNARE complex assembly in vitro. Our earlier studies demonstrated a decreased rate of SNARE assembly (∼4-fold), as monitored by gel filtration chromatography in the presence of Sec6, as well as reduced levels of assembled SNARE complexes using a native gel mobility shift assay (46).
To test whether the Sec9-142 protein is sufficient to remove the inhibition of Sec6 on SNARE assembly, we adapted the previous native gel binary SNARE assembly assay to measure the SNARE assembly rate. Previously, these experiments were run on thin, upright, native gels (pH 7.4). Because of the lack of a stacking layer, both the binary SNARE band and the uncomplexed Sso1 band were often resolved inconsistently. We modified the assay to use a “slab” gel (a thicker, 0.5- to 1.0-cm polyacrylamide gel polymerized in a horizontal gel box) and also lowered the pH of the gel (pH 6.6). These modifications resulted in sharper resolution of the free Sso1 band. The experiment was run with seven time points incubated for up to 72 h, due to the slow nature of in vitro SNARE assembly with the autoinhibited form of Sso1 (11), and the resulting decrease in the uncomplexed Sso1 bands over time was fit to the integrated second order rate equation (11).
This modified assay was used with various combinations of proteins (Sec9 + Sso1, Sec9-142 + Sso1, Sec9 + Sso1 + Sec6, and Sec9-142 + Sso1 + Sec6) to determine whether Sec9-142 affected the rate of SNARE complex assembly. First, we tested whether the Sec9-142 mutant protein has a defect in SNARE assembly by comparing the rates of assembly of the mutant and wild-type Sec9 with Sso1, in the absence of Sec6. After quantification of the free Sso1 band (Fig. 4, A, top left versus top right panel, and B, light blue versus yellow curves), the observed rate (k) of assembly of SNAREs containing Sec9-142 was within 2-fold of those containing wild-type Sec9 (1.66 ± 0.408 m−1s−1 for Sec9-142 versus 0.878 ± 0.318 m−1s−1 for Sec9). We therefore concluded that Sec9-142 does not have a significant effect on the rate of binary SNARE complex assembly.
FIGURE 4.

Sec6 does not affect the rate of SNARE assembly. The cytoplasmic domain of Sso1 and the SNAP-25 domain of Sec9 (WT or Sec9-142) were incubated together at 10 μm for 0–72 h, with or without equimolar Sec6, and resolved on a histidine-MOPS polyacrylamide native gel (pH 6.6). The free Sso1 band was quantified and fit to an integrated second-order rate equation, assuming equal concentrations of reactants as described previously (11). A, representative native gels showing the assembled SNARE complex band and free Sso1 band, which was quantified. The asterisk denotes the non-resolved Sec9 band and nonspecific contaminants from the Sec9-142 prep, and two asterisks denote a contaminant from the Sec9 prep. B, quantification of the native gel assays fit to the integrated second-order rate equation assuming equal concentration of reactants. Data from four replicates were fit globally, and the rate constants are reported as mean ± S.E. of the fit (GraphPad Prism).
We next tested whether Sec6 was able to inhibit the formation of Sec9-142-containing SNARE complexes. Unexpectedly, we did not observe an inhibition of SNARE complex assembly by Sec6 with either the wild type or Sec9-142 despite a potential decrease in the observed intensity of the assembled SNARE complex band (Fig. 4A, bottom left versus top left panel). When the loss of uncomplexed Sso1 over time was quantified (Fig. 4B), there was no significant difference between any of the conditions tested (Sec9, k = 0.878 ± 0.096 m−1s−1; Sec9-142, k = 1.66 ± 0.408 m−1s−1; Sec9+Sec6, k = 0.908 ± 0.273 m−1s−1; Sec9-142+Sec6, k = 1.39 ± 0.415 m−1s−1). Curiously, the extent of the reaction appears to be different under the Sec9 + Sec6 condition: at t = ∞, ∼30% of Sso1 calculated to remain unbound when Sec6 is present, whereas <5% of Sso1 is calculated to remain unbound under the other conditions. Therefore, Sec6 does not appear to affect the rate of observed SNARE assembly but may be affecting the final equilibrium between the proteins. SNARE complex assembly may not be able to proceed to completion (full depletion of Sso1) because of sequestration by Sec6 of a fraction of Sec9 but not Sec9-142. Several explanations could be responsible for the differences between these results and the previous ones, including the fact that the earlier quantifications were performed on samples analyzed by gel filtration chromatography rather than native gels. To understand these conflicting results, we sought to determine whether the differences in the experimental designs were responsible for the discrepancies, and/or whether one or both experiments were misinterpreted.
Gel Filtration Chromatography Cannot Resolve Uncomplexed Sec6 and Sso1 Peaks
To examine differences between the gel filtration and native gel SNARE assembly assays, we repeated the gel filtration assay as described previously (11, 46). In this assay, we monitored SNARE complex assembly by a reduction in the uncomplexed Sso1 A280 peak height. The free Sso1 peak heights in this experiment appeared to be similar to our previous findings (Fig. 5A and Ref. 46). However, when we examined the entirety of the gel filtration profiles, we found that the previous analyses did not account for the contribution of uncomplexed Sec6. Sec6 has a long tail on the gel filtration column and overlaps with the free Sso1 peak, causing an increase in the Sso1 peak height (Fig. 5B, blue trace). When Sec9 is incubated with Sec6 and Sso1, Sec9 binds Sec6, and the Sec6-Sec9 complex migrates as a larger complex that has little to no overlap with the free Sso1 peak (Fig. 5B, red trace). As SNARE complex assembly proceeds, Sec6 is released, and its peak shifts back to the uncomplexed position. This causes an increased overlap with the free Sso1 peak (Fig. 5B, red, orange, and yellow traces) and an increase in the apparent Sso1 peak height at later time points. Additionally, because of a small fraction of Sec6 aggregating over time, a fraction of it can be found in a peak that exits near the void volume of the column. Sso1 and Sec9 are also found in this aggregate peak (data not shown), which affects our ability to quantify the amount of free Sso1. Therefore, quantification of Sso1 peak heights may previously have led to an erroneous interpretation of the rate of binary SNARE assembly in the presence of Sec6.
FIGURE 5.

Sec6 leads to an artificial apparent increase in the free Sso1 peak by gel filtration over time. A, the binary Sec9-Sso1 SNARE complex assembly assay with or without Sec6 as monitored by gel filtration, showing the free Sso1 peak height. As reported previously (46), quantification of this peak led to the conclusion that Sec6 was inhibitory to SNARE complex assembly. B, analysis of the full peaks, including an equimolar Sso1 + Sec6 control, demonstrates that uncomplexed Sec6 leads to an apparent increase in the free Sso1 peak relative to Sso1 at 0 h. C, the three fractions corresponding to the free Sso1 peak (denoted under the Sso1 curve in B) were resolved on SDS-PAGE and stained with Krypton protein stain (Pierce). Shown is a representative gel set from gel filtration experiments in the absence of Sec6. D, the bands in C and the other gels were quantified by densitometry and normalized to the band at t = 0. These values were analyzed together and analyzed as in Fig. 4.
To circumvent the overlapping peaks problem in the gel filtration experiments, we quantified changes in the amount of free Sso1 in the eluted free peak using protein gels. The three fractions that corresponded to the top of the free Sso1 peak from each time point were run on SDS-PAGE gels. All proteins were then detected with the high-sensitivity Krypton fluorescent protein stain (Fig. 5C), and the free Sso1 band was analyzed to determine the rate constant for the loss of free Sso1 (Fig. 5D) (see “Experimental Procedures” for details of the analysis). This rate is faster than that measured by native gel in Fig. 4 (0.488 ± 0.3858 m−1s−1 for Sec9 and 0.583 ± 0.341 m−1s−1 for Sec9 + Sec6), but with larger errors. Despite the differences, these data agree that the presence of Sec6 does not inhibit the rate of SNARE complex assembly.
Sec6 Binds Both the Binary and Ternary SNARE Complexes, and This Interaction Is Reduced Significantly When the Sec9-142 Mutant Is Present
Our results above resolved the difference between our current data and those published previously (46). However, they did not provide insights into the nature of the Sec6-Sec9 interaction in vivo. We therefore tested whether, as observed for other MTC-SNARE complex pairs (44, 45, 47–51), Sec6 could bind to binary Sec9-Sso1 or ternary Sec9-Sso1-Snc2 SNARE complexes. We designed the in vitro binding experiments to maximize the signal from the bound complex (see “Experimental Procedures”). At concentrations much higher or much lower than the equilibrium dissociation concentration (Kd), the signals will not be significantly different between various conditions due to the absence of binding at low concentrations and increased background binding at high concentrations. At concentrations of Sec6 closer to the Kd, differences in signal will be more readily observable. For each binding reaction, the molar ratio of Sec6:GST-protein was normalized to the GST background signal, and all related conditions were tested for significant differences in binding (one-sided analysis of variance with multiple comparisons on non-matched data sets).
We chose to use 0.05, 0.5, and 5.0 μm Sec6 in these experiments because 0.5–1.0 μm was the previously estimated Kd for the Sec6-Sec9 complex (46). These concentrations were added to various SNARE constructs for a final concentration of 25 nm each construct: GST and GST-Sso1 as negative controls, GST-Sso1·Sec9 and GST-Sso1·Sec9-142 to test binding to the binary SNARE complex, and GST-Sso1·Sec9·Snc2 and GST-Sso1·Sec9-142·Snc2 to test for binding to the ternary SNARE complexes. All Sso1-containing constructs were formed using just the SNARE domain (Sso1-CTb) to maximize purification of the assembled SNARE complexes. The binding of Sec6 to SNARE complexes at concentrations 5- to 10-fold above (Fig. 6, 5 μm, left panel) or below (Fig. 6, 0.05 μm, right panel) the approximate Kd of the Sec6-Sec9 interaction show no significant differences over background under any of the conditions tested.
FIGURE 6.
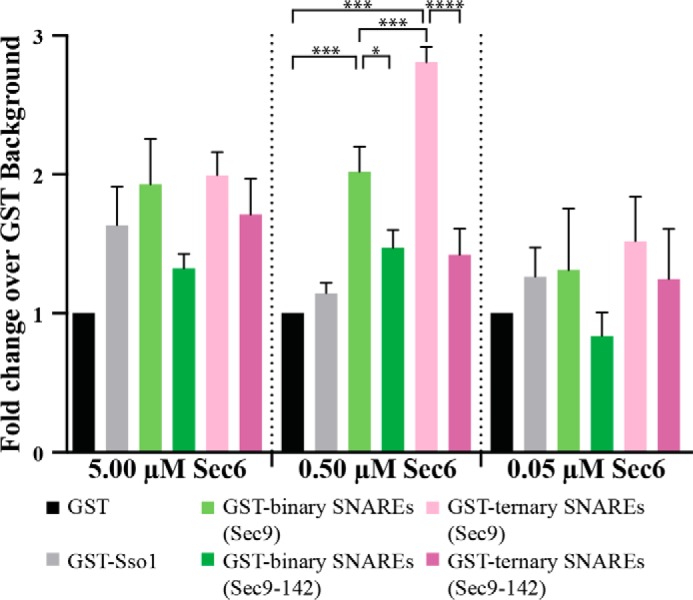
Sec6 binds both the binary and ternary SNARE complex. Sec6 protein, at the listed concentrations, was incubated with 25 nm GST and GST-SNAREs (GST-Sso1CTb, GST-Sso1CTb·Sec9 or binary SNAREs, and GST-Sso1CTb·Sec9·Snc2 or ternary SNAREs). The Sec6 protein that bound to the beads was quantified by densitometry of SDS-PAGE stained with Krypton high-sensitivity protein stain. Each experiment is represented as the fold change over the GST-only background binding, and the differences between the pairwise interactions were determined by a one-way analysis of variance statistical test on non-matched parametric data with Holm-Sidak correction for multiple comparisons in GraphPad Prism. n = 3, graphed as mean ± S.E. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.005; ****, p ≤ 0.001.
When the GST-SNAREs were incubated at concentrations near the Kd (Fig. 6, 0.5 μm, center panel), the amount of Sec6 bound to SNARE complexes was significantly different from GST alone. Sec6 bound both binary GST-Sso1·Sec9 (Fig. 6, light green column) and ternary GST-Sso1·Sec9·Snc2 (Fig. 6, light pink column) SNARE complexes over background (p ≤ 0.005). Additionally, there was a significant increase in Sec6 binding to ternary complexes compared with binary complexes, indicating that Sec6 has a greater affinity for the ternary than the binary complex (Fig. 6, p ≤ 0.005, light green versus light pink columns). Finally, the binding to binary and ternary SNARE complexes containing the Sec9-142 protein was decreased significantly (Fig. 6, p ≤ 0.05 for binary SNARE complexes, light green versus dark green bars, and p ≤ 0.001 for ternary SNARE complexes, light pink versus dark pink bars). Neither binary nor ternary SNARE complexes containing Sec9-142 bound Sec6 significantly over the GST background. These data demonstrate that Sec6 binds both the binary and ternary SNARE complexes. Furthermore, binding of Sec6 to SNARE complexes requires residues in the linker region of Sec9 because the interaction is abrogated in the presence of Sec9-142. Additionally, they demonstrate how the folded and disordered forms of Sec9 vary in their ability to bind Sec6. Although the disordered protein was able to compensate for the loss of the Sec9-142 binding site, the ordered protein in the assembled complex is no longer able to compensate for those mutated residues.
Discussion
Our biochemical studies identified, for the first time, an interaction between a subunit of the yeast exocyst tethering complex and assembled exocytic SNARE complexes (Fig. 6). Our previously published results had concluded that Sec6 was inhibitory to binary Sec9-Sso1 SNARE complex assembly (46), a finding in contrast with other identified MTC-SNARE interactions (44, 45, 47–54). However, this analysis instead suggests that the previous data were misinterpreted. We show here that Sec6 can bind both the binary Sec9-Sso1 and ternary Sec9-Sso1-Snc2 SNARE complexes, and that these interactions are disrupted by the Sec9-142 mutations. We therefore conclude that the synthetic growth defect observed in sec9-142 cells is due to a disruption of the Sec6-SNARE complex interaction rather than a loss of Sec6-mediated SNARE inhibition.
In addition to the linker of Sec9, residues in the v-SNARE Snc2 may be contributing to the Sec6-SNARE complex interaction. Mutations in Snc2 that face outward from the assembled SNARE complex disrupt a direct interaction between Sec6 and Snc2, resulting in defects in localization of the exocyst complex (70). Our studies show that Sec6 has a higher affinity for the Sec9-Sso1-Snc2 ternary complex than the Sec9-Sso1 binary complex (Fig. 6). We therefore suggest that Snc2 is providing additional binding residues on the surface of the assembled SNARE complex for Sec6 binding and that the Snc2 mutant protein may be disrupting Sec6-SNARE complex binding, similar to the disruption seen with the Sec9-142 mutant. Therefore, the role of the Sec6-SNARE complex interaction may be to help maintain proper localization of the exocyst complex.
Most MTCs, including exocyst, interact with their cognate SNARE complexes or with individual SNARE proteins (44–54). However, the function of these interactions is not well understood. The best studied of the MTC-SNARE complex interactions is between HOPS and its cognate SNARE complex. This interaction appears to have several functions. First, HOPS recruitment to the assembled SNARE complex is important for maintaining its localization at sites of vacuole fusion (71). Secondly, HOPS competes with the disassembly machinery (Sec17/Sec18) for binding to the assembled SNARE complex and preferentially binds to trans-SNARE complexes. This likely protects the prefusion trans-SNARE complex from disassembly (44, 72). Finally, HOPS has a higher affinity for properly formed SNARE complexes than for non-cognate SNARE complexes, suggesting that it may “proofread” the SNARE complex prior to fusion (55).
It is interesting to speculate that exocyst-SNARE complex interactions may have similar roles as the HOPS-SNARE interactions. HOPS and exocysts are not structurally homologous to each other, but similarities in their other binding partners suggest that they may be functionally analogous. Both interact with Rab GTPases, lipids, and Sec1/Munc18 proteins. However, the Sec1/Munc18 protein for HOPS, Vps33, is a stoichiometric member of the HOPS complex, whereas the exocyst binds transiently to its Sec1/Munc18 protein, Sec1, through Sec6 (73, 74). Similarly, the COG complex, which is structurally related to exocysts, binds both the Sec1/Munc18 proteins Sly1 and Vps45 through its Cog4 subunit (75, 76). These similar protein-protein interactions suggest that additional interactions and roles may be conserved across trafficking pathways and across species. The regulation of SNARE complex assembly by the tethering factors may be one such commonality.
The studies presented here provide a crucial step toward deciphering the role of exocyst-SNARE interactions in exocytic growth in yeast and suggest future experiments in other model systems. We now show that Sec6 is not a negative regulator of SNARE complex assembly but, rather, that it interacts directly with SNARE complexes. This is a common feature among MTCs from various trafficking pathways in different species. However, many questions remain unanswered. Is the entire exocyst complex involved in the SNARE interaction or just the Sec6 subunit? Is the SNARE complex responsible or required for recruiting the exocyst to the plasma membrane? What roles do other molecules (Rab GTPases, myosin-type motors, lipids, etc.) play in this process? Moving forward, tools developed recently for purifying the yeast exocyst complex and testing the functional consequences of individual subunit loss3 may be adapted to answer some of these questions. For now, these studies provide a crucial first step in identifying a novel, conserved, and required interaction and an allele with which to test these hypotheses in future.
Author Contributions
M. M. and M. L. D. designed the study and wrote the paper. S. A. S. designed the mass spectrometry experiments, which were optimized and performed by S. M. M. L. D. performed all other experiments and completed all data analysis. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank P. Novick (University of California, San Diego, La Jolla, CA) for yeast strains and advice. We also thank F. Morgera for initial observation of the Sec6-ternary SNARE complex interaction and R. Gilmore, S. Ryder, D. Lambright, and members of the Munson laboratory for critical reading of the manuscript and advice.
This work was supported by National Institutes of Health Grant GM068803 (to M. M.). The authors declare they have no conflicts of interest with the contents of this article.
M. R. Heider, submitted for publication.
- MTC
- multisubunit tethering complex
- IDP
- intrinsically disordered protein
- t
- target
- v
- vesicle
- CATCHR
- complexes associated with tethering containing helical rods
- COG
- conserved oligomeric Golgi
- HOPS
- homotypic fusion and vacuole protein sorting
- EDC
- 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride.
References
- 1.Jahn R., and Scheller R. H. (2006) SNAREs: engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643 [DOI] [PubMed] [Google Scholar]
- 2.Yu I.-M., and Hughson F. M. (2010) Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol. 26, 137–156 [DOI] [PubMed] [Google Scholar]
- 3.Heider M. R., and Munson M. (2012) Exorcising the exocyst complex. Traffic 13, 898–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossetto O., Schiavo G., Montecucco C., Poulain B., Deloye F., Lozzi L., and Shone C. C. (1994) SNARE motif and neurotoxins. Nature 372, 415–416 [DOI] [PubMed] [Google Scholar]
- 5.Söllner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., and Rothman J. E. (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324 [DOI] [PubMed] [Google Scholar]
- 6.Brennwald P., Kearns B., Champion K., Keränen S., Bankaitis V., and Novick P. (1994) Sec9 is a SNAP-25-like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell 79, 245–258 [DOI] [PubMed] [Google Scholar]
- 7.Aalto M. K., Ronne H., and Keränen S. (1993) Yeast syntaxins Sso1p and Sso2p belong to a family of related membrane proteins that function in vesicular transport. EMBO J. 12, 4095–4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerst J. E., Rodgers L., Riggs M., and Wigler M. (1992) SNC1, a yeast homolog of the synaptic vesicle-associated membrane protein/synaptobrevin gene family: genetic interactions with the RAS and CAP genes. Proc. Natl. Acad. Sci. U.S.A. 89, 4338–4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber T., Zemelman B. V., McNew J. A., Westermann B., Gmachl M., Parlati F., Söllner T. H., and Rothman J. E. (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 [DOI] [PubMed] [Google Scholar]
- 10.Rice L. M., Brennwald P., and Brünger A. T. (1997) Formation of a yeast SNARE complex is accompanied by significant structural changes. FEBS Lett. 415, 49–55 [DOI] [PubMed] [Google Scholar]
- 11.Nicholson K. L., Munson M., Miller R. B., Filip T. J., Fairman R., and Hughson F. M. (1998) Regulation of SNARE complex assembly by an N-terminal domain of the t-SNARE Sso1p. Nat. Struct. Biol. 5, 793–802 [DOI] [PubMed] [Google Scholar]
- 12.Fasshauer D., Bruns D., Shen B., Jahn R., and Brünger A. T. (1997) A structural change occurs upon binding of syntaxin to SNAP-25. J. Biol. Chem. 272, 4582–4590 [DOI] [PubMed] [Google Scholar]
- 13.Söllner T., Bennett M. K., Whiteheart S. W., Scheller R. H., and Rothman J. E. (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75, 409–418 [DOI] [PubMed] [Google Scholar]
- 14.Hayashi T., McMahon H., Yamasaki S., Binz T., Hata Y., Südhof T. C., and Niemann H. (1994) Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 13, 5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McNew J. A., Parlati F., Fukuda R., Johnston R. J., Paz K., Paumet F., Söllner T. H., and Rothman J. E. (2000) Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature 407, 153–159 [DOI] [PubMed] [Google Scholar]
- 16.Izawa R., Onoue T., Furukawa N., and Mima J. (2012) Distinct contributions of vacuolar Qabc- and R-SNARE proteins to membrane fusion specificity. J. Biol. Chem. 287, 3445–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang B., Gonzalez L. Jr., Prekeris R., Steegmaier M., Advani R. J., and Scheller R. H. (1999) SNARE interactions are not selective: implications for membrane fusion specificity. J. Biol. Chem. 274, 5649–5653 [DOI] [PubMed] [Google Scholar]
- 18.Fasshauer D., Antonin W., Margittai M., Pabst S., and Jahn R. (1999) Mixed and non-cognate SNARE complexes: characterization of assembly and biophysical properties. J. Biol. Chem. 274, 15440–15446 [DOI] [PubMed] [Google Scholar]
- 19.Hohenstein A. C., and Roche P. A. (2001) SNAP-29 is a promiscuous syntaxin-binding SNARE. Biochem. Biophys. Res. Commun. 285, 167–171 [DOI] [PubMed] [Google Scholar]
- 20.Weimbs T., Low S. H., Chapin S. J., Mostov K. E., Bucher P., and Hofmann K. (1997) A conserved domain is present in different families of vesicular fusion proteins: a new superfamily. Proc. Natl. Acad. Sci. U.S.A. 94, 3046–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dietrich L. E., Boeddinghaus C., LaGrassa T. J., and Ungermann C. (2003) Control of eukaryotic membrane fusion by N-terminal domains of SNARE proteins. Biochim. Biophys. Acta 1641, 111–119 [DOI] [PubMed] [Google Scholar]
- 22.Munson M., Chen X., Cocina A. E., Schultz S. M., and Hughson F. M. (2000) Interactions within the yeast t-SNARE Sso1p that control SNARE complex assembly. Nat. Struct. Biol. 7, 894–902 [DOI] [PubMed] [Google Scholar]
- 23.Medine C. N., Rickman C., Chamberlain L. H., and Duncan R. R. (2007) Munc18–1 prevents the formation of ectopic SNARE complexes in living cells. J. Cell Sci. 120, 4407–4415 [DOI] [PubMed] [Google Scholar]
- 24.Zhang X., Bi E., Novick P., Du L., Kozminski K. G., Lipschutz J. H., and Guo W. (2001) Cdc42 interacts with the exocyst and regulates polarized secretion. J. Biol. Chem. 276, 46745–46750 [DOI] [PubMed] [Google Scholar]
- 25.Zhang X., Orlando K., He B., Xi F., Zhang J., Zajac A., and Guo W. (2008) Membrane association and functional regulation of Sec3 by phospholipids and Cdc42. J. Cell Biol. 180, 145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adamo J. E., Rossi G., and Brennwald P. (1999) The Rho GTPase Rho3 has a direct role in exocytosis that is distinct from its role in actin polarity. Mol. Biol. Cell 10, 4121–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He B., Xi F., Zhang X., Zhang J., and Guo W. (2007) Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 26, 4053–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamashita M., Kurokawa K., Sato Y., Yamagata A., Mimura H., Yoshikawa A., Sato K., Nakano A., and Fukai S. (2010) Structural basis for the Rho- and phosphoinositide-dependent localization of the exocyst subunit Sec3. Nat. Struct. Mol. Biol. 17, 180–186 [DOI] [PubMed] [Google Scholar]
- 29.Baek K., Knödler A., Lee S. H., Zhang X., Orlando K., Zhang J., Foskett T. J., Guo W., and Dominguez R. (2010) Structure-function study of the N-terminal domain of exocyst subunit Sec3. J. Biol. Chem. 285, 10424–10433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo W., Roth D., Walch-Solimena C., and Novick P. (1999) The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. EMBO J. 18, 1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H., Turner C., Gardner J., Temple B., and Brennwald P. (2010) The Exo70 subunit of the exocyst is an effector for both Cdc42 and Rho3 function in polarized exocytosis. Mol. Biol. Cell. 21, 430–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunet S., and Sacher M. (2014) Are all multisubunit tethering complexes bona fide tethers? Traffic 15, 1282–1287 [DOI] [PubMed] [Google Scholar]
- 33.Novick P., Field C., and Schekman R. (1980) Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21, 205–215 [DOI] [PubMed] [Google Scholar]
- 34.Grote E., Carr C. M., and Novick P. J. (2000) Ordering the final events in yeast exocytosis. J. Cell Biol. 151, 439–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farré J.-C., and Subramani S. (2011) Rallying the exocyst as an autophagy scaffold. Cell 144, 172–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bodemann B. O., Orvedahl A., Cheng T., Ram R. R., Ou Y.-H., Formstecher E., Maiti M., Hazelett C. C., Wauson E. M., Balakireva M., Camonis J. H., Yeaman C., Levine B., and White M. A. (2011) RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144, 253–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nichols C. D., and Casanova J. E. (2010) Salmonella-directed recruitment of new membrane to invasion foci via the host exocyst complex. Curr. Biol. 20, 1316–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sivaram M. V., Furgason M. L., Brewer D. N., and Munson M. (2006) The structure of the exocyst subunit Sec6p defines a conserved architecture with diverse roles. Nat. Struct. Mol. Biol. 13, 555–556 [DOI] [PubMed] [Google Scholar]
- 39.Wu S., Mehta S. Q., Pichaud F., Bellen H. J., and Quiocho F. A. (2005) Sec15 interacts with Rab11 via a novel domain and affects Rab11 localization in vivo. Nat. Struct. Mol. Biol. 12, 879–885 [DOI] [PubMed] [Google Scholar]
- 40.Dong G., Hutagalung A. H., Fu C., Novick P., and Reinisch K. M. (2005) The structures of exocyst subunit Exo70p and the Exo84p C-terminal domains reveal a common motif. Nat. Struct. Mol. Biol. 12, 1094–1100 [DOI] [PubMed] [Google Scholar]
- 41.Hamburger Z. A., Hamburger A. E., West A. P. Jr., and Weis W. I. (2006) Crystal structure of the S. cerevisiae exocyst component Exo70p. J. Mol. Biol. 356, 9–21 [DOI] [PubMed] [Google Scholar]
- 42.Moore B. A., Robinson H. H., and Xu Z. (2007) The crystal structure of mouse Exo70 reveals unique features of the mammalian exocyst. J. Mol. Biol. 371, 410–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Croteau N. J., Furgason M. L., Devos D., and Munson M. (2009) Conservation of helical bundle structure between the exocyst subunits. PLoS ONE 4, e4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins K. M., Thorngren N. L., Fratti R. A., and Wickner W. T. (2005) Sec17p and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J. 24, 1775–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shestakova A., Suvorova E., Pavliv O., Khaidakova G., and Lupashin V. (2007) Interaction of the conserved oligomeric Golgi complex with t-SNARE Syntaxin5a/Sed5 enhances intra-Golgi SNARE complex stability. J. Cell Biol. 179, 1179–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sivaram M. V., Saporita J. A., Furgason M. L., Boettcher A. J., and Munson M. (2005) Dimerization of the exocyst protein Sec6p and its interaction with the t-SNARE Sec9p. Biochemistry 44, 6302–6311 [DOI] [PubMed] [Google Scholar]
- 47.Diefenbacher M., Thorsteinsdottir H., and Spang A. (2011) The Dsl1 tethering complex actively participates in soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor (SNARE) complex assembly at the endoplasmic reticulum in Saccharomyces cerevisiae. J. Biol. Chem. 286, 25027–25038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arasaki K., Takagi D., Furuno A., Sohda M., Misumi Y., Wakana Y., Inoue H., and Tagaya M. (2013) A new role for RINT-1 in SNARE complex assembly at the trans-Golgi network in coordination with the COG complex. Mol. Biol. Cell 24, 2907–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conibear E., Cleck J. N., and Stevens T. H. (2003) Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol. Biol. Cell 14, 1610–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pérez-Victoria F. J., and Bonifacino J. S. (2009) Dual roles of the mammalian GARP complex in tethering and SNARE complex assembly at the trans-Golgi network. Mol. Cell. Biol. 29, 5251–5263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balderhaar H. J. K., Lachmann J., Yavavli E., Bröcker C., Lürick A., and Ungermann C. (2013) The CORVET complex promotes tethering and fusion of Rab5/Vps21-positive membranes. Proc. Natl. Acad. Sci. U.S.A. 10.1073/pnas.1221785110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lürick A., Kuhlee A., Bröcker C., Kümmel D., Raunser S., and Ungermann C. (2015) The HABC domain of the snare vam3 interacts with the hops tethering complex to facilitate vacuole fusion. J. Biol. Chem. 290, 5405–5413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lobingier B. T., and Merz A. J. (2012) Sec1/Munc18 protein Vps33 binds to SNARE domains and the quaternary SNARE complex. Mol. Biol. Cell. 10.1091/mbc.E12-05-0343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krämer L., and Ungermann C. (2011) HOPS drives vacuole fusion by binding the vacuolar SNARE complex and the Vam7 PX domain via two distinct sites. Mol. Biol. Cell 22, 2601–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Starai V. J., Hickey C. M., and Wickner W. (2008) HOPS proofreads the trans-SNARE complex for yeast vacuole fusion. Mol. Biol. Cell 19, 2500–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu H., Jun Y., Thompson J., Yates J. R., and Wickner W. T. (2010) HOPS prevents the disassembly of trans-SNARE complexes by Sec17p/Sec18p during membrane fusion. EMBO J. 29, 1948–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uversky V. N. (2013) Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 1834, 932–951 [DOI] [PubMed] [Google Scholar]
- 58.Uversky V. N. (2013) A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 10.1002/pro.2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uversky V. N. (2002) Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11, 739–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wright P. E., and Dyson H. J. (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.ROSEN H. (1957) A modified ninhydrin colorimetric analysis for amino acids. Arch. Biochem. Biophys. 67, 10–15 [DOI] [PubMed] [Google Scholar]
- 62.Olsen J. V., Schwartz J. C., Griep-Raming J., Nielsen M. L., Damoc E., Denisov E., Lange O., Remes P., Taylor D., Splendore M., Wouters E. R., Senko M., Makarov A., Mann M., and Horning S. (2009) A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics 8, 2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panchaud A., Singh P., Shaffer S. A., and Goodlett D. R. (2010) xComb: A cross-linked peptide database approach to protein-protein interaction analysis. J. Proteome Res. 9, 2508–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radulovic D., Jelveh S., Ryu S., Hamilton T. G., Foss E., Mao Y., and Emili A. (2004) Informatics platform for global proteomic profiling and biomarker discovery using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 3, 984–997 [DOI] [PubMed] [Google Scholar]
- 65.Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., and Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 66.Gietz R. D., and Woods R. A. (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87–96 [DOI] [PubMed] [Google Scholar]
- 67.Sikorski R. S., and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McLellan T. (1982) Electrophoresis buffers for polyacrylamide gels at various pH. Anal. Biochem. 126, 94–99 [DOI] [PubMed] [Google Scholar]
- 69.Wong C., Sridhara S., Bardwell J. C., and Jakob U. (2000) Heating greatly speeds Coomassie Blue staining and destaining. BioTechniques 28, 426–428, 430,, 432 [DOI] [PubMed] [Google Scholar]
- 70.Shen D., Yuan H., Hutagalung A., Verma A., Kümmel D., Wu X., Reinisch K., McNew J. A., and Novick P. (2013) The synaptobrevin homologue Snc2p recruits the exocyst to secretory vesicles by binding to Sec6p. J. Cell Biol. 202, 509–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang L., Merz A. J., Collins K. M., and Wickner W. (2003) Hierarchy of protein assembly at the vertex ring domain for yeast vacuole docking and fusion. J. Cell Biol. 160, 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Collins K. M., and Wickner W. T. (2007) Trans-SNARE complex assembly and yeast vacuole membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 104, 8755–8760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wiederkehr A., de Craene J.-O., Ferro-Novick S., and Novick P. (2004) Functional specialization within a vesicle tethering complex: bypass of a subset of exocyst deletion mutants by Sec1p or Sec4p. J. Cell Biol. 167, 875–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morgera F., Sallah M. R., Dubuke M. L., Gandhi P., Brewer D. N., Carr C. M., and Munson M. (2012) Regulation of exocytosis by the exocyst subunit Sec6 and the SM protein Sec1. Mol. Biol. Cell. 23, 337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laufman O., Kedan A., Hong W., and Lev S. (2009) Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. EMBO J. 28, 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laufman O., Hong W., and Lev S. (2013) The COG complex interacts with multiple Golgi SNAREs and enhances fusogenic assembly of SNARE complexes. J. Cell Sci. 126, 1506–1516 [DOI] [PubMed] [Google Scholar]
- 77.Rost B., Sander C., and Langridge R. (2004) SOPM: a self-optimized method for protein secondary structure prediction. Proteins 10.1110/ps.03503304/full [DOI] [Google Scholar]