

Background: Interaction between oxidative stress (CSE) and protease (NE) may contribute to COPD pathogenesis.
Results: Although NE enhances CSE-induced IL-8, it suppresses VEGF production, which is due to degradation by uptake of NE into bronchial epithelial cells.
Conclusion: NE contributes to the pathogenesis of COPD by enhancing inflammation and apoptosis.
Significance: This provides a molecular mechanism for understanding COPD pathogenesis.
Keywords: chronic obstructive pulmonary disease (COPD), cigarette smoke, interleukin, neutrophil, vascular endothelial growth factor (VEGF), CXCL-8, cigarette smoke extract, neutrophil elastase
Abstract
Inflammation by IL-8-induced neutrophil recruitment and apoptosis of epithelial cells by decreased expression of VEGF have been suggested as one of the complicated pathogenic mechanisms of chronic obstructive pulmonary disease (COPD). The role of neutrophil elastase (NE) in the development of COPD is also well known. However, little is known about how they interact. The objective of this study was to elucidate the effect of NE on cigarette smoke extract (CSE)-induced IL-8 and VEGF production and its molecular mechanism in bronchial epithelial cells. CSE increased both IL-8 and VEGF production in human bronchial epithelial cells (BEAS-2B). Although NE significantly enhanced CSE-induced IL-8 production, it suppressed VEGF production. This differential regulation was not CSE-specific. The effect of NE on IL-8 production, but not VEGF, was ERK-dependent. Interestingly, in contrast to decreased VEGF protein expression, NE accelerated VEGF transcription by CSE, suggesting post-translational modification. When cells were incubated with purified NE, it was detected in the cytoplasm, suggesting the intracellular translocation of NE. Furthermore, NE fragmented recombinant human VEGF in vitro but not recombinant human IL-8. These results indicate that VEGF down-regulation is due to direct degradation by NE, which is translocated into cells. Similar to in vitro cell experiments, elastase treatment increased CSE-induced IL-8; however, it suppressed VEGF production in bronchoalveolar lavage fluid of CSE-treated mice. Moreover, elastase treatment enhanced CSE-induced emphysema in mice. Considering the actions of IL-8 and VEGF, our results suggest that NE contributes to the pathogenesis of COPD by enhancing inflammation and apoptosis.
Introduction
Chronic obstructive pulmonary disease (COPD)2 is characterized by persistent airflow limitation that is usually progressive. It is one of the leading causes of morbidity and mortality worldwide, and it results in an economic and social burden that is both substantial and increasing (1). Although there are no proven disease-modifying treatments, a variety of agents are under development as a result of advances in the understanding of COPD pathogenesis (2).
Cigarette smoke (CS) plays a major role in the pathogenesis of COPD. However, the mechanism of CS and COPD development is not fully understood and could be explained by different hypotheses including inflammation, protease-antiprotease imbalance, oxidant stress (3–6), apoptosis (7), and accelerated lung aging (8). Among them, inflammation, oxidative stress, and protease-antiprotease imbalance have been suggested as a pathogenic triad in COPD (6). Lung inflammation caused by CS is generally associated with an influx of inflammatory cells such as neutrophils, macrophages, and CD8+ T lymphocytes (9). CS itself contains a high concentration of reactive oxygen species, which causes tissue destruction and DNA damage (10). Moreover, CS triggers generation of additional reactive oxygen species, and reactive oxygen species amplify inflammation, suggesting cross-talk between oxidative stress and inflammation (6, 11). The protease-antiprotease imbalance arises from the activity of inflammatory cells, especially neutrophils recruited by CS. Neutrophils secrete various proteases including neutrophil elastase (NE), proteinase 3, myeloperoxidase, and cathepsin. Most importantly, NE contributes to the pathogenesis of emphysema (3) by the cleavage of extracellular matrix and lung destruction. During the cleavage of extracellular matrix, chemotactic fragments are produced (12, 13), which results in continued accumulation of inflammatory cells and tissue destruction. Therefore, inflammation and proteases are likely to be mutually reinforcing. All these results indicate that inflammation, oxidative stress, and protease-antiprotease imbalance constantly interact with each other.
Accumulating evidence suggests that apoptosis of epithelial cells is important in COPD pathogenesis (14). As vascular endothelial growth factor (VEGF) stimulates endothelial cell and type II epithelial cell growth and survival, decreased expression of VEGF has been suggested to be responsible for apoptosis of epithelial cells in the emphysematous lung. In fact, long term incubation with CS decreases VEGF mRNA expression and production in human primary airway epithelial cells (15). So far, the association of NE with CS-mediated down-regulation of VEGF has not been elucidated.
In this study, we evaluated the effect of NE on cigarette smoke extract (CSE)-induced interleukin (IL)-8 and VEGF production and its mechanism in human bronchial epithelial cells. We discovered that NE differentially regulates CSE-induced IL-8 and VEGF. NE enhanced CSE-induced IL-8 production. In contrast, VEGF production was suppressed by NE, which is due to post-translational modification (degradation) by uptake of NE into cells.
Experimental Procedures
Cells and Reagents
Normal human bronchial epithelial cells (BEAS-2B) were maintained in defined keratinocyte serum-free medium (Gibco by Life Technologies) at 37 °C under 5% CO2. U937 cells were maintained in RPMI 1640 medium (Gibco) containing 10% heat-inactivated FBS, 100 units/ml penicillin, and 100 mg/ml streptomycin at 37 °C under 5% CO2. Human sputum NE was purchased from Elastin Products Co. (Owensville, MO). Elastase was dissolved in 50% glycerol, 50% 0.02 m NaOAc (pH 5). Recombinant human VEGF-165 and IL-8 were from BioLegend (San Diego, CA). Recombinant human tumor necrosis factor-α (TNF-α) and IL-1β were from R&D Systems (Minneapolis, MN). Rabbit polyclonal anti-phosphorylated ERK and anti-hypoxia-inducible factor (HIF)-1α antibodies and PD98059 were obtained from Cell Signaling Technology (Danvers, MA). Rabbit polyclonal anti-VEGF and anti-NE antibodies and goat polyclonal anti-GAPDH antibody were from Santa Cruz Biotechnology (Santa Cruz, CA). Human IL-8 mAb and protein A/G UltraLink Resin were from Thermo Scientific (Rockford, IL). Alexa Fluor 555 goat anti-rabbit IgG (heavy + light) antibody was from Invitrogen. HeLa cell lysates were from BD Biosciences. Chloroquine, hydrogen peroxide, and porcine pancreatic elastase (PPE) were from Sigma-Aldrich. U0126 was from Calbiochem (EMD Millipore Corp., Billerica, MA).
Cigarette Smoke Extract
CSE was prepared as described previously (16, 17). Commercial cigarettes (THIS; 84 mm long with a diameter of 8 mm; purchased from Korea Tomorrow & Global Corp., Republic of Korea) were smoked continuously by a bottle system connected to a vacuum machine. The smoke from 10 cigarettes was bubbled in 50 ml of defined keratinocyte serum-free medium. The large insoluble particles contained in the resulting suspension were removed by filtering the solution through a 0.22-μm filter (16, 17).
Western Blot Analysis
Total cellular proteins were extracted using 1× cell lysis buffer (Cell Signaling Technology). Protein concentration was measured using the Bradford protein assay according to the manufacturer's instructions (Bio-Rad). Proteins were resolved by 4–12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. The membranes were blocked with 5% skim milk, PBS, 0.1% Tween 20 for 1 h before being incubated overnight at 4 °C with primary antibodies in blocking buffer. The membranes were washed in 1× PBS, 0.1% Tween 20 and incubated with secondary antibodies for 1 h. After successive washes, the membranes were developed using an ECL kit.
Immunoprecipitation
Immunoprecipitation of intracellular NE was performed using anti-NE Antibody. After rotating samples at 4 °C overnight, protein A/G UltraLink Resin was added to each tube and rotated at 4 °C for 1 h. The pellets were washed three times in 1× cell lysis buffer. The samples were heated for 10 min in 15 μl of loading sample buffer and subjected to SDS-PAGE and immunoblot analysis.
Fragment Generation of VEGF or IL-8
VEGF-165 or IL-8 was incubated in medium, vehicle control (50% glycerol, 50% 0.02 m NaOAc (pH 5)), 10 units/ml NE, or 10 μg/ml PPE at 37 °C for 30 min. The reaction was stopped by adding Western blot sample buffer, and samples were subjected to Western blot analysis for VEGF and IL-8.
RNA Extraction and Real Time Quantitative Polymerase Chain Reaction (PCR)
Total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA). cDNA synthesis was performed using avian myeloblastosis virus reverse transcriptase (Promega, Madison, WI) according to the manufacturer's instructions. The real time PCR was based on the TaqManTM technology (Applied Biosystems by Life Technologies). The following TaqMan probes were used: VEGF (Hs00900055_m1) and GAPDH (Hs99999905_m1).
Immunofluorescence Staining for NE
Cells grown in 35-mm dishes in the presence or absence of NE for 2 h were fixed in methanol and incubated with rabbit polyclonal anti-NE antibody diluted 1:100 in 3% BSA for 24 h. The cells were subsequently incubated with Alexa Fluor 555 goat anti-rabbit antibody diluted 1:100 in 3% BSA for 30 min. After successful washes, cells were analyzed with a fluorescence microscope (Nikon ECLIPSE TE300).
Mice
Female 8-week-old C57BL/6 wild-type (WT) mice were purchased from OrientBio (Kapyong, Korea). Animal experiments were approved by the Institutional Animal Care and Use Committee (number 15-0121-S1A0(2)) of Seoul National University Hospital, Seoul, Korea.
Intratracheal Administration of Elastase and CSE
C57BL/6 WT mice were anesthetized and instilled intratracheally with vehicle, 50 μg of PPE, 100 μl of CSE, or 50 μg of PPE in 100 μl of CSE. Elastase was instilled once a week for 2 weeks, and CSE was instilled once a week for 3 weeks. Three mice were used in each group. Mice were sacrificed at week 4 after first instillation to collect bronchoalveolar lavage fluid (BALF). To obtain BALF, 1 ml of PBS was instilled and retrieved following tracheostomy. BALF was centrifuged at 2000 rpm for 5 min at 4 °C, and supernatants were used for analysis of cytokines. Mice were sacrificed at week 8 to examine lung histopathology.
Measurement of Emphysema
Mouse lungs were fixed with 4% neutral buffered paraformaldehyde. Fixed lung was dehydrated, embedded with paraffin, sectioned, and stained with hematoxylin and eosin (H&E). Emphysema was quantified by measuring the mean linear intercept (MLI). Four randomly selected ×100 fields per specimen were photographed in a blinded manner. The MLI was measured by placing four 1000-μm lines over each field. The total length of each line divided by the number of alveolar intercepts gives the average distance. The non-parenchymal area was not included.
Determination of Cytokine Release
Cytokine levels in culture supernatants were determined using a commercially available Bio-Plex ProTM cytokine assay kit (Bio-Rad) according to the manufacturer's instructions. VEGF or IL-8 in BALF was measured using the Bio-Plex Pro cytokine assay kit or mouse IL-8 ELISA kit (MyBioSource, San Diego, CA), respectively.
Statistical Analysis
We compared MLI among groups by using post hoc Tukey's multiple comparison testing after analysis of variance using Stata 13.1 (StataCorp, College Station, TX). All in vitro experiments were repeated at least three times, and data were subjected to Student's t test for analysis of statistical significance using Prism (GraphPad). Results are given as the mean ± S.D. A p value <0.05 was considered significant.
Results
CSE Increased Extracellular IL-8 and VEGF Production through ERK Pathway
It has been suggested that mitogen-activated protein kinases, especially p38 and ERK, play a role in CS-induced proinflammatory signaling (18, 19). Therefore, we investigated the role of the mitogen-activated protein kinase pathway in CSE-induced cytokine production. CSE activated ERK in a dose- and time-dependent manner (Fig. 1A). It induced IL-8 and VEGF release with a different dose dependence: IL-8 production was increased by 30% CSE, but not by 10% CSE; in contrast, VEGF production was increased by 10% CSE (Fig. 1B). Blocking ERK activation by chemical inhibitor decreased CSE-mediated production of IL-8 and VEGF completely (Fig. 1B). These data suggest that the ERK pathway is responsible for IL-8 and VEGF release in response to CSE stimulation in lung epithelial cells.
FIGURE 1.
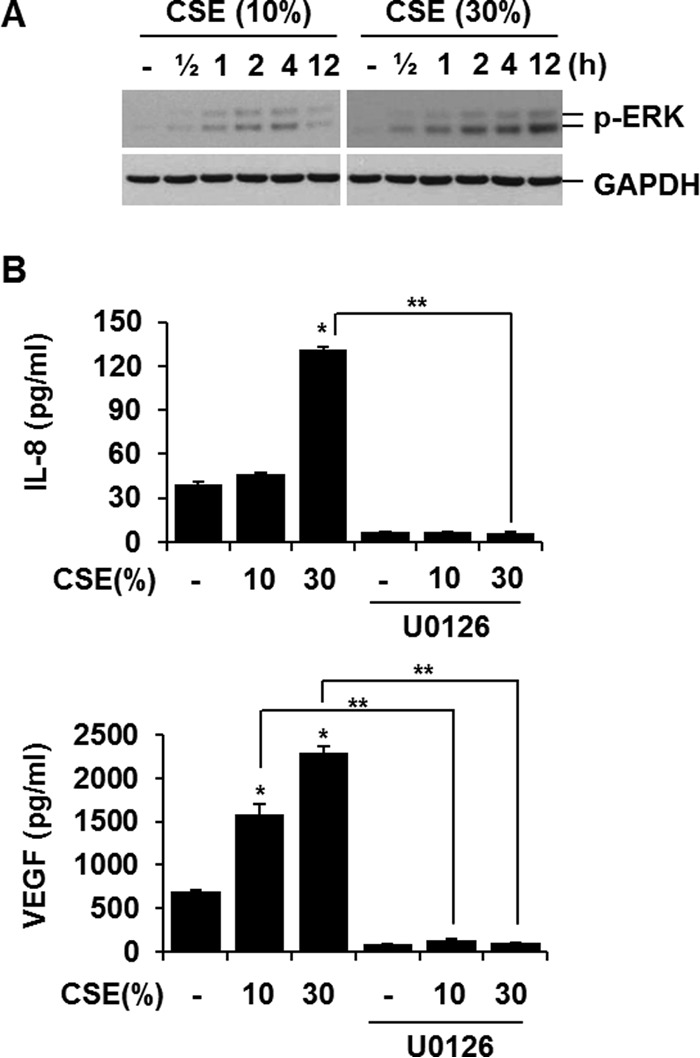
CSE increased extracellular IL-8 and VEGF production through ERK pathway. A, CSE activates ERK. BEAS-2B cells were treated with CSE (10 or 30%) for the indicated times. Total cellular extracts were subjected to Western blot analysis for phosphorylated ERK (p-ERK) and GAPDH. B, blocking ERK activation decreases IL-8 and VEGF production. Cells were pretreated with ERK inhibitor (20 μm U0126) for 1 h and stimulated with CSE in the presence or absence of ERK inhibitors. IL-8 and VEGF concentrations in media were determined by ELISA. ELISA data represent the mean ± S.D. (error bars) of triplicates. *, p < 0.05 versus control; **, p < 0.05. Results are representative of three separate experiments.
CSE-induced IL-8 and VEGF Production Is Differently Regulated by NE
We next evaluated the effect of NE treatment on CSE-induced IL-8 and VEGF production. NE treatment alone slightly increased IL-8 production (Fig. 2). Although 10% CSE treatment alone did not induce IL-8 production, co-treatment with CSE and NE augmented IL-8 production compared with NE treatment alone (Fig. 2A). Treatment with NE after CSE stimulation showed a similar effect, suggesting that enhancement of the effect of NE on IL-8 production is irrespective of the time of treatment (data not shown). Either NE treatment alone or co-treatment with CSE suppressed VEGF production compared with vehicle treatment or CSE treatment alone, respectively (Fig. 2A). To investigate whether this opposite effect of NE on IL-8 and VEGF production is CSE stimulation-specific, we investigated the effect of NE on hydrogen peroxide (H2O2), TNF-α, or IL-1β-induced IL-8 and VEGF production. The differential effects of NE on IL-8 and VEGF production were also observed in both H2O2-, TNF-α-, and IL-1β-stimulated cells (Fig. 2B), indicating that the effect of NE is not stimulus-specific.
FIGURE 2.
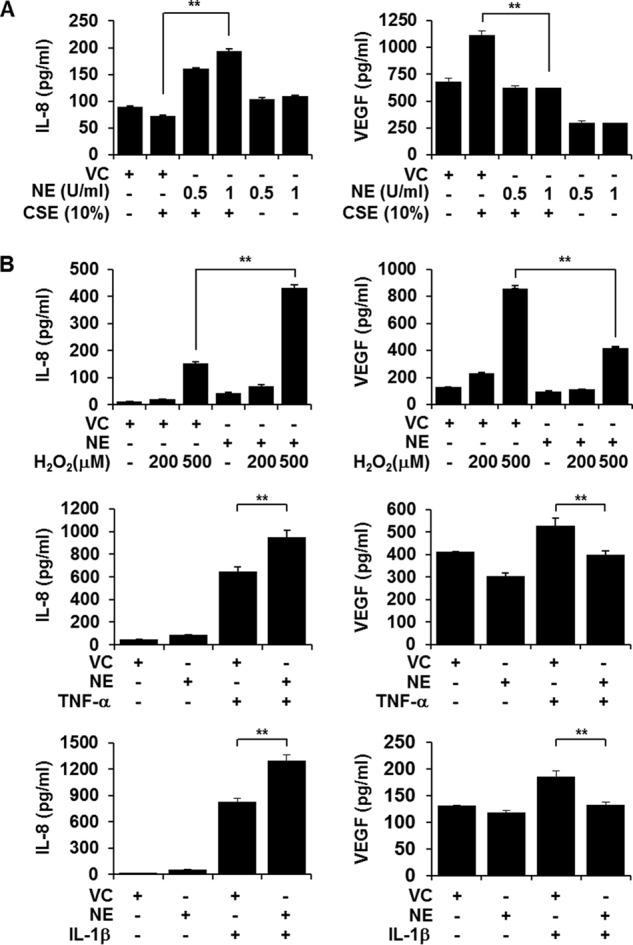
CSE-induced IL-8 and VEGF production is differentially regulated by NE. A, BEAS-2B cells were pretreated with NE (0.5 or 1 unit (U)/ml) or vehicle control (VC) for 4 h and stimulated with CSE for 24 h in the presence or absence of NE or vehicle control. B, cells were treated with NE (1 unit/ml) in the presence or absence of H2O2 (200 or 500 μm), TNF-α (10 ng/ml), or IL-1β (1 ng/ml) for 13 h. IL-8 and VEGF concentrations in media were determined by ELISA. ELISA data represent the mean ± S.D. (error bars) of triplicates. **, p < 0.05. Results are representative of three separate experiments.
Suppression of CSE-induced VEGF Production by NE Is Independent of ERK Pathway compared with NE-induced Augmentation of IL-8 Production, Which Is ERK-dependent
We evaluated the effect of NE treatment on CSE-induced ERK activation. NE treatment accelerated CSE-induced ERK activation (Fig. 3A). Next, the role of ERK activation in mediating the opposite effects of NE on IL-8 and VEGF production was investigated. To evaluate whether accelerated ERK activation by NE is responsible for enhanced IL-8 production, ERK activation was blocked, and IL-8 production was measured after co-treatment with NE and CSE. To block ERK activation, cells were pretreated with two different chemical inhibitors of ERK activation (U0126 and PD98059). Blocking ERK activation inhibited the NE-mediated increase in CSE-induced IL-8 release completely (Fig. 3B), suggesting that the augmenting effect of NE on CSE-induced IL-8 production is mediated through the ERK pathway. In contrast, NE-induced suppression of CSE-mediated VEGF production was not affected by inhibition of ERK activation (Fig. 3C), indicating that NE-induced suppression of CSE-mediated VEGF production is determined by mechanisms other than the ERK pathway.
FIGURE 3.
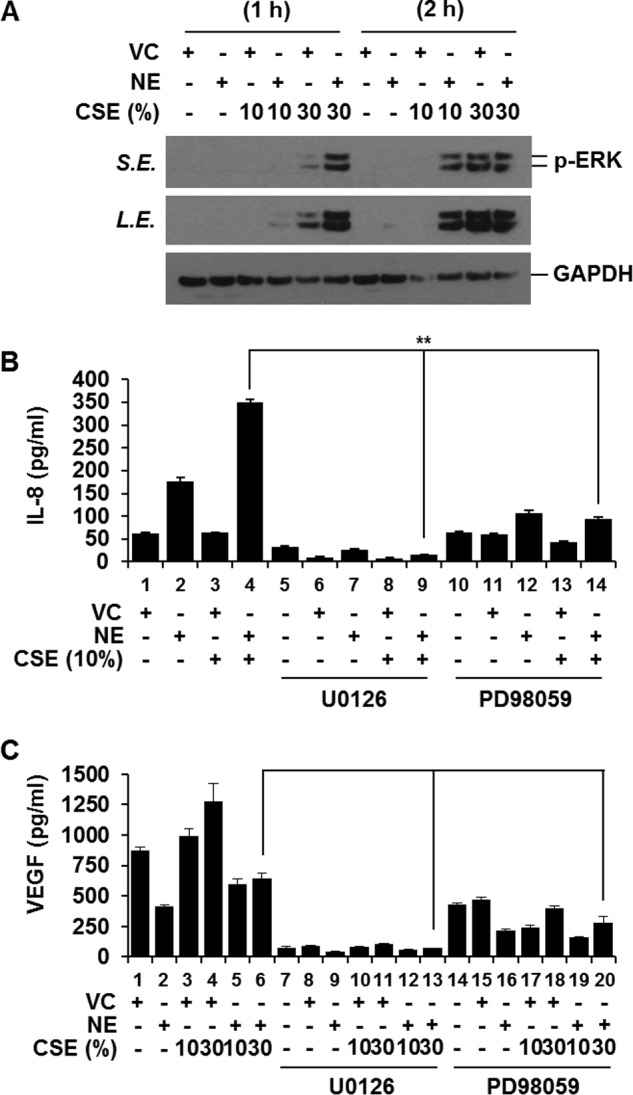
Suppression of CSE-induced VEGF production by NE is independent of ERK pathway; in contrast, NE-induced augmentation of IL-8 production is ERK-dependent. A, co-treatment with NE and CSE further activates ERK. BEAS-2B cells were incubated with vehicle control (VC) or NE in the presence or absence of CSE (10 or 30%) for 1 or 2 h. Total cellular extracts were subjected to Western blot analysis for phosphorylated ERK (p-ERK) and GAPDH. B and C, BEAS-2B cells were pretreated with an ERK inhibitor (20 μm U0126 or 40 μm PD98059) for 1 h and stimulated with CSE and NE in the presence or absence of ERK inhibitors. IL-8 and VEGF concentrations in media were determined by ELISA. ELISA data represent the mean ± S.D. (error bars) of triplicates. **, p < 0.05 (lane 4 versus lane 9 and lane 4 versus lane 14) Results are representative of three separate experiments. S.E., short exposure; L.E., long exposure.
Neither CSE nor NE Affects the Expression of HIF-1α
HIF-1α is a major transcription factor involved in regulation of VEGF gene transcription. Therefore, we tested the effect of CSE and NE on the expression of HIF-1α. HIF-1α was not induced in CSE-stimulated cells (Fig. 4, A and C). NE also did not affect the level of HIF-1α (Fig. 4, B and C), which suggests that another mechanism is involved in NE-induced down-regulation of VEGF.
FIGURE 4.
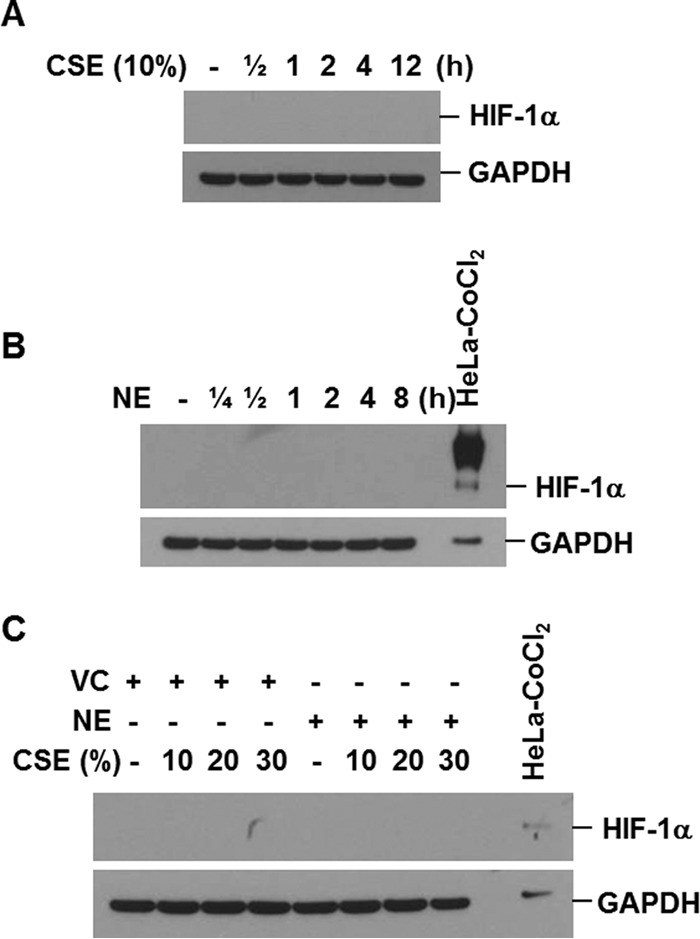
Neither CSE nor NE affects the expression of HIF-1. A and B, BEAS-2B cells were treated with CSE (10%) or NE (1 unit/ml) for the indicated times. C, cells were pretreated with vehicle control (VC) or NE (1 unit/ml) for 4 h and then stimulated with CSE for 4 h. Total cellular extracts were subjected to Western blot analysis for HIF-1α and GAPDH. The extracts of HeLa cells treated with CoCl2 were used as a positive control for HIF-1α expression. Results are representative of three separate experiments.
NE-induced Suppression of Extracellular VEGF Production Is Due to Down-regulation of Intracellular VEGF Protein by Post-translational Modification
Decreased concentration of VEGF in culture supernatant by NE treatment could be due to either decreased intracellular production of VEGF or failure to secrete VEGF. To differentiate these possibilities, we first examined the intracellular VEGF expression after treatment with NE. Intracellular VEGF expression decreased rapidly at 15 min after NE stimulation (Fig. 5A), suggesting that suppression of extracellular VEGF by NE might be due to down-regulation of intracellular VEGF. We next examined the mechanism of intracellular VEGF down-regulation by NE. To evaluate whether down-regulation of intracellular VEGF by NE is regulated at the transcriptional level, mRNA levels of VEGF were assessed by quantitative real time PCR after treatment with CSE, NE, or both. Both CSE and NE treatment increased VEGF mRNA expression. Interestingly, treatment with NE and CSE further enhanced VEGF mRNA expression (Fig. 5B). These results indicate that down-regulation of intracellular VEGF protein by NE may occur by post-translational modification.
FIGURE 5.

NE-induced suppression of extracellular VEGF production is due to down-regulation of intracellular VEGF protein by post-translational modification. A, BEAS-2B cells were treated with NE (1 unit/ml) for the indicated times. Total cellular extracts were subjected to Western blot analysis for VEGF and GAPDH. B, cells were pretreated with NE (1 unit/ml) or vehicle control (VC) for 4 h and then stimulated with CSE (10 or 30%) for 13 h in the presence or absence of NE or vehicle control. RNA isolated from the cells was analyzed by quantitative real time PCR for the expression of VEGF and GAPDH genes. Data represent the mean ± S.D. (error bars) of triplicates. Results are representative of three separate experiments.
Down-regulation of Intracellular VEGF Is Due to Direct Cleavage after Translocation of NE into the Cell
We speculated that NE-induced down-regulation of intracellular VEGF protein expression might be due to intracellular degradation of VEGF protein. Because the lysosome and proteasome are the main sites of intracellular protein degradation, we evaluated whether the down-regulation of intracellular VEGF protein expression by NE treatment is due to degradation via the lysosomal or proteasomal pathway. When lysosomal activity or the proteasome pathway was blocked by chloroquine or MG132, respectively, NE-induced down-regulation of intracellular VEGF was not affected (Fig. 6, A and B). According to the results showing that down-regulation of intracellular VEGF by NE is independent of the lysosomal or proteasomal pathway, the possibility was raised that decreased expression of VEGF was from direct degradation of VEGF by NE in the cytoplasm. To test this possibility, we first examined whether NE enters into normal human lung epithelial cells. BEAS-2B cells were treated with NE, and immunofluorescence staining for internalized NE was performed. Analysis of NE-treated cells showed that NE was introduced into BEAS-2B cells (Fig. 6C). Increased fluorescence intensity after NE treatment indicates that the NE level in cytoplasm had increased. For confirmation, intracellular NE was immunoprecipitated with anti-NE antibody, and the level of NE expression was determined by Western blotting. NE was detected in immunoprecipitates (Fig. 6D), which supports the assumption that lung epithelial cells internalize NE. Another question we had was whether the reduction of VEGF expression at the post-translational level is due to direct cleavage of VEGF by internalized NE. To elucidate this, we assessed whether NE is capable of degrading VEGF in vitro. Recombinant human VEGF-165 was highly sensitive to NE or PPE cleavage. Most of VEGF-165 was degraded after 30 min of incubation with NE or PPE (Fig. 6E). However, interestingly, recombinant human IL-8 was intact (Fig. 6F). IL-8 seems to be resistant to NE cleavage. Taken together, these data suggest that NE enters into the cell to directly cleave VEGF, which results in down-regulation of intracellular VEGF.
FIGURE 6.

Down-regulation of intracellular VEGF is due to direct cleavage after translocation of NE into the cell. A and B, BEAS-2B cells were pretreated with chloroquine (100 μm) or MG132 (10 μm) for 1 h and then stimulated with vehicle control (VC) or NE in the presence or absence of chloroquine or MG132 for the indicated times. Total cellular extracts were subjected to Western blot analysis for VEGF and GAPDH. C, BEAS-2B cells were treated with 20 units/ml NE or vehicle control for 2 h. Immunofluorescence staining for internalized NE was performed. Cells were analyzed using an ECLIPSE TE300 (Nikon) fluorescence microscope. D, cells were treated with NE (10 units/ml) or vehicle control for 1 h. Intracellular NE was immunoprecipitated with anti-NE antibody. The level of NE expression in the immunoprecipitates was determined by Western blot analysis. U937 cell lysates were used as a positive control for NE expression. E and F, recombinant human VEGF-165 (VEGF; 50 ng/ml) or IL-8 (50 ng/ml) was incubated with NE (10 units/ml) in vehicle (50% glycerol, 50% 0.02 m NaOAc (pH 5)) or PPE (10 μg/ml) in media at 37 °C for 30 min. The reaction was stopped by adding 4× sample buffer. Samples were subjected to Western blot analysis for VEGF or IL-8 under reducing conditions. Results are representative of three separate experiments. IP, immunoprecipitation; IB, immunoblotting; IgG LC, IgG light chain; S.E., short exposure; L.E., long exposure; rhVEGF, recombinant human VEGF.
Elastase Treatment Suppresses CSE-induced VEGF, Increases IL-8 in BALF, and Enhances CSE-induced Emphysema in Mice
To confirm the effect of elastase on CSE-induced VEGF/IL-8 production, mice were intratracheally instilled with elastase and CSE as described in Fig. 7A. CSE increased the levels of VEGF and IL-8 in BALF of mice (Fig. 7B). Elastase suppressed CSE-induced VEGF production but increased IL-8 after 4 weeks of intratracheal instillation (Fig. 7B). In addition, when the severity of emphysema was measured by MLI at week 8, mice treated with elastase and CSE had more severe emphysema than mice treated with CSE alone (Fig. 7C). This raises the possibility that the changes of VEGF and IL-8 might precede the histological change.
FIGURE 7.

Effect of elastase on the CSE-induced VEGF and IL-8 production in BALF and emphysema formation in mice. A, experimental protocols for murine model of emphysema. C57BL/6 WT mice were intratracheally instilled with vehicle, elastase, CSE, or elastase plus CSE as described under “Experimental Procedures.” Mice (n = 3 per group) were sacrificed at week 4 after first instillation to collect BALF and at week 8 to isolate lungs for histopathological examination. B, effect of elastase on the levels of VEGF and IL-8 in BALF of CSE-treated mice. The CSE-induced increase in VEGF was suppressed by co-treatment with elastase. In contrast, it increased IL-8. C, effect of elastase on CSE-induced emphysema formation. Paraffin-embedded lung tissue sections were used for H&E staining. The MLIs from four independent sections were evaluated. Elastase enhances CSE-induced emphysema in mice. Data are presented as mean ± S.E. (error bars) (control versus CSE, p = 0.154; control versus elastase, p = 0.118; control versus CSE plus elastase, p < 0.001; CSE versus elastase, p = 0.999; CSE versus CSE plus elastase, p < 0.001; elastase versus CSE plus elastase, p = 0.001).
Discussion
The pathogenesis of COPD is very complex. Inflammation, oxidative stress, proteases, apoptosis, autophagy, and cell senescence are known to be involved in the initiation and progression of COPD. Many studies suggest that all of them are closely interrelated in COPD development, which is not fully understood. To understand the cross-talk among protease, inflammation, and apoptosis, we evaluated the effect of NE on CSE-induced IL-8 production, which has an important role in neutrophilic inflammation, and VEGF, which protects against epithelial cell apoptosis.
The major environmental risk factor for COPD is CS. CS is itself a rich source of oxidants, and it triggers production of oxidants from inflammatory cells (6). CS directly or indirectly through oxidants induces chronic inflammation by releasing inflammatory cytokines such as TNF-α (20) and IL-8 (21). IL-8 released from bronchial epithelial cells by CS recruits neutrophils into the lung and thus further amplifies chronic inflammation. In this study, CSE released IL-8, and it was dependent on ERK activation. This is in accordance with previous reports that showed that the CS-induced inflammation cascade is mediated by activation of the mitogen-activated protein kinase (MAPK) pathway (18, 19). CSE is frequently used as a surrogate for CS in in vitro experiments. CSE is usually generated by the bubbling of cigarette smoke through medium to capture soluble components of the smoke. CS contains more than 4,000 chemicals and additives (22), many of which are volatile. Although in vitro treatment with CSE may not accurately reflect in vivo exposure to CS, reports show that when airway epithelial cells were exposed to volatile CS IL-8 was released into the culture medium via p38 (23). It is well known that neutrophils recruited by the action of IL-8 release NE. The main action of NE is degradation of matrix protein, during which chemotactic fragments are produced (12, 13), resulting in continued accumulation of inflammatory cells and tissue destruction. This implies that NE enhances CS-induced inflammation indirectly via recruitment of inflammatory cells. However, it has not been clear whether NE enhances CS-induced inflammation directly through its effect on the MAPK pathway. In this study, we found that NE augmented CSE-induced IL-8 production via the ERK pathway, suggesting that NE could activate epithelial cells directly via MAPK in addition to its proteolytic activity on extracellular matrix. IL-8 is a well known neutrophil chemotactic factor. CSE-induced IL-8 production mediates recruitment of neutrophils or other granulocytes, and recruited neutrophils secrete several proteinases including elastase. Active elastase enhances IL-8 production by CSE and by other inflammatory cytokines. A vicious cycle causing continued neutrophil accumulation and inflammation seems to exist.
Recently, the importance of apoptosis in COPD pathogenesis has been emphasized. CS induces epithelial cell apoptosis in which decreased VEGF expression is known to be involved. In fact, down-regulation of VEGF is observed in bronchial epithelium of patients with COPD (24). Moreover, lung-targeted VEGF inactivation and VEGF receptor inhibition lead to emphysema and lung cell apoptosis (25, 26), which suggests that VEGF has a protective activity against COPD. Contrary to our expectations, CSE increased VEGF release from bronchial epithelial cells in this study. CSE-induced VEGF release was also observed in human lung fibroblast, suggesting that it is not cell type-specific (27). Then, considering the increased VEGF release from epithelial cells by CSE, how does VEGF down-regulation occur in the lung parenchyma of COPD patients? Interestingly, NE suppressed CSE-induced VEGF release, which implies that the down-regulation of VEGF in COPD patients might be mediated at least partially by NE.
To find the mechanism of down-regulation of VEGF by NE, we initially evaluated HIF-1α. HIF-1α is a major transcription factor involved in regulation of VEGF gene transcription. HIF-1α expression is reduced in the lung tissue of patients with severe COPD, and insufficient HIF-1α has been suggested to cause impaired expression of VEGF (28). However, in this study, HIF-1α was not induced in CSE- or NE-stimulated cells, which suggests that another mechanism(s) is involved in VEGF down-regulation. NE decreased VEGF expression in total cellular protein extract at 15 min after stimulation. Usually, fast degradation of intracellular protein is through the proteasome. However, proteasomal inhibitors (PS-341 and MG132) did not block NE-mediated VEGF degradation. Therefore, we hypothesized that NE that was internalized into epithelial cells is involved in the direct cleavage of VEGF. Immunofluorescence staining and the immunoprecipitation assay for intracellular NE supported this hypothesis. NE was detected in the cytoplasm of normal bronchial epithelial cells after NE incubation. Similar to our data, other neutrophil granule proteins such as PR3 and myeloperoxidase have been reported to be internalized into endothelial cells and to contribute to tissue injury (29). However, the uptake mechanism of NE is still unclear. A recent report suggests that NE enters endosomes of human lung adenocarcinoma cells via a dynamin- and clathrin-dependent, caveolin-1- and flotillin-independent mechanism (30). In addition, we showed that NE fragmented VEGF but not IL-8 in vitro. NE seems to be able to target VEGF in vivo and in vitro for degradation. Although prolonged incubation with NE might damage IL-8 stability, VEGF seems to be more sensitive to NE-mediated proteolytic cleavage. Similar to in vitro cell experiments, elastase treatment increased CSE-induced IL-8; however, it suppressed VEGF production in BALF of CSE-treated mice. These findings suggest that elastase treatment enhances CSE-induced emphysema in mice.
In conclusion, our study reveals a cross-talk between oxidative stress (CSE) and protease (elastase) on IL-8 and VEGF production in lung epithelial cells and in mice. The interactive action of CSE and elastase accelerates the inflammatory response by increasing proinflammatory cytokine production and apoptosis by suppressing survival signaling protein expression, consequently contributing to COPD pathogenesis.
Author Contributions
K.-H. L., C.-H. L., and C.-G. Y. designed the study and wrote the paper. K.-H. L., C.-H. L., J. J., and A.-H. J. performed and analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Jinwoo Lee for critical reading of the manuscript.
The authors declare that they have no conflicts of interest, financial or otherwise, with the contents of this article.
- COPD
- chronic obstructive pulmonary disease
- NE
- neutrophil elastase
- CS
- cigarette smoke
- CSE
- cigarette smoke extract
- HIF
- hypoxia-inducible factor
- PPE
- porcine pancreatic elastase
- BALF
- bronchoalveolar lavage fluid
- MLI
- mean linear intercept.
References
- 1.Vestbo J., Hurd S. S., Agusti A. G., Jones P. W., Vogelmeier C., Anzueto A., Barnes P. J., Fabbri L. M., Martinez F. J., Nishimura M., Stockley R. A., Sin D. D., and Rodriguez-Roisin R. (2012) Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease, GOLD executive summary. Am. J. Respir. Crit. Care Med. 187, 347–365 [DOI] [PubMed] [Google Scholar]
- 2.Gross N. J. (2012) Novel antiinflammatory therapies for COPD. Chest 142, 1300–1307 [DOI] [PubMed] [Google Scholar]
- 3.Abboud R. T., and Vimalanathan S. (2008) Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int. J. Tuberc. Lung Dis. 12, 361–367 [PubMed] [Google Scholar]
- 4.Mak J. C. (2008) Pathogenesis of COPD. Part II. Oxidative-antioxidative imbalance. Int. J. Tuberc. Lung Dis. 12, 368–374 [PubMed] [Google Scholar]
- 5.Roth M. (2008) Pathogenesis of COPD. Part III. Inflammation in COPD. Int. J. Tuberc. Lung Dis. 12, 375–380 [PubMed] [Google Scholar]
- 6.Fischer B. M., Pavlisko E., and Voynow J. A. (2011) Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chron. Obstruct. Pulmon. Dis. 6, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plataki M., Tzortzaki E., Rytila P., Demosthenes M., Koutsopoulos A., and Siafakas N. M. (2006) Apoptotic mechanisms in the pathogenesis of COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 1, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito K., and Barnes P. J. (2009) COPD as a disease of accelerated lung aging. Chest 135, 173–180 [DOI] [PubMed] [Google Scholar]
- 9.O'Donnell R., Breen D., Wilson S., and Djukanovic R. (2006) Inflammatory cells in the airways in COPD. Thorax 61, 448–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deslee G., Adair-Kirk T. L., Betsuyaku T., Woods J. C., Moore C. H., Gierada D. S., Conradi S. H., Atkinson J. J., Toennies H. M., Battaile J. T., Kobayashi D. K., Patterson G. A., Holtzman M. J., and Pierce R. A. (2010) Cigarette smoke induces nucleic-acid oxidation in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 43, 576–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rahman I., and Adcock I. M. (2006) Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 28, 219–242 [DOI] [PubMed] [Google Scholar]
- 12.Hunninghake G. W., Davidson J. M., Rennard S., Szapiel S., Gadek J. E., and Crystal R. G. (1981) Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science 212, 925–927 [DOI] [PubMed] [Google Scholar]
- 13.Weathington N. M., van Houwelingen A. H., Noerager B. D., Jackson P. L., Kraneveld A. D., Galin F. S., Folkerts G., Nijkamp F. P., and Blalock J. E. (2006) A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat. Med. 12, 317–323 [DOI] [PubMed] [Google Scholar]
- 14.Morissette M. C., Parent J., and Milot J. (2009) Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int. J. Chron. Obstruct. Pulmon. Dis. 4, 19–31 [PMC free article] [PubMed] [Google Scholar]
- 15.Thaikoottathil J. V., Martin R. J., Zdunek J., Weinberger A., Rino J. G., and Chu H. W. (2009) Cigarette smoke extract reduces VEGF in primary human airway epithelial cells. Eur. Respir. J. 33, 835–843 [DOI] [PubMed] [Google Scholar]
- 16.Ishii T., Matsuse T., Igarashi H., Masuda M., Teramoto S., and Ouchi Y. (2001) Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L1189–L1195 [DOI] [PubMed] [Google Scholar]
- 17.Lee J. H., Hanaoka M., Kitaguchi Y., Kraskauskas D., Shapiro L., Voelkel N. F., and Taraseviciene-Stewart L. (2012) Imbalance of apoptosis and cell proliferation contributes to the development and persistence of emphysema. Lung 190, 69–82 [DOI] [PubMed] [Google Scholar]
- 18.Moretto N., Facchinetti F., Southworth T., Civelli M., Singh D., and Patacchini R. (2009) α,β-Unsaturated aldehydes contained in cigarette smoke elicit IL-8 release in pulmonary cells through mitogen-activated protein kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, L839–L848 [DOI] [PubMed] [Google Scholar]
- 19.Lau W. K., Chan S. C., Law A. C., Ip M. S., and Mak J. C. (2012) The role of MAPK and Nrf2 pathways in ketanserin-elicited attenuation of cigarette smoke-induced IL-8 production in human bronchial epithelial cells. Toxicol. Sci. 125, 569–577 [DOI] [PubMed] [Google Scholar]
- 20.Sapey E., Wood A. M., Ahmad A., and Stockley R. A. (2010) Tumor necrosis factor-α rs361525 polymorphism is associated with increased local production and downstream inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 182, 192–199 [DOI] [PubMed] [Google Scholar]
- 21.Mio T., Romberger D. J., Thompson A. B., Robbins R. A., Heires A., and Rennard S. I. (1997) Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am. J. Respir. Crit. Care Med. 155, 1770–1776 [DOI] [PubMed] [Google Scholar]
- 22.Baker R. R., Pereira da Silva J. R., and Smith G. (2004) The effect of tobacco ingredients on smoke chemistry. Part 1: flavorings and additives. Food Chem. Toxicol. 42, (suppl.) S3–S37 [DOI] [PubMed] [Google Scholar]
- 23.Beisswenger C., Platz J., Seifart C., Vogelmeier C., and Bals R. (2004) Exposure of differentiated airway epithelial cells to volatile smoke in vitro. Respiration 71, 402–409 [DOI] [PubMed] [Google Scholar]
- 24.Suzuki M., Betsuyaku T., Nagai K., Fuke S., Nasuhara Y., Kaga K., Kondo S., Hamamura I., Hata J., Takahashi H., and Nishimura M. (2008) Decreased airway expression of vascular endothelial growth factor in cigarette smoke-induced emphysema in mice and COPD patients. Inhal. Toxicol. 20, 349–359 [DOI] [PubMed] [Google Scholar]
- 25.Tang K., Rossiter H. B., Wagner P. D., and Breen E. C. (2004) Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J. Appl. Physiol. 97, 1559–1566 [DOI] [PubMed] [Google Scholar]
- 26.Kasahara Y., Tuder R. M., Taraseviciene-Stewart L., Le Cras T. D., Abman S., Hirth P. K., Waltenberger J., and Voelkel N. F. (2000) Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Investig. 106, 1311–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farid M., Kanaji N., Nakanishi M., Gunji Y., Michalski J., Iwasawa S., Ikari J., Wang X., Basma H., Nelson A. J., Liu X., and Rennard S. I. (2013) Smad3 mediates cigarette smoke extract (CSE) induction of VEGF release by human fetal lung fibroblasts. Toxicol. Lett. 220, 126–134 [DOI] [PubMed] [Google Scholar]
- 28.Yasuo M., Mizuno S., Kraskauskas D., Bogaard H. J., Natarajan R., Cool C. D., Zamora M., and Voelkel N. F. (2011) Hypoxia inducible factor-1α in human emphysema lung tissue. Eur. Respir. J. 37, 775–783 [DOI] [PubMed] [Google Scholar]
- 29.Yang J. J., Preston G. A., Pendergraft W. F., Segelmark M., Heeringa P., Hogan S. L., Jennette J. C., and Falk R. J. (2001) Internalization of proteinase 3 is concomitant with endothelial cell apoptosis and internalization of myeloperoxidase with generation of intracellular oxidants. Am. J. Pathol. 158, 581–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregory A. D., Hale P., Perlmutter D. H., and Houghton A. M. (2012) Clathrin pit-mediated endocytosis of neutrophil elastase and cathepsin G by cancer cells. J. Biol. Chem. 287, 35341–35350 [DOI] [PMC free article] [PubMed] [Google Scholar]