

Background: Vibrio cholerae secretes a toxin that forms channels in host cells.
Results: Our study provides a complete structure/function mapping of the toxin membrane-interacting surface.
Conclusion: Membrane binding integrates glycan recognition and direct lipid and cholesterol interactions.
Significance: Targeting membrane interactions may be a viable approach for treating infectious disease.
Keywords: bacterial pathogenesis, bacterial toxin, cholesterol, membrane, structure-function
Abstract
Bacterial pore-forming toxins (PFTs) are structurally diverse pathogen-secreted proteins that form cell-damaging channels in the membranes of host cells. Most PFTs are released as water-soluble monomers that first oligomerize on the membrane before inserting a transmembrane channel. To modulate specificity and increase potency, many PFTs recognize specific cell surface receptors that increase the local toxin concentration on cell membranes, thereby facilitating channel formation. Vibrio cholerae cytolysin (VCC) is a toxin secreted by the human pathogen responsible for pandemic cholera disease and acts as a defensive agent against the host immune system. Although it has been shown that VCC utilizes specific glycan receptors on the cell surface, additional direct contacts with the membrane must also play a role in toxin binding. To better understand the nature of these interactions, we conducted a systematic investigation of the membrane-binding surface of VCC to identify additional membrane interactions important in cell targeting. Through cell-based assays on several human-derived cell lines, we show that VCC is unlikely to utilize high affinity protein receptors as do structurally similar toxins from Staphylococcus aureus. Next, we identified a number of specific amino acid residues that greatly diminish the VCC potency against cells and investigated the interplay between glycan binding and these direct lipid contacts. Finally, we used model membranes to parse the importance of these key residues in lipid and cholesterol binding. Our study provides a complete functional map of the VCC membrane-binding surface and insights into the integration of sugar, lipid, and cholesterol binding interactions.
Introduction
Vibrio cholerae cytolysin (VCC),4 an accessory toxin of the human pathogen V. cholerae, is secreted as an 82-kDa water-soluble monomer and assembles to form heptameric β-barrel channels in cell membranes (1). Structurally and functionally distinct from the “classical” ADP-ribosylating A-B toxin associated with severe diarrheal symptoms of the human cholera disease, mouse models suggest that VCC is one of several factors involved in colonization of the small intestine by defending the bacterium against host immune cells such as neutrophils (2, 3). As an important contributor to the pathogenicity of pandemic cholera, it is important to understand the mechanism by which VCC recognizes and inflicts damage on cells and tissues.
VCC belongs to a group of pore-forming toxins (PFTs) with a similar structural fold that includes the well characterized staphylococcal α-hemolysin (4, 5). High resolution crystal structures of several family members have been determined for water-soluble and fully assembled channels including VCC (1, 6), staphylococcal α- and γ-hemolysins (7–12), and NetB from Clostridium perfringens (13). The channel-forming amphipathic loops of these structures undergo substantial rearrangements from water-soluble to oligomeric states, the latter of which are typically heptameric (with the channel consisting of a 14-stranded β-barrel). In water-soluble monomers, these loops are folded up against the body of the monomer to prevent the energetically costly exposure of hydrophobic surfaces to aqueous solution. How water-soluble monomers assemble to form the final channel is less well understood, but the generally accepted paradigm based on studies of staphylococcal α-hemolysin (14) and applicable to VCC (15) suggests that water-soluble monomers bind individually to membranes via interactions with the membrane-contacting rim domain (Fig. 1A), diffuse and assemble into oligomeric non-lytic “prepores,” and then cooperatively insert their amphipathic loops into the membrane to form the final β-barrel. It is important to note that VCC and structurally related toxins are only one subset of a much larger group of structurally dissimilar classes of PFTs, which have been extensively reviewed elsewhere (16).
FIGURE 1.

VCC-membrane interactions. A, schematic representation of steps in VCC pore formation. Water-soluble monomers bind to cell membranes (1) utilizing both complex N-glycan binding on cell surface receptors (R) by the β-prism lectin domain (yellow) and direct membrane interactions with the rim domain (blue). Monomers diffuse across the cell membrane (2), forming prepore oligomeric intermediates before unfolding amphipathic loops (3) that form the transmembrane β-barrel channel pore. B, schematic representation of the fully assembled heptameric VCC pore structure determined by x-ray crystallography. The transmembrane β-barrel (Stem) is surrounded by putative membrane-contacting loops in the rim (blue). A ring of aromatic amino acids denotes the predicted membrane interface shown in stick representation (orange). A second inactive lectin with a β-trefoil fold is shown in magenta. C, close-up of the three loops that comprise the membrane-contacting rim domain. D, sequence of the loops subjected to alanine-scanning mutagenesis. Residues in green were already alanine or glycine and not mutated, whereas red residues failed quality control specifications and were not tested further. Black underlined residues passed quality control and were included.
There is significant interest in understanding how membrane-active toxins selectively target host cell membranes with high affinity, and it is clear that different PFTs utilize distinct receptors to achieve this goal. With some staphylococcal toxins, membrane recognition utilizes protein receptors, and several of these targets have been identified (17). To the best of our knowledge, no protein receptor has yet been identified for VCC, which is also missing the lipid-binding pockets identified in staphylococcal toxins (6). Unlike the staphylococcal toxins, VCC contains two additional C-terminal domains with β-trefoil and β-prism lectin-like folds (Fig. 1B). The VCC β-prism domain targets complex N-glycans on host cell membranes with nanomolar affinity (18) and increases the affinity of the toxin for cells. The β-trefoil domain in VCC is inactive, but in homologous toxins, it recognizes terminal galactosyl glycan moieties with micromolar affinity (19). In addition to binding glycans, there is evidence that VCC activity is enhanced by cholesterol, sphingolipids, and ceramides in the cell membrane (20), although the mechanism by which this occurs is still not well understood. Any such interactions are likely mediated through the rim domain (Fig. 1B). Close inspection of the VCC oligomeric structure indicates a ring of phenylalanine and tyrosine residues around the barrel that outline the putative membrane-water interface (Fig. 1C). Based on this model, three loops within the VCC rim domain form close interactions with the membrane and may contain potential binding sites for membrane components. Because these loops contain amino acids unlikely to penetrate deeply into the membrane (histidines near the tip of loop 3 for example), it is likely that these loops change in structure or cause a substantial perturbation of the lipid bilayer. For this reason, one might expect some degree of flexibility in these loops in the membrane-embedded state.
To systematically probe the membrane-interacting surface of VCC, we conducted alanine-scanning mutagenesis of these three loops and used a cell-based assay to measure effects on the potency of the toxin (Fig. 1D). Other investigations have uncovered important sites within the VCC rim domain (21, 22). This study builds upon the previous findings by offering a systematic scan of the entire VCC rim domain, an analysis of how membrane and glycan interactions are related, and additional insights into the mechanism of pore assembly. Similar methods have been instrumental in uncovering membrane interactions for other toxins, most notably the cholesterol-dependent cytolysin perfringolysin O (PFO) (23) and the Cry4Aa mosquitocidal toxin from Bacillus thuringiensis (24). This approach was instrumental in identifying a dipeptide (threonine-leucine) TL sequence responsible for cholesterol recognition in PFO (23). In addition to the TL site, other possible cholesterol binding patterns have been identified in membrane-interacting proteins such as the CRAC (-(L/V)X1–5YX1–5(R/K)-) and inverse CARC motifs (-(K/R)X1–5(Y/F)X1–5(L/V)-) first found in the benzodiazepine receptor (25, 26).
In terms of protein receptors, mutagenesis studies of staphylococcal LukE and LukS-PV suggest that interactions with their cellular receptors CXCR1/CXCR2 (27) and C5aR (28), respectively, occur via the toxin rim domain and that some flexibility of rim domain loops is essential for proper recognition of the protein receptor (27, 29, 30). Activity against membranes is likely to be more complex than simply recognizing receptors as lipid composition, clustering, and lipid phase all affect the ability of monomers in model systems to assemble into lytic pores (31, 32).
To differentiate between true loss of function and misfolding or aggregation, we used a rigorous quality control filter to discard mutant proteins that displayed anomalous behavior. Our comprehensive mapping of the entire rim domain interaction surface identified 10 mutations that reduced the hemolytic activity of VCC by over 10-fold (a loss of >90.0% activity). To explore the relationship between direct membrane interactions by the rim domain and sugar receptor binding by the β-prism domain, we coupled the most severe rim domain mutants with a second mutation (D617K) to knock out glycan binding in the β-prism domain. Finally, to investigate the underlying cause of the mutation effects, we measured their activity in a purified liposome system (unlikely to contain protein or sugar receptors) by oligomerization assays and surface plasmon resonance (SPR). We found that the most severe rim domain mutations do not diminish the activity of wild-type protein in competition assays, indicating that the deficiency in activity is due to initial membrane binding rather than later steps in oligomerization and channel formation. We hypothesize that these mutations affect specific interactions with lipids and cholesterol and suggest several known cholesterol-binding motifs as potential candidates for their effects.
Experimental Procedures
Construction of VCC Mutants
Site-directed mutations were introduced into the full-length VCC gene (hlyA) from V. cholerae O1 El Tor strain 8731 previously cloned into the pHis-Parallel2 vector (33) using an overlapping primer protocol (34). Briefly, complementary 5′ and 3′ primers 39–45 bases long were designed with the desired mutation introduced at the center of the primer. The entire vector was amplified with these primers in a PCR using PfuUltra DNA polymerase (Agilent), and the parental strands were removed by digestion with DpnI. DNA was transformed into competent NEB5α cells (New England Biolabs), and individual colonies were grown, miniprepped, and sequenced to confirm the introduction of the mutation.
Protein Expression and Purification
VCC constructs (WT and mutants) were transformed into SHuffle Express T7 (New England Biolabs) competent cells and cultured overnight at 37 °C in 20 ml of Luria broth supplemented with 100 μg/ml ampicillin. The overnight culture was added to 1 liter of Luria broth/ampicillin and incubated with constant shaking at 37 °C until the A600 reached ∼0.6. At this point, 1 mm isopropyl thio-β-galactoside was added to induce VCC expression, and the culture was incubated with shaking for another 3 h at 30 °C. Following expression, cells were pelleted in a swinging bucket centrifuge at 3,900 × g for 30 min, and the cell pellet was resuspended in 10 ml of TBS buffer (20 mm Tris, pH 7.5, 150 mm NaCl) supplemented with 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride, and 10 mm imidazole. Following passage three times through an EmulsiFlex C5 high pressure homogenizer (Avestin, Inc.), the lysate was spun at 40,000 × g for 30 min at 4 °C in a Lynx 6000 centrifuge (F20–12x50 LEX rotor, Thermo Scientific). Following centrifugation, the supernatant was collected and loaded onto a nickel-chelating column and washed with TBS and TBS + 40 mm imidazole. Purified VCC was eluted with TBS containing 250 mm imidazole. The protein was further purified over an S200 10/300 size exclusion column (GE Healthcare) in TBS buffer, and the purity was estimated by running on a sodium dodecyl sulfate (SDS)-polyacrylamide gel. Mutants that failed to produce a monodisperse elution profile on the size exclusion column were excluded from further analysis.
Isolation of Primary Human Polymorphonuclear Leukocytes and Culture of Primary Human Immune Cell Lines
All human immune cell lines used in this study (THP-1, Jurkat, and HUT-R5) were maintained in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. Human blood samples were obtained from normal healthy donors from the New York Blood Center the morning of each experiment. The New York Blood Center obtains written informed consent from all participants, and all identifiers are stripped from material prior to shipment. Polymorphonuclear leukocytes were isolated from the peripheral blood of two independent donors according to methods described previously (35). Briefly, packed leukocytes were resuspended in Hanks' balanced salt solution and overlaid onto Ficoll-Paque PLUS (GE Healthcare) followed by centrifugation at 2,000 rpm for 30 min at room temperature. After centrifugation, the peripheral blood mononuclear cell layer was removed as well as any remaining Ficoll. The lower fraction containing red blood cells and polymorphonuclear leukocytes was resuspended in sterile endotoxin-free 0.9% NaCl + 3% dextran and placed at 25 °C for 25 min to allow for sedimentation of red blood cells. The upper layer containing polymorphonuclear leukocytes was removed, and residual contaminating red blood cells were lysed in 9 ml of sterile ammonium-chloride-potassium lysing solution (Life Technologies) for 2–3 min. Cells were resuspended in RPMI medium + 10% FBS, filtered through a sterile 70-μm filter, and used immediately in VCC cytotoxicity assays.
VCC Cytotoxicity Assays
Human immune cell lines (THP-1 monocytes, Jurkat T cells, and HUT-R5 T cells) or primary human polymorphonuclear leukocytes were seeded into a 96-well plate at 100,000 cells/well in 90 μl of RPMI medium + 10% FBS. A 2-fold serial dilution of VCC in phosphate-buffered saline (PBS) was added to cells for 1 h at 37 °C, and viability was assessed after addition of 10 μl of CellTiter colorimetric viability indicator (Promega) followed by incubation at 37 °C according to the manufacturer's instructions. All measurements were made on an EnVision 2103 plate reader (PerkinElmer Life Sciences).
Hemolysis Assays
Purified VCC was activated by proteolytic cleavage with a 1:350 (w/w) ratio of α-chymotrypsin for 30 min at room temperature. Mutants that failed to show a clean proteolysis profile were excluded from further analysis. A serial dilution of activated VCC was added to individual wells of a 96-well clear bottom plate containing a ∼0.5% solution (normalized to 1 A595) of defibrinated rabbit blood in blood dilution buffer (20 mm NaH2PO4, 150 mm NaCl, 1 mg/ml BSA, pH 7.4). Absorbance data were collected at room temperature in an iMark microplate reader (Bio-Rad) at 595 nm every 15 s for 60 min with 1 s of shaking between measurements. A measure of percent lysis after 30 min was calculated from the data at the two time points using the following equation.
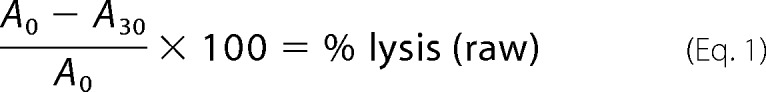 |
where A0 and A30 are the absorbances at time = 0 min and time = 30 min, respectively, at 595 nm. The raw percent hemolysis value at time i was normalized to a maximum value of 100% using the following equation.
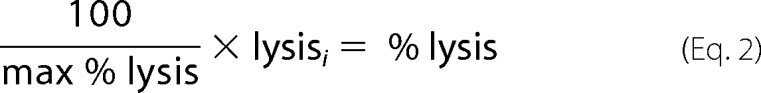 |
The average percent lysis at each concentration of VCC after 30 min was plotted and fit to a sigmoidal function for calculating the hemolytic dose at 50% lysis (HD50) using KaleidaGraph v.4.1.3 (Synergy Software). Data points were collected in triplicate and plotted as the mean with error bars denoting the S.E.
Isothermal Titration Calorimetry (ITC)
Purified VCC was dialyzed for 24 h against TBS buffer using SnakeSkin dialysis tubing (Pierce) with a 10-kDa molecular mass cutoff. VCC sugar binding activity was assayed by measuring binding to asialofetuin, a glycoprotein shown previously to bind to VCC with ∼1 μm affinity (36). A 110 μm asialofetuin solution was made by dissolving lyophilized asialofetuin (Sigma) into the dialysate buffer. ITC data were collected using a MicroCal VP-ITC (Microcal, LLC). The sample chamber (1.44 ml) was loaded with dialyzed VCC (23 μm) and titrated with 30 injections of 9.3 μl of asialofetuin (110 μm). Blank-subtracted ITC data were processed using Nitpic v.1.1.2 (37), fit to a two-site binding model using SEDPHAT v.12.1b (38), and displayed with GUSSI v.1.0.8.
Analytical Ultracentrifugation
Purified wild-type VCC (VCCWT) protein and mutants were concentrated to 1 mg/ml and dialyzed against TBS buffer. Samples were loaded into double sector synthetic boundary cells equipped with sapphire windows allowing matching of the sample and reference menisci. Sedimentation velocity analysis was conducted using interference optics with a Beckman-Coulter XL-I analytical ultracentrifuge. The rotor was equilibrated under vacuum at 20 °C, and after a period of ∼1 h, the rotor was accelerated to 40,000 rpm. Interference scans were acquired at 60-s intervals for ∼7 h. Physical constants were calculated for the protein (molecular weight and partial specific volume) and buffer (density and viscosity) using the program Sednterp (39). Ultracentrifugation data were fit using the g(s*) method as implemented in the DCDT+ software package (v.2.4.0) (40, 41).
Liposome Preparation
To prepare liposomes, 50 mg of dried lipid was dissolved in 2-methyl-2-propanol at 45 °C for soy asolectin (Sigma-Aldrich) or 55 °C for total porcine brain extract lipids (Avanti Polar Lipids) in a 50-ml round bottom flask. Cholesterol (10 mg/ml in ethanol) was added at a 30% molar ratio, and the lipids were dried under a nitrogen flow. The resulting film was vacuum-dried overnight in the dark and resuspended in 10 ml of TBS buffer to a final concentration of 5 mg/ml. Following resuspension, the mixtures were subjected to 20 freeze-thaw cycles in alternating liquid nitrogen and 45/55 °C water baths. The resulting unilamellar liposome solutions were passed 11 times through a 100-nm membrane using a miniextruder (Avanti Polar Lipids).
SDS-PAGE-based Oligomerization Assays
Toxins were activated by α-chymotrypsin proteolysis (as described previously) and incubated with liposome solutions (1:1 lipid:protein by weight) at room temperature for 1 h. Samples were run on a 5% polyacrylamide gel and stained with Coomassie dye. After destaining, gels were scanned using a Typhoon TRIO scanner (GE Healthcare), and images quantified by densitometric analysis using the ImageQuant TL software package (version 7.0). For competition gels, all bands were normalized to the VCCWT band after subtracting the background. Similarly, for assembly kinetics gels, bands were normalized using the overnight VCCWT assembly band after background correction. All data were plotted using KaleidaGraph v.4.1.3.
Surface Plasmon Resonance
All SPR data were collected on a Biacore T200 system using an L1 lipophilic chip (GE Healthcare). The chip surface was preconditioned with 10 μl of 20 mm CHAPS (solubilized in HBS-N buffer containing 10 mm HEPES, pH 7.4, 150 mm NaCl) followed by 10 μl of 50 mm NaOH. Asolectin and brain lipid liposomes were diluted to 0.5 mm stocks and injected at a flow rate of 10 μl/min for 15 min. Typical loading densities were 3,000 and 7,000 resonance units for asolectin and brain lipid liposomes, respectively. The surface was washed with 50 mm NaOH for 1 min and blocked with 0.1 mg/ml BSA for 1 min. Liposome surfaces were stable for the duration of the experiment using this procedure. VCCWT and mutant proteins were dialyzed into HBS-N buffer and cut with α-chymotrypsin before analysis. To test toxin activity, eight consecutive 5-min injections (50 μg/ml) were flowed over the chip with 2.5-min wash phases in between. To regenerate the chip, 3-min injections of 20 mm CHAPS were followed by 1-min injections of 50 mm NaOH until no further loss in signal was detected.
Results
Activity of VCC against Primary and Derived Cell Lines
Previous studies have demonstrated that VCC is active against a wide range of cell types including erythrocytes (42), intestinal cells (43), neutrophils (44), and other nucleated cell lines (43). Related toxins from S. aureus display tropism due to the presence of specific receptors on different cell types (17). To assess whether VCC shows similar differences in activity across different human cell lines, we tested purified VCC against several human primary and derived cell lines including HUT-R5 and Jurkat human T cell lines, the THP-1 human monocyte-like cell line, and primary human neutrophils (Fig. 2A). VCCWT was titrated in a dose-response experiment (15 pm to 120 nm) against roughly 100,000 cells, and toxin-dependent killing was measured after 1.5 h. Cell killing was confirmed by measuring ethidium bromide uptake into treated cells, which began at ∼30 min and plateaued at around 60 min. All four cell types displayed a similar susceptibility within a narrow window with the half-point of toxin activity ranging between 200 and 800 pm. These results suggest that VCC does not display selectivity for any one of these four immune cell types.
FIGURE 2.
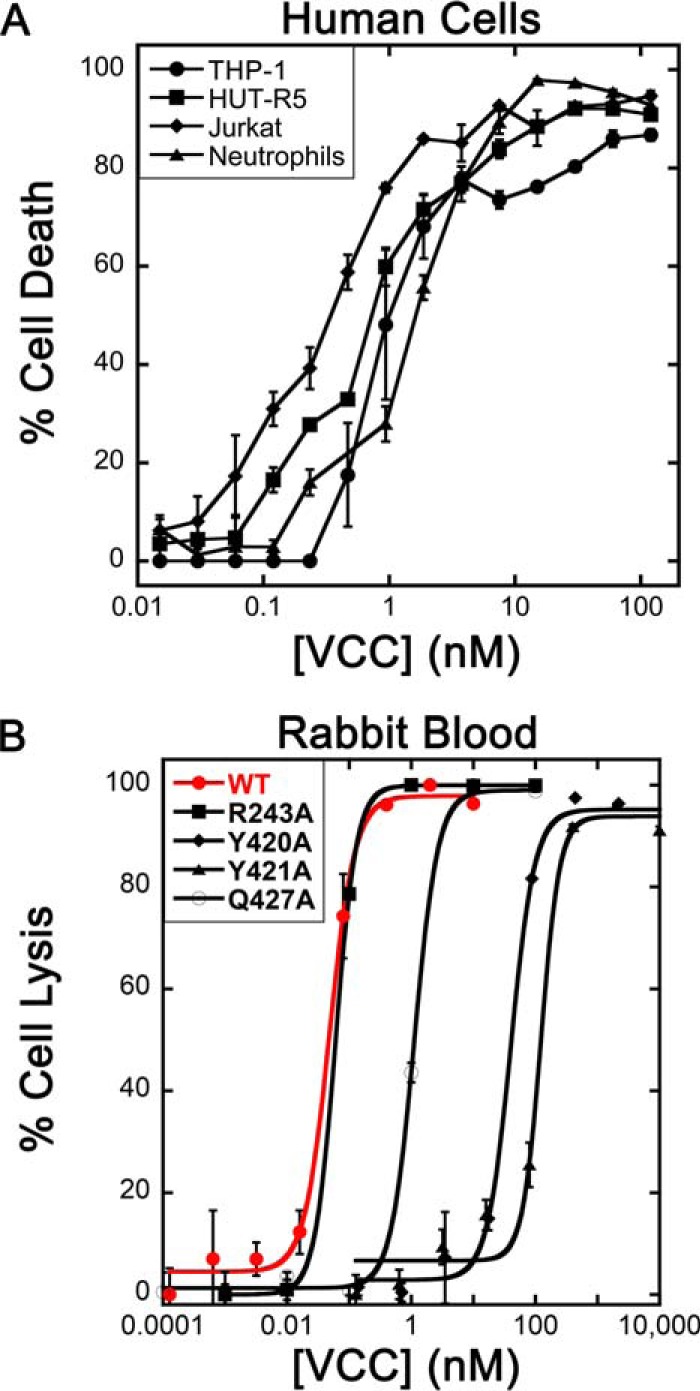
Cytotoxicity and hemolysis data. A, VCCWT exhibits similar levels of potency against four human cell types. Error bars indicate the S.E. calculated from three repetitions (six for neutrophils representing two donors). B, VCCWT is equally effective against rabbit blood. Hemolysis curves for VCCWT (red) and several rim domain mutants are displayed with overlaid sigmoidal fits. Error bars represent the S.E. from three repetitions.
It is known that VCC prefers rabbit erythrocytes over human, sheep, and chicken (36, 42) perhaps due to the overabundance of complex type N-glycans on the cell surface (45). To ascertain whether the efficacy of VCC toward rabbit erythrocytes is comparable with the human immune cell types tested, we tested VCC against a similar titer of washed rabbit erythrocytes (∼100,000 cells) in a microtiter plate lysis assay using optical density at 595 nm as a measure of cell lysis. The HD50 by wild-type activated VCC was ∼100 pm (Fig. 2B). This number is not largely different from the efficacy of VCC toward the human immune cells we tested as would be expected if a receptor were not involved. These results suggest that rabbit erythrocytes are a suitable model for understanding VCC-membrane interactions and that VCC does not exhibit tropic behavior indicative of a selective receptor on neutrophils or other human cell lines tested.
Construction and Quality Control of VCC Rim Domain Alanine-scanning Mutations
Based on the oligomeric structures of VCC and related pore-forming toxins, we expected significant direct steric interactions between the rim domain loops and the outer leaflet of the plasma membrane. We used a complete systematic alanine-scanning mutagenesis of the three primary rim domain loops predicted to be membrane-proximal to uncover specific interactions necessary for lytic activity of the VCC toxin. If a receptor-binding site exists within the rim domain or if nonspecific interactions are necessary for monomer binding to the membrane we might uncover these hot spots through a mutagenesis strategy. In all, 30 individual mutants were made by site-directed mutagenesis encompassing residues 234–243, 356–365, and 417–430.
The transition of VCC from water-soluble monomer to oligomeric transmembrane channel involves several steps including glycan receptor binding, rim domain interactions with the membrane, oligomerization into a prepore, and channel insertion. To guard against unanticipated factors that might affect other steps in the process, we used a four-tier quality control procedure to filter out mutants that displayed aberrant behavior. First, mutants were purified over a size exclusion column and only passed if they displayed a monodisperse elution profile (to address possible solution aggregation). Second, only mutants that could be cleanly cleaved by α-chymotrypsin (necessary to activate the protoxin by removing the prodomain) were included with the assumption that misfolded protein would be more susceptible to spurious cutting by the protease. Third, a subset of mutants with varying losses in activity was subjected to analytical ultracentrifugation (Fig. 3) to confirm their monomeric state in solution. And fourth, the glycan binding activity of mutants with varying losses in activity was measured against a model glycoprotein (asialofetuin) using ITC (Fig. 4) to ensure that observed losses in hemolytic activity of rim mutants were not due to peripheral effects. In all, of 30 mutations, 25 expressed and purified mutants passed our specifications and were included in our analysis.
FIGURE 3.
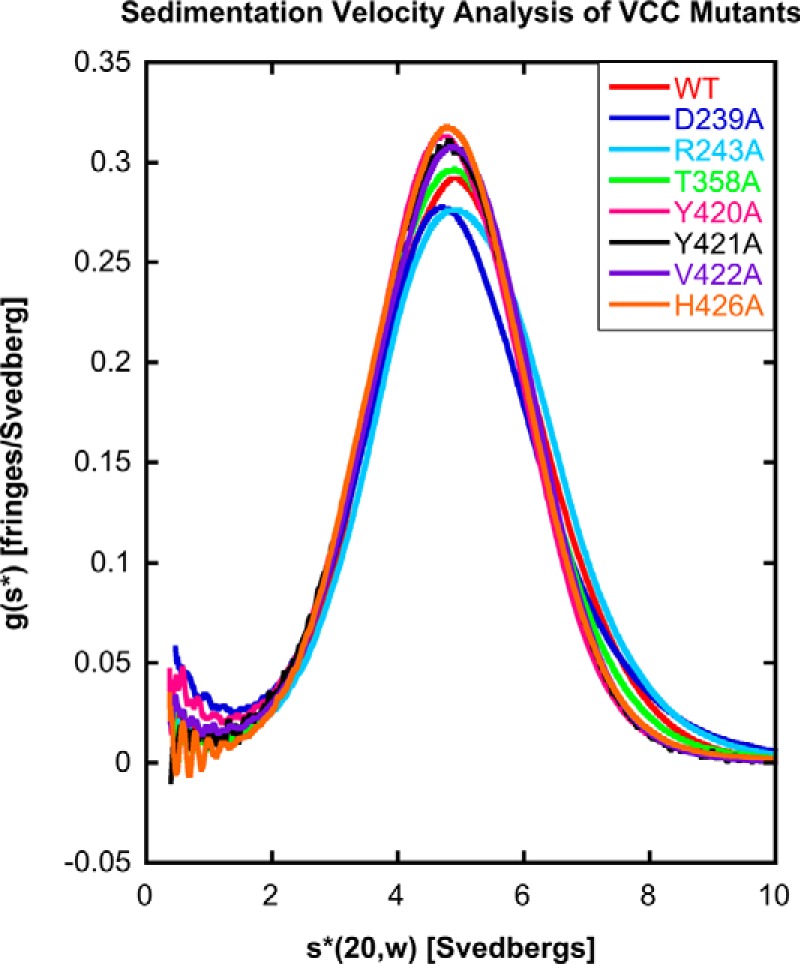
Analytical ultracentrifugation of selected VCC mutants. VCCWT and seven representative mutants were subjected to sedimentation velocity analytical ultracentrifugation to determine whether introduced mutations have any effect on the oligomerization state of the protein. All proteins displayed a similar profile when analyzed using g(s*) analysis, suggesting that the mutations did not lead to any observable change in the hydrodynamic properties of the protein.
FIGURE 4.

ITC experiments measuring VCC binding to asialofetuin. Mutations introduced into the rim domain do not negatively affect the sugar binding ability of the VCC β-prism domain. The glycoprotein asialofetuin was used as a substrate for assaying the activity of the VCC lectin. Data fit best when two glycans per asialofetuin were included in the fit, although some glycosylation heterogeneity is expected in the protein. Five representative mutants were tested including Y421A, which exhibited the greatest loss of activity in hemolysis assays. Representative data from one of three experiments are shown. Error bars represent the S.E. of three repetitions. DP, differential power.
Hemolysis Assays of VCC Rim Domain Alanine-scanning Mutants
To assess the effects of rim domain mutants on toxin-mediated cell lysis, purified and activated VCCWT and mutant proteins were added to washed rabbit blood in microtiter plates, and cell lysis was monitored spectrophotometrically. The degree of cell lysis for VCCWT and mutants was plotted against toxin concentration after 30 min at room temperature (Fig. 2B). The HD50 was calculated based on a sigmoidal fit of the dose-response data and compared with the HD50 of VCCWT (Fig. 5A and Table 1). For all mutations, the shape and slope of the hemolysis curve were not significantly affected, merely the position of the half-point of lysis.
FIGURE 5.
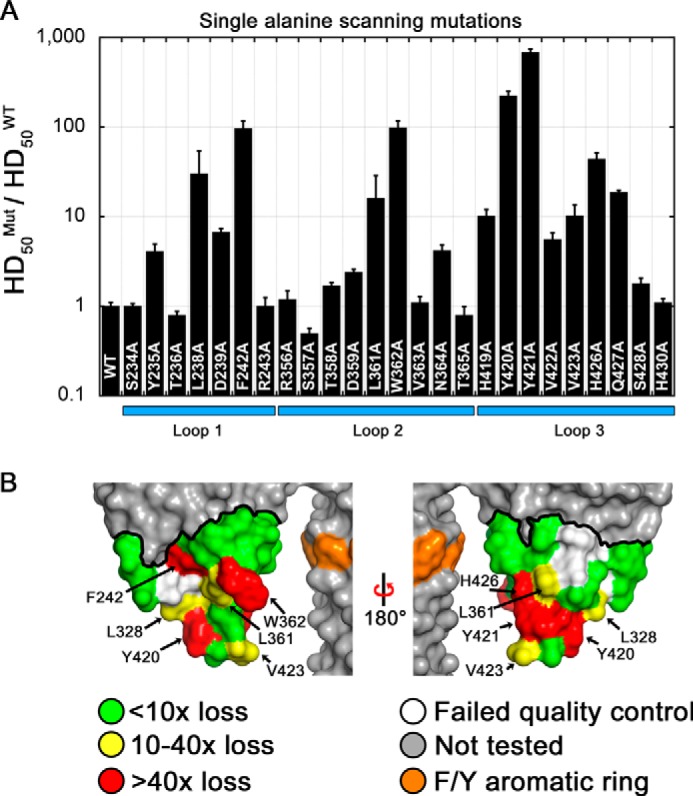
Alanine-scanning mutagenesis. A, mutagenesis data from 25 alanine mutations are displayed as the ratio of HD50 values for each mutant (Mut) compared with VCCWT. The HD50 is defined as the concentration of VCC required to elicit 50% lysis of rabbit blood in 30 min at 25 °C. Ten mutants exhibited greater than 10-fold decreases in lytic activity, two mutants were near 100-fold (F242A and W362A), and two mutants (Y420A and Y421A) lost more than 100-fold activity. Loop 3 was the most susceptible overall to activity loss upon mutagenesis. Numerical data are presented in Table 1. Error bars represent the S.E. B, surface representation of the VCC rim domain with the mutagenesis boundary outlined with a thick black line. Residues are color-coded by loss in activity: green for less than 10-fold, yellow for 10–40-fold, and red for greater than 40-fold. Residues that failed to meet quality control specifications are shown in white. The Q427A mutant is completely buried in the structure and therefore does not contribute to the protein surface.
TABLE 1.
HD50 values for all mutants described in this study
Hemolysis data were fit using a sigmoidal function to determine the amount of protein required to elicit 50% lysis (HD50) for wild type and 25 mutants (left three columns). For comparison, the ratio of the mutant versus wild-type HD50 is displayed along with the percent wild type activity exhibited by each mutant. All values are significant to p ≤ 0.01 except for those marked with an asterisk based on a single factor analysis of variance test (n = 3). Hemolysis data for additional mutations involving L238A, Y420A, Y421A, and glycan/rim domain double mutants are shown in the right three columns. Mutants without measurable activity are denoted with NA, and mutants with less than 10% wild type activity are highlighted in bold type.

Of the impaired mutations, 10 displayed a greater than 10-fold loss in hemolytic activity (loss of >90.0% activity) compared with VCCWT (L238A, F242A, L361A, W362A, H419A, Y420A, Y421A, V423A, H426A, and Q427A). Two neighboring mutants, Y420A and Y421A, exhibited the largest loss in activity with 225- (loss of 99.5% activity) and 684-fold (loss of 99.9% activity) losses, respectively. The six least active mutants involve the removal of hydrophobic and aromatic side chains that form a band around the lower tip of the rim domain (Fig. 5B). The most severe mutations are spread across the three rim domain loops with two coming from loop 1 (L238A and F242A), one coming from loop 2 (W362A), and three coming from loop 3 (Y420A, Y421A, and H426A). A second histidine mutation (H419A from loop 3) had a more modest effect with a roughly 10-fold loss in hemolytic activity. The six aforementioned mutations represent most of the aromatic residues below the stem Phe/Tyr aromatic ring, which likely demarcates the lipid-water membrane interface. One additional tyrosine mutation (Y235A) near the membrane interface was relatively unaffected in activity (∼4-fold), and another tyrosine mutation (Y417A) did not pass quality control. Three hydrophobic residues below the Phe/Tyr line also had modest effects on hemolytic activity including L361A (37-fold loss) and V423A (10-fold loss). Lastly, one polar residue located at the predicted membrane interface (Q427A) resulted in a 19-fold loss, although this buried residue contributes no exposed surface area on the rim domain based on VCC crystal structures.
Additional Mutagenesis of Rim Domain Residues
Several sequence-specific motifs have been identified in membrane-active toxins that facilitate membrane interactions via cholesterol binding. We performed additional targeted mutagenesis beyond our initial scan to investigate these motifs in VCC and used the same quality control procedure to ensure that mutations did not have unintended effects. The first we considered is the threonine-leucine (TL) sequence first identified in PFO. VCC has a single TL motif within loop 1 (Thr237-Leu238) and a second similar AL motif within loop 2 (Ala360-Leu361). Mutation of L238A and L361A led to a 70- and 37-fold loss in activity, respectively. This degree of loss is not as high as in PFO where mutation of the leucine to alanine led to a 100-fold loss in activity (23). Mutating both leucine residues in VCC (L238A and L361A) at the same time had a much more dramatic effect with a total loss of over 1,000-fold activity (Fig. 6A and Table 1). This suggests that the two putative motifs may compensate for each other's loss of function. Mutation of Leu238 to isoleucine and valine resulted in a 3- and 7-fold loss, respectively, whereas in the case of PFO, mutation of the leucine to isoleucine and valine led to a loss of 4.4- and 5.6-fold, respectively. These data suggest that leucine residues play an important role in membrane interactions possibly via cholesterol binding. However, the fact that VCC has other residues that when mutated to alanine exhibit an even greater loss in activity (such as Tyr420 and Tyr421) demonstrates that a single TL motif alone is not responsible for membrane recognition as is seen in PFO.
FIGURE 6.
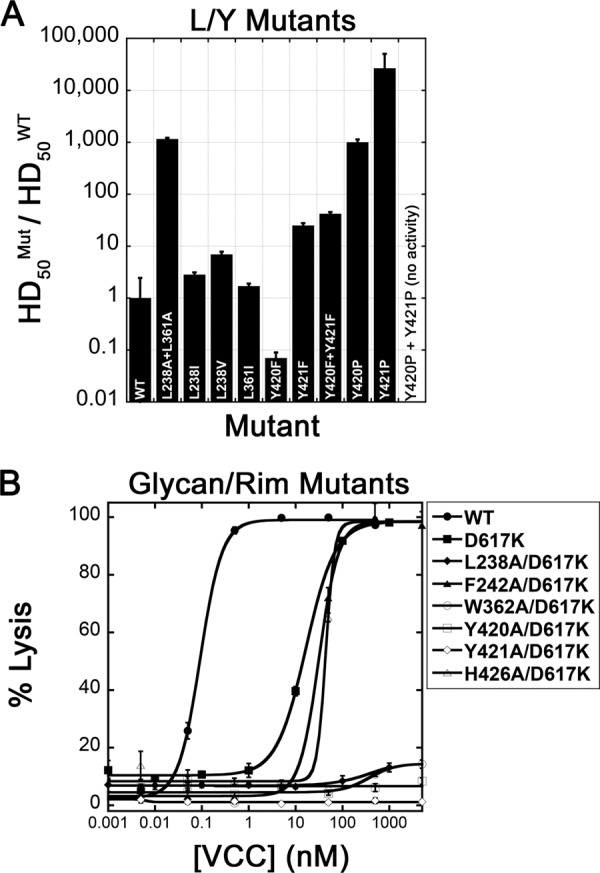
Leu/Tyr mutants and glycan double mutants. A, additional rim domain mutations were made to investigate the possibility of TL, CRAC, or CARC type cholesterol-binding motifs. The proline double mutant did not result in any measurable cell lysis. Numerical data are presented in Table 1. B, rim domain mutations were combined with a mutation in the β-prism domain (D617K) that blocks glycan binding. Most double mutants, with the exception of F242A/D617K and H426A/D617K, resulted in a nearly complete loss of hemolytic activity up to a 5 μm concentration. Error bars represent the S.E. of three repetitions.
A second set of motifs we investigated included the CRAC and inverse CARC sites. Mutagenesis and modeling studies suggest that in these motifs cholesterol stacks against the key central aromatic residue through CH-π stacking interactions (46). Inspection of the VCC rim domain indicates a number of overlapping putative CRAC and CARC sequences, although only one sequence contains a central aromatic residue that led to a significant loss in hemolytic activity when mutated to alanine. This putative CARC motif, containing the sequence 418KHYYVV423, included the two residues most sensitive to mutagenesis in the VCC rim domain. If this sequence were a functional CARC motif, mutation of the central tyrosine to phenylalanine would be tolerated, whereas mutation to other amino acids would not. Because there are two tyrosine residues in the center of the sequence, we mutated each independently to phenylalanine and proline and created double phenylalanine and proline mutants. Interestingly, the activity of Y420F actually improved upon mutation, whereas the activity of Y421F decreased 25-fold (Fig. 6A and Table 1). The Y420F/Y421F double mutant displayed a slightly more substantial loss than the Y421F mutation alone (>97.6% loss in activity). Furthermore, mutation of either residue to proline almost completely abolished activity (to less than 0.1% VCCWT activity), and mutation of both tyrosines to proline resulted in protein that passed quality control but had no measurable hemolytic activity. These results are consistent with VCC containing a CARC motif within the rim domain. CRAC and CARC motifs typically reside on α-helical segments, but the inherent flexibility in the membrane-interacting loops may allow a productive cholesterol-interacting conformation to occur.
The Interplay between Sugar Binding Mutants and Rim Domain Mutants
In addition to direct rim domain interactions, lectin-glycan interactions play an important role in membrane targeting by VCC (18, 36). To ensure that rim domain mutants do not have unintended effects on the β-prism domain lectin activity, we measured binding of VCCWT and a selection of single alanine mutants to the glycoprotein asialofetuin by ITC. Previous studies indicate that asialofetuin contains glycans that mimic membrane sugar receptors and is an indicator of glycan binding activity (36, 47). Our ITC experiments confirm that unaffected (S357A), moderately affected (H419A and H426A), and severely affected (Y420A and Y421A) rim domain mutations all retain nearly wild-type levels of asialofetuin binding activity (Fig. 4). Mutation of a key residue necessary for glycan binding (D617K) or complete removal of the VCC β-prism domain completely abolished measurable binding of asialofetuin (data not shown).
To investigate the extent to which rim domain mutations and glycan binding mutations collectively contribute to hemolytic activity, we created double mutations of selected alanine-scanning mutations on a D617K background. D617K alone leads to a roughly 180-fold loss in hemolytic activity against rabbit blood (Fig. 6B and Table 1). Introduction of the F242A and H426A mutations on the D617K background led to a larger loss in activity than any of the single mutations alone. Introduction of L238A, W362A, Y420A, and Y421A (all on the D617K background) led to mutant protein with little or no measurable lytic activity up to a concentration of 5 μm (Fig. 6B and Table 1). These results demonstrate that glycan binding and rim domain interactions are independent and additive, each contributing to toxin activity. Mutations within the rim domain are made worse with the removal of glycan interactions to the point where four of the double mutants tested are essentially dead.
Effect of Rim Domain Mutations on Activity against Liposomes
To further explore the mechanism by which rim domain mutations affect hemolytic activity, we used a liposome system to control individual membrane components. Previous studies show that VCC maintains a fairly high activity of pore formation in liposomes made of asolectin lipids (a crude extract of soy lipids) supplemented with a 30% molar ratio of cholesterol (48). Asolectin liposomes are not expected to contain complex N-glycans targeted by VCC or specific protein receptors, thereby simplifying interpretation of oligomerization results. If the rim domain mutants affect binding to protein receptors or glycan receptors, then we should see no difference in the pore-forming activity of VCCWT and mutant proteins on soybean liposomes. This system also allowed us to directly test the influence of cholesterol on membrane activity.
We tested the pore-forming activity of a subset of rim domain alanine mutations to assess whether their loss in activity is specific to cell membranes or a more general phenomenon of lipid membrane interactions. Rim domain mutations that exhibited deficient activity against rabbit blood also displayed reduced pore-forming activity on asolectin liposomes when assessed by SDS-PAGE (Fig. 7A). It is important to note that these assays are conducted at much higher protein concentrations (μm) than cell assays (pm to nm), and so the degree of loss in activity may not be as severe as that measured on cells. Still, the mutants exhibited substantially less pore-forming activity on liposomes with and without added cholesterol with VCCWT also exhibiting less activity against liposomes without cholesterol. This suggests that rim domain membrane binding is enhanced by cholesterol, although cholesterol is not absolutely necessary for toxin activity at high enough concentrations.
FIGURE 7.

Liposome gel-based assays. A, SDS-PAGE assembly gel showing VCCWT and a panel of additional glycan and rim domain mutations assembled on asolectin liposomes ±30% cholesterol. Monomer (M) and oligomeric pore (P) bands are designated by black arrows. All toxins were cut by chymotrypsin prior to assembly, and samples were not boiled before loading on the gel. B, time course of VCC assembly. VCC assembly reactions were stopped at various time points, and only the oligomer band is shown. Bands were quantified and plotted normalized to the maximum amount of VCCWT oligomer. Error bars denote the S.E. of two experiments. ON, overnight. C, competition assay showing that mutant toxins do not interfere with VCCWT activity. Numbers in boxes and under the bar graph indicate the molar ratio of VCCWT:mutant (Mut) protein. D, competition assay performed on rabbit blood. Curves result from a mixture of VCCWT protein (concentration on the x axis) and a 10 nm constant concentration (∼100 times the VCCWT HD50) of each designated mutant protein. Mutant proteins did not reduce the potency of VCCWT against cells. The β-prism lectin domain from VCC was used as a control to show that cell surface glycan sites are not saturated at this concentration.
The loss in activity of rim domain mutants could be caused by a reduced ability of the monomer to productively associate with the cell membrane but could also be attributed to a disruption in later stages of oligomerization and pore formation. To address the possibility that mutations are merely affecting the kinetics of pore formation, we performed a time course assay to look at the magnitude and rate of SDS-resistant oligomer formation. Fully assembled β-barrel channels remain heptameric in SDS as long as the samples are not boiled. This allows for visualization of the degree of pore formation on SDS-polyacrylamide gels, and quantification of the extent of VCC oligomerization can be accomplished by gel densitometry of assembled species. Focusing on the two most significant mutations (Y420A and Y421A), we found that the magnitude of pore formation, rather than the rate, is most affected by the mutations (Fig. 7B). Even after an overnight incubation, the mutants do not reach the amount of VCCWT oligomer seen on the gel, suggesting that the concentration of monomer on the membrane is not sufficient to promote oligomerization.
If the mutations are affecting the mechanism of oligomerization or pore insertion, we would predict that formation of heterooligomers between mutants and VCCWT would result in less channel formation. If the mutations merely caused a decrease in the ability to bind to the cell membrane, the mutants should not interfere with the ability of the VCCWT protein to bind and assemble on liposomes. We observed that adding up to 1 molar eq. of the mutant had no effect on the ability of VCCWT to form SDS-resistant oligomers (Fig. 7C) and did not reduce the activity of VCCWT against rabbit blood in competition assays even when added at a ∼100-fold excess of the HD50 of VCCWT (Fig. 7D). These data suggest that rim domain mutants primarily affect the ability of VCC to adhere to the lipid membrane rather than subsequent steps in toxin oligomerization and channel insertion.
SPR Studies of Membrane Binding by VCCWT and Alanine-scanning Mutants
To further investigate the effects of rim domain mutations on membrane interactions, we used SPR to look at VCC binding and assembly on liposomes. Liposomes were first captured on an SPR chip containing a lipophilic surface and blocked using BSA. Analysis of SPR sensorgrams allowed us to quantify the relative amount of monomer bound to the membrane during the binding phase and the resulting amount of oligomer assembled on the liposome following the wash step.
To first ensure that asolectin liposomes do not interact with the glycan-binding domain of VCC, we tested the D617K mutant on the chip. We saw a similar amount of both membrane binding and oligomerization of the D617K mutant as VCCWT on asolectin liposomes containing 30% cholesterol (Fig. 8A). A number of the most significant VCC rim domain mutations including L238A, F242A, Y420A, and Y421A all exhibited less signal during the binding phase and consequently less oligomer formation measured following the wash phase (Fig. 8A). Remarkably, the difference between VCCWT and the mutants in both membrane association and oligomer formation nearly disappeared when cholesterol was excluded from asolectin liposomes (Fig. 8B). To see whether this effect is specific to soy lipids, we also tested several mutants on liposomes made with total porcine brain lipids, which contain residual cholesterol (20). In brain lipids supplemented with 30% cholesterol, we saw an even more dramatic difference between VCCWT and rim domain mutant toxin (liposome capture was more efficient as well) in both membrane binding and oligomerization (Fig. 8C). Removal of cholesterol in this case did result in less efficient binding and oligomerization by VCCWT but not to the same degree as observed with cholesterol-free asolectin lipids (Fig. 8D). The activity that VCCWT retains may be attributable to the residual cholesterol in crude brain lipid preparations or additional lipid interactions in brain versus soy lipids that rim domain mutations disrupt such as those involving ceramides or sphingolipids.
FIGURE 8.

SPR analysis of rim domain mutations. A, activity of VCCWT and rim domain mutants on asolectin liposomes containing 30% cholesterol. Scatter plot points indicate the residual number of resonance units (RU) measured on liposomes after each injection and wash. The total mass of toxin flowed over the chip is designated on the x axis. Resonance units on the y axis are normalized to the total amount of liposomes captured in each experiment. The bar graph (inset) shows the highest resonance unit signal measured during the binding phase of the first injection point in each series. B, same analysis shown for asolectin liposomes without any added cholesterol. C and D show similar data for brain lipid liposomes ±30% added cholesterol.
Discussion
For pathogens to inflict harm on host cells, secreted PFTs need to tightly adhere to membranes and efficiently oligomerize into transmembrane channels. This requires some level of specificity or toxins may permeabilize the pathogen's own membrane. PFTs have evolved to recognize a diverse set of membrane receptors and requirements for cholesterol or host-specific glycans to aid in cell targeting.
This study illustrates how VCC utilizes two types of interactions to achieve high affinity binding to cell membranes. The first is mediated through a dedicated β-prism lectin domain that targets host-specific glycan molecules on the surface of susceptible cells. This interaction appears to be necessary, but not sufficient, for VCC to target cells with high affinity in the picomolar range. The second interaction occurs via direct lipid- and cholesterol-mediated interactions involving three loops that comprise the VCC rim domain. Although cholesterol is not absolutely necessary for SDS-stable pores to form, it appears to substantially increase the ability of the toxin to bind and permeabilize cell membranes. This is distinct from cholesterol-dependent cytolysins, which have a strict requirement for cholesterol (49) and utilize protein receptors (50). In fact, cholesterol is also used by cholesterol-dependent cytolysins as a trigger for initiating conformational changes necessary for pore formation (51), a feature that has not been observed in VCC.
In the past several years, a number of protein and lipid receptors have been identified for leukocidins and hemolysins from S. aureus that share structural similarity with the core domain of VCC. This is usually reflected in a higher activity toward certain cell types, which present these receptors versus those missing the specific lipid or protein. Although human neutrophils have been suggested as a target of VCC in infectious models, we see no preference for neutrophils over other immune cells such as T cells and monocytes. In fact, VCC lyses rabbit erythrocytes as well as (or better than) it kills human immune cells in our assays. This absence of tropism suggests that VCC is unlikely to use a cell-specific protein receptor in targeting cells. So unlike staphylococcal toxins, which bind proteins and lipids, the acquisition of lectin domains by Vibrio toxins may be an alternative mechanism for targeting cells that also leads to a broader efficacy against various human cell lines. The slight preference for rabbit erythrocytes, as we have observed before (18), may arise by a higher density of complex N-glycans on these cells.
It has been proposed that the β-prism domain acts as a regulatory switch to trigger pore formation and that rim domain mutations would prevent this structural rearrangement from happening (22, 47). Our double rim/glycan mutations indicate that membrane binding by the rim domain and glycan binding by the β-prism domain provide independent and additive contributions to membrane activity. Rim domain mutants are adversely affected by β-prism domain mutations and vice versa. This behavior can also be explained by a shift in the membrane-bound monomer equilibrium that can also be overcome by increasing the solution toxin concentration.
Our complete mapping of the VCC rim domain uncovered a number of residues that when mutated to alanine sharply decrease the ability of VCC to lyse rabbit erythrocytes. Not surprisingly, the most substantial mutations affect either hydrophobic or aromatic amino acids, which are more likely to interact with the hydrophobic membrane interior or the more polar membrane-water interfacial region. The fact that these mutations affect cells in both the model hemolysis assay and liposomes without sugar and protein components lends further support to the notion that VCC does not utilize a protein receptor to attack cells. As we have shown, several of the most significant residues fall into motifs that bind cholesterol in other toxins and membrane-embedded proteins. The first type, ubiquitous in cholesterol-dependent cytolysins (23), is a two-amino acid TL sequence that forms specific interactions with membrane cholesterol. We saw two potential instances of this motif (Thr237-Leu238 and the similar Ala360-Leu361) and measured a 37–70-fold loss in activity when either leucine was individually mutated to alanine (Fig. 5A and Table 1). In VCC, compensation of one motif for the second could lessen the effect observed when these sites are removed individually.
A recent study (22) examined the role of rim domain loops in VCC activity by replacing the end of each loop with a glycine-alanine linker and by mutating one or two positions on each loop (namely Leu238, Trp362, and Tyr421). By measuring activity at a single toxin concentration, they saw a significant loss in activity due to these three mutations (and a lesser amount at a fourth position, Thr237, that failed our quality control test). Similarly, we also noted a significant loss in activity at these three sites in addition to the seven other sites that we identified with a loss of >90% wild-type activity. In their study, they conclude that although rim domain mutants do affect membrane binding these mutants also lead to a nonspecific membrane-associated state that is deficient in membrane oligomerization and channel insertion. We did not see evidence for such a state, particularly in our SPR experiments where the degree of channel formation was roughly proportional to the amount of toxin that we observed in the membrane-binding phase (Fig. 8). We argue that a simple shift in the equilibrium of monomer binding to the membrane can account for all of the reduced hemolytic activity observed in both studies. This is most evident in our concentration-dependent hemolytic assays and liposome experiments where we could compensate for the loss in activity by merely increasing the concentration of toxin. Therefore, subsequent steps in oligomerization and pore formation are solely dependent on the local concentration of monomer on the membrane surface, which we believe is not deficient in oligomerization ability. This also explains our observation that rim domain mutations shift the HD50 to a higher value but do not change the shape of the lysis curve.
The previous study also observed that severe mutations (L238A, W362A, and Y421A) lead to no detectible SDS-stable pores formed in gel-based liposome assays (22). We were able to observe these pores by increasing the concentration of toxin. We saw SDS-resistant pores formed in all of our single alanine mutants although at differing degrees of efficiency. If mutations were affecting the ability of toxins to oligomerize or insert channels, forming mixed oligomers between mutant and wild-type toxins should have a negative effect on the activity of the toxin. We did not see this behavior even when adding 10-fold molar excesses of mutants to wild-type toxin at low concentrations and equimolar amounts at higher concentrations (Fig. 7, C and D).
The two most substantial alanine mutants are Y420A and Y421A, which are found on the tip of loop 3, the longest and most central loop in the rim domain. Tyrosine residues are often found near the membrane-water interface and can form ring-stacking interactions with cholesterol as well as hydrogen bonds with lipid headgroups. The sequence surrounding the YY motif is consistent with the so-called CARC sequence, indicating the possibility of a third potential cholesterol-binding motif in VCC. If this were the case, all three rim domain loops could involve themselves in cholesterol binding interactions. It may be more complicated than this, however, as other lipids such as ceramide and sphingolipids may also be required for full activity against membranes (20). In our SPR studies, we observed a roughly 5-fold increase in pore-forming activity of VCCWT against brain lipids, which contain sphingomyelin and ceramides, than in soybean asolectin, which does not. Although we now know the effects of individual mutations on the overall activity against cells and liposome membranes, more work will be necessary to understand whether specific lipid and cholesterol interactions can be attributed to individual or groups of residues on the rim domain surface. TL, CARC, or other sites may be completely modular in their interactions or work together to form a network of interactions that contribute to productive binding to the cell membrane.
Flexibility of rim domain loops is another feature likely to play a role in VCC membrane interactions. The crystal structure of the VCC heptameric pore (1) shows loop 3 extending nearly halfway through the lipid membrane. This structure was determined in detergent micelles so the positions of all three rim domain loops may not be indicative of their final position when assembled on membrane bilayers. Mutations uncovered in our study could lead to alterations in flexibility, causing additional effects on toxin activity. We observed that mutation of Tyr420 or Tyr421 to proline led to an almost complete loss of hemolytic activity against rabbit erythrocytes with the double mutant having no measurable activity. Proline residues are expected to severely affect backbone flexibility and also disrupt α-helices. Interestingly, studies of the homologous Panton-Valentine leukocidin S toxin uncovered a similar need for flexibility in rim domain loops that was also disrupted by mutation (29). In the leukocidin case, this flexibility was required to make specific interactions with a protein receptor, but this idea may be extended to lipid interactions as well.
In conclusion, rim domain interactions with the membrane play an important role in the ability of VCC to productively anchor to cells. Glycan binding interactions increase the potency of the wild-type toxin against membranes, possibly additively or multiplicatively through avidity effects with rim domain interactions. The types of membrane interactions are complex with multiple candidates for cholesterol- and lipid-binding sites, and further work with defined lipid mixtures may shed light on the involvement of individual residues in specific lipid or cholesterol interactions. This work indicates the possibility that antibodies or small molecule compounds that block rim domain interactions could have a prophylactic activity against VCC as well as related PFTs that target the membrane through similar aromatic residue-rich loops.
Author Contributions
R. O. designed the study and wrote the paper. S. D., A. B., and J. H. designed and carried out all mutagenesis and hemolysis studies. S. D. and R. O. performed the SPR experiments. F. A. and V. J. T. designed and carried out cell killing assays. J. W. L. and J. L. C. designed and carried out centrifugation experiments.
Acknowledgment
We thank George Korza for assistance with collecting SPR data.
This work was supported by NIAID, National Institutes of Health Grant R15 AI101977. The authors declare that they have no conflicts of interest with the contents of this article.
- VCC
- V. cholerae cytolysin
- PFT
- pore-forming toxin
- PFO
- perfringolysin O
- SPR
- surface plasmon resonance
- ITC
- isothermal titration calorimetry
- HD50
- concentration resulting in 50% lysis
- CRAC
- cholesterol recognition/interaction amino acid consensus.
References
- 1.De S., and Olson R. (2011) Crystal structure of the Vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc. Natl. Acad. Sci. U.S.A. 108, 7385–7390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olivier V., Haines G. K. 3rd, Tan Y., and Satchell K. J. (2007) Hemolysin and the multifunctional autoprocessing RTX toxin are virulence factors during intestinal infection of mice with Vibrio cholerae El Tor O1 strains. Infect. Immun. 75, 5035–5042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Queen J., and Satchell K. J. (2012) Neutrophils are essential for containment of Vibrio cholerae to the intestine during the proinflammatory phase of infection. Infect. Immun. 80, 2905–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhakdi S., and Tranum-Jensen J. (1991) α-Toxin of Staphylococcus aureus. Microbiol. Rev. 55, 733–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berube B. J., and Bubeck Wardenburg J. (2013) Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins 5, 1140–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson R., and Gouaux E. (2005) Crystal structure of the Vibrio cholerae cytolysin (VCC) pro-toxin and its assembly into a heptameric transmembrane pore. J. Mol. Biol. 350, 997–1016 [DOI] [PubMed] [Google Scholar]
- 7.Song L., Hobaugh M. R., Shustak C., Cheley S., Bayley H., and Gouaux J. E. (1996) Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science 274, 1859–1866 [DOI] [PubMed] [Google Scholar]
- 8.Olson R., Nariya H., Yokota K., Kamio Y., and Gouaux E. (1999) Crystal structure of staphylococcal LukF delineates conformational changes accompanying formation of a transmembrane channel. Nat. Struct. Biol. 6, 134–140 [DOI] [PubMed] [Google Scholar]
- 9.Pédelacq J. D., Maveyraud L., Prévost G., Baba-Moussa L., González A., Courcelle E., Shepard W., Monteil H., Samama J. P., and Mourey L. (1999) The structure of a Staphylococcus aureus leucocidin component (LukF-PV) reveals the fold of the water-soluble species of a family of transmembrane pore-forming toxins. Structure 7, 277–287 [DOI] [PubMed] [Google Scholar]
- 10.Guillet V., Roblin P., Werner S., Coraiola M., Menestrina G., Monteil H., Prévost G., and Mourey L. (2004) Crystal structure of leucotoxin S component: new insight into the staphylococcal β-barrel pore-forming toxins. J. Biol. Chem. 279, 41028–41037 [DOI] [PubMed] [Google Scholar]
- 11.Yamashita K., Kawai Y., Tanaka Y., Hirano N., Kaneko J., Tomita N., Ohta M., Kamio Y., Yao M., and Tanaka I. (2011) Crystal structure of the octameric pore of staphylococcal γ-hemolysin reveals the β-barrel pore formation mechanism by two components. Proc. Natl. Acad. Sci. U.S.A. 108, 17314–17319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foletti D., Strop P., Shaughnessy L., Hasa-Moreno A., Casas M. G., Russell M., Bee C., Wu S., Pham A., Zeng Z., Pons J., Rajpal A., and Shelton D. (2013) Mechanism of action and in vivo efficacy of a human-derived antibody against Staphylococcus aureus α-hemolysin. J. Mol. Biol. 425, 1641–1654 [DOI] [PubMed] [Google Scholar]
- 13.Savva C. G., Fernandes da Costa S. P., Bokori-Brown M., Naylor C. E., Cole A. R., Moss D. S., Titball R. W., and Basak A. K. (2013) Molecular architecture and functional analysis of NetB, a pore-forming toxin from Clostridium perfringens. J. Biol. Chem. 288, 3512–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker B., Braha O., Cheley S., and Bayley H. (1995) An intermediate in the assembly of a pore-forming protein trapped with a genetically-engineered switch. Chem. Biol. 2, 99–105 [DOI] [PubMed] [Google Scholar]
- 15.Löhner S., Walev I., Boukhallouk F., Palmer M., Bhakdi S., and Valeva A. (2009) Pore formation by Vibrio cholerae cytolysin follows the same archetypical mode as β-barrel toxins from Gram-positive organisms. FASEB J. 23, 2521–2528 [DOI] [PubMed] [Google Scholar]
- 16.Los F. C., Randis T. M., Aroian R. V., and Ratner A. J. (2013) Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 77, 173–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DuMont A. L., and Torres V. J. (2014) Cell targeting by the Staphylococcus aureus pore-forming toxins: it's not just about lipids. Trends Microbiol. 22, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levan S., De S., and Olson R. (2013) Vibrio cholerae cytolysin recognizes the heptasaccharide core of complex N-glycans with nanomolar affinity. J. Mol. Biol. 425, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaus K., Lary J. W., Cole J. L., and Olson R. (2014) Glycan specificity of the Vibrio vulnificus hemolysin lectin outlines evolutionary history of membrane targeting by a toxin family. J. Mol. Biol. 426, 2800–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zitzer A., Zitzer O., Bhakdi S., and Palmer M. (1999) Oligomerization of Vibrio cholerae cytolysin yields a pentameric pore and has a dual specificity for cholesterol and sphingolipids in the target membrane. J. Biol. Chem. 274, 1375–1380 [DOI] [PubMed] [Google Scholar]
- 21.Paul K., and Chattopadhyay K. (2012) Single point mutation in Vibrio cholerae cytolysin compromises the membrane pore-formation mechanism of the toxin. FEBS J. 279, 4039–4051 [DOI] [PubMed] [Google Scholar]
- 22.Rai A. K., and Chattopadhyay K. (2015) Revisiting the membrane interaction mechanism of a membrane-damaging β-barrel pore-forming toxin Vibrio cholerae cytolysin. Mol. Microbiol. 97, 1051–1062 [DOI] [PubMed] [Google Scholar]
- 23.Farrand A. J., LaChapelle S., Hotze E. M., Johnson A. E., and Tweten R. K. (2010) Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. U.S.A. 107, 4341–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howlader M. T., Kagawa Y., Miyakawa A., Yamamoto A., Taniguchi T., Hayakawa T., and Sakai H. (2010) Alanine scanning analyses of the three major loops in domain II of Bacillus thuringiensis mosquitocidal toxin Cry4Aa. Appl. Environ. Microbiol. 76, 860–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H., and Papadopoulos V. (1998) Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 139, 4991–4997 [DOI] [PubMed] [Google Scholar]
- 26.Epand R. M., Thomas A., Brasseur R., and Epand R. F. (2010) Cholesterol interaction with proteins that partition into membrane domains: an overview. Subcell. Biochem. 51, 253–278 [DOI] [PubMed] [Google Scholar]
- 27.Reyes-Robles T., Alonzo F. 3rd, Kozhaya L., Lacy D. B., Unutmaz D., and Torres V. J. (2013) Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe. 14, 453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spaan A. N., Henry T., van Rooijen W. J., Perret M., Badiou C., Aerts P. C., Kemmink J., de Haas C. J., van Kessel K. P., Vandenesch F., Lina G., and van Strijp J. A. (2013) The staphylococcal toxin Panton-Valentine leukocidin targets human C5a receptors. Cell Host Microbe 13, 584–594 [DOI] [PubMed] [Google Scholar]
- 29.Laventie B.-J., Guérin F., Mourey L., Tawk M. Y., Jover E., Maveyraud L., and Prévost G. (2014) Residues essential for Panton-Valentine leukocidin S component binding to its cell receptor suggest both plasticity and adaptability in its interaction surface. PLoS One 9, e92094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spaan A. N., Schiepers A., de Haas C. J., van Hooijdonk D. D., Badiou C., Contamin H., Vandenesch F., Lina G., Gerard N. P., Gerard C., van Kessel K. P., Henry T., and van Strijp J. A. (2015) Differential interaction of the staphylococcal toxins Panton-Valentine leukocidin and γ-hemolysin CB with human C5a receptors. J. Immunol. 195, 1034–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valeva A., Hellmann N., Walev I., Strand D., Plate M., Boukhallouk F., Brack A., Hanada K., Decker H., and Bhakdi S. (2006) Evidence that clustered phosphocholine head groups serve as sites for binding and assembly of an oligomeric protein pore. J. Biol. Chem. 281, 26014–26021 [DOI] [PubMed] [Google Scholar]
- 32.Schwiering M., Brack A., Stork R., and Hellmann N. (2013) Lipid and phase specificity of α-toxin from S. aureus. Biochim. Biophys. Acta 1828, 1962–1972 [DOI] [PubMed] [Google Scholar]
- 33.Sheffield P., Garrard S., and Derewenda Z. (1999) Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15, 34–39 [DOI] [PubMed] [Google Scholar]
- 34.Braman J., Papworth C., and Greener A. (1996) Site-directed mutagenesis using double-stranded plasmid DNA templates. Methods Mol. Biol. 57, 31–44 [DOI] [PubMed] [Google Scholar]
- 35.DuMont A. L., Yoong P., Surewaard B. G., Benson M. A., Nijland R., van Strijp J. A., and Torres V. J. (2013) Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect. Immun. 81, 1830–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saha N., and Banerjee K. K. (1997) Carbohydrate-mediated regulation of interaction of Vibrio cholerae hemolysin with erythrocyte and phospholipid vesicle. J. Biol. Chem. 272, 162–167 [DOI] [PubMed] [Google Scholar]
- 37.Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C. A., and Schuck P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Houtman J. C., Brown P. H., Bowden B., Yamaguchi H., Appella E., Samelson L. E., and Schuck P. (2007) Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 16, 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laue T. M., Shah B. H., Ridgeway T. M., and Pelletier S. L. (1992) Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S., and Rowe A., eds), pp. 90–125, Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 40.Stafford W. F., 3rd (1992) Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal. Biochem. 203, 295–301 [DOI] [PubMed] [Google Scholar]
- 41.Philo J. S. (2006) Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal. Biochem. 354, 238–246 [DOI] [PubMed] [Google Scholar]
- 42.Honda T., and Finkelstein R. A. (1979) Purification and characterization of a hemolysin produced by Vibrio cholerae biotype El Tor: another toxic substance produced by cholera vibrios. Infect. Immun. 26, 1020–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zitzer A., Walev I., Palmer M., and Bhakdi S. (1995) Characterization of Vibrio cholerae El Tor cytolysin as an oligomerizing pore-forming toxin. Med. Microbiol. Immunol. 184, 37–44 [DOI] [PubMed] [Google Scholar]
- 44.Valeva A., Walev I., Weis S., Boukhallouk F., Wassenaar T. M., and Bhakdi S. (2008) Pro-inflammatory feedback activation cycle evoked by attack of Vibrio cholerae cytolysin on human neutrophil granulocytes. Med. Microbiol. Immunol. 197, 285–293 [DOI] [PubMed] [Google Scholar]
- 45.Sutton-Smith M., Wong N. K., Khoo K. H., Wu S. W., Yu S. Y., Patankar M. S., Easton R., Lattanzio F. A., Morris H. R., Dell A., and Clark G. F. (2007) Analysis of protein-linked glycosylation in a sperm-somatic cell adhesion system. Glycobiology 17, 553–567 [DOI] [PubMed] [Google Scholar]
- 46.Fantini J., and Barrantes F. J. (2013) How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 4, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rai A. K., Paul K., and Chattopadhyay K. (2013) Functional mapping of the lectin activity site on the β-prism domain of Vibrio cholerae cytolysin: implications for the membrane pore-formation mechanism of the toxin. J. Biol. Chem. 288, 1665–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zitzer A., Harris J. R., Kemminer S. E., Zitzer O., Bhakdi S., Muething J., and Palmer M. (2000) Vibrio cholerae cytolysin: assembly and membrane insertion of the oligomeric pore are tightly linked and are not detectably restricted by membrane fluidity. Biochim. Biophys. Acta 1509, 264–274 [DOI] [PubMed] [Google Scholar]
- 49.Flanagan J. J., Tweten R. K., Johnson A. E., and Heuck A. P. (2009) Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48, 3977–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giddings K. S., Zhao J., Sims P. J., and Tweten R. K. (2004) Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 11, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 51.Heuck A. P., Savva C. G., Holzenburg A., and Johnson A. E. (2007) Conformational changes that effect oligomerization and initiate pore formation are triggered throughout perfringolysin O upon binding to cholesterol. J. Biol. Chem. 282, 22629–22637 [DOI] [PubMed] [Google Scholar]