

Background: Although oligomers are considered more important, mature fibrils also show evidence as cytotoxic agents in neurodegenerative diseases.
Results: Oligomers and fibrils both kill PC12 cells albeit mechanistically differently. In vivo, only oligomers inhibit hippocampal long term potentiation.
Conclusion: Protein aggregates, even those irrelevant to disease, are capable of inducing different toxic actions in neuronal cells.
Significance: Understanding these toxic mechanisms is vital in improving amyloidosis therapy.
Keywords: amyloid, apoptosis, cell death, fibril, neurodegenerative disease, oligomer, protein aggregation, protein misfolding, long term potentiation
Abstract
Despite significant advances, the molecular identity of the cytotoxic species populated during in vivo amyloid formation crucial for the understanding of neurodegenerative disorders is yet to be revealed. In this study lysozyme prefibrillar oligomers and fibrils in both mature and sonicated states have been isolated through an optimized ultrafiltration/ultracentrifugation method and characterized with various optical spectroscopic techniques, atomic force microscopy, and transmission electron microscopy. We examined their level and mode of toxicity on rat pheochromocytoma (PC12) cells in both differentiated and undifferentiated states. We find that oligomers and fibrils display cytotoxic capabilities toward cultured cells in vitro, with oligomers producing elevated levels of cellular injury toward undifferentiated PC12 cells (PC12undiff). Furthermore, dual flow cytometry staining experiments demonstrate that the oligomers and mature fibrils induce divergent cellular death pathways (apoptosis and secondary necrosis, respectively) in these PC12 cells. We have also shown that oligomers but not sonicated mature fibrils inhibit hippocampal long term potentiation, a form of synaptic plasticity implicated in learning and memory, in vivo. We conclude that our in vitro and in vivo findings confer a level of resistance toward amyloid fibrils, and that the PC 12-based comparative cytotoxicity assay can provide insights into toxicity differences between differently aggregated protein species.
Introduction
The failure of specific proteins to correctly fold and adopt their native functional structures has been correlated with a vast range of debilitating diseases including Alzheimer and Parkinson diseases (1–3). Such proteinaceous fibrillar aggregates known as amyloid fibrils are currently implicated in scores of degenerative diseases that affect a variety of peripheral tissues as well as the central nervous system (4, 5).
Although the dysfunctional assembly of peptides does not always carry a negative consequence, as seen with the tumoricidal molten globule-oleic acid complex HAMLET (Human Alpha-Lactalbumin Made LEthal to Tumor cells (6)), a group of ∼25 unrelated proteins have been shown to be causative agents in the formation of a number of clinically distinct conditions (7). However, recent evidence suggests the ability to aggregate may be a generic property of possibly all polypeptide chains under specific denaturing conditions. Hen egg white lysozyme (HEWL)2 can be engineered to aggregate at acidic pH and elevated temperatures (8) making it a very useful model to study protein misfolding and disease.
Despite the advancements in the field of protein aggregation and disease pathology, very little is known about the exact in vivo mechanisms of formation and cytotoxicity. Various studies involving animal models (9) and cell lines (10, 11) provided early evidence that supported the idea that mature fibrils were solely cytotoxic. However, this hypothesis has been the subject of intense scrutiny in recent years, and a large number of experiments have shown that the prefibrillar aggregates, known as oligomers, are intrinsically involved in and may even be the sole and direct cause of cell damage observed in various disease models (7, 12–15). The growth in literature surrounding the role of oligomers in neurodegenerative disease pathology has coincided with increasing levels of controversy surrounding mature fibrils in these disorders. One argument suggests that fibrils may possess no cytotoxic abilities at all (13, 15, 16), whereas there is a substantial body of experimental evidence that fully demonstrates that amyloid fibrils are capable of causing cellular death in numerous situations (17–19).
A related point of interest is whether cellular differentiation confers any significant biological resistance against amyloid fibrils and oligomers. Currently there is a substantial lack of knowledge regarding cell susceptibility and resistance to protein aggregates. Studies have presented evidence that suggests that toxic oligomers and fibrils display differing levels of toxicity toward particular cell lines (20, 21). It has also been shown that differentiated cells exhibit elevated levels of resistance against amyloid injury (22).
In this article we investigate the neurotoxic effects of HEWL oligomers, sonicated fibrils, and mature fibrils on the rat neuronal cell line PC12 in both differentiated and undifferentiated states. Mature amyloid fibrils and prefibrillar aggregates were isolated and characterized using a battery of techniques including atomic force microscopy (AFM), thioflavin T (ThT) fluorescence, and Congo Red (CR) birefringence. Here, we report for the first time that both mature amyloid fibrils and prefibrillar oligomeric species, isolated after 21 days from wild type HEWL, are highly toxic to PC12 cells in vitro, and in addition, the separated fractions elicit different cellular death pathways, one apoptosis, the other a form of cell death associated with necrotic cell death or a late apoptotic mechanism. Differentiated PC12 cells (PC12diff) showed an elevated level of resistance toward oligomers only, while no such resistance was found for mature amyloid fibrils. We also investigated the effects of HEWL aggregates on long term potentiation (LTP), a model of cellular mechanisms underlying learning and memory formation. Interestingly, we found that LTP in vivo was inhibited by lysozyme oligomers but not sonicated fibrils.
These results indicate that although produced from the same original protein, amyloid bodies in different aggregated states represent individual cytotoxic entities possessing unique properties, potentially providing insight into the true injurious events observed in neurodegenerative diseases.
Experimental Procedures
Proteins and Reagents
All reagents were of analytical grade or the highest purity available. HEWL, propidium iodide (PI), 1-anilinonaphthalene-8-sulfonic acid (ANS), and Tris were purchased from Sigma. Alamar Blue and all cell culturing equipment were purchased from Invitrogen. RPMI and PBS were purchased from GIBCO. Congo Red and ThT were obtained from Acros. FITC-labeled annexin-V was purchased from Promokine.
HEWL Aggregation
HEWL was prepared in distilled water adjusted to pH 2 with HCl. The final protein concentration was 1 mm (molecular mass of HEWL = 14.3 kDa, extinction coefficient ϵ1% = 26.4 at 280 nm). Aliquots were incubated at 65 °C for 21 days to allow sufficient fibrillization to occur (8). Control HEWL was prepared fresh at room temperature at pH 2.
HEWL Separation
Desired fractions of lysozyme were separated in relationship to particle mass after 21 days of incubation. Samples were ultracentrifuged for 1 h at 100,000 × g using a Beckman Optima TLX Ultracentrifuge. The pellet, containing mature amyloid fibrils, was resuspended in pH 2 distilled water to a concentration of 1 mm. At this point sonicated samples were subjected to ultrasound power for 30 s periodically for 5 min. The supernatant was spun down via ultrafiltration, utilizing 100-kDa cutoff filters (Amicon Ultra-4 Centrifugal Filter Devices). The resulting filtrate was further separated by repeating the ultracentrifugation process with 30-kDa cutoff filters. The resulting retentate (<100 kDa and >30 kDa) constituted the oligomer solution. Aggregated samples were prepared fresh on the day diluted and a final concentration of 1 mm was determined using a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific) averaging three readings for use in cytotoxic assays.
Thioflavin T Assay
ThT was carried out as described previously (23). HEWL samples were measured using a Jasco FP 6200 spectrofluorimeter. Fluorescence intensity was measured by excitation at 440 nm (slit width 5 nm) and emission at 482 nm (slit width 10 nm), averaging over 30 s.
Congo Red Spectroscopic and Birefringence Assay
Both Congo Red assays was carried out as described (23). UV-visible absorbance was measured using a NanoDrop ND-1000 Spectrophotometer between wavelengths 400 and 700 nm. A maximal spectral difference at 505 nm is evincive of amyloid fibrils. For the birefringence assay the Congo Red/HEWL solutions were analyzed using a Nikon Eclipse E400 POL with the polarizers crossed at a 90° angle to each other. Amyloid birefringence was identified by the presence of apple green coloration.
ANS Binding
ANS was carried out as described (24) using excitation at 390 nm, and emission between 410 and 600 nm.
AFM Imaging
HEWL samples were prepared for AFM analysis by depositing the fibril suspension onto freshly cleaved mica by spin coating. The spin recipe encompassed a dispersion routine (750 rpm for 45 s followed by 1000 rpm for 45 s) allowing the fibril suspension to be spread evenly across the mica surface. Samples were then dried (4000 rpm for 15 s) to remove any excess solvent. AFM measurements were performed in ambient conditions on an Asylum MFP-3D atomic force microscope (Asylum Research, Santa Barbara, CA). The probe normal spring constant was between 1.2 and 2.0 newton/m with a tip apex <5 nm. High resolution acoustically driven cantilevers (Nanosensors, SSS-FM) operating at a resonance frequency of 60–80 KHz in AC-mode were used with a scan resolution of 1024 × 1024 and scan frequency of 0.6 Hz. Structures were analyzed using MFP-3D IGOR PRO software.
Transmission Electron Microscopy
Protein samples were visualized using a JEOL 2100 Transmission Electron Microscope operating at 200 kV with a lanthanum hexaboride (LaB6) emission source. Prior to microscopy, 10 μl of HEWL sample was deposited onto carbon-coated grids. The solution was displaced with 0.5% glutaraldehyde and subsequently washed with dH2O. The sample was stained using uranyl acetate and allowed to air dry before transferring to microscope.
Cell Culture and Differentiation
Undiffereniated PC12 (PC12undiff) cells were maintained in RPMI 1640 medium/GlutaMAXTM-1 supplemented with 10% FCS, and 1% penicillin/streptomycin. Culture medium was replaced every 3 days. For cell viability assays, cells were seeded at a density of 106 cells/well in 96-well plates. For NGF-induced differentiation, PC12 cells were seeded onto collagen-coated 96-well plates at a cell density of 1.5 × 104 cells/well in RPMI 1640 medium/GlutaMAXTM-1. Wells were supplemented with 1% FCS, 1% penicillin/streptomycin, and 0.01% NGF every 2 days for a period of 7 days to allow differentiation. Immediately prior to the experiment the 1 mm stock of the fibrillar and prefibrillar aggregates were diluted with distilled water to produce concentrations varying from 20 to 300 μm. The cells, lysozyme aggregates, and media (volume 100 μl) were added and incubated overnight prior to treatment with Alamar Blue. All tests were conducted in triplicate. All cells were maintained in a 95% air, 5% CO2 humidified atmosphere at 37 °C.
Alamar Blue Cell Viability Assay
Alamar Blue fluorescence measurements were carried out as described previously (25). Emission levels were measured using a SPECTRAmax Gemini XS Microplate Spectrofluorometer. Cell viability was expressed as a percentage of fluorescence absorbance in the aggregate-exposed cells in relationship to untreated cells.
Flow Cytometry
Cells were seeded at 106 cells/ml/well the day before analysis in 12-well suspension plates. Wells were treated with 25 μm of the aggregated solution and left for 24 h overnight at 37 °C. Cells (1 ml) were removed and spun down at 1600 rpm for 5 min prior to washing in a 1× annexin binding buffer (10.9 mm Hepes, 140 mm NaCl, and 2.5 mm CaCl2, pH 7.4). Cells were spun down and stained with 10 μl of anti-annexin V antibody and left for 15 min on ice in the dark. Cells were spun down, washed, and resuspended in 500 μl of 1× annexin binding buffer. Immediately prior to FACS analysis, cells were treated with 10 μl of 50 μg/ml of PI. Flow cytometry was carried out using a FACSCalibur flow fluorocytometer (BD Biosciences). Compensation was carried out using control samples for untreated PI/annexin V only, double stained, and non-stained.
Western Blot Analysis of PARP Cleavage
Cells were seeded at appropriate densities (PC12undiff at 1 × 106 cells/well) (PC12diff at 5 × 105 cells/well) and incubated with HEWL fractions for 24 h at 37 °C. Cells were washed and lysed using 100 μl of Laemmli buffer (4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromphenol blue, 0.125 m Tris-HCl). Protein (20 μg) was subjected to Western blot analysis with anti-poly(ADP-ribose) polymerase (PARP) (Thermo Fisher Scientific Inc.) and anti-β-actin (Thermo Fisher Scientific Inc.) antibodies. Cells were washed and tagged with anti-mouse infrared secondary antibody and developed using an Odyssey® Infrared Imaging System (LI-COR).
Electrophysiological Techniques
In vivo electrophysiology was performed using techniques described previously (26). Animal experiments were licensed by the Department of Health and Children, Ireland. Adult male Wistar rats were anesthetized with urethane (1.5 g/kg, intraperitoneally). Single pathway recordings of field excitatory postsynaptic potentials (EPSPs) were made from the stratum radiatum in the CA1 area of the dorsal hippocampus in response to stimulation of the ipsilateral Schaffer collateral/commissural pathways. Test EPSPs were evoked at a frequency of 0.033 Hz and at a stimulation intensity was adjusted to give an EPSP amplitude of 50% of maximum. The high-frequency stimulation (HFS) protocol for inducing LTP consisted of 10 trains of 20 stimuli, inter-stimulus interval of 5-ms, and inter-train intervals of 2 s. The intensity was increased to give an EPSP of 75% of maximum amplitude during the HFS. LTP is expressed as the mean ± S.E. % baseline field EPSP amplitude recorded over at least a 30-min baseline period. Similar results were obtained when the EPSP slope rather than amplitude was measured.
For statistical analysis, EPSP amplitudes were grouped into 10-min epochs. Standard one-way analysis of variance was used to compare the magnitude of LTP between multiple groups followed by post hoc Tukey's tests. Unpaired Student's t tests were used for two-group comparisons. A p < 0.05 was considered statistically significant.
To inject samples into the rat brain a cannula was implanted in the lateral cerebral ventricle (coordinates: 1 mm lateral to the midline and 4 mm below the surface of the dura) just prior to electrode implantation. Injections (15 μl over 10 min) were made via a Hamilton syringe connected to the internal cannula.
Results
Isolation and Characterization of Prefibrillar Aggregates and Mature Amyloid Fibrils of Hen Egg White Lysozyme
HEWL samples incubated for 21 days were separated through a series of ultracentrifugation and ultrafiltration steps (see “Experimental Procedures”). To eliminate any potential diffusion-limitation issues that could arise from using mature fibrils, samples were sonicated post-isolation to produce shorter fragments of mature fibrils that still maintain their general fibrillar structure (27). To validate the presence of amyloid structures conclusively and to avoid false negative results, newly isolated samples were subjected to a combination of Congo Red spectroscopic and birefringence assays as well as ThT (23). Both oligomers and sonicated fibrils showed no signs of birefringence, however, mature HEWL fibrils are clearly birefringent at ×4 magnification (Fig. 1A). For Congo Red spectral analysis all samples showed a shift in the maximum absorbance from 495 to 505 nm (Fig. 1B). HEWL was also examined daily for 21 days using the cationic benzothiazole dye ThT, which exhibits enhanced fluorescence upon binding to amyloid fibrils. HEWL at 65 °C showed a marked daily increase in fluorescence intensity values when compared with values exhibited by control HEWL at room temperature (Fig. 1C). Furthermore, when treated with ANS ultracentrifuged/ultrafiltrated oligomers, sonicated fibrils, and mature fibrils all showed a blue shift in spectral maxima (Fig. 1D).
FIGURE 1.

Probing the aggregation of HEWL ultracentrifuged/ultrafiltrated fractions. A, samples were analyzed in bright field (left) and using crossed polarizers at 90° (right) with a Nikon Eclipse E400 POL microscope at ×4 magnification. B, UV-visible absorbance spectra for Congo Red alone and HEWL-separated fractions. All spectra were obtained from solutions containing 5 mm KH2PO4, 150 mm NaCl, pH 7.4. C, ThT absorbance readings of HEWL grown for 21 days at 65 °C in pH 2 conditions. HEWL control made fresh on the day and subject to no incubation can be also be seen. D, fluorescence emission spectra of HEWL samples upon binding the hydrophobic dye ANS. A and B are representative of 3 separate experiments. C and D, n = 1.
Microscopy Imaging of Amyloid Aggregates
As a direct visual confirmation of the existence of oligomeric and fibrils species, oligomers, sonicated fibrils, and mature fibrils were analyzed using AFM and TEM in conjunction with the above spectroscopic measurements (Fig. 2A). In contrast to the mature fibrils, both the oligomeric and sonicated samples exhibited a scattered array of spots providing direct evidence of the successful size-based fractionation through the combined ultracentrifugation/ultrafiltration methodology. For the mature fibril, the three-dimensional structure and the α z-height line section was measured 3.26 ± 0.65 nm, whereas the β z-height line section at the crossover was 4.23 ± 0.46 nm, indicative of helical twists in the mature fibril (Fig. 2, C and D) as classically found in typical cases (28). Oligomers, sonicated fibrils, and mature lysozyme fibrils were also visualized using TEM. All samples were negatively stained with the dye uranyl acetate (Fig. 2E). All samples show distinct differences in shape and size as a result of the destructive nature of sonication.
FIGURE 2.

AFM images of amyloid aggregates on mica formed from hen egg lysozyme. A, oligomers, sonicated fibrils, and amyloid mature fibrils visualized using AFM. B, shows a typical z-height AFM image of a fibril using a 1-μm scan range. Fibril fine structure is evident from the periodic steps indicated by the white circles. The periodic steps can also be seen in the three-dimensional image found in C. D, represent z-height line sections on areas of the fibril that are between the step of the periodic structure (α) and through the step itself (β). E, transmission electron microscopy of HEWL samples. Samples represented are oligomers, sonicated fibrils, and mature fibrils. Samples were deposited onto carbon-coated grids and stained with 2% uranyl acetate prior to visualization. These images are representative of three separate experiments.
Oligomers and Fibrils Are Cytotoxic to PC12 Cells of Both Undifferentiated and Differentiated Morphologies
The toxicities of oligomeric and fibrillar species of both sonicated and mature states were assessed using the PC12 cell line. Cultured PC12 cells have the propensity to continuously divide and provide a model for tumor research. Upon continued treatment with NGF, PC12undiff cells transform from a neoplastic morphology and begin to extend branching varicose processes (Fig. 3A). Different fibrillar and prefibrillar aggregates were added to both cell lines at concentrations in the 20 to 300 μm range and incubated at 37 °C for 24 h. Percentage cell death was calculated using the Alamar Blue viability assay. All lysozyme species show significant efficacy toward PC12diff cells with oligomers yielding LC50 values of 44 μm and fibrils values of 91 μm (Table 1). PC12undiff cells were treated with identical concentrations. The LC50 values for mature amyloid fibrils (94 μm) showed no discernible difference in susceptibility. Interestingly, oligomer-treated PC12undiff cells give an LC50 value of 29 μm, considerably lower than that observed with the differentiated cell type. Sonicated fibrils, whereas not as potent as oligomers on both cell lines, were more potent than mature full fibrils. The monomeric state of HEWL was additionally examined and showed no cytotoxicity toward either cell line (Fig. 3, B and C).
FIGURE 3.
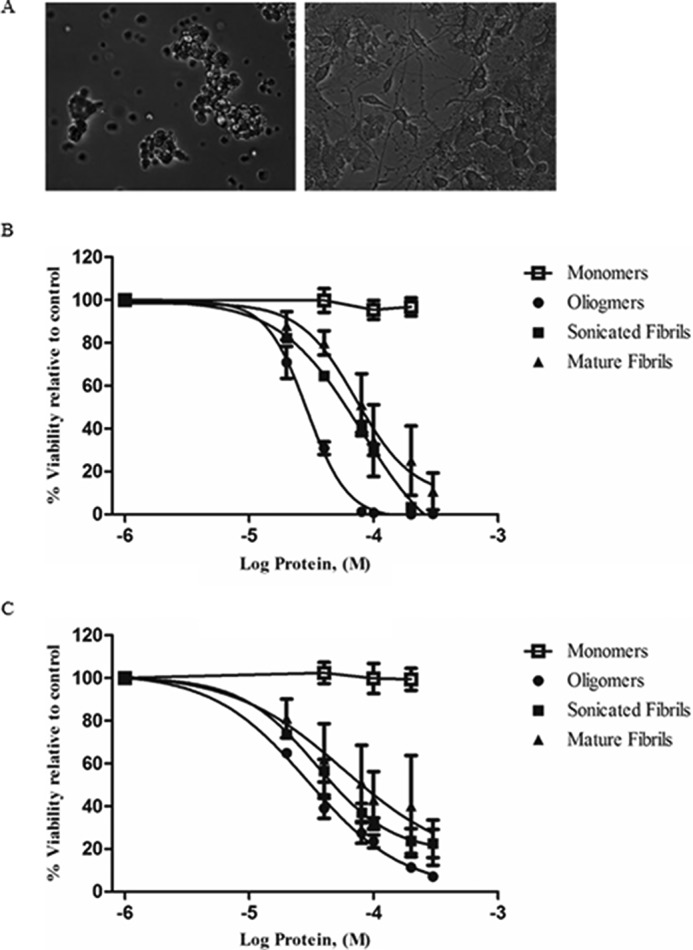
Effects of NGF treatment on cultured PC12 cells. A, light microscopic image of undifferentiated (left) and 100 ng/ml of treated differentiated cells (right) (scale bar: 100 μm). B and C, Alamar Blue emission viability results for PC12 cell lines treated with HEWL-separated fractions (Log scale). Undifferentiated (B) and differentiated (C) PC12 cells were exposed to varying concentrations of prefibrillar, and fibrillar aggregates then assayed using the fluorescent staining compound Alamar Blue. Monomeric HEWL was also examined and displayed no decrease in viability. The error bar indicates the values of mean ± S.E. of three experiments and were calculated using GraphPad software.
TABLE 1.
LC50 values (μm) of amyloid oligomers, fibrils, and sonicated fibrils on PC12 undifferentiated and differentiated cells
LC50 values are mean ± S.D. of three independent experiments carried out.
| Cell line | Oligomers | Sonicated fibrils | Fibrils |
|---|---|---|---|
| LC50 μm | |||
| PC12undiff | 28 ± 0.5 | 68 ± 1.7 | 94 ± 5.5 |
| PC12diff | 44 ± 1.1 | 78 ± 17.2 | 92 ± 10.2 |
HEWL Oligomers and Amyloid Fibrils Induce Cellular Death via Apoptotic and Secondary Necrotic Pathways, Respectively
During apoptosis, cells undergo highly specific morphological changes including plasma membrane blebbing, pyknosis, and exposure of the negatively charged phospholipid phosphatidylserine (PS) on the inner leaflet of the cell membrane (29, 30). Necrotic cells undergo a different form of cell death characterized by swelling and sudden deflation of the dying cell, resulting in leakage of internal cellular contents into the surrounding milieu (31). PC12undiff/diff cell death was analyzed by FACS using a combined stain for FITC-labeled annexin-V (apoptosis) and PI (necrosis or non-apoptotic). This experiment demonstrates that both fibrils and oligomers induce high levels of cell death within PC12undiff/diff cells. Oligomer-treated PC12undiff cells (Fig. 4B) show elevated annexin V-FITC staining when compared against the control group (Fig. 4A). Fibrils show increased staining for annexin V-FITC and PI (Fig. 4D). Similarly, PC12diff cells also show differences in cell death mechanisms. Oligomeric treated cells show a propensity for binding annexin V-FITC staining (Fig. 4F), whereas fibrillar treated PC12diff cells showed increased annexin V/PI double positive staining (Fig. 4H) when compared with control (Fig. 4D). PC12undiff/diff cells were also analyzed using sonicated fibrils. PC12undiff cells showed increased staining for annexin-V (Fig. 4C), whereas PC12diff cells showed approximately equal staining for annexin-V and dual annexin-V/PI (Fig. 4G). In addition cellular death in PC12undiff/diff cells treated with HEWL samples was examined using Western blot analysis probing for PARP. In both cell lines native HEWL and the monomeric fraction showed no apoptotic death. Sonicated fibrils and mature fibrils elicited a slight apoptotic response although oligomers appeared to induce the highest level of apoptotic cell death (Fig. 5). These results suggest that PC12undif/diff cells treated with HEWL-isolated oligomers, sonicated fibrils, and mature results in the cleavage of the apoptotic marker protein PARP, indicating that apoptosis has been activated in response to these proteinaceous fractions, which is in agreement with our flow cytometry data.
FIGURE 4.

Death pathway of PC12undiff/diff cells treated with HEWL oligomers, sonicated fibrils, and mature fibrils was assessed using duel staining for annexin V-FITC/PI and flow cytometry. Cells were treated with a fraction for 24 h and then stained for the presence of both fluorescent markers. The x axis indicates the numbers of annexin V-FITC-stained cells. The y axis indicates the numbers of PI-strained cells. Nontreated PC12undiff cells (A) and PC12diff cells (E) are shown. PC12undiff cells treated with 25 μm oligomer (B)-sonicated fibrils (C), and fibrils (D) show increased staining for both annexin V and PI. PC12diff cells were treated with identical concentrations of oligomers (F), sonicated fibrils (G), and mature fibrils (H). All samples were gated according to untreated cells, and compensated using untreated cells stained for both dyes. Data are representative of one of three similar experiments.
FIGURE 5.
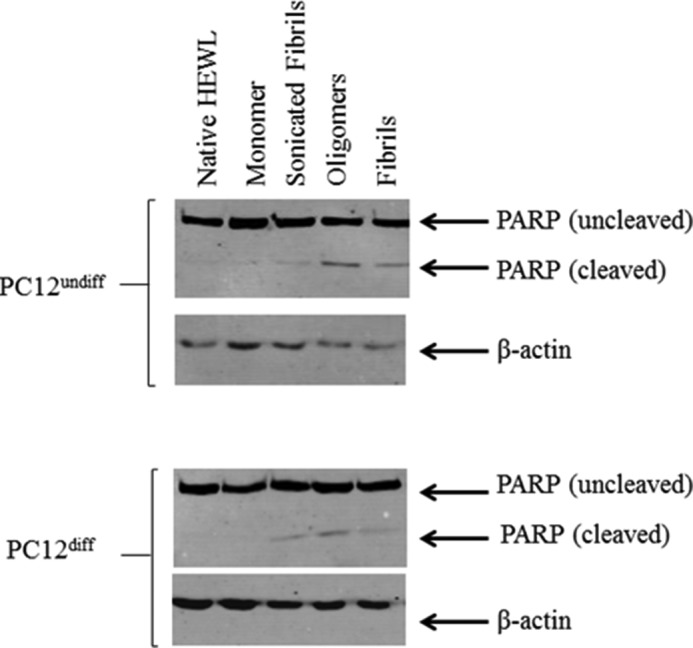
Western blot analysis of PARP cleavage in cell lysates obtained from PC12undiff/diff cells. Cells were cultured for 24 h after treatment with 30 μm HEWL native protein, monomers, oligomers, sonicated fibrils, and mature full-length fibrils. Both intact PARP (116 kDa) and the apoptotic marker PARP cleavage fragment (86 kDa) are shown. β-Actin used as a loading control. Results are representative of two separate experiments.
Inhibition of LTP by Lysozyme Oligomers
Amyloid neurodegenerative diseases commonly lead to cognitive decline and memory deterioration. To determine whether HEWL aggregates alter normal brain functions in vivo we examined their effects on hippocampal LTP, a well established correlate of learning and memory, which is potently inhibited by amyloid β-protein (Aβ) (26). Because we had previously found that fibril preparations of Aβ are inactive in this model we compared the actions of oligomers and sonicated fibrils in the present experiments. Fig. 6 shows that in anesthetized control rats HFS induced robust and long lasting potentiation (>3 h) of excitatory synaptic transmission in the CA1 area of the hippocampus after intracerebroventricular injection of either 15 μl of distilled water (n = 3) or 1 mm non-aggregated HEWL (15 nmol, n = 4) (combined group, 151 ± 15%, n = 7, p < 0.05, compared with pre-HFS baseline). In contrast, injection of the same volume of a 350 μm HEWL oligomer sample completely inhibited LTP at 3 h post-HFS (5.25 nmol, 103 ± 5%, n = 7, p > 0.05, compared with baseline, p < 0.05, compared with control LTP) without affecting baseline synaptic transmission (102 ± 9%, n = 4, p > 0.05, compared with pre-injection recording, Fig. 7, A and B). We chose this concentration of HEWL oligomers based on the in vitro toxicity data and pilot in vivo experiments. Furthermore, a 10-fold lower dose of HEWL oligomers (35 μm in 15 μl, 0.525 nmol) did not inhibit LTP (145 ± 10%, n = 4, p < 0.05, compared with baseline, p > 0.05, compared with control LTP, Fig. 7C). Interestingly, short fibrillar fragments generated by sonication of mature fibrils failed to inhibit LTP even when a much higher dose was injected (1.5 mm in 15 μl, 22.5 nmol, 137 ± 10%, n = 4, p > 0.05 compared with control LTP, Fig. 6). Of note, prior to HFS there was no significant difference in baseline excitability between any groups shown in Fig. 6 as measured by EPSP amplitude (2.7 ± 0.2, 2.5 ± 0.3, and 2.4 ± 0.3 mV for vehicle, HEWL oligomers, and sonicated fibrils, respectively) or stimulation intensity (6.6 ± 0.4, 7.0 ± 0.5, and 7.2 ± 0.6 mA for vehicle, HEWL oligomers, and sonicated fibrils, respectively).
FIGURE 6.
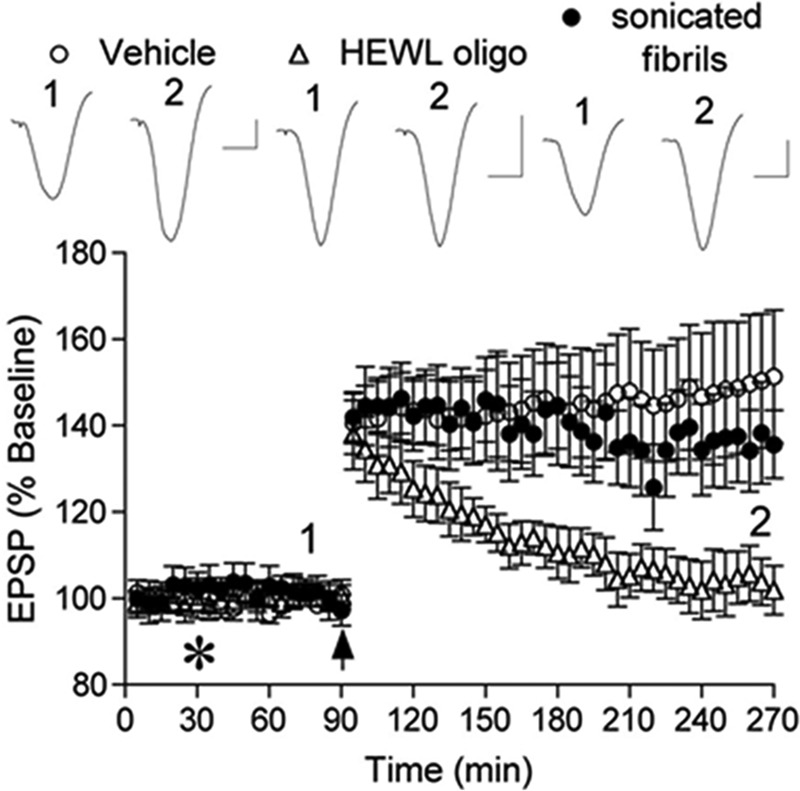
HEWL oligomers but not mature fibrillar fragments inhibit hippocampal LTP in vivo. Intracerebroventricular injection (asterisk) of vehicle (distilled water or non-aggregated HEWL) 1 h before high frequency conditioning stimulation (arrow) did not affect LTP (open circles). In contrast, animals injected with HEWL oligomers were unable to maintain LTP (open triangles). Intracerebroventricular injection of sonicated mature fibrils had no effect on LTP (closed circles). Insets show representative EPSP traces at the time indicated. Horizontal bar, 10 ms; vertical bar, 1 mV. Results are the means of three separate experiments.
FIGURE 7.
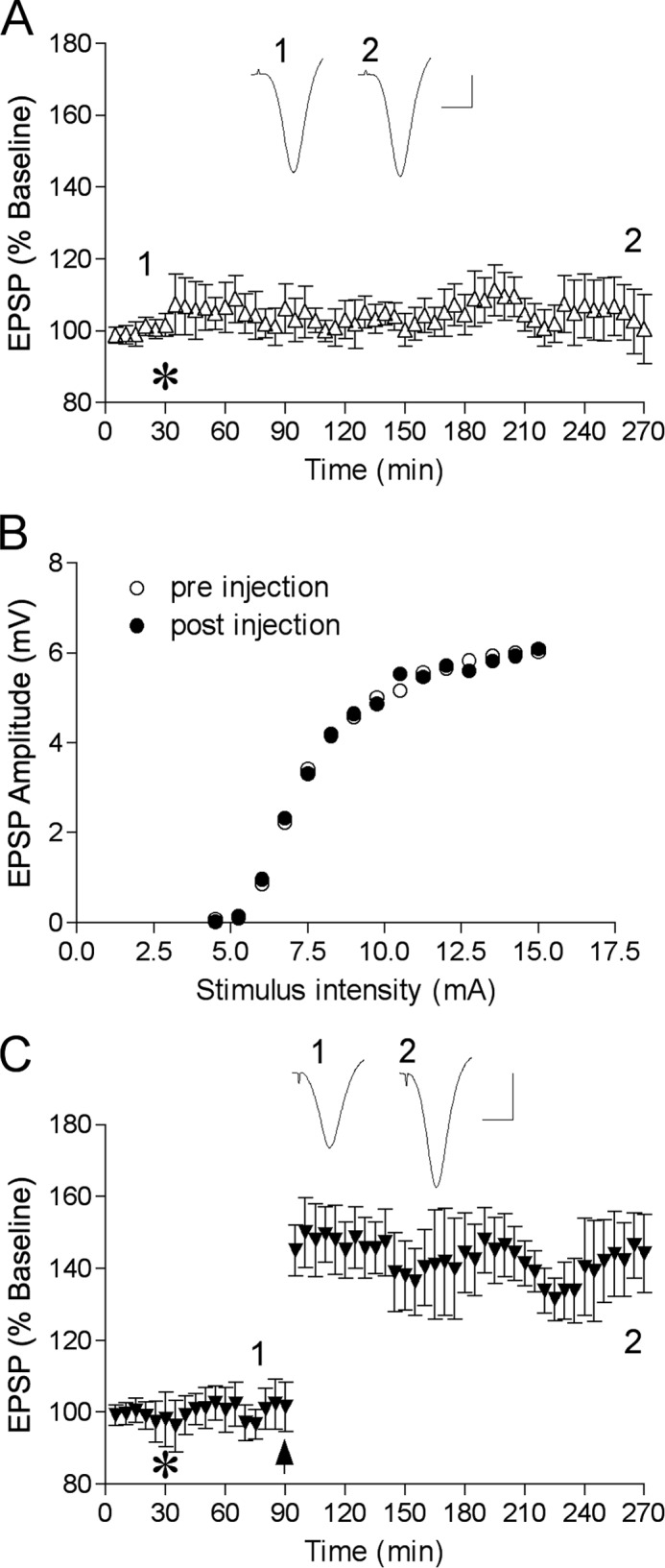
Lack of effect of HEWL oligomers at the dose of 5.25 nmol on baseline synaptic transmission (A and B) and at 0.525 nmol on LTP (C). A, time course study of baseline transmission after injection of HEWL oligomers (5.25 nmol). The test pulse evoked a 50% of maximum EPSP amplitude. B, single example of stimulus-response (input-output) curve before and 4 h after injection of HEWL oligomers (5.25 nmol). C, LTP time course in animals injected with 0.525 nmol of HEWL oligomers. Insets show representative EPSP traces at the time indicated. Horizontal bar, 10 ms; vertical bar, 1 mV.
Discussion
The current study demonstrates that HEWL amyloid fibrils and oligomers, judiciously isolated via ultracentrifugation and ultrafiltration, are highly toxic to cultured PC12 cells of both differentiated and undifferentiated states and that their cell death responses are specific to each. Both prefibrillar soluble aggregates and mature fibrillar aggregates displayed cytotoxicity toward rat PC12 cells, with differentiated cells showing an enhanced level of resistance against oligomers. Isolated fractions also induce differing cell death pathways in PC12undiff/diff with oligomers activating an apoptotic response, whereas fibrils generate secondary necrotic, non-apoptotic death.
The prolonged incubation of native HEWL protein resulted in the formation of oligomers, which preceded the conversion of mature fibrils, consistent with current knowledge on amyloid formation (18, 32). When compared with the oligomers, the mature amyloid fibrils isolated in this experiment showed all of the hallmark spectroscopic signatures, relatively more pronounced Congo Red birefringence, increased ThT binding, and a shift in UV-visual Congo Red (Fig. 1, A–C). All fractions also showed differences in their hydrophobic region exposure when measured using ANS (Fig. 1D). These results indicate that the transition from native HEWL protein toward fibrillar aggregate coincides with a change in the protein environment, whereby the protein loses its native three-dimensional structural packing and displays increased exposure of hydrophobic residues.
We also examined the separate fractions using high-resolution AFM and TEM (Fig. 2, A–D). Mature fibrils appear as long single-stranded protofilaments, displaying a characteristic left handed “twist.” Both sonicated and oligomer samples appear smaller in size yet distinctively unique from each other proving that no further elongation of the aggregate was achieved and that the size-based purification procedure was successful.
There is considerable debate regarding the true pathogenic role of oligomers and mature fibrils in neurodegenerative diseases. Early studies suggested that mature amyloid fibrils were the main pathogenic entities involved in amyloidosis (9, 33). In contrast there is equally strong evidence that oligomeric species may also have a pathogenic role in these disorders (15, 34, 35). The perception of the role for amyloid fibrils has changed over recent years and it has been suggested that their true function may be that of an evolutionary protection mechanism employed by the body after the fundamental damage was carried out at an earlier phase by the soluble oligomers (36).
Our study demonstrates that HEWL oligomers and fibrils display toxicity toward PC12undiff/diff cells and that soluble oligomers may not be the only toxic component involved in neurodegenerative disease. The higher levels of HEWL needed to induce cell death are representational of the normally non-toxic nature of the protein while in the native fold. Native HEWL at 200 μm had no effect on cell viability (results not shown) suggesting that the aggregated state of the protein is responsible for inducing cell death. The ability of PC12 cells to transpose between neoplastic and differentiated states makes PC12 a very useful model for the study of “tumor versus healthy” environments in cells (37). PC12undiff cells display a lower LC50 value than the PC12diff cell when treated with HEWL oligomers (28 versus 44 μm) (Table 1). When treated with mature fibrils both undifferentiated and differentiated PC12 cells displayed similar LC50 values, suggesting that although the differentiated state of PC12 cells seems to confer no level of resistance against mature HEWL fibrils, the undifferentiated state appears to be more susceptible to the cytotoxic proficiencies displayed by the oligomeric species of the same protein. LC50 values for PC12undiff/diff cells treated with sonicated fibrils reside between those of the two proteinaceous samples.
This protective role is consistent with earlier work examining amyloid injury in differentiated and undifferentiated human neurotypic SH-SY5Y cells (22). A possible reason for this is that phenotypic alterations that are a consequence of differentiation modify aggregate binding to the cell surface (38).
Using dual apoptotic and necrotic staining, we have shown that oligomeric treated PC12undiff/diff cells display increased annexin V staining indicative of apoptosis, although these cells treated with mature fibrils show annexin V/PI double positive staining, suggesting a late apoptotic or secondary necrotic death. FACS was also conducted on the sonicated fibrillar samples, using identical settings. Sonicated fibrils used to treat PC12undiff cells showed increased apoptotic staining, whereas the PC12diff cells showed equal amounts of staining for both apoptosis and late necrosis, suggesting that these cells are entering the later stages of the apoptotic cycle of cell death (Fig. 4, A–H). PC12undiff/diff cells exhibited cleaved PARP, an apoptotic indicator, when treated with oligomers, sonicated fibrils, and mature fibrils (Fig. 5). Monomeric treated cells show no signs of apoptotic death, which is supported by the evidence that these samples are not cytotoxic to cells. HEWL oligomers produced the highest level of apoptotic death, whereas sonicated fibrils and mature fibrils gave slightly lower levels of cleaved PARP. This data in collaboration with FACS analysis suggests cells treated with HEWL fractions, in particular oligomers, are dying through an apoptotic cell death pathway. The lower levels of apoptotic cell death seen in fibrillar treated cells suggest that in addition to apoptosis, non-apoptotic cell death is also occurring.
We found that HEWL oligomers inhibited hippocampal LTP in vivo (Fig. 6), a process linked with memory loss and function. Despite the fact that these oligomers are not as potent at inhibiting LTP as, for example, Aβ oligomers (39), the data provide new insight into a role of protein misfolding in neurodegenerative diseases as well as potential benefits of immunotherapies using conformation selective antibodies. We have previously found that insoluble amyloid fibrils formed by Aβ did not affect LTP in vivo (40). One reason for the lack of effect is that, due to diffusional restrictions, such large aggregates are not reaching hippocampal parenchyma from the injection site in the ventricle. To address this issue we used a sonication approach to produce short fragments of mature fibrils whereas maintaining general fibrillar structure (27). The observed difference in the abilities of oligomers and fibrillar fragments to inhibit LTP in vivo (Fig. 6) is in accord with our cell death findings (Fig. 3, B and C), where sonicated fibrils were less toxic than oligomers in vitro, and probably reflects the different nature of the interaction between neurons and these different HEWL aggregates. Indeed, it has been demonstrated that the pro-apoptotic signaling pathway is involved in inhibition of LTP by Aβ oligomers (41). Alternatively, intracerebroventricular injected sonicated fibrils may not be reaching hippocampal parenchyma and rather interact with cells at the ventricular surface.
It is important to note that whereas oligomers have been shown to universally display inherent toxicity through a shared membrane permeabilization mechanism of pathogenesis (34, 42), fibrils exhibit much greater diversity with regard to pathogenicity and unlike oligomers may employ a number of various mechanisms of cytotoxicity (43, 44). Fibrils formed from differing proteins and indeed from the same peptides have shown dramatic differences in terms of conformational variation and cytotoxicity (18, 45–47). This may be responsible for the variations observed in terms of cytotoxic capability and could be a reason for the inconsistency displayed by numerous experiments examining the cytotoxic ability of fibrils.
The true toxic component involved in the cell/tissue damage symptomatic of neurodegenerative diseases is still a proverbial gray area in the world of amyloid pathology (7, 48). Our results demonstrate that HEWL aggregates exhibit toxicity toward PC12 cells, and that the exact method of toxicity may be unique for individual fractions as well as dependent on the differentiated state of the cell. Oligomers inhibit LTP in rats in vivo, although sonicated fibrils appear to have no effect. Although both oligomers and fibrils have been shown to be damaging in vitro, in vivo experiments have yet to show that fibrils possess the ability to disrupt LTP, further highlighting the complicated matrix that surrounds the efficacy of these protein species. Knowledge of these subtle differences would be very important when designing anti-fibrillogenesis therapeutics. Despite this, our findings confirm that innocuous hen lysozyme can be engineered to produce both cytotoxic soluble prefibrillar aggregates and mature amyloid fibrils, further strengthening the claim that fibrillar conformation, and not the identity of the protein, is key to cellular toxicity and the underlying specific cell death mechanism.
Author Contributions
N. P. H., I. K., M. J. R., and K. H. M. conceived and coordinated the study. N. P. H. and K. H. M. wrote the paper, with I. K. and M. J. R. contributing certain specific sections. N. P. H., I. K., and E. K. McC. performed and analyzed the experiments. S. A. G. performed preliminary experiments that led up to this study. S. M. and Y. X. provided technical assistance. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Barry Moran and Sandra Bright for assistance with the FACS analysis.
This work was supported in part by grants from the Health Research Board of Ireland (to I. K.) and Science Foundation Ireland (to M. J. R). The authors declare that they have no conflicts of interest with the contents of this article.
- HEWL
- hen egg white lysozyme
- PC12
- pheochromocytoma 12
- ANS
- 1-anilinonaphthalene-8-sulfonic acid
- ThT
- thioflavin T
- PI
- propidium iodide
- AFM
- atomic force microscopy
- TEM
- transmission electron microscopy
- LTP
- long term potentiation
- EPSPs
- excitatory postsynaptic potentials
- HFS
- high-frequency stimulation
- Aβ
- amyloid β-protein
- PARP
- poly(ADP-ribose) polymerase.
References
- 1.Pepys M. B. (2006) Amyloidosis. Annu. Rev. Med. 57, 223–241 [DOI] [PubMed] [Google Scholar]
- 2.Dobson C. M. (2001) The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stefani M. (2004) Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 1739, 5–25 [DOI] [PubMed] [Google Scholar]
- 4.Merlini G., Seldin D. C., and Gertz M. A. (2011) Amyloidosis: pathogenesis and new therapeutic options. J. Clin. Oncol. 29, 1924–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Min S., Meehan J., Sullivan L. M., Harte N. P., Xie Y., Davey G. P., Svanborg C., Brodkorb A., and Mok K. H. (2012) Alternatively folded proteins with unexpected beneficial functions. Biochem. Soc. Trans. 40, 746–751 [DOI] [PubMed] [Google Scholar]
- 7.Stefani M. (2010) Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 277, 4602–4613 [DOI] [PubMed] [Google Scholar]
- 8.Krebs M. R., Wilkins D. K., Chung E. W., Pitkeathly M. C., Chamberlain A. K., Zurdo J., Robinson C. V., and Dobson C. M. (2000) Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain. J. Mol. Biol. 300, 541–549 [DOI] [PubMed] [Google Scholar]
- 9.Shimada K., Maeda S., Murakami T., Nishiguchi S., Tashiro F., Yi S., Wakasugi S., Takahashi K., and Yamamura K. (1989) Transgenic mouse model of familial amyloidotic polyneuropathy. Mol. Biol. Med. 6, 333–343 [PubMed] [Google Scholar]
- 10.Yankner B. A., Duffy L. K., and Kirschner D. A. (1990) Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250, 279–282 [DOI] [PubMed] [Google Scholar]
- 11.Iversen L. L., Mortishire-Smith R. J., Pollack S. J., and Shearman M. S. (1995) The toxicity in vitro of β-amyloid protein. Biochem. J. 311, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim H.-J., Chae S.-C., Lee D.-K., Chromy B., Lee S. C., Park Y.-C., Klein W. L., Krafft G. A., and Hong S.-T. (2003) Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J. 17, 118–120 [DOI] [PubMed] [Google Scholar]
- 13.Reixach N., Deechongkit S., Jiang X., Kelly J. W., and Buxbaum J. N. (2004) Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. U.S.A. 101, 2817–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oddo S., Caccamo A., Tran L., Lambert M. P., Glabe C. G., Klein W. L., and LaFerla F. M. (2006) Temporal profile of amyloid-β (Aβ) oligomerization in an in vivo model of Alzheimer disease. J. Biol. Chem. 281, 1599–1604 [DOI] [PubMed] [Google Scholar]
- 15.Vieira M. N., Forny-Germano L., Saraiva L. M., Sebollela A., Martinez A. M., Houzel J.-C., De Felice F. G., and Ferreira S. T. (2007) Soluble oligomers from a non-disease related protein mimic Aβ-induced Tau hyperphosphorylation and neurodegeneration. J. Neurochem. 103, 736–748 [DOI] [PubMed] [Google Scholar]
- 16.Giannakopoulos P., Herrmann F. R., Bussière T., Bouras C., Kövari E., Perl D. P., Morrison J. H., Gold G., and Hof P. R. (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology 60, 1495–1500 [DOI] [PubMed] [Google Scholar]
- 17.Yoshiike Y., Akagi T., and Takashima A. (2007) Surface structure of amyloid-β fibrils contributes to cytotoxicity. Biochemistry 46, 9805–9812 [DOI] [PubMed] [Google Scholar]
- 18.Mossuto M. F., Dhulesia A., Devlin G., Frare E., Kumita J. R., de Laureto P. P., Dumoulin M., Fontana A., Dobson C. M., and Salvatella X. (2010) The non-core regions of human lysozyme amyloid fibrils influence cytotoxicity. J. Mol. Biol. 402, 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue W. F., Hellewell A. L., Gosal W. S., Homans S. W., Hewitt E. W., and Radford S. E. (2009) Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem. 284, 34272–34282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cecchi C., Baglioni S., Fiorillo C., Pensalfini A., Liguri G., Nosi D., Rigacci S., Bucciantini M., and Stefani M. (2005) Insights into the molecular basis of the differing susceptibility of varying cell types to the toxicity of amyloid aggregates. J. Cell Sci. 118, 3459–3470 [DOI] [PubMed] [Google Scholar]
- 21.Bucciantini M., Rigacci S., Berti A., Pieri L., Cecchi C., Nosi D., Formigli L., Chiti F., and Stefani M. (2005) Patterns of cell death triggered in two different cell lines by HypF-N prefibrillar aggregates. FASEB J. 19, 437–439 [DOI] [PubMed] [Google Scholar]
- 22.Cecchi C., Pensalfini A., Liguri G., Baglioni S., Fiorillo C., Guadagna S., Zampagni M., Formigli L., Nosi D., and Stefani M. (2008) Differentiation increases the resistance of neuronal cells to amyloid toxicity. Neurochem. Res. 33, 2516–2531 [DOI] [PubMed] [Google Scholar]
- 23.Nilsson M. R. (2004) Techniques to study amyloid fibril formation in vitro. Methods 34, 151–160 [DOI] [PubMed] [Google Scholar]
- 24.Mossberg A.-K., Mok K. H., Morozova-Roche L. A., and Svanborg C. (2010) Structure and function of human α-lactalbumin made lethal to tumor cells (HAMLET)-type complexes. FEBS J. 277, 4614–4625 [DOI] [PubMed] [Google Scholar]
- 25.O'Brien J., Wilson I., Orton T., and Pognan F. (2000) Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 267, 5421–5426 [DOI] [PubMed] [Google Scholar]
- 26.Klyubin I., Ondrejcak T., Hayes J., Cullen W. K., Mably A. J., Walsh D. M., and Rowan M. J. (2014) Neurotransmitter receptor and time dependence of the synaptic plasticity disrupting actions of Alzheimer's disease Aβ in vivo. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chatani E., Lee Y.-H., Yagi H., Yoshimura Y., Naiki H., and Goto Y. (2009) Ultrasonication-dependent production and breakdown lead to minimum-sized amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 106, 11119–11124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fändrich M., Schmidt M., and Grigorieff N. (2011) Recent progress in understanding Alzheimer's β-amyloid structures. Trends Biochem. Sci. 36, 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vermes I., Haanen C., Steffens-Nakken H., and Reutelingsperger C. (1995) A novel assay for apoptosis: flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J. Immunol. Methods 184, 39–51 [DOI] [PubMed] [Google Scholar]
- 30.Wang X. (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15, 2922–2933 [PubMed] [Google Scholar]
- 31.Fiers W., Beyaert R., Declercq W., and Vandenabeele P. (1999) More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene 18, 7719–7730 [DOI] [PubMed] [Google Scholar]
- 32.Gharibyan A. L., Zamotin V., Yanamandra K., Moskaleva O. S., Margulis B. A., Kostanyan I. A., and Morozova-Roche L. A. (2007) Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J. Mol. Biol. 365, 1337–1349 [DOI] [PubMed] [Google Scholar]
- 33.Novitskaya V., Bocharova O. V., Bronstein I., and Baskakov I. V. (2006) Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J. Biol. Chem. 281, 13828–13836 [DOI] [PubMed] [Google Scholar]
- 34.Glabe C. G. (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 27, 570–575 [DOI] [PubMed] [Google Scholar]
- 35.Sakono M., and Zako T. (2010) Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 277, 1348–1358 [DOI] [PubMed] [Google Scholar]
- 36.Kirkitadze M. D., Bitan G., and Teplow D. B. (2002) Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J. Neurosci. Res. 69, 567–577 [DOI] [PubMed] [Google Scholar]
- 37.Oberdoerster J., and Rabin R. A. (1999) NGF-differentiated and undifferentiated PC12 cells vary in induction of apoptosis by ethanol. Life Sci. 64, 267–272 [DOI] [PubMed] [Google Scholar]
- 38.Olesen O. F., Dagø L., and Mikkelsen J. D. (1998) Amyloid β neurotoxicity in the cholinergic but not in the serotonergic phenotype of RN46A cells. Brain Res. Mol. Brain Res. 57, 266–274 [DOI] [PubMed] [Google Scholar]
- 39.Klyubin I., Wang Q., Reed M. N., Irving E. A., Upton N., Hofmeister J., Cleary J. P., Anwyl R., and Rowan M. J. (2011) Protection against Aβ-mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiol. Aging 32, 614–623 [DOI] [PubMed] [Google Scholar]
- 40.Klyubin I., Mably A., Minogue A., Hu N.-W., Farrell M., Walsh D., and Rowan M. (2012) Alzheimer's disease human brain β-amyloid-containing extracts inhibit hippocampal long-term potentiation in rats in vivo: Relationship between soluble and insoluble preparations. Alzheimers Dement. 8, P648 [Google Scholar]
- 41.Jo J., Whitcomb D. J., Olsen K. M., Kerrigan T. L., Lo S. C., Bru-Mercier G., Dickinson B., Scullion S., Sheng M., Collingridge G., and Cho K. (2011) Aβ(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 14, 545–547 [DOI] [PubMed] [Google Scholar]
- 42.Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., and Stefani M. (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511 [DOI] [PubMed] [Google Scholar]
- 43.Conway K. A., Harper J. D., and Lansbury P. T. Jr. (2000) Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry 39, 2552–2563 [DOI] [PubMed] [Google Scholar]
- 44.Marzban L., Park K., and Verchere C. B. (2003) Islet amyloid polypeptide and type 2 diabetes. Exp. Gerontol. 38, 347–351 [DOI] [PubMed] [Google Scholar]
- 45.Aguzzi A., and Haass C. (2003) Games played by rogue proteins in prion disorders and Alzheimer's disease. Science 302, 814–818 [DOI] [PubMed] [Google Scholar]
- 46.Collinge J. (2001) Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24, 519–550 [DOI] [PubMed] [Google Scholar]
- 47.Prusiner S. B., Scott M. R., DeArmond S. J., and Cohen F. E. (1998) Prion protein biology. Cell 93, 337–348 [DOI] [PubMed] [Google Scholar]
- 48.Lansbury P. T., and Lashuel H. A. (2006) A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443, 774–779 [DOI] [PubMed] [Google Scholar]