

Background: Sustained Cx43 hemichannel opening is detrimental to bone cells; however, the mechanism underlying the closure of hemichannels was unknown.
Results: Extracellular prostaglandin E2 released by hemichannels activates MAPK, leading to Cx43 phosphorylation and hemichannel closure.
Conclusion: Osteocytic Cx43 hemichannels is regulated by a feedback inhibition mechanism.
Significance: This study uncovers a novel pathway in fine-tuning Cx43 hemichannels in response to mechanical stimulation.
Keywords: channel activation, connexin, mitogen-activated protein kinase (MAPK), osteocyte, phosphorylation, Hemichannels, Mechanical stimulation
Abstract
Cx43 hemichannels serve as a portal for the release of prostaglandins, a critical process in mediating biological responses of mechanical loading on bone formation and remodeling. We have previously observed that fluid flow shear stress (FFSS) opens hemichannels; however, sustained FFSS results in hemichannel closure, as continuous opening of hemichannels is detrimental to cell viability and bone remodeling. However, the mechanism that regulates the closure of the hemichannels is unknown. Here, we show that activation of p44/42 ERK upon continuous FFSS leads to Cx43 phosphorylation at Ser279-Ser282, sites known to be phosphorylated sites by p44/42 MAPK. Incubation of osteocytic MLO-Y4 cells with conditioned media (CM) collected after continuous FFSS increased MAPK-dependent phosphorylation of Cx43. CM treatment inhibited hemichannel opening and this inhibition was reversed when cells were pretreated with the MAPK pathway inhibitor. We found that prostaglandin E2 (PGE2) accumulates in the CM in a time-dependent manner. Treatment with PGE2 increased phospho-p44/42 ERK levels and also Cx43 phosphorylation at Ser279-Ser282 sites. Depletion of PGE2 from CM, and pre-treatment with a p44/42 ERK pathway-specific inhibitor, resulted in a complete inhibition of ERK-dependent Cx43 phosphorylation and attenuated the inhibition of hemichannels by CM and PGE2. Consistently, the opening of hemichannels by FFSS was blocked by PGE2 and CM and this blockage was reversed by U0126 and the CM depleted of PGE2. A similar observation was also obtained in isolated primary osteocytes. Together, results from this study suggest that extracellular PGE2 accumulated after continuous FFSS is responsible for activation of p44/42 ERK signaling and subsequently, direct Cx43 phosphorylation by activated ERK leads to hemichannel closure.
Introduction
Osteocytes are mechanosensory cells in the bone. Osteocytes respond to mechanical stimulation by release of signaling molecules, such as prostaglandins, nitric oxide, and ATP (1–4). Such biochemical signals generated in osteocytes are conveyed to other bone cells, such as osteoblasts and osteoclasts, thereby, facilitating bone formation and remodeling. We and others have previously shown that prostaglandin E2 (PGE2)3 released by Cx43 hemichannels acts in an autocrine/paracrine manner via activation of EP2/EP4 receptors to promote gap junction communication through transcriptional regulation of Cx43 (5, 6) and osteocytic survival (7).
Cx43 is post-translationally modified by phosphorylation, which has been reported to be involved in gap junction and hemichannel activity, protein assembly, trafficking and turnover, etc. (8, 9). Several kinases are known to phosphorylate the C terminus of the Cx43 protein and multiple phosphorylation sites have been identified (9). A study shows that phosphorylation at Ser368 on Cx43 by protein kinase C reduces hemichannel permeability in an in vitro reconstituted system (10). Mitogen-activated protein kinase (MAPK)-dependent phosphorylation of Cx43 at Ser279-Ser282 leads to reduced gap junction communication and plaque formation, and also increases internalization of Cx43 from the cell surface (11–13). Casein kinase 1 activity leads to Cx43 phosphorylation at Ser325-Ser328-Ser330 and an increase in gap junction formation (14). Cx43 trafficking to the cell surface increases as a result of protein kinase A activation coincident with an increase in Cx43 phosphorylation at Ser364 (15).
FFSS is known to influence cellular physiology by activating mechanosensors and signaling pathways, especially in endothelial cells and osteocytes (16, 17). PGE2 is an essential molecule that mediates anabolic function of mechanical loading on the bone (18). It has been observed that PGE2 is released by Cx43 hemichannels when osteocytic MLO-Y4 cells are subjected to FFSS (2). We have recently shown that activation of PI3K-Akt signaling by FFSS plays an essential role in activating Cx43 hemichannels (19). Also, Cx43 hemichannels were adaptive as a gradual decrease in hemichannel dye uptake was observed when MLO-Y4 cells were subjected to FFSS for a long period of time (20). Regulated closure of hemichannels is equally as important as the opening of these channels because persistent opening of these big channels would lead to membrane depolarization and disruption of electrical and chemical gradient. Additionally, persistent high concentrations of PGE2 exhibits adverse, inhibitory effect on bone formation (21).
In this paper, experiments were conducted to understand if closure of hemichannels is mediated by the factors released by hemichannels as a result of sustained FFSS and if the factor(s) functions in a feedback inhibitory mechanism in closing hemichannels. We observed that PGE2 released through Cx43 hemichannels induced by mechanical stimulation leads to Cx43 phosphorylation at Ser279-Ser282 by activated MAPK, and this phosphorylation is responsible for closure of Cx43 hemichannels in response to long term, continuous FFSS.
Experimental Procedures
Materials
Culturing media, α-MEM and S-MEM, Lucifer yellow (LY), and rhodamine dextran (RD) used for dye uptake experiments were purchased from Life Technologies. Antibodies against p44/42 ERK, total p44/42 ERK, and PD98059 were obtained from Cell Signaling Laboratories (Boston, MA). Anti-Cx43 phospho-specific Ser279-Ser282 antibody was generated as previously described (22). U0126 was purchased from LC Laboratories (Woburn, MA). An ELISA-based PGE2 kit and PGE2 affinity column were obtained from Cayman Chemicals (Ann Arbor, MI).
Cell Culture, FFSS, and Mechanical Loading by Fluid Dropping
MLO-Y4 cells cultured in α-MEM at low cell density (3 × 104 cells/cm2) on sheets coated with collagen were used for FFSS experiments as described previously (2). Fluid flow was generated by parallel plate flow chambers separated by a gasket of defined thickness with gravity-driven fluid flow using a peristaltic pump. The thickness of the gasket determines the channel height. By adjusting the channel height and flow rate, stress levels of 4 dynes/cm2 were generated. Cells were plated in the flow chamber with the surface area of 5 cm2. Controls consisted of MLO-Y4 cells in S-MEM not subjected to FFSS. Each test was conducted for the respective time as indicated. The circulating medium was S-MEM. The entire flow system was encased within a large walk-in CO2 incubator at 5% CO2 and 37 °C. The mechanical loading was also conducted by dropping 50 μl of S-MEM from 5.7 cm as previously reported (23).
Preparation of Primary Osteocytes
The preparation of enriched primary osteocytes from bone chips was modified from a previously published protocol (24). Briefly, long bones were dissected from 3–4-week-old mice and bone marrow was removed by flushing with PBS. The bones were cut into pieces about 2 mm in length and digested by alternate uses of collagenase type I and EDTA on a rotating shaker in a CO2 incubator at 37 °C. After multiple treatments with collagenase type I and EDTA to remove other bone cells, the bone chips were plated on collagen-coated dish in α-MEM with 2.5% FBS + 2.5% bovine calf serum and left untouched for 9 days. The osteocytes from the bone pieces were remove by trypsinization and seeded in collagen-coated glasses to perform FFSS in α-MEM with 2.5% FBS + 2.5% bovine calf serum.
Dye Uptake Assay
MLO-Y4 cells cultured at low density with minimal cell-cell contacts on glass slides coated with collagen were subjected to FFSS or fluid dropping. The slides were incubated with a mixture containing 0.4% LY (molecular mass 547 Da) and 0.4% RD (molecular mass 10 kDa) for 5 min and washed five times with 1 × phosphate-buffered saline. These cells were later fixed in 1% paraformaldehyde for 10 min. RD is used a tracer and cells that uptake RD are considered as dead cells. Fluorescence images were obtained using a Zeiss epifluorescence microscope. Both LY and RD fluorescence cells were counted and a ratio of cells uptaking LY (minus cells taking RD) to total cells per image was calculated and plotted.
CM Collection, PGE2 Quantification, and Depletion of PGE2 from CM
MLO-Y4 cells cultured on collagen-coated sheets were subjected to continuous FFSS (4 dynes/cm2) for 0.5, 2, 4, 8, and 24 h and cultured media (condition media (CM)) that was used for flow was collected. Control media was collected from parallel cultures without FF treatment. PGE2 in the CM was measured using an ELISA-based PGE2 kit (Cayman Chemicals) based on the manufacturer's instructions. CM was applied to a PGE2 affinity column (Cayman Chemicals) to deplete PGE2 as previously described (25).
Western Blotting
MLO-Y4 cell lysates were lysed in lysis buffer (5 mm Tris, pH 7.8, 5 mm EDTA, 5 mm EGTA) plus a cocktail of protease and phosphatase inhibitors and resolved on SDS-PAGE gels. After transferring the protein onto the nitrocellulose membrane, the membranes were probed with specific antibodies. HRP-conjugated secondary antibodies were used and the blots were developed using an ECL kit (Amersham Biosciences) and the signal was detected with Alexa Fluor 680 goat anti-rabbit (Invitrogen) or Alexa Fluor 800 donkey anti-mouse (Invitrogen) using an Odyssey infrared imager (Li-Cor, Omaha, NE).
Statistical Analysis
All data were analyzed using one-way analysis of variance and Student-Newman-Keul's test was applied for comparison using GraphPad Prism software (GraphPad Prism, San Diego, CA). The significance of the data are implicated with an asterisk (***, p < 0.001; **, p < 0.01; *, p < 0.05).
Results
Sustained FFSS Activates MAPK Signaling, but Inhibition of MAPK Has No Effect on the Opening of Hemichannels
To study the effect of FFSS on activation of the MAPK pathway, continuous FFSS was applied to MLO-Y4 osteocytic cells for 0.5, 2, 4, and 24 h. There is a slight increase in activated, phospho-p44/42 ERK (pERK) levels after 30 min of FFSS and a gradual decrease by 2 and 4 h. Interestingly, after 24 h of FFSS there is a significant increase in pERK levels when compared with control (no FFSS) and 30 min of FFSS (Fig. 1A). Quantification of band intensity confirmed this observation (Fig. 1A, lower panel). To understand if MAPK activation is involved in regulation of the Cx43 hemichannel opening, we pretreated cells with PD98059, a MAPK inhibitor, and stimulated the hemichannel opening either by FFSS (Fig. 1B) or dropping culture media onto the cells (Fig. 1C). Hemichannel opening was analyzed through LY dye uptake where the PD98059-treated cells (FF + PD; DP + PD) showed a similar degree of dye uptake when compared with that of the untreated cells (FF; DP). Untreated, unstimulated cells (C) and PD98059-treated cells (C + PD) were used as controls, which showed minimal dye uptake. These results suggest that the MAPK pathway is activated in response to FFSS after 24 h. However, inhibition of the MAPK pathway does not affect the opening of Cx43 hemichannels.
FIGURE 1.

Continuous FFSS for 24 h activates p44/42 MAPK, but inhibition of activated MAPK does not affect initial Cx43 opening of hemichannels by FFSS. A, MLO-Y4 cells were subjected to FFSS for 0.5, 2, 4, and 24 h, and the lysates collected were immunoblotted using anti-phospho-p44/42 ERK or p44/42 ERK (total ERK) antibody. The ratio of mean band intensities of phospho-p44/42 ERK to total p44/42 ERK was plotted as showing a significant increase in phospho-p44/42 ERK levels after 24-h FFSS. 24 h versus unstimulated cells; ***, p < 0.001, n = 3, and the data are presented as mean ± S.E. B, effect of PD98059, an inhibitor of MAPK, on Cx43 hemichannel opening was determined by treating MLO-Y4 cells with 50 μm PD98059 for 16 h and subjecting the cells to FFSS of 16 dynes/cm2 for 30 min. Untreated control (C) and untreated plus PD98059 cells (C+PD) were used as controls. An increase in dye uptake was observed in PD98059-treated cells when subjected to FFSS (FF+PD) and the percentage of dye uptake was similar to that of the untreated cells subjected to FFSS (FF). C, a similar observation was obtained when cells were mechanically stimulated by dropping medium onto the cells followed by dye uptake analyses. PD98509-treated (DP+PD) cells and untreated cells (DP) showed an increase in dye uptake percentage after stimulation when compared with that of the controls (C+PD and C). All the data are presented as mean ± S.E., n = 3.
Long Exposure of FFSS Increases Cx43 Phosphorylation at Ser279-Ser282
Cx43 Ser279 and Ser282 residues were previously shown to be phosphorylated by MAPK and this phosphorylation is correlated with a decrease of Cx43 gap junction coupling (26). Lysates of MLO-Y4 cells after 0.5, 2, 4, and 24 h of continuous FFSS were immunoblotted using antibodies specifically against Cx43 phospho-Ser279-Ser282 (22) or total Cx43 (Fig. 2A). The ratio of band intensities of MAPK-phosphorylated Cx43 versus total Cx43 was plotted as a graph (Fig. 2B). A significant increase in Cx43 phosphorylation at Ser279-Ser282 was observed after 24 h of FFSS when compared with control (0 h). Also a slight increase in Cx43 phosphorylation was obtained at 30 min of FFSS, which corresponds to a minor increase of active, phosphorylated ERK levels during the initial period of time, possibly due to the effect of fresh culture media.
FIGURE 2.
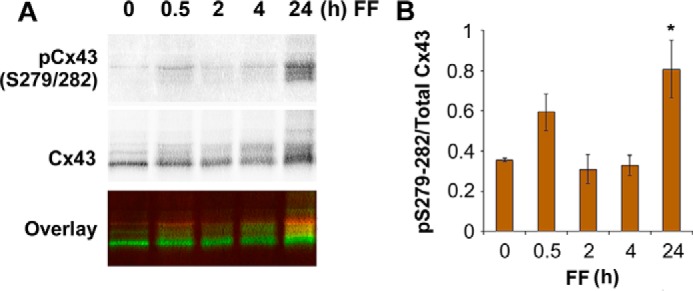
Activation of p44/42 ERK and increase of Cx43 phosphorylation at Ser279-Ser282 residues after continuous FFSS for 24 h. A, MLO-Y4 cell lysates were collected after continuous FFSS for 24 h and immunoblotted using phospho-specific Cx43 Ser279-Ser282 antibody. pCx43S279/282 is indicated in red and total Cx43 in green (bottom panel). B, the ratio of mean band intensities of Cx43 phospho-Ser279-Ser282 and total Cx43 was calculated and was plotted as a graph. All data are presented as mean ± S.E. Non-treated (0 h) versus 24 h, *, p < 0.05, n = 5.
CM from FFSS-treated Cells Increases Phosphorylation of Cx43 at Ser279-Ser282 and Closure of Cx43 Hemichannels
We previously reported that Cx43 hemichannel closure occurs after 24 h of continuous FFSS (20). To determine whether the activation of MAPK and subsequent phosphorylation of Cx43 are mediated by the factor(s) released by continuous FFSS, we treated de novo MLO-Y4 cells with CM collected after subjecting MLO-Y4 cells to 24-h FFSS for different periods of time (0.5, 2, 4, and 24 h). The cell lysates obtained after incubation with CM were immunoblotted using phospho-Ser279-Ser282 antibody and the ratio of band intensities obtained from phospho-Ser279-Ser282 to total Cx43 were quantified (Fig. 3A, right panel). We also examined the phosphorylation of cell surface-expressed Cx43 by ERK after CM treatment for various periods of time and observed that cell surface Cx43 phosphorylation increased after 30 min when compared with the control. A sustained treatment of cells with CM for 2 and 4 h showed a decrease in ERK-dependent Cx43 phosphorylation (Fig. 3B). Interestingly, the level of total Cx43 protein was not altered up to 4 h. These results suggest that the CM is able to induce phosphorylation of Cx43 through activation of the MAPK pathway and the phosphorylation peaks at 30 min after CM treatment for both total Cx43 and also cell surface Cx43.
FIGURE 3.
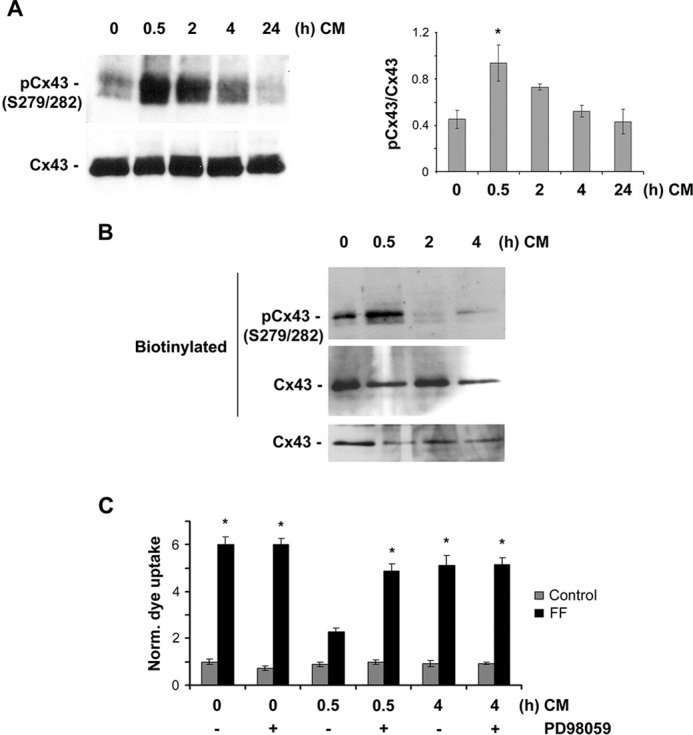
CM collected after 24-h FFSS promotes Cx43 phosphorylation at Ser279-Ser282 residues and inhibits FFSS-induced opening of Cx43 hemichannels. A, MLO-Y4 cells were treated with CM (collected after continuous FFSS for 24 h) for 30 min. Cell lysates were immunoblotted with antibody against Cx43 phospho-specific Ser279-Ser282 or total Cx43 protein. Band intensity was quantified and the graph was plotted using ratio of Cx43 phospho-Ser279-Ser282 and total Cx43. The data are presented as mean ± S.E. CM-treated versus untreated cells: *, p < 0.05; n = 3. B, MLO-Y4 cells were treated with CM (collected after continuous FFSS for 24 h) for 30 min and a cell surface biotinylation assay was performed. Biotinylated samples and total lysates were immunoblotted with antibody against Cx43 phospho-specific Ser279-Ser282 or total Cx43 protein. C, MLO-Y4 cells were pretreated with CM (collected after continuous FFSS for 24 h) for 0.5 and 4 h in the absence or presence of PD98059, subjected to FFSS for 30 min, and assayed for LY dye uptake. Treatment with CM for 30 min significantly blocked dye uptake induced by FFSS, whereas preincubation with CM for 4 h did not have such effect. Pretreatment of MLO-Y4 cells with 50 μm PD98059, a MAPK inhibitor, resulted in a significant reversal of inhibition of dye uptake by the pretreatment of CM for 30 min, not 4 h. With FF, 0.5 h treatment versus all other conditions, *, p < 0.05, n = 3.
To determine whether this phosphorylation leads to closure of Cx43 hemichannels, a dye uptake experiment was performed by incubating fresh MLO-Y4 cells with CM collected from 24-h FFSS and subjecting the cells to 30 min of FFSS. Cells treated with 24-h FF CM showed a significant decrease in dye uptake when compared with that of the untreated cells (Fig. 3C). This inhibition of dye uptake was reversed when the cells were preincubated with the MAPK inhibitor PD98059. As a control for this experiment, cells were treated with FF CM for 4 h and subjected to FFSS. These cells showed similar dye uptake levels as that of the control FFSS-subjected cells. Similarly, cells preincubated with PD98059 and treated with CM for 4 h also had no effect on dye uptake. Results from this experiment suggest that CM obtained from 24-h FF increases Cx43 phosphorylation at Ser279-Ser282 and phosphorylation at these sites leads to closure of Cx43 hemichannels.
PGE2 Released in the CM under Continuous FFSS Is Responsible for MAPK-mediated Cx43 Phosphorylation
The above results suggest that the factor(s) released into the CM during FFSS could be responsible for Cx43 phosphorylation and hemichannel closure. We previously reported that FFSS induces PGE2 release through Cx43 hemichannels (2). In an attempt to identify the factor(s) responsible for Cx43 phosphorylation and subsequent channel closure, the dynamics of PGE2 release was studied during continuous FFSS because PGE2 is one of the factors released by Cx43 in response to FFSS. PGE2 levels were measured in the CM collected from different time points after continuous FFSS (0.5, 2, 4, and 24 h). A time-dependent increase in PGE2 levels in the CM collected from continuous FFSS was observed (Fig. 4A). These results suggest that FFSS induces PGE2 release and PGE2 accumulates over time in the CM collected from continuous FFSS.
FIGURE 4.

Extracellular PGE2 is accumulated during continuous FFSS and PGE2 increases phosphorylation of p44/42 ERK and Cx43 at Ser279-Ser282 residues. A, the concentrations of PGE2 in CM collected from different periods of continuous FFSS were determined using PGE2 EIA kit. 4, 8, and 24 h versus 0 min (un-stimulated); ***, p < 0.001. B and C, MLO-Y4 cells were treated with 1 or 10 nm PGE2 for 0.5, 2, 4, and 24 h and cell lysates were immunoblotted with anti-phospho-p44/42 ERK and p44/42 ERK antibodies (B) or anti-Cx43 Ser279-Ser282 and Cx43 antibody (C). The band intensities were quantified and graphed using the ratios of phospho- versus total p44/42 ERK or Cx43 levels. All the data are presented as mean ± S.E., n = 3.
PGE2 is known to activate MAPK signaling through EP2 or EP4 receptors (27, 28). Previous studies from our laboratory and others have shown that PGE2 signaling occurs through EP2 and EP4 receptors in MLO-Y4 osteocytes (5, 7). To test whether PGE2 mediate Cx43 phosphorylation through activation of the MAPK pathway, MLO-Y4 cells were treated with 1 and 100 nm PGE2 for various periods of time. A time-dependent increase in ERK phosphorylation was observed when compared with total ERK after treatment (Fig. 4B) and higher concentrations of PGE2 appear to require less time to reach full activation. A similar time-dependent increase in Cx43 phosphorylation was observed (Fig. 4C) and this data were quantified (Fig. 4C, lower panel). These results suggest that PGE2 activates the p44/42 ERK pathway, leading to phosphorylation of Cx43 at Ser279-Ser282.
Depletion of PGE2 from CM and Inhibition of ERK 1/2 Kinase Results in Prevention of Cx43 Phosphorylation at Ser279-Ser282
To determine whether PGE2 is responsible for the effect of CM on p44/42 ERK activation and subsequent Cx43 phosphorylation, PGE2 was depleted from the CM collected after continuous FFSS. CM depleted of PGE2 (CM(−PGE2)) was used to treat MLO-Y4 cells for 30 min. CM treatment resulted in an increase in phosphorylation of p44/42 ERK and Cx43 when compared with untreated cells (C) (Fig. 5A). Importantly, depletion of PGE2 from CM (CM(−PGE2)) failed to activate p44/42 ERK and increase Cx43 phosphorylation.
FIGURE 5.
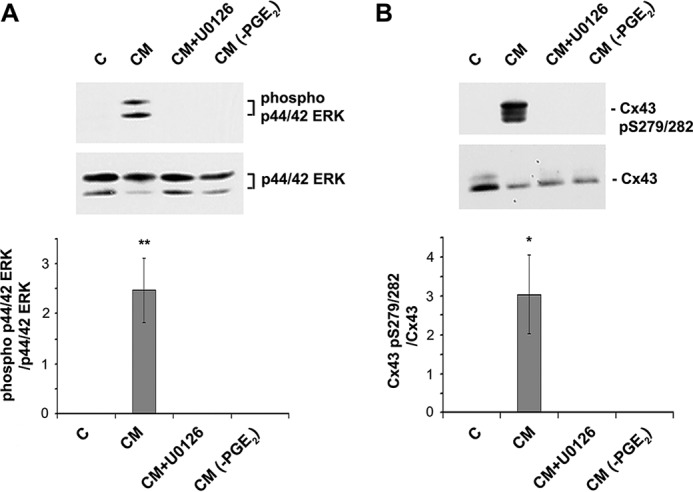
FF-CM depleted of PGE2 and U0126 treatment results in inhibition of p44/42 ERK and Cx43 phosphorylation. MLO-Y4 cell lysates were treated with CM, PGE2-depleted CM, or pretreated with U0126 prior to CM treatment and cell lysates were immunoblotted with anti-phospho-p44/42 ERK and total ERK (A) or anti-Cx43 Ser279-Ser282 and Cx43 antibodies (B). The band intensity was quantified and the ratio of phospho- versus total p44/42 ERK and Cx43 was calculated (A and B, lower panels). All the data are presented as mean ± S.E., n = 3. CM versus C, CM+U0126 and CM-PGE2; **, p < 0.01. *, p < 0.05.
To understand if Cx43 phosphorylation at Ser279-Ser282 is a result of activation of p44/42 ERK pathway, cells were treated with U0126, an ERK1/2-specific inhibitor, prior to incubating with conditioned media (CM + U0126). This treatment led to a complete inhibition of p44/42 ERK phosphorylation as well as Cx43 phosphorylation at Ser279-Ser282 (Fig. 5B), suggesting that the ERK1/2 pathway is responsible for phosphorylation of Cx43.
Extracellular PGE2 Released by Osteocytes and Activated MAPK Are Responsible for Hemichannel Closure by Sustained FFSS
We further examined the role of extracellular PGE2 and activated MAPK signaling in regulating hemichannel activity in response to FFSS. The opening of hemichannels by LY uptake by 30 min FFSS was significantly blocked with the addition of extracellular PGE2 in MLO-Y4 cell and primary osteocytes (Fig. 6, A and B). However, this channel blockage by extracellular PGE2 was attenuated by an ERK1/2 specific inhibitor, U0126 (Fig. 6, A and B). Furthermore, pretreatment of de novo MLO-Y4 cells with CM collected after 24 h exposure to FFSS inhibited the hemichannel opening by FFSS given for 30 min and this inhibition was not observed in CM depleted of PGE2 (Fig. 6C). Together, these data suggest that extracellular PGE2 released by the Cx43 hemichannel in osteocytes functions as a feedback inhibitor that activates MAPK signaling, phosphorylates Cx43, and closes hemichannels.
FIGURE 6.

Inhibition of MAPK and depletion of PGE2 from FF-CM attenuates the inhibitory effect of PGE2 or FF-CM on hemichannel opening in response to FFSS. A, MLO-Y4 cells were treated with 1 nm PGE2 for 24 h and then with or without U0126 for 30 min before subjecting cells to FFSS for 30 min or non-FFSS control. B, primary mouse osteocytes were treated with 1 or 100 nm PGE2 for 24 h and then with or without U0126 for 30 min before subjecting cells to FFSS for 30 min or non-FFSS control. C, MLO-Y4 cells were treated with FF-CM (collected after 24 h of FFSS) or FF-CM depleted of PGE2 (CM(-PGE2)) for 24 h before subjecting the cells to FFSS for 30 min or non-FF control. LY dye uptake assay was performed and quantified. All the data are presented as mean ± S.E., n = 3. ***, p < 0.001; **, p < 0.01; *, p < 0.05.
Discussion
In this study, we demonstrate the molecular basis of Cx43 hemichannel regulation in response to mechanical stimulation. p44/42 ERK signaling invoked by accumulative PGE2 released by continuous FFSS is responsible for the inhibition of Cx43 hemichannel opening. A long exposure of FFSS results in phosphorylation of Cx43 by activated p44/42 ERK, which leads to the closing of Cx43 hemichannels. PGE2 is observed to accumulate during continuous FFSS and found to activate p44/42 ERK, which subsequently leads to phosphorylation of Cx43. Importantly, PGE2 in the CM appears to be the key regulator of p44/42 ERK activation and phosphorylation of Cx43 at Ser279-Ser282, which leads to hemichannel closure. As illustrated in Fig. 7, our results suggest that PGE2 released through osteocytic Cx43 hemichannels in an autocrine/paracrine fashion acts as a negative feedback factor that activates MAPK signaling leading to Cx43 phosphorylation and closure of hemichannels. Controlled closing of osteocytic hemichannels is crucial for modulating the anabolic function of mechanical stimulation on bone tissue, reducing the catabolic effect by higher concentrations of extracellular prostaglandins and protecting osteocytes from detrimental effects caused by sustained opening of hemichannels.
FIGURE 7.

A schematic model showing the hemichannel closure after long FFSS is regulated by Cx43 phosphorylation by MAPK, which is activated by extracellular PGE2 released by hemichannels via feedback inhibition mechanism. During short term FFSS, Cx43 hemichannels (HC) in osteocytes are induced open, which allows the release of bone modulating factors such as PGE2. A sustained mechanical stimulation causes Cx43 hemichannel closure due to accumulation of PGE2 in the extracellular environment. The accumulated PGE2, likely through the EP2/EP4 receptor, activates p44/42 MAPK and increases Cx43 phosphorylation at Ser279-Ser282 residues thereby leading to closure of Cx43 hemichannels.
In response to mechanical stimulation, osteocytes are assumed to sense stress and convey mechanical signals to other bone cell types, such as osteoblasts and osteoclasts (29). Osteocytes respond to mechanical stimuli by releasing bone remodeling factors, such as prostaglandins, ATP, NO, etc. (1, 3, 4, 25). These factors elicit downstream signaling pathways in osteocytes as well as other bone cells to ultimately modulate bone formation and remodeling. We have previously shown that PGE2 is released by osteocyte-like cells, MLO-Y4 cells through Cx43 hemichannels in response to FFSS (2). We have also shown that FFSS initially leads to opening of Cx43 hemichannels, but these channels gradually close with time and no hemichannel activity was detected after 24 h FFSS (20). Due to addition of fresh medium, we observed that ERK activation and Cx43 phosphorylation at Ser279-Ser282 were slightly increased after 0.5 h. However, there was no impact on Cx43 hemichannel closure. This is likely caused by the activation of PI3K-Akt at the early stage of FFSS. We recently showed that the PI3K-Akt pathway activated by FFSS is involved in opening the Cx43 hemichannels (19). AKT phosphorylation on Cx43 directly regulates Cx43 binding to α5 integrin, leading to hemichannel opening in response to mechanical stimulation. AKT activation by fluid flow occurs in the first couple of hours and becomes completely inactive after 24 h. Cx43 phosphorylation totally agrees with the AKT activation pattern. At a later time MAPK activation and phosphorylation are dominant as a result of extracellular PGE2 released by osteocytes. Therefore, at the 30-min time point, even if there is a low level of MAPK phosphorylation, the predominant regulator is AKT and integrin binding, and the ultimate effect results in having hemichannels in the open state.
Regulation of Cx43 hemichannel opening is likely to play a critical role in maintaining a balance of the levels of extracellular bone remodeling factors, such as PGE2. PGE2 is known to have biphasic effects on bone remodeling; PGE2 induces bone formation when present at lower concentrations by inducing osteoblasts differentiation into osteocytes (30). Higher concentrations and long term exposure of bone to PGE2 result in stimulation of osteoclasts, which leads to bone resorption (21, 31). In addition, lower concentrations of PGE2 are known to inhibit osteoclast formation (32), and prolonged PGE2 treatment results in reduced mineralization (33). Therefore, controlled release of autocrine/paracrine factors like PGE2 by regulating the Cx43 hemichannel opening could be important to maintain bone architecture and facilitate bone remodeling.
The activation of ERK appears to depend on the concentration of PGE2 released. It has to reach a certain threshold to activate ERK. ERK activation by PGE2 is unique because it has slow, but sustained activation and even has activated ERK after 24 h of PGE2 treatment. This activation is different from ERK activation by growth factors/hormones, which tend to be quick and short-lived. In addition to concentrations of extracellular PGE2, this activation also depends on the abundance of PGE2 receptors on the cell surface. PGE2 receptors, EP2/EP4, which are known to activate ERK are present in MLO-Y4 osteocytes. At this stage, it is not certain if PGE2 receptors directly activate ERK or indirectly through activation of other signaling mechanisms and the involvement of the latter possibility is likely given that this activation is slow and long-lasting.
Phosphorylation of Cx43 at Ser279-Ser282 by p44/42 MAPK is known to inhibit gap junction communication (9, 26). The question was whether this phosphorylation could similarly lead to hemichannel closure. Another study has also suggested that Cx43 phosphorylation at Ser279-Ser282 sites is a signal for retrieval of this protein from the cell surface (11). The effect of Cx43 phosphorylation by MAPK on hemichannels could either lead to channel closure and/or on reduced surface expression of Cx43. Additionally, phosphorylation at these two residues inhibits the formation of gap junctional plaques and induces clathrin-dependent internalization of hemichannels, thereby fine-tuning endocytosis and gap junction assembly of Cx43 (13). Both of these events could lead to attenuation of hemichannel function with decreased PGE2 release. Here we showed that 30 min treatment with CM collected after 24 h of FFSS dramatically increased Cx43 phosphorylation by MAPK on the cell surface and this is well correlated with the closure of hemichannels. This experimental evidence suggests that channel gating, not surface expression of Cx43, is primarily affected by MAPK signaling. However, after 2 and 4 h treatment with CM, there is no alteration of total Cx43 on the cell surface, but phosphorylated Cx43 by MAPK on the cell surface was greatly reduced. This is consistent with our previous observation that after 24 h of continuous FFSS Cx43, the cell surface is reduced to the similar level as non-FFSS controls (20). These data suggest that longer exposure to FFSS and PGE2 reduces the Cx43 protein on the cell surface, which is likely a result of increased retrieval of Cx43 by MAPK phosphorylation.
Cx43 hemichannels are important portals for prostaglandin release and the controlled PGE2 level is crucial for bone formation and remodeling in response to mechanical loading. Here we revealed a feedback inhibition mechanism via direct phosphorylation of Cx43 by MAPK in regulating hemichannel function and extracellular PGE2 levels. Elucidation of the role of signaling pathways in regulation of the Cx43 hemichannel regulation could lead to a better understanding of biological function of mechanical loading in bone formation and remodeling process.
Author Contributions
M. A. R. designed, performed, and analyzed the experiments. S. B. designed, performed, and analyzed the experiments and contributed to the writing. P. D. L. performed and analyzed the experiments and contributed antibodies against Cx43 pS279/S282. R. K. designed, performed, and analyzed the experiments. J. X. J. conceived and coordinated the study, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Eugene Sprague and Jian Luo in assisting in the fluid flow experiments and members of Dr. Jiang's laboratory for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants AR46798 and GM55632 and Welch Foundation Grant AQ-1507. We declare that they have no conflicts of interest with the contents of this article.
- PGE2
- prostaglandin E2
- LY
- Lucifer yellow
- RD
- rhodamine dextran
- MEM
- minimal Eagle's medium
- CM
- condition medium
- FFSS
- fluid flow shear stress
- MAPK
- mitogen activated protein kinase
- ERK
- extracellular signal regulated kinases
- Cx
- connexin.
References
- 1.Ajubi N. E., Klein-Nulend J., Nijweide P. J., Vrijheid-Lammers T., Alblas M. J., and Burger E. H. (1996) Pulsating fluid flow increases prostaglandin production by cultured chicken osteocytes: a cytoskeleton-dependent process. Biochem. Biophys. Res. Commun. 225, 62–68 [DOI] [PubMed] [Google Scholar]
- 2.Cherian P. P., Siller-Jackson A. J., Gu S., Wang X., Bonewald L. F., Sprague E., and Jiang J. X. (2005) Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol. Biol. Cell 16, 3100–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genetos D. C., Kephart C. J., Zhang Y., Yellowley C. E., and Donahue H. J. (2007) Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J. Cell. Physiol. 212, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein-Nulend J., Semeins C. M., Ajubi N. E., Nijweide P. J., and Burger E. H. (1995) Pulsting fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts-correlation with prostaglandin upregulation. Biochem. Biophys. Res. Commun. 217, 640–648 [DOI] [PubMed] [Google Scholar]
- 5.Cherian P. P., Cheng B., Gu S., Sprague E., Bonewald L. F., and Jiang J. X. (2003) Effects of mechanical strain on the function of gap junctions in osteocytes are mediated through the prostaglandin EP2 receptor. J. Biol. Chem. 278, 43146–43156 [DOI] [PubMed] [Google Scholar]
- 6.Xia X., Batra N., Shi Q., Bonewald L. F., Sprague E., and Jiang J. X. (2010) Prostaglandin promotion of osteocyte gap junction function through transcriptional regulation of connexin 43 by glycogen synthase kinase 3/β-catenin signaling. Mol. Cell. Biol. 30, 206–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitase Y., Barragan L., Qiang H., Kondoh S., Jiang J. X., Johnson M. L., and Bonewald L. F. (2010) Mechical induction of PGE2 in osteocytes blocks glucocorticoid-induced apoptosis through both the β-catenin and PKA pathways. J. Bone Miner. Res. 25, 2657–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Márquez-Rosado L., Solan J. L., Dunn C. A., Norris R. P., and Lampe P. D. (2012) Connexin43 phosphorylation in brain, cardiac, endothelial and epithelial tissues. Biochim. Biophys. Acta 1818, 1985–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solan J. L., and Lampe P. D. (2009) Connexin 43 phosphorylation: structural changes and biological effects. Biochem. J. 419, 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bao X., Lee S. C., Reuss L., and Altenberg G. A. (2007) Change in permeant size selectivity by phosphorylation of connexin 43 gap-junction hemichannels by PKC. Proc. Natl. Acad. Sci. U.S.A. 104, 4919–4924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruch R. J., Trosko J. E., and Madhukar B. V. (2001) Inhibition of connexin 43 gap junctional intercellular communication by TPA requires ERK activation. J. Cell. Biochem. 83, 163–169 [DOI] [PubMed] [Google Scholar]
- 12.Pahujaa M., Anikin M., and Goldberg G. S. (2007) Phosphorylation of connexin43 induced by Src: regulation of gap junctional communication between transformed cells. Exp. Cell Res. 313, 4083–4090 [DOI] [PubMed] [Google Scholar]
- 13.Johnson K. E., Mitra S., Katoch P., Kelsey L. S., Johnson K. R., and Mehta P. P. (2013) Phosphorylation on Ser-279 and Ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol. Biol. Cell 24, 715–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper C. D., and Lampe P. D. (2002) Cascein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 277, 44962–44968 [DOI] [PubMed] [Google Scholar]
- 15.TenBroek E. M., Lampe P. D., Solan J. L., Reynhout J. K., and Johnson R. G. (2001) Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J. Cell Biol. 155, 1307–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Traub O., and Berk B. C. (1998) Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler. Thromb. Vasc. Biol. 18, 677–685 [DOI] [PubMed] [Google Scholar]
- 17.Klein-Nulend J., Bakker A. D., Bacabac R. G., Vatsa A., and Weinbaum S. (2013) Mechanosensation and transduction in osteocytes. Bone 54, 182–190 [DOI] [PubMed] [Google Scholar]
- 18.Harada S. I., Balena R., Rodan G. A., and Rodan S. B. (1995) The role of prostaglandins in bone formation. Connect. Tissue Res. 31, 279–282 [DOI] [PubMed] [Google Scholar]
- 19.Batra N., Riquelme M. A., Burra S., Kar R., Gu S., and Jiang J. X. (2014) Direct regulation of osteocytic connexin 43 hemichannels through AKT kinase activated by mechanical stimulation. J. Biol. Chem. 289, 10582–10591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siller-Jackson A. J., Burra S., Gu S., Xia X., Bonewald L. F., Sprague E., and Jiang J. X. (2008) Adaptation of connexin 43-hemichannel prostaglandin release to mechanical loading. J. Biol. Chem. 283, 26374–26382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian X. Y., Zhang Q., Zhao R., Setterberg R. B., Zeng Q. Q., Iturria S. J., Ma Y. F., and Jee W. S. (2008) Continuous PGE2 leads to net bone loss while intermittent PGE2 leads to net bone gain in. Bone 42, 914–920 [DOI] [PubMed] [Google Scholar]
- 22.Solan J. L., and Lampe P. D. (2008) Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun. Adhes. 15, 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burra S., Nicolella D. P., Francis W. L., Freitas C. J., Mueschke N. J., Poole K., and Jiang J. X. (2010) Dendritic processes of osteocytes are mechanotransducers that induce the opening of hemichannels. Proc. Natl. Acad. Sci. U.S.A. 107, 13648–13653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stern A. R., Stern M. M., Van Dyke M. E., Jähn K., Prideaux M., and Bonewald L. F. (2012) Isolation and culture of primary osteocytes from the long bones of skeletally mature and aged mice. BioTechniques 52, 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng B., Kato Y., Zhao S., Luo J., Sprague E., Bonewald L. F., and Jiang J. X. (2001) Prostaglandin E2 is essential for gap junction-mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain. Endocrinology 142, 3464–3473 [DOI] [PubMed] [Google Scholar]
- 26.Warn-Cramer B. J., Cottrell G. T., Burt J. M., and Lau A. F. (1998) Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 273, 9188–9196 [DOI] [PubMed] [Google Scholar]
- 27.Yu L., Wu W. K., Li Z. J., Li H. T., Wu Y. C., and Cho C. H. (2009) Prostaglandin E2 promotes cell proliferation via protein kinase C/extracellular signal regulated kinase pathway-dependent induction of c-Myc expression in human esophageal squamous cell carcinoma cells. Int. J. Cancer 125, 2540–2546 [DOI] [PubMed] [Google Scholar]
- 28.Rao R., Redha R., Macias-Perez I., Su Y., Hao C., Zent R., Breyer M. D., and Pozzi A. (2007) Prostaglandin E2-EP4 receptor promotes endothelial cell migration via ERK activation and angiogenesis in vivo. J. Biol. Chem. 282, 16959–16968 [DOI] [PubMed] [Google Scholar]
- 29.Bonewald L. F. (2006) Mechanosensation and transduction in osteocytes. Bonekey Osteovision 3, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ke H. Z., Jee W. S., Mori S., Li X. J., and Kimmel D. B. (1992) Effects of long-term daily adminstration of prostaglandin-E2 on maintaining elevated promimal tibial metaphyseal cancellous bone mass in male rats. Calcif. Tissue Int. 50, 245–252 [DOI] [PubMed] [Google Scholar]
- 31.Okuda A., Taylor L. M., and Heersche J. N. (1989) Prostaglandin E2 initially inhibits and then stimulates bone resportion in isolated rabbit osteoclast cultures. Bone Miner. 7, 255–266 [DOI] [PubMed] [Google Scholar]
- 32.Ono K., Akatsu T., Murakami T., Kitamura R., Yamamoto M., Shinomiya N., Rokutanda M., Sasaki T., Amizuka N., Ozawa H., Nagata N., and Kugai N. (2002) Involvement of cyclo-oxygenase-2 in osteoclast formation and bone destruction in bone metastasis of mammary carcinoma cell lines. J. Bone Miner. Res. 17, 774–781 [DOI] [PubMed] [Google Scholar]
- 33.Kajii T., Suzuki K., Yoshikawa M., Imai T., Matsumoto A., and Nakamura S. (1999) Long-term effects of prostaglandin E2 on the mineralization of a clonal osteoblastic cell line (MC3T3-E1). Arch. Oral Biol. 44, 233–241 [DOI] [PubMed] [Google Scholar]