

Introduction
This Reflections article tells the story of the early work in my laboratory at the University of Washington that led to discovery of the sodium and calcium channel proteins, followed by a briefer description of the structure and function of these remarkable membrane proteins that has emerged from research in my laboratory and others over many years. I began my scientific career as an undergraduate chemistry major at Brown University, where my senior thesis research with Dr. Joseph Steim introduced me to the mysteries of membrane proteins in 1967–1968. Little was known about membrane proteins in those days, and the techniques to investigate them were crude by modern standards. Nevertheless, my efforts to solubilize and separate membrane proteins from red blood cells kindled a life-long research interest. As a graduate student in physiological chemistry with Dr. Peter L. Pedersen at Johns Hopkins Medical School in 1968–1972, my thesis research focused on the F1-ATPase, the peripheral membrane component of the ATP synthase of mitochondria. Our work joined the results from other laboratories to establish the complex nine-subunit architecture of that key protein using the newly developed SDS-PAGE method. We also determined some of the substrate binding and functional properties of F1-ATPase. A seminar class on neurotransmitters and their receptors with Dr. Solomon Snyder, then an assistant professor of pharmacology at Johns Hopkins, began an interest in neurobiology, which I have continued to pursue. As a postdoctoral fellow with Dr. Marshall Nirenberg at the Laboratory of Biochemical Genetics in the National Heart, Lung, and Blood Institute (NHLBI) at the National Institutes of Health, I moved into neurobiology and molecular pharmacology. Following his Nobel Prize-winning research on the genetic code, Nirenberg had pioneered studies of molecular and cellular neurobiology using cultured neuroblastoma cells as an experimental system. My initial postdoctoral research project was to develop biochemical methods to investigate the voltage-gated sodium channels of neuroblastoma cells. It was a challenging project that led directly to my continuing research interest in the voltage-gated ion channel proteins.
Voltage-gated ion channels are responsible for action potential generation in neurons, myocytes, and other excitable cells. They participate in many forms of regulation in other cell types. In excitable cells, action potentials typically are initiated by activation of voltage-gated sodium channels, which conduct sodium rapidly into the cell and depolarize the cell membrane potential. Depolarization activates voltage-gated calcium channels, which conduct calcium into the cell. Calcium entry sustains the depolarization of the cell membrane and generates intracellular calcium transients that initiate many intracellular events, including contraction, secretion, synaptic transmission, regulation of enzymes, and gene expression. Action potentials are terminated by activation of voltage-gated potassium channels, which conduct potassium out of the cell, repolarize the membrane, and contribute to setting the resting membrane potential.
Finding Channels
The modern era of research on voltage-gated ion channels began with the insightful use of the voltage-clamp method to analyze action potential generation by Drs. Alan Hodgkin and Andrew Huxley at Cambridge University. This key method allowed the investigator to control the voltage across the membrane with a feedback circuit while recording the ionic current passing through ion channels. With this revolutionary technique, Hodgkin and Huxley dissected the functional roles of sodium and potassium channels in generating the action potential in the giant axon of the squid, and they developed a complete model to explain action potential generation (1). Detailed studies with the voltage-clamp technique through the 1960s and 1970s led to conceptual models for the key functional properties of sodium channels: voltage-dependent activation, selective ion conductance, and fast inactivation (2, 3). These conceptual models have been remarkably enduring and have greatly influenced the molecular and structural studies that have followed them. However, in the late 1970s, a key gap in understanding of voltage-gated ion channels was the complete lack of information about the channel molecules themselves, which were widely assumed to be transmembrane proteins unique to excitable cells.
Neurotoxins as Molecular Probes
Identifying ion channel molecules was a challenging task, as removing them from excitable membranes would prevent measurement of their functional activity and therefore prevent purification by the available biochemical approaches of the time. Neurotoxin labeling provided a solution to this problem. During my research as a postdoctoral fellow with Dr. Marshall Nirenberg and as a staff member in the Laboratory of Biochemical Genetics at the National Heart, Lung, and Blood Institute (NHLBI) at the National Institutes of Health, from 1972 to 1977, I studied the actions of multiple classes of neurotoxins on sodium channels using ion flux and neurotoxin binding methods on neuroblastoma cells. These cultured cells were electrically excitable and generated action potentials similar to those in nerve and muscle. They provided a valuable experimental preparation to begin the process of applying biochemical techniques to electrical excitability. My work on toxins and neuroblastoma cells led to a model of sodium channels with three neurotoxin receptor sites (reviewed in Ref. 4). Site 1 bound the pore-blocking toxins tetrodotoxin and saxitoxin (5–7). Site 2 bound the lipid-soluble activators of sodium channels, batrachotoxin, veratridine, and aconitine. Site 3 bound the α-scorpion toxins and sea anemone toxins, which slow the fast inactivation process of sodium channels. Work in the laboratory of François Couraud and Hervé Rochat at the University of Aix-Marseille defined neurotoxin Site 4, which bound the β-scorpion toxins that enhance activation of sodium channels (8), and work in the laboratory of Michel Lazdunski at the University of Nice (9) added further information on the actions of neurotoxins at these multiple receptor sites on sodium channels. The neurotoxins acting on sodium channels fell cleanly into two classes: on the one hand, pore blockers like tetrodotoxin and saxitoxin acting at Site 1, and on the other hand, gating modifier toxins that act at the other three neurotoxin receptor sites. This fundamental dichotomy in ion channel toxins has remained to the present day, with many more pore-blocking and gating-modifier toxins of voltage-gated sodium, calcium, and potassium channels having been discovered and characterized in the ensuing years. My early work on neuroblastoma cells also showed that the gating modifier toxins act via an allosteric mechanism to persistently activate sodium channels; that toxins acting at Sites 2 and 3 acted synergistically in channel activation; and that the binding of α-scorpion toxins at Site 3 was voltage-dependent, as though these toxins interacted directly with the voltage sensor of the channels (4).
I established my new laboratory in the Department of Pharmacology at the University of Washington in 1977. The Department of Pharmacology was directed at that time by Dr. Edwin G. Krebs, who received the Nobel Prize for his discovery of protein phosphorylation as a regulatory process in 1992. He moved the department in the direction of cell signaling and molecular pharmacology, and my new research interests fit well with those research areas. The first goal of my new laboratory was to use neurotoxins to identify the sodium channel protein and discover its molecular properties. Only two lab members made the trip west from Maryland to Seattle: postdoctoral fellow Daniel Beneski and lab technician Cynthia Morrow. We were fortunate to be able to make rapid progress on our goal of understanding sodium channels at the molecular level despite the small size of our group.
Photoaffinity Labeling Sodium Channels
Dan Beneski made the first breakthrough in “finding channels” by identifying the protein subunits of sodium channels by photoaffinity labeling with a photoreactive derivative of an α-scorpion toxin that we purified from the venom of the North African scorpion Leiurus quinquestriatus, a highly toxic species whose venom was commercially available at the time. He discovered two polypeptides, a large α subunit with a molecular mass of ∼260 kDa and a smaller β subunit of ∼36 kDa in neuroblastoma cells and in rat brain synaptosomes (Fig. 1) (10). The photoaffinity labeling of these two protein subunits was specific and was fully blocked by pre-saturation of the toxin receptor site with unlabeled toxin. Moreover, scorpion toxin binding and photoaffinity labeling of the α and β subunits were blocked by membrane depolarization, supporting direct interaction of the toxin with the voltage-gated channel protein itself, and photoaffinity labeling was observed in neuroblastoma cell lines that were electrically excitable but not in electrically inexcitable neuroblastoma cell lines (10). These characteristics of the photoaffinity labeling studies gave us confidence that we had labeled the protein subunits of sodium channels. We were very excited about this first glimpse of the sodium channel protein. In fact, late on a Wednesday night as he developed his first successful autoradiogram from his photoaffinity labeling experiments, Dan Beneski stared in disbelief at the two clear subunit bands that were revealed, and it was difficult for him to go home and sleep through the night. I was equally excited when we looked at his new result together the next morning. I imagine that I said something trite like “Wow! Let's do it again!” Fortunately, we were able to repeat the results with no difficulty. After completing our series of experiments, we submitted our manuscript for review in August, 1979. It was accepted in revised form, sent to PNAS for publication in October, 1979, and eventually published in January, 1980.
FIGURE 1.
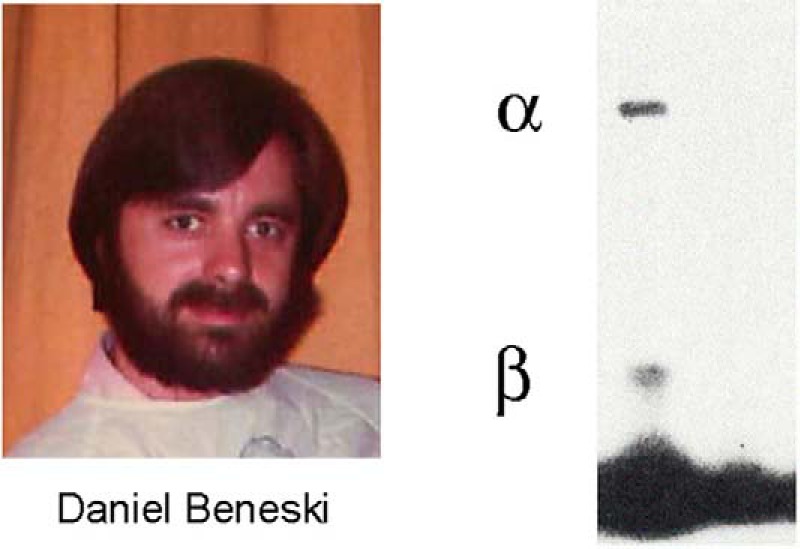
Left, Daniel Beneski, ca. 1980. Right, α and β subunits covalently labeled with [125I]-labeled scorpion toxin (Beneski and Catterall (10)). Lane 1, specific labeling; lane 2, nonspecific labeling.
The work on identification of the sodium channel subunits by photoaffinity labeling was presented for the first time at a Taniguchi Symposium on Brain Science in Tsukuba, Japan in November 1979, where it received an enthusiastic response. The Taniguchi Symposia were aimed at newly emerging research areas in all branches of science and emphasized an interdisciplinary approach with active participation of junior scientists. The Taniguchi Symposium on Brain Science in 1979 was an exceptional opportunity for me to meet scientists interested in ion channels with a worldwide perspective and to learn from them. The program also included generous time to experience Japanese culture, which was new to most of the participants. We visited sites of interest in Tokyo, Osaka, Kyoto, and Nara, and we enjoyed traditional Japanese food and entertainment presented by geisha. On one memorable evening in Osaka, we were taken to a restaurant specializing in the prized Japanese fugu, the puffer fish that produces the potent sodium channel blocker tetrodotoxin, which we all had used in the laboratory. Almost all of us dared to eat the fugu sashimi, and we snapped photos of each other as we feigned dying from the poison. In the end, we all survived and declared that fugu was just as excellent for dinner as its toxin was excellent for inhibiting sodium channels in the laboratory.
Purification of Sodium Channels
The next step in our quest to understand sodium channels at the molecular level required purification of the channel protein. We expected this to be a difficult task as no membrane protein with subunits as large as 260 kDa had been purified before. As if to confirm our concerns, binding of the gating-modifier scorpion toxins was lost upon solubilization of sodium channels using a wide range of detergents and conditions, which we viewed as a serious obstacle to progress. However, high-affinity binding of the pore-blocking toxins tetrodotoxin and saxitoxin could be retained by careful solubilization in mild detergents mixed with phospholipids (11). We resolved to purify the sodium channel based on its saxitoxin binding activity. The story of radiolabeled saxitoxin as a probe for sodium channels is an interesting sidelight in itself, involving the U.S. Senate, the Central Intelligence Agency, and tons of Alaska butter clams.1
My first graduate student, Robert Hartshorne, took up the challenge of purifying the sodium channel from rat brain based on saxitoxin binding. After optimizing our protocol for solubilization and stabilization of the protein in detergent/phospholipid mixtures, Bob was soon able to isolate a complex of α and β subunits by purification ∼3000-fold from rat brain membranes using affinity chromatography on a wheat germ agglutinin-Sepharose column, ion exchange chromatography, and size separation by sucrose density gradient sedimentation (12). Bob's work was aided greatly by our prior knowledge of the subunit composition of sodium channels through our photoaffinity labeling studies with scorpion toxins, because we knew that prevention of proteolysis to retain the 260-kDa α subunit was essential for success. We developed exceptionally detailed protocols to prevent proteolytic cleavage of the α subunit during our purification procedures. Analysis by SDS-PAGE of these purified preparations with new graduate student Donald Messner and technician Jeff Coppersmith revealed a complex of three subunits: a large α subunit of 260 kDa that was noncovalently associated with a β1 subunit of 36 kDa and disulfide-linked to a β2 subunit of 33 kDa (Fig. 2) (12–14). This subunit composition of sodium channels as a complex of the principal pore-forming α subunit with one or two auxiliary β subunits has withstood the test of time and remains the basis of our understanding of sodium channel biochemistry and cell biology today.
FIGURE 2.

Subunit structure of voltage-gated sodium channels. A and B, sodium channel purified from rat brain showing the α, β1, and β2 subunits and their molecular weights (13). As illustrated, the α and β2 subunits are linked by a disulfide bond. C, model of the sodium channel from biochemical data. Tetrodotoxin, saxitoxin, and scorpion toxins bind to the α subunits of sodium channels as indicated and were used as molecular tags to identify and purify the sodium channel protein (10, 13, 14). D, single channel currents conducted by a single purified sodium channel incorporated into a planar bilayer (20).
The sodium channel is a minor brain protein. Relatively large amounts of sodium channel protein were required for biochemical and functional analysis. Our standard preparation of the time started with 100 freshly isolated rat brains. The sodium channel group of 4–6 people, led by technician Carl Baker, would form an assembly line for the weekly tissue preparation phase of the purification procedure. Great care was required throughout the purification procedure to keep the sample ice-cold, in an appropriate mixture of detergent and phospholipid, and in the continuous presence of freshly prepared protease inhibitors to prevent degradation. Successful preparations of functionally intact sodium channels required nearly three days and nights of constant work in the cold room, a challenge that the graduate students and prep technician in the group carried out with dedication and enthusiasm. With these precautions, highly purified preparations with 1:1:1 stoichiometry of α, β1, and β2 subunits could be obtained (Fig. 2) (14). All three subunits were later shown to be glycosylated and to have hydrophobic domains consistent with transmembrane segments (15).
Functional Reconstitution of Sodium Channels
Our purified preparation of brain sodium channels was well defined biochemically, but was it a functional sodium channel? We knew that the binding of gating-modifier toxins like the scorpion toxins was lost upon detergent solubilization, so my second graduate student, Michael Tamkun, had already taken up the challenge of designing methods to restore voltage-dependent binding of α-scorpion toxins as a surrogate marker for recovery of voltage-dependent gating function. Mike found that reconstitution of purified sodium channels into phospholipid vesicles consisting of phosphatidylcholine and mixed brain lipids was effective in restoring voltage-dependent scorpion toxin binding, as demonstrated by generating a membrane potential across the reconstituted vesicle membrane using ion gradients and artificial ionophores (16, 17). His work gave us confidence that we had isolated both the pore and the gating apparatus of sodium channels, as labeled by saxitoxin and α-scorpion toxins, respectively, so we forged ahead to measure ion conductance activity of purified sodium channels. Postdoctoral fellow Jane Talvenheimo established methods to measure sodium channel ion conductance activity in reconstituted membrane vesicles using 22Na+ as a tracer and the gating modifier toxins batrachotoxin and α-scorpion toxin to synergistically activate the channel (18). She and Mike Tamkun succeeded in fully reconstituting sodium channel function from purified components (17). By carefully titrating the protein-lipid mixture, they were able to show that the majority of purified sodium channel molecules could be functionally reconstituted, further nailing down the conclusion that only the purified α and β subunits were required for channel function.
While we were working hard to purify and reconstitute the sodium channel protein, the newly developed giga-seal patch clamp electrophysiological recording technique was applied to sodium channels for the first time. These studies revealed a single channel conductance of 18 picosiemens and voltage-dependent probability of pore opening and mean open time of the single sodium channels (19). To determine the single channel properties of purified sodium channels, Bob Hartshorne moved for his postdoctoral studies to the laboratory of Dr. Mauricio Montal at the University of California at San Diego, a leader in studies of ion channels in planar phospholipid bilayers. Jane Talvenheimo purified and reconstituted sodium channels in Seattle, shipped them to San Diego, and Bob, Bernhard Keller, and Mauricio Montal recorded the functional activity of pure batrachotoxin-activated sodium channels after fusion of reconstituted vesicles with planar bilayers for the first time (Fig. 2D) (20). To our delight, single sodium channel currents appeared with the conductance and voltage dependence expected for sodium channels activated by batrachotoxin. It was a great feeling to know that we had succeeded in purification and reconstitution of a fully functional voltage-gated sodium channel, and we were ready to celebrate. One such celebration took place at the home of Mike Tamkun, and a photo has survived to record the event (Fig. 3). Bob Hartshorne, Mike Tamkun, Don Messner, and Jane Talvenheimo (unfortunately with her face shaded by her hand) were all present, beer mugs in hand, along with other lab members and myself in the upper right corner.
FIGURE 3.

Celebrating the sodium channel. Photo from a lab party at the home of Michael and Kim Tamkun, ca. 1984. Front row from left, Michael Tamkun (graduate student), Benson Curtis (graduate student), Max Willow (postdoctoral fellow), Jane Talvenheimo (postdoctoral fellow, shielding her eyes), John Schmidt (graduate student), Cynthia Morrow (lab technician), and Tohru Gonoi (postdoctoral fellow). Second row from left, Scott Sherman (M. D./Ph. D student), Don Messner (graduate student), William Downey (lab technician), and Bob Hartshorne (graduate student). Standing from left, Sidney Postma (anesthesiology resident), Richard Sharkey (postdoctoral fellow), and Bill Catterall.
In parallel with our studies on sodium channels from brain, Michael Raftery and his postdoctoral colleagues William Agnew and S. Rock Levinson at California Institute of Technology showed that sodium channels from the electroplaque of the electric eel, purified on the basis of their binding of tetrodotoxin, had a major molecular component of ∼250 kDa (21). Similarly, Robert Barchi at the University of Pennsylvania found that sodium channels purified from skeletal muscle based on their saxitoxin binding activity were composed of proteins with similar molecular properties to the α and β1 subunits of brain sodium channels (22). It was not possible to reconstitute sodium channel function from these purified channel preparations at the time, but it seemed likely to all of us that these studies also had successfully purified the sodium channel protein.
At this time, in 1984–1985, our success in purifying the sodium channel, defining its subunit structure, and reconstituting its function from purified components was seen as an important advance in understanding the molecular basis of electrical excitability. To my great surprise at the time, I was invited to give lectures at some major international meetings. I particularly recall my plenary lecture at the Annual Congress of the Japanese Pharmacological Society in Kyoto in the spring of 1984, where I made the first presentation of the complete story of discovery, purification, and functional reconstitution of the sodium channel (Fig. 4), and I gave a later plenary lecture at the International Congress of Pharmacology in London in the summer of that same year. Our research group felt honored that our progress on molecular analysis of sodium channels was recognized by these invitations for presentations at these major international congresses.
FIGURE 4.

Presenting the discovery, purification, and functional reconstitution of the sodium channel. The author (with much more hair and much redder hair than currently!) is shown presenting a plenary lecture at the Annual Congress of the Japanese Pharmacological Society in Kyoto in March, 1984.
Purification and Reconstitution of Calcium Channels
In the midst of our efforts to reconstitute sodium channels, graduate student Benson Curtis joined the lab and embarked on a bold project to purify and reconstitute calcium channels. He singlehandedly developed methods to label skeletal muscle calcium channels with [3H]nitrendipine, a high-affinity calcium antagonist drug, solubilize the labeled calcium channels with the mild detergent digitonin, and purify them using a combination of wheat germ agglutinin affinity chromatography, ion exchange chromatography, and sucrose density gradient centrifugation, similar to our preparation of sodium channels (23, 24). His initial analysis by SDS-PAGE revealed a large α subunit, which migrated as a diffuse band below 200 kDa, a β subunit of 50 kDa, and a γ subunit of 33 kDa. Reconstitution of this preparation in phospholipid vesicles confirmed that it was functional in calcium conductance (25). Reconstitution of a similar calcium channel preparation in planar phospholipid bilayers by Franz Hofmann and Wolfgang Trautwein at University of Saarlandes in Germany revealed single channel currents with the conductance and voltage dependence of bona fide calcium channels (26). We celebrated that a second class of ion channels had been characterized at the molecular level.
Subunit Architecture of Calcium Channels
Although the purified calcium channel protein was well behaved in many respects, the behavior of the α subunit band in SDS-PAGE analysis under different experimental conditions was unpredictable. New postdoctoral fellows Dr. Masami Takahashi from Japan and Dr. Michael Seagar from France discovered the reason for that unpredictability. They found that the original protein band designated as the α subunit actually contained two distinct calcium channel components: an α1 subunit with a molecular mass of 200–220 kDa that contained the binding site for nitrendipine and other calcium antagonist drugs plus a second component composed of a disulfide-linked complex of two proteins, an α2 subunit of ∼145 kDa disulfide linked to a δ subunit of ∼30 kDa (Fig. 5) (27). The disulfide-linked α2δ complex migrated with an unexpectedly high apparent molecular mass of 190–200 kDa, indistinguishable from the α1 subunit when disulfide bonds were not reduced. In early preparations, reduction of disulfide bonds had caused this band to disappear in SDS-PAGE, probably due to inadequate control of proteolysis.
FIGURE 5.

Subunit structure of voltage-gated calcium channels. A, summary of the biochemical properties of purified skeletal muscle Ca2+ channels. Lanes 1 and 2, silver stain of polypeptides; lane 3, staining with an antibody against the α1 subunit; lane 4, staining with concanavalin A (ConA), a lectin binding high-mannose N-linked carbohydrate chains; lane 5, staining with wheat germ agglutinin (WGA), a lectin staining N-linked complex carbohydrate chains; lane 6, photoaffinity labeling with azidopine, a photoreactive dihydropyridine (DHP); lane 7, photoaffinity labeling with 3-trifluoromethyl-3-(m-iodophenyl)diazirine (TID), a hydrophobic probe of the transmembrane regions of proteins; lane 8, phosphorylation by cAMP-dependent protein kinase. Adapted from Ref. 27. NEM, N-ethylmaleimide. B, the subunit architecture of Ca2+ channels purified from skeletal muscle is illustrated. The model is updated from the original description of the subunit structure of skeletal muscle Ca2+ channels (27). This model also fits biochemical and molecular biological results for neuronal Ca2+ channels. P, sites of phosphorylation by cAMP-dependent protein kinase and protein kinase C. Y, sites of N-linked glycosylation. Insets, Michael Seagar (left) and Masami Takahashi (right).
These new results were very surprising in their complexity. Masami and Mike, joined by postdoctoral fellow Dr. Bernhard Reber and lab technician Jean Jones, set out to characterize the calcium channel complex using a battery of methods to label drug-binding sites, sites of N-linked glycosylation, sites of cAMP-dependent protein phosphorylation, and hydrophobic regions that might be transmembrane segments (Fig. 5A). Their comprehensive analysis led to a model of calcium channel subunit architecture that has stood the test of time with only minor modifications (Fig. 5B) (27). They found that the α1 subunit contained the binding sites for calcium antagonist drugs and was glycosylated, hydrophobic, and phosphorylated by cAMP-dependent protein kinase. Weconcluded that it was the principal transmembrane, pore-forming subunit of the channel. In contrast, the α2 subunit was heavily glycosylated but not labeled by drugs, phosphorylated, nor hydrophobic, whereas the disulfide-linked δ subunit was both glycosylated and hydrophobic. We concluded that the α2 subunit is an extracellular glycoprotein disulfide-linked to a transmembrane δ subunit, but more recent work shows that this subunit is further processed by cleavage of its apparent transmembrane segment and addition of a glycosylphosphatidylinositol anchor, a form of membrane association that was unknown in 1987.2 The β subunit was phosphorylated but neither glycosylated nor hydrophobic, so we designated it as a cytoplasmic subunit in the calcium channel complex. Finally, the γ subunit was both hydrophobic and glycosylated, so we placed it in a transmembrane position in the complex. We were stunned by the complexity of this calcium channel architecture, but we were quickly reassured that the results and model were correct as parallel work from the laboratories of Kevin Campbell (University of Iowa), Joerg Striessnig and Hartmut Glossmann (University of Innsbruck), Michel Lazdunski, and Franz Hofmann provided independent support for specific elements of the overall calcium channel model (28–32). The team of scientists in my laboratory that identified, purified, reconstituted, and defined the subunit structure of sodium and calcium channels was small and close-knit. They moved on to an interesting array of careers and family activities.3
Auxiliary Subunits of Sodium and Calcium Channels
The pore-forming subunits of sodium and calcium channels were expected, but the auxiliary subunits were not anticipated and were initially controversial. However, studies with subunit-specific antibodies confirmed that the α and β subunits of sodium channels were stoichiometrically associated in brain neurons (33–35). Moreover, studies of biosynthesis and assembly of sodium channels in developing neurons in cell culture revealed that subunit assembly occurs during intracellular synthesis and processing of the subunit polypeptides, and association with β subunits favors insertion of mature sodium channel complexes into the plasma membrane (35). These results further supported the significance of these auxiliary subunits in the sodium channel complex. They also presaged much subsequent work with cloned sodium and calcium channel subunits, which confirms a chaperone-like function for the sodium channel β subunits in cell surface insertion of mature sodium channel complexes and similar roles for the β and α2δ subunits of calcium channels (36–42).
Sodium Channel Structure in Two and Three Dimensions
Purification and reconstitution of the sodium and calcium channel proteins were the main research focus of my laboratory from its inception in 1977 until 1987 when the calcium channel subunit architecture was defined. Increasingly toward the end of this decade of work, we re-focused on structure-function studies based on determination of the primary structures of the principal subunits of sodium and calcium channels. This work has consumed most of our research effort from 1987 to the present, as well as the research effort of many other laboratories in the field. We have been fortunate in this phase of our work to achieve atomic-level answers to the three key functional questions about sodium and calcium channels: How does transmembrane voltage activate and inactivate these channel proteins? How are sodium and calcium conducted rapidly and selectively? How do drugs and toxins modify these key functional properties of sodium and calcium channels? Some highlights of this work are briefly summarized below, following a description of the structure of sodium channels in two and three dimensions.
Cloning and Expression
Cloning and expression of the sodium and calcium channels opened the way to extensive work in many laboratories on the structure and function of these key cell-signaling proteins. Shosaku Numa at Kyoto University and his large team of collaborators were first to clone cDNA encoding the sodium channel principal subunit and determine its primary structure (43, 44). Other laboratories followed their lead (45–47), including a collaborative team effort that we forged to clone brain sodium channels using specific antibodies (48, 49). Functional expression of cloned sodium channels in Xenopus oocytes further confirmed that the cloned cDNAs encoded fully functional sodium channels and opened the way to structure-function studies through mutagenesis (48, 50). The structure of sodium channel α subunits was the most complex of any membrane protein at the time it was established. This protein of ∼2,000 amino acid residues is organized in four homologous domains where each domain contains six transmembrane segments (S1–S6) and an additional membrane reentrant segment (Fig. 6). Structure-function studies revealed that each homologous domain containing six transmembrane segments consists of two distinct functional modules, a voltage-sensing module composed of the S1–S4 segments and a pore-forming module composed of the S5 and S6 segments and the membrane-reentrant pore loop (P) between them (reviewed in Ref. 51). This two-dimensional map of the sodium channel provided the template for many years of structure-function studies as described below.
FIGURE 6.

The primary structures of the subunits of the voltage-gated sodium channels. Cylinders represent α helical segments. Bold lines represent the polypeptide chains of each subunit with length approximately proportional to the number of amino acid residues in the brain sodium channel subtypes. The extracellular domains of the β1 and β2 subunits are shown as immunoglobulin-like folds. Ψ, sites of probable N-linked glycosylation; P in red, sites of demonstrated protein phosphorylation by PKA (circles) and PKC (diamonds); green, pore-lining segments; white circles, the outer (EEEE) and inner (DEKA) rings of amino residues that form the ion selectivity filter and the tetrodotoxin-binding site; yellow, S4 voltage sensors; h in blue circle, inactivation particle in the inactivation gate loop; blue circles, sites implicated in forming the inactivation gate receptor (51). Sites of binding of α- and β-scorpion toxins and a site of interaction between α and β1 subunits are also shown. Tetrodotoxin is a specific blocker of the pore of sodium channels (3), whereas the α- and β-scorpion toxins block fast inactivation and enhance activation, respectively, and thereby generate persistent sodium current that causes depolarization block of nerve conduction (111). Tetrodotoxin has been used as a tool to probe the pore of the sodium channel, whereas the scorpion toxins have been valuable as probes of voltage sensor function. Inset, structure of the fast inactivation gate of mammalian Nav1.2 channels in solution determined by NMR (91).
X-ray Crystallography
Because of their complexity, mammalian sodium channels are especially challenging for high-level expression, purification, and crystallization; analysis of their three-dimensional structure has eluded scientists in our field for over twenty-five years. In contrast, the members of the NaChBac family of ancestral bacterial sodium channels have the structure of a single domain of their eukaryotic descendants with no large intracellular or extracellular linking segments (52). They function as symmetrical homotetramers, providing additional simplicity of structural design. We screened numerous bacterial sodium channels and selected the voltage-gated sodium channel from Arcobacter butzleri (NavAb) as the best candidate for structural studies (53). The x-ray crystal structure of NavAb at 2.7 Å revealed a central pore surrounded by the four pore-forming modules (S5-P-S6) lining its walls. The four voltage-sensing modules composed of S1–S4 segments are located in nearly symmetrical positions on the periphery of the pore (53). A side view shows the arrangement of the 24 transmembrane segments (Fig. 7A). The voltage-sensing module of one subunit interacts most closely with the pore-forming module of its nearest neighbor (Fig. 7A, highlighted in colors). This domain-swapped arrangement interlaces the subunits and may contribute to concerted opening of the central pore.
FIGURE 7.

Structure of NavAb. A, side view of NavAb channels with the transmembrane segments of one subunit highlighted in colors. B, side view of the structure of the NavAb voltage sensor in an activated state (53). ENC, extracellular negative cluster (red); HCS, hydrophobic constriction site (green); INC, intracellular negative cluster (red). C, structural transitions during activation of the voltage sensor. Note the steady outward movement of the S4 gating charges from left to right (75).
Analysis of Sodium Channel Structure and Function
Site-directed Mutagenesis and Functional Analysis
Introduction of site-directed mutations into mammalian sodium channels and analysis of their functional effects by electrophysiological recording have yielded a remarkably detailed view of the components of the sodium channel that are required for its physiological activity. In early work, wild-type and mutant sodium channels were expressed from mRNA transcribed in vitro, injected into Xenopus oocytes, and studied by two-microelectrode voltage-clamp (50). With help from our colleague Dr. Bertil Hille (Department of Physiology and Biophysics, University of Washington), postdoctoral fellow Dr. Tohru Gonoi established the whole-cell patch clamp method in my laboratory for studies of sodium channels in cultured cells (54). The addition of postdoctoral fellow, and later research professor, Dr. Todd Scheuer to the group cemented our skills in electrophysiology and membrane biophysics. We used the more precise whole-cell patch clamp method extensively for biophysical studies of sodium channel mutants and for studies of ion channel regulation (55). The combination of site-directed mutagenesis with functional analysis by expression in Xenopus oocytes or mammalian cell lines has been used in dozens of laboratories to give key insights into sodium channel structure and function.
Antibody Mapping
Knowledge of the primary structure of the principal subunits of sodium and calcium channels also opened the way to structure-function studies using sequence-directed antibodies to identify functional domains and sites of ligand binding on sodium and calcium channels in our laboratory (56–58). In this experimental approach, we raised polyclonal antibodies against specific short peptide sequences (12–16 residues) predicted to be available on the surface of the sodium channel. These site-directed antibodies that recognize specific amino acid sequences were used to probe sodium channel function, identify covalently labeled channel fragments with bound toxins or drugs, and define the level of expression of the different subtypes of sodium channel proteins.
Voltage-dependent Activation
The exquisite sensitivity of voltage-gated sodium channels to transmembrane voltage is unique among membrane proteins, approached only by their close molecular cousins, the voltage-gated calcium and potassium channels. For that reason, the mechanism of voltage sensing has been a holy grail for many in the field. Hodgkin and Huxley proposed that voltage sensitivity arises because “charged particles” associated with sodium channels move across the membrane under the influence of the membrane electric field and cause opening of the channel (1). This charge movement implied that the conformational change that activates the sodium channel protein would cause a capacitive current as the charged particles (now termed gating charges) moved across the membrane. This gating current was first measured by Dr. Clay Armstrong and Dr. Francisco (Pancho) Bezanilla in the squid giant axon, the experimental preparation that Hodgkin and Huxley had used so effectively in their pioneering work (2, 59). Modern methods show that 3–4 positive charges move outward across the membrane during activation of each of the four voltage sensors in sodium channels (60, 61).
The earliest analysis of the amino acid sequence of the sodium channel α subunit revealed predicted S4 α helices with positively charged amino acid residues, usually arginines, positioned at intervals of three with hydrophobic residues surrounding them (43). Because of their high concentration of charge, these S4 helices were proposed to project into the cytosol in a helical hairpin structure in the initial model of sodium channels (43). However, in this position, the charged amino acid residues would be unable to sense and react to changes in the transmembrane potential, which extends only across the membrane itself. To connect this concentration of positive charge to the voltage-sensing process, I proposed the sliding helix model of voltage sensor function (62, 63) based on thermodynamic considerations. First, in order that the array of positive charges in the S4 segments could participate in voltage sensing, they were placed in a transmembrane position. Second, to stabilize them in their transmembrane position, they were proposed to form ion pairs with negatively charged residues from other transmembrane segments. Third, to allow them to move gating charge outward upon depolarization without destabilizing the structure, they were proposed to move outward along a spiral pathway by exchange of ion pair partners. A similar helical screw model was proposed based on structural modeling from the amino acid sequence (64).
All of the predictions of the sliding-helix model have now been verified through work on sodium channels in many laboratories (65). Mutations of the S4 arginine gating charges strongly affect the voltage dependence of activation (66). The S4 segments are in a transmembrane position in both resting and activated states, as judged by neurotoxin binding to the S3-S4 linkers on the extracellular side of the cell (67–69). The S4 segments move outward through the voltage-sensing module upon depolarization, as detected by covalent reaction of substituted cysteine residues with intracellular or extracellular cysteine reagents (70, 71). As they move outward, the S4 segments exchange ion pair partners with negatively charged amino acid residues in the S1–S3 segments, as demonstrated by disulfide locking of the voltage sensor with pairs of substituted cysteine residues (72–75).
This model of voltage sensing by sodium channels has been placed in three-dimensional context by molecular modeling and x-ray crystallography (Fig. 7, B and C) (53, 75). The voltage sensor in the NavAb structure is in an activated state, with three gating charges (R1–R3) interacting with the extracellular negative cluster on the extracellular side of the membrane permeability barrier formed by a hydrophobic constriction site (HCS) (Fig. 7B) (53). The fourth gating charge (R4) interacts with the intracellular negative cluster on the intracellular side of the HCS.
The structure of the resting state of the sodium channel voltage sensor has not been determined, because of the difficulty to generate and stabilize the resting state in the absence of the large negative resting membrane potential in which it exists in cells. We determined the most likely structure of the resting state of the voltage sensor using the Rosetta Membrane ab initio structural modeling program, developed by Dr. David Baker and his colleagues (Department of Biochemistry, University of Washington), which is the most successful paradigm for prediction of membrane protein structure (75–77). We tested the predictions of the molecular models of the resting and activated states of the voltage sensor in state-dependent disulfide-crosslinking experiments. These studies defined a series of resting and activated states of the voltage sensor (Fig. 7C) (75). In response to depolarization, the S4 segment moves outward through a gating pore exchanging ion pair partners and moving gating charges through the HCS. Movement of the R3 gating charge to the extracellular side of the HCS generates Activated State 1, the first state of the voltage sensor that leads to pore opening. Further depolarization leads to Activated State 2, which is similar to the state captured in the NavAb crystal structure, then to Activated State 3 (Fig. 7C), and finally to one further activated state captured in the NavRh crystal structure (78).
The voltage-driven outward and rotational movements of the S4 segment induce corresponding counter-rotation of the S1–S3 segments as ion pair partners are exchanged. These movements exert a torque in the plane of the intracellular membrane surface on the S5 and S6 segments in the pore module through their covalent connection via the S4-S5 linker and their noncovalent interactions (75). Subtle rotational and bending motions of the pore-lining S6 segments open the pore at its intracellular end in an iris-like motion. These motions can be most easily seen in movies that we constructed by linking the six states of the voltage-sensing module, as defined in disulfide-locking experiments, with concerted pore opening (see online movies in Ref. 75).
Scorpion Toxins as Probes of Voltage Sensor Structure
In addition to their fundamental role in identification of the sodium channel subunits by photoaffinity labeling (10), scorpion toxins have proven to be excellent tools in structure-function studies. Scorpion toxins are peptides of ∼60–70 amino acid residues that are tightly folded and locked in place by disulfide bonds (79). They have a wedge-shaped structure, as determined by x-ray crystallography and NMR (79). Through a combination of extensive mutagenesis and molecular modeling of sodium channel states with the Rosetta Membrane algorithm, we have found that these toxins fit into the wedge-shaped aqueous cleft in the voltage sensor formed by the S1-S2 and S3-S4 extracellular loops (Fig. 7B). Binding in this position prevents the voltage-driven conformational changes in the voltage sensor (Fig. 7C), effectively locking the voltage sensor in specific states. α- and β-scorpion toxins work together to modify sodium channel function. β-Scorpion toxins trap the voltage sensor in domain II in an activated conformation, thereby favoring activation of the channel at negative membrane potentials because the work of activating that voltage sensor has already been done by toxin binding (80, 81). α-Scorpion toxins bind to the voltage sensor in domain IV in an intermediate resting state and prevent coupling of channel activation to fast inactivation, thereby working together with the β-scorpion toxin bound to the voltage sensor in domain II to produce persistently activated sodium channels (82). The two bound toxins form a two-fold pseudosymmetric complex with the sodium channel voltage sensors in domains II and IV (81). These structural models of the voltage sensors with scorpion toxins bound give insight into the conformational changes that take place during the function of mammalian voltage sensors.
Ion Conductance
The structure of NavAb reveals a central pore with an outer vestibule, a narrow selectivity filter, a large water-filled central cavity, and an intracellular activation gate in the closed position (Fig. 8A) (53). The ion selectivity filter consists of three sodium interaction sites: a high-field-strength site formed by a nearly square array of the side chains of four glutamate residues followed by two additional nearly square sites formed by the backbone carbonyls of leucine and threonine residues (Fig. 8, B and C) (53). Molecular dynamics simulations reveal that these sites accommodate partially dehydrated sodium ions, rather than fully dehydrated ones, during the permeation process (83), as if the selectivity filter “feels” the identity of the conducted ion indirectly through its inner hydration shell. As observed in single channel recordings, up to 107 sodium ions moved through the selectivity filter per second at 0 mV in the presence of a physiological sodium gradient in these molecular dynamics simulations (83). The presence of a high-field-strength site and the conductance of hydrated sodium ions clearly distinguish the sodium channel permeation process from potassium channels, in which fully dehydrated potassium ions interact directly with backbone carbonyls without intermediary waters of hydration and without any charged amino acid side chains (84). This distinct chemistry of ion permeation had evolved even in prokaryotes, illustrating a truly ancient division of pore structure, function, and chemistry in the ion channel protein superfamily.
FIGURE 8.

Structure of the pore and selectivity filter of NavAb and CavAb. A, architecture of the NavAb pore. Purple, Glu177 side chains; gray, pore volume. The S5 and S6 segments and the P loop from two lateral subunits are shown (53). B, top view of the ion selectivity filter. Symmetry-related molecules are colored white and yellow; P-helix residues are colored green. Hydrogen bonds between Thr175 and Trp179 are indicated by gray dashes. Electron densities from Fo − Fc omit maps are contoured at 4.0 σ (blue and gray), and subtle differences can be appreciated (small arrows) (53). C, side view of the ion selectivity filter. Glu177 (purple) interactions with Gln172, Ser178, and the backbone of Ser180 are shown in the far subunit. Fo − Fc omit map at 4.75 σ (blue) and putative cations or water molecules (red spheres, IonEX) are shown. Electron density around Leu176 (gray; Fo − Fc omit map at 1.75 σ) and a putative water molecule are shown (gray sphere). Na+ coordination sites: SiteHFS, SiteCEN, and SiteIN (53). D, CavAb selectivity filter. Electron density at the selectivity filter of 175TLDDWSN181 is shown. The 2Fo − Fc electron density map (contoured at 2 σ) of select residues in the selectivity filter with two diagonally opposed subunits is shown in stick format, the Ca2+ ions along the ion pathway are shown in green spheres, and water molecules are shown in red spheres. Note that it is likely that adjacent calcium-binding sites are not occupied simultaneously because of electrostatic repulsion. Our model is that the site at Asp181 in the outer vestibule and the high-field-strength site at Asp177 are occupied together, the sites at Asp178 and the carbonyl sites at Leu176/Thr175 are occupied together, and the selectivity filter and vestibule oscillate between these two ion occupancies to mediate rapid conductance.
Fast Inactivation
As Hodgkin and Huxley showed, fast inactivation terminates the sodium current within a few milliseconds of its activation (1). This second gating process has distinct voltage and time dependence from activation. Early studies with proteases showed that fast inactivation is a separate molecular process because it could be destroyed by proteolytic enzyme treatment of the intracellular surface of the channel without any effects on activation or ion conductance (85). We used the site-directed antibody method to identify the intracellular loop connecting domains III and IV of the sodium channel α subunit as the fast inactivation gate (56, 86). Our results supported the idea that this loop folds into the channel pore and blocks it like a hinged lid, because the inactivation gate was protected from antibody binding when the channel was already inactivated (56, 86). Identification of the inactivation gate by site-directed antibodies was confirmed by mutagenesis studies (66), and we found that a hydrophobic motif (Ile-Phe-Met, IFM) served as the latch that holds the gate closed (87). The IFM motif moves into the pore and becomes inaccessible to chemical reagents during fast inactivation (88). Single channel recording showed that the IFM motif holds the inactivation gate shut because mutations in it cause repetitive re-openings (89). In contrast, a preceding pair of glycine residues serves as a hinge, and mutations of those residues slow entry to the inactivated state (90). The structure of the fast inactivation gate was determined by NMR, revealing a scaffold of an α helix preceded by two β turns on which the IFM motif is arrayed (Fig. 6, inset) (91). This structure gives the only high-resolution view of a functional component of mammalian sodium channels. The hinged-lid model of fast inactivation fits well with classical concept of an intracellular fast inactivation gate that is sensitive to proteolysis (2).
Slow Inactivation
Eukaryotic sodium channels also have a distinct slow-inactivation process that proceeds on the time scale of hundreds of milliseconds and is reversed in seconds (92, 93). Although fast inactivation is not observed in bacterial sodium channels because they have no intracellular linkers, they do have a slow inactivation process (94). The slow-inactivated state of NavAb is its most stable state at 0 mV. A crystal structure has captured wild-type NavAb in two potentially slow-inactivated states (95). The hallmark structural change during slow inactivation is collapse of the pore from a nearly square configuration to a striking dimer-of-dimers organization in which two S6 segments have moved toward the pore axis and two have moved away (95). This conformational change to a dimer-of-dimers structure is observed at the selectivity filter, central cavity, and intracellular activation gate. A similar collapsed-pore structure was observed for another bacterial sodium channel that is also thought to be in a slow-inactivated state (78).
Ion Conductance and Selectivity in Calcium Channels
Bacterial sodium channels are equally similar in amino acid sequence to mammalian sodium and calcium channels, and they are the likely ancestors of both of these ion channel protein families (96). Voltage-dependent activation of calcium channels is likely to be similar to sodium channels in its fundamental mechanism, because this gating process is an ancient and conserved feature of these ion channels. Voltage-dependent inactivation of calcium channels may also resemble slow inactivation of sodium channels. On the other hand, the mechanism of calcium conductance must have fundamentally different chemistry to allow rapid and highly selective calcium entry and yet avoid entry of the 70-fold more abundant sodium ions in extracellular fluids. Sodium and calcium ions have similar molecular diameters (∼2 Å), so selective conductance must involve specific chemical interactions rather than molecular sieving. The selectivity filter sequence in NavAb is -TLESWSM-. The -TLE- residues form the high-field-strength site and two backbone carbonyl sites that interact sequentially with sodium ions as they move through the pore; the -SWSM- residues form the outer vestibule that leads ions from the extracellular solution into the pore. The NavAb selectivity filter binds a single calcium ion in the 10 mm concentration range, which blocks the pore. We found that the addition of a second negative charge to give the sequence -TLEDWSM- creates a second calcium-binding site in the selectivity filter (97). This small molecular change has a dramatic effect on ion permeation and selectivity, converting NavAb from 33-fold selective for sodium to 30-fold selective for calcium, nearly a 1,000-fold change (97). Two additional substitutions of Asp residues to give the sequence -175TLDDWSD181- create a third calcium-binding site at the extracellular opening of the vestibule and increase the calcium selectivity to 400-fold, which overall is a 12,000-fold change in ion selectivity from NavAb, to yield a value similar to a mammalian calcium channel (97). This construct, which we named CavAb, has one calcium-binding site in the vestibule formed by Asp181, two calcium-binding sites in the ion selectivity filter formed by the carboxylate side chains of Asp177 and Asp178, and a fourth binding site in the selectivity filter formed by the backbone carbonyls of Leu176 and Thr175 (Fig. 8D). These four binding sites work in tandem. Calcium ions bind to the first and third sites or to the second and fourth sites. Occupancy of adjacent sites is prevented by electrostatic repulsion. When the second and fourth sites are occupied, calcium binding to the first site from the extracellular fluid knocks the innermost calcium ion into the cell and returns occupancy of the first and third site. This “knock-off” mechanism yields high selectivity by blocking entry of sodium ions in the pore, yet it allows high conductance by repulsive interactions between bound calcium ions (97), as proposed in biophysical models of calcium permeation (98–100). This fundamental mechanism allows electrical signals in the cell surface membrane to be transduced into an intracellular calcium transient, which initiates all of the physiological events that are triggered by electrical signals, including secretion, contraction, neurotransmission, gene transcription, and more. Development of eukaryotes to metazoans required this coupling of cell surface electrical signaling to intracellular calcium signaling; therefore, the emergence of voltage-gated sodium and calcium channels was one of the key events in metazoan evolution.
Looking Back and Forward
Molecular studies of voltage-gated ion channels were a new idea when we started our experiments in 1977. With good luck and the exceptional efforts of many colleagues, first sodium channels, and then calcium channels, yielded to biochemical approaches aimed at purification and functional characterization. It has been gratifying to see these channels become among the best understood membrane proteins in terms of the details of their structure and function. Sodium channels were the first of the superfamily of 143 related ion channels of the voltage-gated chanome (96) in the human genome to be characterized at the molecular level, so the early work from my laboratory and others has had an important impact beyond the advances on sodium and calcium channels themselves. Work in my laboratory has also focused on the molecular pharmacology of sodium and calcium channels (recently reviewed in Ref. 101) and their regulation by hormones and neurotransmitters acting through second messenger signaling pathways whose members are organized in ion channel signaling complexes (reviewed in Refs. 102–105). These studies have yielded atomic-level views of drug action on sodium and calcium channels as well as insights into physiological regulation of these channels by multi-protein signaling complexes in the physiological contexts of electrical signaling, synaptic transmission, and excitation-contraction coupling.
In parallel with the work on the structure and function of sodium and calcium channels, human geneticists, neurologists, and cardiologists have made a series of unexpected, groundbreaking discoveries of ion channelopathies that are caused by mutations in sodium and calcium channels, including periodic paralysis, epilepsy, migraine, chronic pain, and cardiac arrhythmia (106–110). Looking to the future, research in my laboratory will increasingly apply the advances in understanding the structure, function, and molecular pharmacology of sodium and calcium channels to better define the pathophysiology and to accelerate novel therapies for periodic paralysis (111–113), epilepsy (114, 115), autism (116), and heart failure (117, 118). We look forward to substantial advances in treatment of these diseases based on new understanding of their underlying functional defects and the emerging novel approaches to therapeutics.
Acknowledgments
Research in my laboratory at the University of Washington on the biochemistry, structure, and function of sodium and calcium channels has been generously funded by three long-running research grants from the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health (R01 NS15751, R01 NS22625, and R01 NS25704) and by additional grants from the National Institutes of Health, the National Science Foundation, the Muscular Dystrophy Association, and the American Heart Association. Special thanks go to long-time collaborators Ruth Westenbroek (Research Associate Professor of Pharmacology) and Todd Scheuer (Research Professor Emeritus of Pharmacology) for their crucial insights and key technical contributions to our research progress. Special thanks go as well to my structural biology collaborators at the University of Washington, Rachel Klevit (Professor of Biochemistry), David Baker (Professor of Biochemistry and Investigator of the Howard Hughes Medical Institute), and Ning Zheng (Professor of Pharmacology and Investigator of the Howard Hughes Medical Institute), whose generosity and expertise have made it possible for us to pursue our structure-function studies to the atomic level. Finally, it has been a pleasure to write this Reflections article for the JBC. I joined the American Society for Biological Chemistry as a graduate student member in 1969, and I published my thesis research in the JBC, as well as most of the early biochemical studies of sodium channels that are featured here. The American Society for Biochemistry and Molecular Biology and the JBC continue as major forces in science, with a purview that has broadened from biochemistry to encompass much of modern biomedical science, including my favorite topics of cell signaling, membrane biology, neurobiology, and molecular pharmacology.
Footnotes
The story of saxitoxin as a molecular tool is a remarkable sidebar to this account of the early biochemical studies of sodium channels. Saxitoxin is produced by dinoflagellates that bloom as “red tide” off the coast of the Pacific Northwest, Western Canada, and Alaska. The toxin is concentrated by filter-feeding mollusks such as clams and mussels. It is a public health risk for people eating those shellfish. Isolating saxitoxin in quantities useful for research would be an exceptional effort for a single laboratory. However, in the late 1960s and early 1970s, the Central Intelligence Agency collected biological toxins to study how to defend against their use in biological warfare and potentially to learn how to use them in covert operations. In a famous episode, these activities were revealed to the U. S. Senate Intelligence Committee chaired by Senator Frank Church in a review of CIA operations, and they were deemed to be inappropriate in light of international agreements against chemical and biological warfare. This led to a requirement for the CIA to divest its stocks of biological toxins, including saxitoxin, and plans were laid for destruction of these materials. After reading about these plans in the Washington Post in the mid-1970s while a staff scientist at the National Institutes of Health, I wrote to the Senate Committee urging that saxitoxin and other precious research resources in these CIA stockpiles be transferred to the National Institutes of Health for research purposes. In parallel, and with greater impact, Professor J. Murdoch Ritchie of Yale University also urged re-purposing these stockpiles of saxitoxin for research (119). As a result, saxitoxin that had been isolated by harvesting tons of Alaskan butter clams with bulldozers and then extracted and purified in grand scale at the U. S. Department of Defense Laboratories at Frederick, Maryland became available to researchers for studies of sodium channels. This stockpile of saxitoxin, and the methods developed by Ritchie and colleagues to radiolabel it with 3H (6, 7) were essential for the eventual purification and analysis of the sodium channel protein in my laboratory and others.
The α2δ subunits of calcium channels are encoded by the same gene, whose protein product was found to be proteolytically cleaved and disulfide-linked to yield the mature α2δ subunits in early biochemical studies of purified skeletal muscle calcium channels (120). Recent work shows that this post-translational processing reaction proceeds further to delete the transmembrane segment of the δ subunit and add a glycophosphatidylinositol anchor in neurons and other transfected cells (121). It is not known at present whether this additional post-translational processing occurs for the skeletal muscle calcium channel; however, it would not have been possible to characterize this form of post-translational processing in the original biochemical studies of skeletal muscle calcium channels, as this mode of post-translational processing had not been discovered at that time.
Where are they now? Dan Beneski earned an M. D. degree at Jefferson Medical College and led a large practice in anesthesiology in Philadelphia. After his postdoctoral research at the University of California at San Diego, Bob Hartshorne became Assistant and then Associate Professor of Pharmacology at Oregon Health & Science University, but Bob died tragically in a hiking accident near Glacier Peak in the Washington Cascades. Don Messner moved to Washington University in St. Louis for postdoctoral studies, became a faculty member at the University of Rochester, and then returned to Seattle to join Bastyr University where he is Research Associate Professor. Mike Tamkun pursued postdoctoral studies at Johns Hopkins University, thrived as a faculty member at the Department of Physiology at Vanderbilt University, and now heads the Molecular, Cellular, & Integrative Neurosciences Program as Professor of Biomedical Sciences at Colorado State University. Jane Talvenheimo pursued her research career at Amgen in Thousands Oaks, California, contributing to their drug discovery efforts. Ben Curtis was a postdoctoral fellow at the Laboratory of Molecular Biology at the University of Cambridge, United Kingdom and then at the California Institute of Technology. We have lost track of Ben after a move to New Zealand. Masami Takahashi returned from his postdoctoral research in Seattle to a successful career at Mitsubishi-Kasei Research Institute in Japan and then became Chair of Biochemistry at Kitasato University near Tokyo. Mike Seagar became Director of the INSERM Unit on Neurobiology of Ion Channels at the University of Aix-Marseille in France, where he has recently retired from his administrative role. Bernie Reber was a faculty member in the Department of Pharmacology at the University of Bern, Switzerland and now leads a research laboratory at the Swiss Army Research Institute. Jeff Coppersmith moved on to a career in audiology in Seattle. Cynthia Morrow and Jean Jones retired from research and raised their families in the Seattle area. Carl Baker continued his work on the biochemical properties of sodium and calcium channels with us for many years and then moved on to genetic research at the University of Washington.
References
- 1.Hodgkin A. L., and Huxley A. F. (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong C. M. (1981) Sodium channels and gating currents. Physiol. Rev. 61, 644–683 [DOI] [PubMed] [Google Scholar]
- 3.Hille B. (2001) Ionic Channels of Excitable Membranes, 3rd Ed, Sinauer Associates Inc., Sunderland, MA [Google Scholar]
- 4.Catterall W. A. (1980) Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu. Rev. Pharmacol. Toxicol. 20, 15–43 [DOI] [PubMed] [Google Scholar]
- 5.Benzer T. I., and Raftery M. A. (1972) Partial characterization of a tetrodotoxin-binding component from nerve membrane. Proc. Natl. Acad. Sci. U.S.A. 69, 3634–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson R., Ritchie J. M., and Strichartz G. R. (1973) The binding of labelled saxitoxin to the sodium channels in nerve membranes. J. Physiol. 235, 783–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritchie J. M., Rogart R. B., and Strichartz G. R. (1976) A new method for labelling saxitoxin and its binding to non-myelinated fibres of the rabbit vagus, lobster walking leg, and garfish olfactory nerves. J Physiol 261, 477–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Couraud F., Jover E., Dubois J. M., and Rochat H. (1982) Two types of scorpion toxin receptor sites, one related to the activation, the other to the activation of the action potential sodium channel. Toxicon 20, 9–16 [DOI] [PubMed] [Google Scholar]
- 9.Vincent J. P., Balerna M., Barhanin J., Fosset M., and Lazdunski M. (1980) Binding of sea anemone toxin to receptor sites associated with gating system of sodium channel in synaptic nerve endings in vitro. Proc. Natl. Acad. Sci. U.S.A. 77, 1646–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beneski D. A., and Catterall W. A. (1980) Covalent labeling of protein components of the sodium channel with a photoactivable derivative of scorpion toxin. Proc. Natl. Acad. Sci. U.S.A. 77, 639–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catterall W. A., Morrow C. S., and Hartshorne R. P. (1979) Neurotoxin binding to receptor sites associated with voltage-sensitive sodium channels in intact, lysed, and detergent-solubilized brain membranes. J. Biol. Chem. 254, 11379–11387 [PubMed] [Google Scholar]
- 12.Hartshorne R. P., and Catterall W. A. (1981) Purification of the saxitoxin receptor of the sodium channel from rat brain. Proc. Natl. Acad. Sci. U.S.A. 78, 4620–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartshorne R. P., Messner D. J., Coppersmith J. C., and Catterall W. A. (1982) The saxitoxin receptor of the sodium channel from rat brain: evidence for two nonidentical β subunits. J. Biol. Chem. 257, 13888–13891 [PubMed] [Google Scholar]
- 14.Hartshorne R. P., and Catterall W. A. (1984) The sodium channel from rat brain: purification and subunit composition. J. Biol. Chem. 259, 1667–1675 [PubMed] [Google Scholar]
- 15.Reber B. F., and Catterall W. A. (1987) Hydrophobic properties of the β1 and β2 subunits of the rat brain sodium channel. J. Biol. Chem. 262, 11369–11374 [PubMed] [Google Scholar]
- 16.Tamkun M. M., and Catterall W. A. (1981) Reconstitution of the voltage-sensitive sodium channel of rat brain from solubilized components. J. Biol. Chem. 256, 11457–11463 [PubMed] [Google Scholar]
- 17.Tamkun M. M., Talvenheimo J. A., and Catterall W. A. (1984) The sodium channel from rat brain: reconstitution of neurotoxin-activated ion flux and scorpion toxin binding from purified components. J. Biol. Chem. 259, 1676–1688 [PubMed] [Google Scholar]
- 18.Talvenheimo J. A., Tamkun M. M., and Catterall W. A. (1982) Reconstitution of neurotoxin-stimulated sodium transport by the voltage-sensitive sodium channel purified from rat brain. J. Biol. Chem. 257, 11868–11871 [PubMed] [Google Scholar]
- 19.Sigworth F. J., and Neher E. (1980) Single Na+ channel currents observed in cultured rat muscle cells. Nature 287, 447–449 [DOI] [PubMed] [Google Scholar]
- 20.Hartshorne R. P., Keller B. U., Talvenheimo J. A., Catterall W. A., and Montal M. (1985) Functional reconstitution of the purified brain sodium channel in planar lipid bilayers. Proc. Natl. Acad. Sci. U.S.A. 82, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agnew W. S., Moore A. C., Levinson S. R., and Raftery M. A. (1980) Identification of a large molecular weight peptide associated with a tetrodotoxin binding proteins from the electroplax of Electrophorus electricus. Biochem. Biophys. Res. Commun. 92, 860–866 [DOI] [PubMed] [Google Scholar]
- 22.Barchi R. L. (1983) Protein components of the purified sodium channel from rat skeletal muscle sarcolemma. J Neurochem 40, 1377–1385 [DOI] [PubMed] [Google Scholar]
- 23.Curtis B. M., and Catterall W. A. (1983) Solubilization of the calcium antagonist receptor from rat brain. J. Biol. Chem. 258, 7280–7283 [PubMed] [Google Scholar]
- 24.Curtis B. M., and Catterall W. A. (1984) Purification of the calcium antagonist receptor of the voltage-sensitive calcium channel from skeletal muscle transverse tubules. Biochemistry 23, 2113–2118 [DOI] [PubMed] [Google Scholar]
- 25.Curtis B. M., and Catterall W. A. (1986) Reconstitution of the voltage-sensitive calcium channel purified from skeletal muscle transverse tubules. Biochemistry 25, 3077–3083 [DOI] [PubMed] [Google Scholar]
- 26.Flockerzi V., Oeken H. J., Hofmann F., Pelzer D., Cavalié A., and Trautwein W. (1986) Purified dihydropyridine-binding site from skeletal muscle t-tubules is a functional calcium channel. Nature 323, 66–68 [DOI] [PubMed] [Google Scholar]
- 27.Takahashi M., Seagar M. J., Jones J. F., Reber B. F., and Catterall W. A. (1987) Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 84, 5478–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leung A. T., Imagawa T., and Campbell K. P. (1987) Structural characterization of the 1,4-dihydropyridine receptor of the voltage-dependent Ca2+ channel from rabbit skeletal muscle: evidence for two distinct high molecular weight subunits. J. Biol. Chem. 262, 7943–7946 [PubMed] [Google Scholar]
- 29.Hosey M. M., Barhanin J., Schmid A., Vandaele S., Ptasienski J., O'Callahan C., Cooper C., and Lazdunski M. (1987) Photoaffinity labelling and phosphorylation of a 165 kilodalton peptide associated with dihydropyridine and phenylalkylamine-sensitive calcium channels. Biochem. Biophys. Res. Commun. 147, 1137–1145 [DOI] [PubMed] [Google Scholar]
- 30.Sieber M., Nastainczyk W., Röhrkasten A., and Hofmann F. (1987) Reconstitution of the purified receptor for calcium channel blockers. Biomed. Biochim. Acta 46, S357–S362 [PubMed] [Google Scholar]
- 31.Sieber M., Nastainczyk W., Zubor V., Wernet W., and Hofmann F. (1987) The 165-kDa peptide of the purified skeletal muscle dihydropyridine receptor contains the known regulatory sites of the calcium channel. Eur. J. Biochem. 167, 117–122 [DOI] [PubMed] [Google Scholar]
- 32.Striessnig J., Knaus H. G., Grabner M., Moosburger K., Seitz W., Lietz H., and Glossmann H. (1987) Photoaffinity labelling of the phenylalkylamine receptor of the skeletal muscle transverse-tubule calcium channel. FEBS Lett. 212, 247–253 [DOI] [PubMed] [Google Scholar]
- 33.Wollner D. A., Messner D. J., and Catterall W. A. (1987) β2 subunits of sodium channels from vertebrate brain: studies with subunit-specific antibodies. J. Biol. Chem. 262, 14709–14715 [PubMed] [Google Scholar]
- 34.Sutkowski E. M., and Catterall W. A. (1990) β1 subunits of sodium channels: studies with subunit-specific antibodies. J. Biol. Chem. 265, 12393–12399 [PubMed] [Google Scholar]
- 35.Schmidt J. W., and Catterall W. A. (1986) Biosynthesis and processing of the α subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46, 437–444 [DOI] [PubMed] [Google Scholar]
- 36.Isom L. L., De Jongh K. S., Patton D. E., Reber B. F. X., Offord J., Charbonneau H., Walsh K., Goldin A. L., and Catterall W. A. (1992) Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science 256, 839–842 [DOI] [PubMed] [Google Scholar]
- 37.Isom L. L., Ragsdale D. S., De Jongh K. S., Westenbroek R. E., Reber B. F., Scheuer T., and Catterall W. A. (1995) Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83, 433–442 [DOI] [PubMed] [Google Scholar]
- 38.Isom L. L., De Jongh K. S., and Catterall W. A. (1994) Auxiliary subunits of voltage-gated ion channels. Neuron 12, 1183–1194 [DOI] [PubMed] [Google Scholar]
- 39.Singer D., Biel M., Lotan I., Flockerzi V., Hofmann F., and Dascal N. (1991) The roles of the subunits in the function of the calcium channel. Science 253, 1553–1557 [DOI] [PubMed] [Google Scholar]
- 40.Brackenbury W. J., and Isom L. L. (2011) Na Channel β subunits: overachievers of the ion channel family. Front Pharmacol. 2, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoppa M. B., Lana B., Margas W., Dolphin A. C., and Ryan T. A. (2012) α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486, 122–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dolphin A. C. (2012) Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat. Rev. Neurosci. 13, 542–555 [DOI] [PubMed] [Google Scholar]
- 43.Noda M., Shimizu S., Tanabe T., Takai T., Kayano T., Ikeda T., Takahashi H., Nakayama H., Kanaoka Y., Minamino N., et al. (1984) Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312, 121–127 [DOI] [PubMed] [Google Scholar]
- 44.Noda M., Ikeda T., Kayano T., Suzuki H., Takeshima H., Kurasaki M., Takahashi H., and Numa S. (1986) Existence of distinct sodium channel messenger RNAs in rat brain. Nature 320, 188–192 [DOI] [PubMed] [Google Scholar]
- 45.Rogart R. B., Cribbs L. L., Muglia L. K., Kephart D. D., and Kaiser M. W. (1989) Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc. Natl. Acad. Sci. U.S.A. 86, 8170–8174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trimmer J. S., Cooperman S. S., Tomiko S. A., Zhou J. Y., Crean S. M., Boyle M. B., Kallen R. G., Sheng Z. H., Barchi R. L., Sigworth F. J., et al. (1989) Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron 3, 33–49 [DOI] [PubMed] [Google Scholar]
- 47.Salkoff L., Butler A., Wei A., Scavarda N., Giffen K., Ifune C., Goodman R., and Mandel G. (1987) Genomic organization and deduced amino acid sequence of a putative sodium channel gene in Drosophila. Science 237, 744–749 [DOI] [PubMed] [Google Scholar]
- 48.Goldin A. L., Snutch T., Lübbert H., Dowsett A., Marshall J., Auld V., Downey W., Fritz L. C., Lester H. A., Dunn R., Catterall W. A., and Davidson N. (1986) Messenger RNA coding for only the α subunit of the rat brain Na channel is sufficient for expression of functional channels in Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 83, 7503–7507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Auld V. J., Goldin A. L., Krafte D. S., Marshall J., Dunn J. M., Catterall W. A., Lester H. A., Davidson N., and Dunn R. J. (1988) A rat brain sodium channel α subunit with novel gating properties. Neuron 1, 449–461 [DOI] [PubMed] [Google Scholar]
- 50.Noda M., Ikeda T., Suzuki H., Takeshima H., Takahashi T., Kuno M., and Numa S. (1986) Expression of functional sodium channels from cloned cDNA. Nature 322, 826–828 [DOI] [PubMed] [Google Scholar]
- 51.Catterall W. A. (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25 [DOI] [PubMed] [Google Scholar]
- 52.Ren D., Navarro B., Xu H., Yue L., Shi Q., and Clapham D. E. (2001) A prokaryotic voltage-gated sodium channel. Science 294, 2372–2375 [DOI] [PubMed] [Google Scholar]
- 53.Payandeh J., Scheuer T., Zheng N., and Catterall W. A. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonoi T., Hille B., and Catterall W. A. (1984) Voltage clamp analysis of sodium channels in normal and scorpion toxin-resistant neuroblastoma cells. J. Neurosci. 4, 2836–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheuer T., Auld V. J., Boyd S., Offord J., Dunn R., and Catterall W. A. (1990) Functional properties of rat brain sodium channels expressed in a somatic cell line. Science 247, 854–858 [DOI] [PubMed] [Google Scholar]
- 56.Vassilev P. M., Scheuer T., and Catterall W. A. (1988) Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 241, 1658–1661 [DOI] [PubMed] [Google Scholar]
- 57.Gordon D., Merrick D., Auld V., Dunn R., Goldin A. L., Davidson N., and Catterall W. A. (1987) Tissue-specific expression of the RI and RII sodium channel subtypes. Proc. Natl. Acad. Sci. U.S.A. 84, 8682–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gordon D., Merrick D., Wollner D. A., and Catterall W. A. (1988) Biochemical properties of sodium channels in a wide range of excitable tissues studied with site-directed antibodies. Biochemistry 27, 7032–7038 [DOI] [PubMed] [Google Scholar]
- 59.Armstrong C. M., and Bezanilla F. (1973) Currents related to movement of the gating particles of the sodium channels. Nature 242, 459–461 [DOI] [PubMed] [Google Scholar]
- 60.Kuzmenkin A., Bezanilla F., and Correa A. M. (2004) Gating of the bacterial sodium channel, NaChBac: voltage-dependent charge movement and gating currents. J. Gen. Physiol. 124, 349–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hirschberg B., Rovner A., Lieberman M., and Patlak J. (1995) Transfer of twelve charges is needed to open skeletal muscle Na+ channels. J. Gen. Physiol. 106, 1053–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Catterall W. A. (1986) Voltage-dependent gating of sodium channels: correlating structure and function. Trends Neurosci. 9, 7–10 [Google Scholar]
- 63.Catterall W. A. (1986) Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem. 55, 953–985 [DOI] [PubMed] [Google Scholar]
- 64.Guy H. R., and Seetharamulu P. (1986) Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. U.S.A. 83, 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Catterall W. A. (2010) Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 67, 915–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stuhmer W., Conti F., Suzuki H., Wang X. D., Noda M., Yahagi N., Kubo H., and Numa S. (1989) Structural parts involved in activation and inactivation of the sodium channel. Nature 339, 597–603 [DOI] [PubMed] [Google Scholar]
- 67.Catterall W. A. (1979) Binding of scorpion toxin to receptor sites associated with sodium channels in frog muscle: correlation of voltage-dependent binding with activation. J. Gen. Physiol. 74, 375–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rogers J. C., Qu Y., Tanada T. N., Scheuer T., and Catterall W. A. (1996) Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. J. Biol. Chem. 271, 15950–15962 [DOI] [PubMed] [Google Scholar]
- 69.Cestèle S., Qu Y., Rogers J. C., Rochat H., Scheuer T., and Catterall W. A. (1998) Voltage sensor-trapping: enhanced activation of sodium channels by β-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 21, 919–931 [DOI] [PubMed] [Google Scholar]
- 70.Yang N., and Horn R. (1995) Evidence for voltage-dependent S4 movement in sodium channel. Neuron 15, 213–218 [DOI] [PubMed] [Google Scholar]
- 71.Yang N., George A. L. Jr., and Horn R. (1996) Molecular basis of charge movement in voltage-gated sodium channels. Neuron 16, 113–122 [DOI] [PubMed] [Google Scholar]
- 72.DeCaen P. G., Yarov-Yarovoy V., Zhao Y., Scheuer T., and Catterall W. A. (2008) Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc. Natl. Acad. Sci. U.S.A. 105, 15142–15147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DeCaen P. G., Yarov-Yarovoy V., Sharp E. M., Scheuer T., and Catterall W. A. (2009) Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 106, 22498–22503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DeCaen P. G., Yarov-Yarovoy V., Scheuer T., and Catterall W. A. (2011) Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 108, 18825–18830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yarov-Yarovoy V., DeCaen P. G., Westenbroek R. E., Pan C. Y., Scheuer T., Baker D., and Catterall W. A. (2012) Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc. Natl. Acad. Sci. U.S.A. 109, E93–E102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yarov-Yarovoy V., Schonbrun J., and Baker D. (2006) Multipass membrane protein structure prediction using Rosetta. Proteins 62, 1010–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yarov-Yarovoy V., Baker D., and Catterall W. A. (2006) Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc. Natl. Acad. Sci. U.S.A. 103, 7292–7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X., Ren W., DeCaen P., Yan C., Tao X., Tang L., Wang J., Hasegawa K., Kumasaka T., He J., Wang J., Clapham D. E., and Yan N. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gurevitz M. (2012) Mapping of scorpion toxin receptor sites at voltage-gated sodium channels. Toxicon 60, 502–511 [DOI] [PubMed] [Google Scholar]
- 80.Zhang J. Z., Yarov-Yarovoy V., Scheuer T., Karbat I., Cohen L., Gordon D., Gurevitz M., and Catterall W. A. (2011) Structure-function map of the receptor site for β-scorpion toxins in domain II of voltage-gated sodium channels. J. Biol. Chem. 286, 33641–33651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang J. Z., Yarov-Yarovoy V., Scheuer T., Karbat I., Cohen L., Gordon D., Gurevitz M., and Catterall W. A. (2012) Mapping the interaction site for a β-scorpion toxin in the pore module of domain III of voltage-gated Na+ channels. J. Biol. Chem. 287, 30719–30728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J., Yarov-Yarovoy V., Kahn R., Gordon D., Gurevitz M., Scheuer T., and Catterall W. A. (2011) Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 108, 15426–15431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chakrabarti N., Ing C., Payandeh J., Zheng N., Catterall W. A., and Pomès R. (2013) Catalysis of Na+ permeation in the bacterial sodium channel NavAb. Proc. Natl. Acad. Sci. U.S.A. 110, 11331–11336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou Y., Morais-Cabral J. H., Kaufman A., and MacKinnon R. (2001) Chemistry of ion coordination and hydration revealed by a potassium channel-Fab complex at 2.0 Å resolution. Nature 414, 43–48 [DOI] [PubMed] [Google Scholar]
- 85.Armstrong C. M., Bezanilla F., and Rojas E. (1973) Destruction of sodium conductance inactivation in squid axons perfused with Pronase. J. Gen. Physiol. 62, 375–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vassilev P., Scheuer T., and Catterall W. A. (1989) Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc. Natl. Acad. Sci. U.S.A. 86, 8147–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.West J. W., Patton D. E., Scheuer T., Wang Y., Goldin A. L., and Catterall W. A. (1992) A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc. Natl. Acad. Sci. U.S.A. 89, 10910–10914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kellenberger S., Scheuer T., and Catterall W. A. (1996) Movement of the Na+ channel inactivation gate during inactivation. J. Biol. Chem. 271, 30971–30979 [DOI] [PubMed] [Google Scholar]
- 89.Kellenberger S., West J. W., Scheuer T., and Catterall W. A. (1997) Molecular analysis of the putative inactivation particle in the inactivation gate of brain type IIA Na+ channels. J. Gen. Physiol. 109, 589–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kellenberger S., West J. W., Catterall W. A., and Scheuer T. (1997) Molecular analysis of potential hinge residues in the inactivation gate of brain type IIA Na+ channels. J. Gen. Physiol. 109, 607–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rohl C. A., Boeckman F. A., Baker C., Scheuer T., Catterall W. A., and Klevit R. E. (1999) Solution structure of the sodium channel inactivation gate. Biochemistry 38, 855–861 [DOI] [PubMed] [Google Scholar]
- 92.Adelman W. J. Jr., and Palti Y. (1969) The effects of external potassium and long duration voltage conditioning on the amplitude of sodium currents in the giant axon of the squid, Loligo pealei. J. Gen. Physiol. 54, 589–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rudy B. (1978) Slow inactivation of the sodium conductance in squid giant axons: Pronase resistance. J. Physiol. 283, 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pavlov E., Bladen C., Winkfein R., Diao C., Dhaliwal P., and French R. J. (2005) The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys. J. 89, 232–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Payandeh J., Gamal El-Din T. M., Scheuer T., Zheng N., and Catterall W. A. (2012) Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu F. H., and Catterall W. A. (2004) The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004, re15. [DOI] [PubMed] [Google Scholar]
- 97.Tang L., Gamal El-Din T. M., Payandeh J., Martinez G. Q., Heard T. M., Scheuer T., Zheng N., and Catterall W. A. (2014) Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 505, 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hess P., and Tsien R. W. (1984) Mechanism of ion permeation through calcium channels. Nature 309, 453–456 [DOI] [PubMed] [Google Scholar]
- 99.Almers W., and McCleskey E. W. (1984) The nonselective conductance due to calcium channels in frog muscle: calcium-selectivity in a single file pore. J. Physiol. 353, 585–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Almers W., McCleskey E. W., and Palade P. T. (1984) A nonselective cation conductance in frog muscle membrane blocked by micromolar external calcium ions. J. Physiol. 353, 565–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Catterall W. A., and Swanson T. M. (2015) Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol. Pharmacol. 88, 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cantrell A. R., and Catterall W. A. (2001) Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat. Rev. Neurosci. 2, 397–407 [DOI] [PubMed] [Google Scholar]
- 103.Catterall W. A. (2000) Structure and regulation of voltage-gated calcium channels. Annu. Rev. Cell Dev. Biol. 16, 521–555 [DOI] [PubMed] [Google Scholar]
- 104.Catterall W. A. (2010) Signaling complexes of voltage-gated sodium and calcium channels. Neurosci. Lett. 486, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Catterall W. A., and Few A. P. (2008) Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901 [DOI] [PubMed] [Google Scholar]
- 106.Venance S. L., Cannon S. C., Fialho D., Fontaine B., Hanna M. G., Ptacek L. J., Tristani-Firouzi M., Tawil R., Griggs R. C., and CINCH investigators (2006) The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain 129, 8–17 [DOI] [PubMed] [Google Scholar]
- 107.Kass R. S. (2005) The channelopathies: novel insights into molecular and genetic mechanisms of human disease. J. Clin. Invest. 115, 1986–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Heron S. E., Scheffer I. E., Berkovic S. F., Dibbens L. M., and Mulley J. C. (2007) Channelopathies in idiopathic epilepsy. Neurotherapeutics 4, 295–304 [DOI] [PubMed] [Google Scholar]
- 109.Dib-Hajj S. D., Cummins T. R., Black J. A., and Waxman S. G. (2010) Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347 [DOI] [PubMed] [Google Scholar]
- 110.van den Maagdenberg A. M., Pietrobon D., Pizzorusso T., Kaja S., Broos L. A., Cesetti T., van de Ven R. C., Tottene A., van der Kaa J., Plomp J. J., Frants R. R., and Ferrari M. D. (2004) A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41, 701–710 [DOI] [PubMed] [Google Scholar]
- 111.Sokolov S., Scheuer T., and Catterall W. A. (2007) Gating pore current in an inherited ion channelopathy. Nature 446, 76–78 [DOI] [PubMed] [Google Scholar]
- 112.Sokolov S., Scheuer T., and Catterall W. A. (2008) Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc. Natl. Acad. Sci. U.S.A. 105, 19980–19985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sokolov S., Scheuer T., and Catterall W. A. (2010) Ion permeation and block of the gating pore in the voltage sensor of NaV1.4 channels with hypokalemic periodic paralysis mutations. J. Gen. Physiol. 136, 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yu F. H., Mantegazza M., Westenbroek R. E., Robbins C. A., Kalume F., Burton K. A., Spain W. J., McKnight G. S., Scheuer T., and Catterall W. A. (2006) Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149 [DOI] [PubMed] [Google Scholar]
- 115.Catterall W. A., Kalume F., and Oakley J. C. (2010) NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Han S., Tai C., Westenbroek R. E., Yu F. H., Cheah C. S., Potter G. B., Rubenstein J. L., Scheuer T., de la Iglesia H. O., and Catterall W. A. (2012) Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fu Y., Westenbroek R. E., Scheuer T., and Catterall W. A. (2014) Basal and β-adrenergic regulation of the cardiac calcium channel CaV1.2 requires phosphorylation of serine 1700. Proc. Natl. Acad. Sci. U.S.A. 111, 16598–16603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fu Y., Westenbroek R. E., Scheuer T., and Catterall W. A. (2013) Phosphorylation sites required for regulation of cardiac calcium channels in the fight-or-flight response. Proc. Natl. Acad. Sci. U.S.A. 110, 19621–19626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Archives (Fall-Winter, 2000–2001) The case of the CIA and butter clam toxin. in Yale Medicine, Vol. 35, No. 1, Yale School of Medicine, New Haven, CT [Google Scholar]
- 120.De Jongh K. S., Warner C., and Catterall W. A. (1990) Subunits of purified calcium channels: α2 and δ are encoded by the same gene. J. Biol. Chem. 265, 14738–14741 [PubMed] [Google Scholar]
- 121.Davies A., Kadurin I., Alvarez-Laviada A., Douglas L., Nieto-Rostro M., Bauer C. S., Pratt W. S., and Dolphin A. C. (2010) The α2δ subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc. Natl. Acad. Sci. U.S.A. 107, 1654–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]