

Background: Neurogenesis, the process of generation of new neurons, is reduced in Alzheimer disease (AD).
Results: Ethosuximide (ETH), an anticonvulsant drug, increased neurogenesis, reduced neurodegeneration, and reversed cognitive impairments in a rat model of AD-like phenotypes.
Conclusion: ETH induces neurogenesis, thus reversing AD-like phenotypes.
Significance: ETH could be used as a therapeutic molecule to enhance neurogenesis.
Keywords: cell differentiation, cell proliferation, hippocampus, neural stem cell (NSC), neurodegeneration, neurodegenerative disease, neurogenesis, neuron, neuroprotection, ethosuximide
Abstract
Neurogenesis involves generation of new neurons through finely tuned multistep processes, such as neural stem cell (NSC) proliferation, migration, differentiation, and integration into existing neuronal circuitry in the dentate gyrus of the hippocampus and subventricular zone. Adult hippocampal neurogenesis is involved in cognitive functions and altered in various neurodegenerative disorders, including Alzheimer disease (AD). Ethosuximide (ETH), an anticonvulsant drug is used for the treatment of epileptic seizures. However, the effects of ETH on adult hippocampal neurogenesis and the underlying cellular and molecular mechanism(s) are yet unexplored. Herein, we studied the effects of ETH on rat multipotent NSC proliferation and neuronal differentiation and adult hippocampal neurogenesis in an amyloid β (Aβ) toxin-induced rat model of AD-like phenotypes. ETH potently induced NSC proliferation and neuronal differentiation in the hippocampus-derived NSC in vitro. ETH enhanced NSC proliferation and neuronal differentiation and reduced Aβ toxin-mediated toxicity and neurodegeneration, leading to behavioral recovery in the rat AD model. ETH inhibited Aβ-mediated suppression of neurogenic and Akt/Wnt/β-catenin pathway gene expression in the hippocampus. ETH activated the PI3K·Akt and Wnt·β-catenin transduction pathways that are known to be involved in the regulation of neurogenesis. Inhibition of the PI3K·Akt and Wnt·β-catenin pathways effectively blocked the mitogenic and neurogenic effects of ETH. In silico molecular target prediction docking studies suggest that ETH interacts with Akt, Dkk-1, and GSK-3β. Our findings suggest that ETH stimulates NSC proliferation and differentiation in vitro and adult hippocampal neurogenesis via the PI3K·Akt and Wnt·β-catenin signaling.
Introduction
Neurogenesis, the process of generation of new neurons, occurs throughout life, with the multipotent neural stem cells (NSCs)6 in the neurogenic brain regions, such as the dentate gyrus (1) of the hippocampus and the subventricular zone (SVZ), differentiating into neurons (2–4). This involves proliferation, migration, and differentiation of NSC and integration of newly generated neurons into existing neuronal circuitry (2–4). Neurogenesis plays an important role in the regulation of learning and memory processes and odor discrimination (2, 5). It is negatively modulated by stress, aging, and certain drugs, whereas it is positively affected by neurotrophic factors, environmental enrichment, and exercise (4). Identifying small molecules that induce neurogenesis and understanding the associated cellular and molecular mechanism(s) may prove valuable in the development of regenerative therapies for neurodegenerative and neurological disorders. Alterations in hippocampal neurogenesis lead to impaired learning and memory and other pathophysiological neurodegenerative disease states. Several studies have suggested decreased neurogenesis in neurodegenerative and neurological disorders, such as Parkinson disease, Alzheimer disease (AD), Huntington disease, depression, epilepsy, schizophrenia, and other age-related disorders (2, 6–14). Early onset of AD is characterized by the accumulation of amyloid-β (Aβ) peptides, hippocampal hyperactivity, and alteration in neuronal networks, leading to increased incidence of epileptic seizures and cognitive and synaptic deficits in patients (15–18). Antiepileptic drugs (AEDs) like levetiracetam and lamotrigine are known to reduce overexcitation and synaptic and cognitive deficits in animal models of AD (15, 18). However, a possible role of AEDs in neurogenesis and associated functions has not been fully explored.
Ethosuximide (ETH) is an AED used commonly in the treatment of absence seizures, a type of idiopathic generalized epilepsy in children (19). It effectively attenuates the spike-wave discharges that characterize absence seizures (20, 21), by blocking T-type voltage-gated Ca2+ channels and reducing low threshold Ca2+ currents (22). ETH delays age-related changes and extends life span in Caenorhabditis elegans, suggesting that it may be a potential therapeutic candidate for neurodegenerative disorders and age-related diseases (23). ETH reduces age-dependent toxicity in C. elegans motor neurons against mutant TAR DNA-binding protein-43, involved in pathogenesis of amyotrophic lateral sclerosis (24). It has been found to reduce infarct volume in an ischemic brain injury rat model (25) and to protect neurons in the hippocampal slice cultures against oxygen and glucose deprivation (25). Also, it promotes neuronal differentiation of rat muscle-derived stem cells in vitro (26) and differentiation of neonatal rat forebrain stem cells to microtubule-associated protein-2-positive neural cells (27). Similarly, another AED, valproic acid, reported to enhance proliferation of neural progenitor cells (NPCs) in the DG (27, 28).
In the present study, we hypothesized that ETH may affect the process of adult neurogenesis, and we assessed its effects on NSC proliferation, neuronal differentiation, and survival in vitro and in the hippocampal region of the rat brain. We found that ETH potently induces neurogenesis in vitro and in the adult hippocampus. Furthermore, we found that the drug reverses learning and memory deficits in an Aβ-induced model of AD-like phenotypes by enhancing neurogenesis. We next elucidated the molecular mechanisms underlying ETH-induced neurogenesis and found that the drug acts by activating the PI3K·Akt/Wnt-β·catenin signaling in the rat brain.
Experimental Procedures
Materials
ETH, Aβ, Aβ scrambled peptide, Tris, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, alamar blue, 5-bromo-2-deoxyuridine (BrdU), basic fibroblast growth factor, epidermal growth factor (EGF), serum-free neurobasal medium, N-2 supplement, B-27 supplement, and TRIzol reagent were obtained from Gibco (Rockville, MD). Bovine serum albumin (BSA), succinimide (inactive analog of ETH), poly-l-lysine, rabbit anti-cleaved caspase-3, mouse anti-S100-β, rabbit anti-nestin, mouse anti-S100-β, and rabbit anti-glial fibriliary acidic protein (GFAP) antibodies were procured from Sigma-Aldrich (29). Lipofectamine LTX was procured from Invitrogen. Cell culture products were purchased from Gibco. Mouse anti-BrdU primary antibody was procured from Santa Cruz Biotechnology (29). Monoclonal mouse anti-neuronal nuclei (NeuN) and nitrocellulose membrane were obtained from Chemicon (Millipore, Billerica, MA). Rabbit anti-Sox-2, mouse anti-β-actin, rabbit anti-CNPase, mouse anti-BrdU, rabbit anti-doublecortin (DCX), rabbit anti-TUJ-1, rabbit anti-phospho-PI3K, rabbit anti-PI3K, rabbit anti-phospho AKT, rabbit anti-AKT, mouse anti-phosphohistone H3, rabbit anti-GSK-3β, rabbit anti-phospho-GSK-3α/β, rabbit anti-β-catenin, and rabbit anti-phospho-β-catenin primary antibodies were obtained from Cell Signaling Technology (Danvers, MA). Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies were purchased from Molecular Probes. Primers were procured from Integrated DNA Technologies (Coralville, IA), and SYBR Green was from Applied Biosystems. Anti-fade mounting medium with DAPI was obtained from Vector Laboratories (Vectashield, Vector Laboratories, Burlingame, CA). Culture wares were procured from Nunc (Roskilde, Denmark).
Animals and ETH Treatment
Adult Wistar rats were obtained from the Animal Breeding Colony of the CSIR-Indian Institute of Toxicology Research, Lucknow, and housed under a 12-h light/dark cycle and temperature of 25 ± 2 °C. Animals were provided ad libitum access to the drinking water and pellet diet and handled according to the guidelines laid down by the Institute's Ethical Committee for Animal Experiments.
Adult male rats received a daily single intraperitoneal injection of ETH (125 mg/kg of body weight) for 3 consecutive days from PND 42 to 45 (acute treatment) or 2 weeks from PND 42 to 56 (chronic treatment). This dose was selected on the basis of therapeutically or pharmacologically relevant levels of ETH, as described earlier (25, 30). Rats in the control group received normal saline as vehicle. To analyze the effects of ETH on cell proliferation, rats from both control and ETH-treated groups received daily single intraperitoneal injection of BrdU (50 mg/kg of body weight) for five consecutive days (PND 52–56). Animals were sacrificed 4 h after the last BrdU injection at PND 56, and brains were dissected for immunohistochemical detection of proliferating cells. To evaluate the effect on NSC survival and their differentiation potential in neuron and glial cells, a set of animals was allowed to survive for an additional 3 weeks (up to PND 77) after the last BrdU injection. Animals were sacrificed at PND 77 for double immunofluorescence analysis of BrdU and neuronal or glial markers.
Preparation of Aβ-induced Rat Model of AD-like Phenotypes and ETH Treatment
A rat model of an Aβ-induced AD-like phenotype was prepared as described earlier (31). Rats were anesthetized with ketamine (intraperitoneally, 100 mg/kg of body weight) and xylazine (30 mg/kg of body weight) and fixed in a stereotaxic apparatus. Rats were given a stereotaxic injection of 2 μl of Aβ(1–42) (0.2 μg/μl dissolved in 0.9% normal saline), into both sides of the hippocampus at AP −3.3, L 2.0, V 4.0 (coordinates in mm with respect to bregma), using an autoinjector pump. Two weeks after Aβ administration, rats were treated with ETH (125 mg/kg of body weight intraperitoneally) for 2 weeks (PND 42–56) as per the following experimental plan.
Experimental Groups
Experimental groups were as follows: Group I (sham control) received stereotaxic 2 μl of normal saline as vehicle; Group II (Aβ group) received a stereotaxic intrahippocampal 2-μl Aβ injection; Group III (Aβ + ETH) consisted of Aβ-injected rats that received ETH; Group IV received ETH; Group V (inactive analog of ETH; succinimide) received succinimide (125 mg/kg of body weight intraperitoneally) for 2 weeks (PND 42–56); Group VI (Aβ scrambled peptide) received stereotaxic intrahippocampal injection of 2 μl of Aβ scrambled peptide; Group VII (Aβ scrambled peptide + ETH) received scrambled peptide and ETH; Group VIII (succinimide + ETH) received succinimide and ETH.
A set of rats was sacrificed at PND 56 for the cell proliferation study, whereas another set was sacrificed at PND 77 for the cell survival/neuronal differentiation study in the DG region and SVZ, as described earlier (31).
Primary Hippocampal NSC Culture
NSC was isolated from the hippocampal tissue of embryonic day 12 rat fetuses following our earlier study (32). Hippocampal tissues were dissected in HBSS, minced, and incubated in 0.1% trypsin for 30 min at 37 °C. A single cell suspension was prepared by gentle trituration, and cell viability was determined by trypan blue dye. Cells were plated in serum-free neurobasal medium containing N-2 supplement (1%), B-27 supplement (2%), EGF (10 ng/ml), basic fibroblast growth factor (10 ng/ml), and 1% antibiotic-antimycotic solution. The NSCs were allowed to grow as neurospheres.
BrdU and Alamar Blue Cell Proliferation Assay
BrdU and alamar blue cell proliferation assays were carried out to see the effects of ETH on cell proliferation. NSCs were plated in black bottom 96-well microplates at a density of 10,000 cells/well. The cells were treated with different concentrations of ETH (25, 50, 100, 200, and 400 μm) dissolved in DMSO for 48 h. Vehicle-treated cells served as a control. After treatment, alamar blue was added, and cells were incubated at 37 °C for 2 h. Background florescence was measured by adding 10 μl of alamar blue solution/well in medium having no cells. Alamar blue dye reduction in term of fluorescence intensity was measured at 530-nm excitation and 590-nm emission. Values of background florescence were subtracted from experimental values. Experiments were performed three times, with three replicates per experiment. Relative cell proliferation = (absorbancesample/absorbancecontrol) × 100. Results were expressed in terms of alamar blue reduction as a percentage of control. Similarly, to assess cell proliferation, control and ETH-treated NSC cultures were labeled with BrdU (20 μm) for 12 h before fixing cells with 4% paraformaldehyde and processing for immunofluorescent analysis.
Neurosphere Growth Kinetics Assay
The neurosphere growth kinetics assay was carried out to study the effects of ETH on cell proliferation and neurosphere formation. A hippocampal single cell suspension was plated in a 12-well plate at a density of 50,000 cells/well in neurobasal medium containing B-27, N-2, basic fibroblast growth factor, and EGF. Cultures were treated with ETH (100 μm). The number and size of neurospheres were analyzed using a Nikon Eclipse Ti-S inverted fluorescent microscope equipped with a Nikon Digital Sight Ds-Ri1 CCD camera and NIS Elements BR imaging software (Nikon, Tokyo, Japan) as described earlier (31).
Immunocyto-/Histochemical Analysis of Proliferating Cells in NSC Cultures and in the Brain
Rats were sacrificed at PND 56 by transcardial perfusion, and serial coronal brain section of 40-μm thickness beginning at bregma −3.14 to −5.20 mm through the dorsal hippocampus (dentate gyrus) and 0.70 to −0.80 mm through the SVZ was taken for immunohistochemical analysis of proliferating cells as described earlier (31). The sections/cultures were treated with 2 n HCl for 30 min, followed by neutralization with borate buffer. The endogenous peroxidase activity was inhibited by H2O2, and nonspecific binding sites were blocked with 3% NGS, 0.5% BSA, and 0.1% Triton X-100. The sections were subsequently incubated with mouse anti-BrdU primary antibody (1:500) followed by incubation in peroxidase-linked secondary antibody (1:200). Color was developed with 3–3′-diaminobenzidine and visualized under a light microscope. For immunofluorescence analysis, paraformaldehyde-fixed sections/cultures were incubated with mouse anti-phosphohistone H3 (1:500) and anti-BrdU primary antibodies (1:500), followed by Alexa Fluor-conjugated secondary antibody, and visualized under an inverted fluorescent microscope.
Neuronal Differentiation and Cell Fate Analysis by Double Co-immunofluorescence in the Brain and NSC Cultures
The effects of ETH on neuronal and glial differentiation and cell phenotype of BrdU+ proliferating cells in the hippocampus and SVZ (at PND 77) and NSC cultures were studied by double immunofluorescence analysis following an earlier published method (31). NSC cultures/coronal sections were co-labeled with BrdU/nestin (NSC marker), BrdU/DCX (immature neuron marker), BrdU/NeuN (mature neuron marker), BrdU/GFAP (glial cell marker), Sox-2/GFAP (glial and neural progenitor cell markers), S100-β (glial cell marker), and CNPase (oligodendrocyte marker). Sections were incubated in mouse anti-BrdU (1:500), rabbit anti-DCX (1:200), rat anti-NeuN (1:500), mouse anti-nestin (1:500), rabbit anti-GFAP (1:100), and mouse anti-phosphohistone H3 primary antibodies (1:500), rabbit anti-Sox-2 (1:200), mouse anti-S100-β (1:200), and rabbit anti-CNPase (1:250) for 24 h at 4 °C. Secondary antibodies used were anti-mouse and anti-rabbit Alexa Fluor 488 (1:200) and anti-rabbit, anti-mouse, and anti-rat Alexa Fluor 594 (1:200). Sections were mounted with DAPI containing Hard SetTM anti-fade mounting medium (Vectashield, Vector Laboratories). Slides/cultures were analyzed for fluorescence co-labeling under a Nikon Eclipse Ti-S inverted fluorescent microscope equipped with a Nikon Digital Sight Ds-Ri1 CCD camera and NIS Elements BR imaging software.
Cell Quantification
Unbiased quantification of proliferating and differentiating cells in the DG region of the hippocampus and SVZ was carried out by a person blind to the experimental groups (1, 32, 33). These brain regions were identified at low magnification (10×), and a contour was drawn. Quantification was done in every sixth section, apart by at least 240 μm in one of six series, with a total of 6 sections/rat analyzed as described earlier (1, 32, 33). Each and every BrdU/phosphohistone H3+, BrdU/DCX+, BrdU/NeuN, BrdU/GFAP, BrdU/DCX, BrdU/nestin, Sox-2/GFAP, and nestin/phosphohistone H3 co-labeled cell was counted in each section. BrdU+ cells were counted for BrdU/NeuN, BrdU/GFAP, BrdU/DCX, BrdU/nestin, Sox-2/GFAP, and nestin/phosphohistone H3 co-labeling at a magnification of ×600 in the DG and SVZ. The number of labeled cells was determined by bilaterally counting cells in a total of six sections and averaged for each rat. The total number of labeled cells was determined by multiplying the total number of BrdU+ cells from six sections by the section periodicity (i.e. 6) and reported as the total number of BrdU+ cells co-labeled with phenotype marker per rat.
Gene Expression Analysis by Quantitative RT-PCR
In order to study the expression of genes involved in neurogenesis and PI3K/Akt/Wnt/β-catenin pathways, quantitative RT-PCR analysis was carried out as described earlier (31, 34, 35). The sequences for primers are available upon request.
Western Immunoblot Analysis
After the respective treatments, NSC cultures were lysed with CelLyticTM MT mammalian tissue lysis/extraction reagent. Western blot analysis was performed as described earlier (31, 36). Membranes were blocked and incubated with phospho-PI3K (1:1000), PI3K (1:1000), phospho-Akt (1:500), Akt (1:1000), GSK-3β (1:1000), phospho-GSK-3α/β (1:500), β-catenin (1:1000), phospho-β-catenin (1:1000), and β-actin (1:10,000) primary antibodies. Protein bands were quantified using Scion Image for Windows (National Institutes of Health).
Treatment of NSC with Wnt·β-Catenin Pathway Inhibitor and PI3K·Akt Inhibitor in the Presence and Absence of ETH
To evaluate the specific involvement of ETH-induced activation of the PI3K/Akt/Wnt pathways in NSC proliferation and neuronal differentiation, hippocampus-derived NSC cultures were treated with potent Wnt antagonist recombinant human protein Dkk-1 (100 nm) and PI3K·Akt inhibitor LY294002 (10 nm) in the presence and absence of ETH. After the respective treatments, NSC proliferation was assessed by a BrdU incorporation assay, and neuronal differentiation was assessed by immunocytochemistry. Further, Western blotting was performed to study protein levels of GSK-3α/β, phospho-GSK-3α/β, β-catenin, and phospho-β-catenin.
In Vivo and in Vitro β-Catenin Knockdown
β-catenin siRNA was stereotaxically injected into the hippocampus. Knockdown was done with a pool of target-specific siRNA sequences for β-catenin and scrambled sequences. After knockdown, rats were treated with ETH, and effects on neuronal differentiation were assessed using immunohistochemistry.
Similarly, transient transfection with β-catenin siRNA was carried out in NSC cultures. Transient transfection was performed with 10 nm β-catenin siRNA at a cell confluence of ∼70% using Lipofectamine LTX transfection reagent. After knockdown, effects on neuronal differentiation were assessed using immunohisto-/cytochemistry.
Conditioned Avoidance Response
The effect of ETH on learning and memory ability in an Aβ-induced rat model of AD-like phenotypes was measured following assessment of two-way conditioned avoidance behavior using a shuttle box as described earlier (37, 38).
In Silico Target Prediction Studies
To study the mechanism of the Akt/Wnt/β-catenin pathway-mediated regulation of hippocampal neurogenesis, we performed molecular docking studies with key regulatory enzymes of these pathways as described earlier (31). The structure of ETH was retrieved from the PubChem server and submitted into the PharmMapper for potential human target identification. Protein Data Bank codes of all of the targets were converted into UniProtKB IDs using the UniProt ID Mapping tool. In order to map the molecular targets and predict the pathway components associated with the proteins targeted by ETH, pathway component analysis was carried out using PANTHER (Protein Analysis through Evolutionary Relationship). Molecular docking studies were conducted to identify the ETH orientation within the binding pockets of the human targets involved in various signaling pathways related to cellular differentiation in the brain. A docking study was performed using the CDOCKER module available in the Discovery Studio molecular simulation package (version 3.5; Accelrys, San Diego, CA). The virtual hits were selected based on two scoring functions. Scoring is made by adding the usual intraligand energy terms (bonded and non-bonded contributions) and protein-ligand non-bonded interactions. Each of the compounds was minimized with a CharmM force field to allow for generation and analysis of a wide range of molecular simulations.
Statistical Analysis
The mean significant difference in the experimental groups was determined using one-way analysis of variance. Comparison of least significant differences among two groups was determined by taking t values for error and keeping degrees of freedom at the 5% level of significance and 95% confidence interval. p values of 0.05 were considered to be statistically significant.
Results
ETH Enhances Hippocampus-derived NSC Proliferation and Neurosphere Formation in Vitro
We carried out an alamar blue cell proliferation assay, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, and neurosphere growth kinetics in order to assess the effects of ETH on proliferation and viability of NSC derived from the hippocampus. NSC cultures were incubated with different concentrations of ETH (25, 50, 100, 150, 200, and 400 μm) for 48 h. ETH significantly enhanced NSC proliferation at 50, 100, and 150 μm in the alamar blue assay (Fig. 1A). Among the concentrations used, 100 μm ETH showed the highest NSC proliferation, whereas 25 μm had no effect on proliferation. The higher doses (200 and 400 μm) of ETH caused significantly decreased NSC proliferation. Similarly, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay also suggested that lower concentrations of ETH (50 and 100 μm) significantly increased NSC proliferation/viability, whereas higher doses (>150 μm) caused a decrease in NSC proliferation (Fig. 1B). The effects of ETH on NSC proliferation were further confirmed by BrdU immunocytochemistry. Quantification of BrdU+ cells suggested a significant increase in NSC proliferation by 100 μm ETH (Fig. 1C), whereas 200 and 400 μm ETH decreased cell proliferation (data not shown). These findings showed that the low concentration of ETH induces proliferation of NSC, whereas higher concentrations of ETH are cytotoxic. Further, we performed a neurosphere formation assay in order to assess whether ETH has any effect on the number of hippocampal multipotent NSCs. Neurospheres are free floating spherical clusters of several multipotent NSCs, formed in the presence of specific mitotic growth factors in vitro. The number and size of clonal neurospheres in vitro is the measure of absolute putative NSC in vivo. Thus, we analyzed the gross morphology, size, and number of neurospheres in control and ETH-treated NSC cultures (Fig. 1D). ETH significantly increased the number and size of primary and secondary clonal neurospheres as compared with control (Fig. 1D). These results suggest that ETH treatment increases the number of multipotent NSCs and hence neurosphere formation.
FIGURE 1.

ETH enhances proliferation of the hippocampus-derived NSC and increases the number and size of primary neurospheres. A and B, NSCs derived from the hippocampus were cultured and treated with various concentrations of ETH for 48 h, and cell proliferation was studied. Values are expressed as mean ± S.E. (error bars) (n = 3). *, p < 0.05 versus control. C, BrdU immunocytochemistry in NSC cultures and BrdU-immunopositive cell quantification in the NSC cultures treated with ETH. Shown is a graphical representation of the number of BrdU+ cells labeled with the nuclear stain DAPI. Scale bar, 20 μm. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control. D, representative phase-contrast photomicrographs of primary neurospheres derived from the hippocampus NSC treated with 100 μm ETH. Shown is a quantification of the number of neurospheres and area of neurospheres. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control. E, representative immunofluorescence images of hippocampus-derived NSCs co-labeled with BrdU and nestin (neural stem cell marker). Shown is a graphical representation of the number of nestin/BrdU+-co-labeled cells with the nuclear stain DAPI. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control. Scale bar, 20 μm. F, quantitative real-time PCR analysis was performed for relative mRNA expression of nestin and normalized to β-actin. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control.
We next studied co-expression of BrdU with nestin in NSC culture. Nestin, an intermediate filament protein, is a molecular marker for multipotent NSC. Nestin is required for proliferation and self-renewal of NSC (39). The number of BrdU and nestin-co-labeled cells was significantly up-regulated by ETH treatment in NSC culture (Fig. 1E). ETH significantly enhanced the mRNA expression of nestin as compared with control (Fig. 1F). These findings demonstrated the NSC proliferation-enhancing potential of ETH in vitro.
ETH Enhances NSC Proliferation and Neuronal Differentiation in the Hippocampus of the Adult Rats
In order to study the effects of chronic ETH treatment on NSC proliferation, we counted BrdU+ cells in the SVZ and DG region of the hippocampus. The BrdU-labeled proliferating cells were visualized by immunohistochemistry. BrdU+ cells were of varying shape, densely stained, and mostly located in the small groups of two or more in the DG region of the hippocampus and SVZ (Fig. 2, A and B). BrdU+ cells were found located in the granular cell layer (GCL) and hilus regions of the DG. The number of BrdU+ cells in the DG and SVZ was significantly increased in rats chronically treated with ETH for 2 weeks, compared with control (Fig. 2, A–C). The total number of BrdU+ cells in the DG and SVZ was not significantly altered by acute treatment of ETH for 3 days, compared with control (data not shown).
FIGURE 2.

ETH enhances proliferation and self-renewal of NSC in the hippocampus and SVZ of adult rats. A, photomicrographs showing immunostaining of BrdU+ cells in the dentate gyrus (1) region of the hippocampus. BrdU+ cells were mostly evident in the hilus and GCL regions, whereas a few BrdU+ cells were also present in the molecular layer (ML) of the DG in both control and ETH treated groups. Arrows, BrdU+ cells. Inset, typical morphology of darkly stained, irregular shaped, and compact BrdU+ nuclei. SPB, suprapyramidal blade; IPB, infrapyramidal blade. Scale bar, 100 μm (A) and 10 μm (inset). B, BrdU+ cells in the SVZ of control and ETH-treated rats. ETH treatment increased the number of BrdU+ cells and thickening of the SVZ lining containing NSC. C, quantitative analysis of the number of BrdU+ cells in the hippocampus and SVZ. Values are expressed as mean ± S.E. (error bars) (n = 6 rats/group). *, p < 0.05 versus control. D, photomicrographs showing immune co-labeling of the NSC marker (green; nestin) with the mitosis-specific marker (red; phosphohistone H3) and DAPI in the hippocampus. Scale bar, 100 μm. E, quantitative analysis of hippocampal sections suggested a significant increase of nestin/phosphohistone H3+ cells in ETH-treated rats as compared to control. Values are expressed as mean ± S.E. (n = 6 rats/group). *, p < 0.05 versus control.
Next, we studied the effects of ETH on phosphorylation of histone H3, a nucleosomal core protein. Phosphorylation of histone H3 (Ser-10) is a crucial mitotic event and a marker of proliferating cells (40). Thus, we carried out immune co-labeling of NESTIN with phosphohistone. We found that the number of nestin/phosphohistone H3+ cells in the DG region was significantly enhanced by chronic ETH treatment, suggesting the presence of cells undergoing mitosis (Fig. 2, D and E). ETH also significantly enhanced nestin mRNA expression in the hippocampus (data not shown). These studies suggested that ETH treatment increases cell proliferation in the rat brain.
Further, the effects of ETH on the pool of NSCs and its fate/phenotypes were studied by co-labeling of neuronal marker (NeuN) with BrdU and the NPC marker (Sox-2) with GFAP. ETH treatment enhanced the number of Sox-2+ cells; however, no significant difference was observed in the Sox-2/GFAP+-co-labeled cell population (Fig. 3, A and B). These results suggest that ETH significantly induced the population of NPCs, whereas it showed no effect on glial population. Effects of ETH on the population of neuron (NeuN), glial cells (S100-β) and oligodendrocytes (CNPase) were studied. The number of BrdU/NeuN+ cells was enhanced by ETH treatment (Fig. 3, C and D), but no significant changes were observed in the number of S100-β+ (Fig. 3, E and F) and CNPase+ cells (Fig. 3, G and H) in the hippocampus. These results suggest that ETH treatment significantly enhanced neuronal differentiation but had no significant effects on glial and oligodendrocyte differentiation of proliferating cells.
FIGURE 3.

ETH enhances the NPC pool and neuronal differentiation in the hippocampus of adult rats. A, photomicrographs showing immunostaining of Sox-2+ cells (red) co-labeled with GFAP (green) and DAPI (blue) in the dentate gyrus region of the hippocampus. Most of the Sox-2/GFAP+ cells were found in the hilus and GCL regions, whereas a few Sox-2/GFAP+ cells were also present in the molecular layer (ML) of the DG. Arrows, Sox-2/GFAP+-co-labeled cells. Inset, higher magnification of Sox-2+ cells and Sox-2/GFAP-co-labeled cells. Scale bar, 100 μm (A) and 10 μm (inset). B, quantitative analysis of the number of Sox-2+ cells and Sox-2/GFAP+-co-labeled cells. Values are expressed as mean ± S.E. (error bars) (n = 6 rats/group). *, p < 0.05 versus control. C, double immunofluorescence images of matured neurons co-labeled with NeuN (red; mature neuronal nuclei marker) and BrdU (green) in the DG region of the hippocampus. Inset, higher magnification images. Scale bar, 100 μm. D, quantitative analysis of NeuN/BrdU-co-labeled cells in the hippocampus. E, images showing immunostaining of the astrocyte-specific marker (S100-β) in the hippocampus. Scale bar, 100 μm. F, quantitative analysis of S100-β+ cells in the hippocampus. G, images showing immunostaining of the oligodendrocyte-specific marker (CNPase) in the hippocampus. Scale bar, 100 μm. H, quantitative analysis of CNPase+ cells in the hippocampus.
ETH Induces Neuronal Differentiation and Reverses Aβ-mediated Inhibitory Effects on the Hippocampal Neurogenesis
BrdU+-proliferating new born cells in the DG have different fates, where they may differentiate into neurons or glia. Thus, we studied the effects of ETH treatment on cell fate/phenotypes of newly born cells in the DG, by co-labeling cells with BrdU/DCX (a marker for immature newborn neurons), BrdU/NeuN (a marker of mature neurons) and BrdU/GFAP (a marker for glial cells). Several studies have implicated reduced adult hippocampal neurogenesis in the pathogenesis of AD. We and others have shown previously that the pathogenic protein in AD brain, Aβ, reduces adult hippocampal neurogenesis in rodents (9, 31, 41). Next, we studied the effects of ETH treatment on Aβ-mediated inhibitory effects on the hippocampal neurogenesis in an Aβ-induced rat model of AD like phenotypes. We found that stereotaxic intrahippocampal injection of Aβ caused significantly decreased proliferation of NSC in the DG (Fig. 4, A and B). The number of BrdU-labeled cells was significantly reduced by Aβ in the hippocampus. ETH treatment significantly enhanced the number of BrdU+ cells (Fig. 4, A and B). Interestingly, Aβ scrambled peptide and succinimide; an inactive analog of ETH showed no significant effect on the number of BrdU-labeled cells (Fig. 4, A and B). Further, we studied the effects of ETH on neuronal differentiation in the hippocampus. We found that most of the BrdU/DCX co-localized cells were present in the GCL of the DG in all of the groups. A significant inducing effect of ETH treatment on newborn neuron population was observed (data not shown). ETH significantly increased the number of BrdU/DCX-co-labeled cells as compared with control rats (data not shown). These results indicated that ETH increases neuronal differentiation of newborn proliferating cells. Interestingly, we observed that intrahippocampal injection of Aβ significantly inhibited neuronal differentiation (data not shown). The number of BrdU/DCX-co-labeled cells was significantly reduced in Aβ-injected rats as compared with the control group. ETH treatment caused significantly increased neuronal differentiation, as evident from the enhanced number of BrdU/DCX-co-labeled cells in the DG in the rat model of AD-like phenotypes (data not shown). Thus, ETH treatment was found to significantly enhance neuronal differentiation and reduce Aβ-mediated inhibitory effects on neurogenesis.
FIGURE 4.

ETH ameliorates Aβ-mediated impaired hippocampal neurogenesis. A, photomicrographs showing immunostaining of BrdU+ cells in the DG region of the hippocampus. Rats were given a single stereotaxic injection of Aβ in the hippocampus. After 1 week of Aβ injection, rats were treated with ETH. ML, molecular layer. Scale bar, 100 μm. B, quantification of BrdU+ cells in the hippocampus. Values are expressed as mean ± S.E. (error bars) (n = 6 rats/group). *, p < 0.05 versus control. C, double immunofluorescence analysis of immature neurons co-labeled with DCX (red; marker for immature neurons) and BrdU (green) in the DG region of the hippocampus. Scale bar, 20 μm. D, quantitative analysis of DCX/BrdU-co-labeled cells suggests that ETH significantly enhanced neuronal differentiation in the Aβ + ETH group, which was inhibited by Aβ. Values are expressed as mean ± S.E. (n = 6 rats/group). *, p < 0.05 versus control. E, double immunofluorescence analysis of matured neurons co-labeled with NeuN (red; mature neuronal nuclei marker) and BrdU (green) in the DG region of the hippocampus. Scale bar, 100 μm. F, quantitative analysis of NeuN/BrdU-co-labeled cells in the hippocampus. Values are expressed as mean ± S.E. (n = 6 rats/group). *, p < 0.05 versus control.
To study the effects of ETH treatment on long term survival of newborn neurons, we carried out co-localization of BrdU with NeuN. We found that most of the BrdU+ cells were co-localized with NeuN mainly in the GCL (Fig. 4, C and D). The number of BrdU/NeuN-co-labeled cells was significantly increased in ETH-treated rats as compared with control rats, suggesting that most of the cells that incorporated BrdU in the GCL differentiated into mature neurons (Fig. 4, C and D). Aβ significantly reduced the number of BrdU/NeuN-positive cells, that was prevented by ETH treatment (Fig. 4, C and D). Aβ scrambled peptide and succinimide showed no significant effect on neuronal differentiation (Fig. 4, C and D). These results suggested that ETH treatment induces neuronal differentiation and long term survival of new born neurons.
ETH Reduces Aβ-induced Neuronal Degeneration in the Hippocampus
Next, we studied the effects of ETH on Aβ-induced hippocampal neuronal degeneration/apoptosis by fluoro-jade B staining and activated caspase-3 co-labeling with BrdU. ETH significantly reduced the number of Aβ-mediated enhanced fluoro-jade B+ cells in the hippocampus (Fig. 5, A and B). Further, ETH also reduced the number of Aβ-mediated enhanced apoptotic cells co-labeled with BrdU (Fig. 5, C and D). More interestingly, we found that scrambled peptide of Aβ and inactive analog of ETH (succinimide) had no significant effects on fluoro-jade B+ and BrdU/CC3+ cells (Fig. 5, A–D). These results suggest that ETH ameliorates Aβ-induced neuronal degeneration and apoptosis in the hippocampus.
FIGURE 5.

ETH provides neuroprotection against Aβ-induced neurodegeneration. A, photomicrographs showing labeling of degenerating neuronal cells by fluoro-jade B (green) in the DG region of the hippocampus. Rats were given a single stereotaxic injection of Aβ and scrambled Aβ in the hippocampus. After 2 weeks of Aβ injection, rats were treated with ETH and an inactive analog of ETH (succinimide). ML, molecular layer. Scale bar, 20 μm. B, quantification of fluoro-jade B+ cells in the hippocampus. Values are expressed as mean ± S.E. (error bars) (n = 6 rats/group). *, p < 0.05 versus control. C, double immunofluorescence analysis of apoptotic cells co-labeled with activated cleaved caspase-3 (CC-3) (red; marker for apoptotic cells) and BrdU (green) in the hippocampus. Scale bar, 20 μm. D, quantitative analysis of BrdU/cleaved caspase-3-co-labeled cells suggests that ETH significantly reduced apoptotic cells that were enhanced due to Aβ.
ETH Enhances Expression of Neurogenic and the Wnt·β-catenin Pathway Genes in the Hippocampus of a Rat Model of AD-like Phenotypes
The Wnt·β-catenin signaling plays an important role in the regulation of adult hippocampal neurogenesis and self-renewal of NSC/progenitor cells (42–44). The canonical Wnt·β-catenin signaling has been implicated in the pathophysiology of AD and other neurodegenerative diseases (31). We assessed the effects of ETH treatment on mRNA expression of neurogenic and the Wnt pathway genes in the hippocampus of Aβ-injected rats, by quantitative RT-PCR. Several neurogenic genes and transcription factors, such as DCX, β-tubulin, nestin, neurogenin, neuroligin, Pax-6, Stat-3, and Neuro-D1, regulate neurogenesis (31). ETH treatment significantly up-regulated the expression of these genes in the hippocampus (Fig. 6A). Interestingly, Aβ caused a decrease in the expression of these neurogenic genes, and this effect of Aβ was reversed by ETH (Fig. 6A).
FIGURE 6.

ETH increases the expression of neurogenic and Wnt·β-catenin pathway genes in the hippocampal region of Aβ treated rats. A and B, quantitative real-time PCR analysis was performed for relative mRNA expression of neurogenic genes and Wnt·β-catenin pathway genes in the hippocampus. β-Actin served as a housekeeping gene for normalization. Values are expressed as mean ± S.E. (error bars) (n = 6 rats/group). *, p < 0.05 versus control.
Next, we assessed the mRNA expression of the regulatory molecules, receptors, and transcription factors of the Wnt pathway. ETH enhanced the expression of Wnt1, Wnt3, Dishevelled, Wnt receptors (Frizzled 1 and LRP-6), Axin-1, LEF-1, β-catenin, APC, and cyclin-D1, as compared with control (Fig. 6B). However, the expression of Wnt5 was not affected (data not shown). On the other hand, ETH caused significantly decreased expression of negative regulatory genes of the Wnt pathway, Dkk-1 and GSK-3β, whereas the expression of Wif-1 was not changed. Dkk-1 is a negative regulator of the Wnt pathway and acts through inhibition of LRP-5/6. Aβ treatment was found to inhibit the expression of the genes involved in regulation of the Wnt pathway in the hippocampus (Fig. 6B). Interestingly, ETH significantly blocked the Aβ-mediated inhibitory effects on the expression of these genes in the hippocampus. These results suggest that ETH inhibits the Aβ-mediated alterations in the expression of the Wnt pathway genes.
ETH Induces Phosphorylation of GSK-3β and Inhibits β-Catenin Phosphorylation, and Inhibition of the Wnt Pathway Blocks ETH-induced NSC Proliferation and Differentiation
β-Catenin and GSK-3β are critical regulators of the Wnt·β-catenin canonical pathway. In the absence of Wnt ligands, GSK-3β phosphorylates cytoplasmic β-catenin for ubiquitin-mediated proteasomal degradation. In the presence of Wnt ligands, cell surface receptors become activated, leading to inhibition of GSK-3β and decreased phosphorylation of cytoplasmic β-catenin. Decreased phosphorylation of β-catenin results in its increased nuclear translocation and activation of the Wnt pathway. We found that a non-cytotoxic concentration of ETH (100 μm) significantly increased GSK-3β phosphorylation and inhibited β-catenin phosphorylation in a time-dependent manner in a hippocampus-derived NSC culture (Fig. 7, A–D). Inhibition of the Wnt pathway through Dkk-1 (which inhibits interaction of Wnt with LRP-5/6) reversed ETH-induced effects on GSK-3β and β-catenin phosphorylation (Fig. 8, A–C).
FIGURE 7.

ETH activates the Wnt·β-catenin pathway time-dependently in NSCs. A–D, after treatment of NSC culture with ETH (100 μm) at various time points (0, 3, 6, 12, 24, 48, and 72 h), protein levels of GSK-3β, phospho-GSK-3β, β-catenin, and phospho-β-catenin were studied by Western blot. Values are expressed as mean ± S.E. (error bars) (n = 3). *, p < 0.05 versus control.
FIGURE 8.

Pharmacological inhibition of the Wnt pathway reduces ETH-mediated stimulatory effects on NSC proliferation and neuronal differentiation in culture. NSC cultures were treated with the Wnt pathway inhibitor Dkk-1 in the presence and absence of ETH. A–C, protein levels of β-catenin, phospho-β-catenin, GSK-3α/β, and phospho-GSK-3α/β in the hippocampus. Values are expressed as mean ± S.E. (error bars) (n = 3). *, p < 0.05 versus control. D and E, Dkk-1 protein reduced the number of BrdU+ cells, thus inhibiting NSC proliferation that was induced by ETH treatment. F and G, Dkk-1 protein blocked the neuronal differentiation (β-tubulin+ cells)-enhancing potential of ETH in the hippocampus-derived NSC culture. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control.
We next studied the role of the Wnt·β-catenin pathway inhibition in the ETH-mediated increase in the proliferation and neuronal differentiation of hippocampus-derived NSC in vitro. The NSC cultures were treated with Dkk-1 protein in the presence and absence of ETH, and the effects on proliferation and differentiation were studied through immunocytochemistry. Dkk-1 significantly reduced the number of BrdU-positive cells and β-tubulin-positive neurons in cultures, suggesting reduced NSC proliferation (Fig. 8, D and E) and neuronal differentiation (Fig. 8, F and G). Interestingly, co-treatment of Dkk-1 significantly inhibited NSC proliferation and neuronal differentiation-enhancing effects of ETH via the Wnt·β-catenin pathway.
ETH Activates the PI3K·Akt Signal Transduction Pathway in Adult Hippocampal NSCs in Vitro
The PI3K·Akt pathway is involved in the regulation of neurogenesis in the hippocampus and other regions of the brain (45–47). Further, GSK-3β inhibition and the Wnt·β-catenin pathway activation through PI3K·Akt is very well established (45, 48). Therefore, to determine whether ETH induces hippocampal neurogenesis through the PI3K·Akt pathway, mRNA expression and phosphorylation of PI3K (Tyr-458) and Akt (Thr-308) were analyzed in an adult hippocampal NSC culture in vitro. ETH significantly enhanced mRNA expression of PI3K and Akt (Fig. 9A). Next, the basal levels of PI3K and Akt and their phosphorylation were evaluated following ETH treatment. Basal levels of PI3K and Akt were not altered by ETH in cultured hippocampal NSC (Fig. 9, B–D). Interestingly, levels of phosphorylated PI3K and Akt were significantly up-regulated in NSC following ETH treatment. These results suggested that the PI3K·Akt pathway is activated by ETH in cultured hippocampal NSCs (Fig. 9, B and D).
FIGURE 9.

ETH activates the PI3K and Akt pathway and enhances NSC proliferation and differentiation. A, ETH treatment enhanced the mRNA expression of PI3K and Akt genes in NSC cultures. Values are expressed as mean ± S.E. (error bars) (n = 3). *, p < 0.05 versus control. B–D, Western blot analysis indicated that ETH enhanced the phosphorylation of PI3K and Akt as compared with control. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control E, NSC cultures were treated with PI3K·Akt inhibitor (LY294002) in the presence and absence of ETH. F, graphical representation of the number of Tuj-1-positive neuronal cells co-labeled with the nuclear stain DAPI. LY294002 inhibited the ETH-induced neuronal differentiation. Scale bar, 20 μm. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control. G and H, Western blot analysis of GSK-3α/β and phospho-GSK-3α/β protein levels. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control.
Blockade of the PI3K·Akt Pathway Inhibits ETH-induced Hippocampal NSC Neuronal Differentiation
We further studied whether specific activation of the PI3K·Akt signaling pathway mediates the action of ETH on neuronal differentiation in hippocampal NSCs in culture. NSC cultures were treated with ETH in the presence and absence of LY294002, an inhibitor of the PI3K·Akt pathway. ETH enhanced the number of Tuj-1-positive neurons in NSC cultures (Fig. 9, E and F). Pretreatment with the Akt inhibitor resulted in a significantly reduced number of Tuj-1-positive neurons. Interestingly, Akt inhibitor significantly blocked the neuronal differentiation-enhancing potential of ETH by reducing the number of Tuj-1-positive neurons (Fig. 9, E and F). Further, ETH significantly enhanced the phosphorylation of GSK-3β, which was significantly blocked by LY294002 (Fig. 9, G and H). These results suggested that the PI3K·Akt pathway is involved in the stimulatory effects of ETH on neural differentiation of NSC.
Knockdown of β-Catenin in the Hippocampus and in NSC Culture Reduces ETH-induced Neuronal Differentiation
To examine ETH-mediated neuronal differentiation via activation and specific involvement of the Wnt·β-catenin pathway, genetic knockdown of β-catenin was carried out both in vitro and in vivo. β-Catenin-mediated Wnt signaling is involved in NSC proliferation, fate determination, and neuronal differentiation during development (49, 50). After stereotaxic injection of β-catenin siRNA in the hippocampus, the fate of NSC was studied immunohistochemically by co-labeling of BrdU with NeuN (Fig. 10A). We found that knockdown of β-catenin caused an inhibitory effect on the ETH-induced increase in the number of BrdU/NeuN-co-labeled cells (Fig. 10B). Similarly, in vitro studies indicated that β-catenin knockdown suppresses neuronal differentiation in ETH-treated NSC cultures (Fig. 10, C and D). Scrambled β-catenin siRNA showed no significant effect on neuronal differentiation. Together, the in vitro and in vivo results suggested that ETH promotes neuronal differentiation via the β-catenin-mediated signaling.
FIGURE 10.

Genetic inhibition of the Wnt pathway reduces ETH-mediated stimulatory effects of neuronal differentiation in the hippocampus and NSC cultures, and ETH reverses learning and memory deficits in an Aβ-induced model of AD-like phenotypes. A and B, β-catenin was knocked down by stereotaxic injection of β-catenin siRNA directly into the hippocampus of control and ETH-treated rats. β-Catenin knockdown significantly reduced the number of BrdU/NeuN+ cells in ETH-treated rats. Scale bar, 100 μm. Values are expressed as mean ± S.E. (error bars) (n = 6). *, p < 0.05 versus control. C and D, NSC cultures were transiently transfected with β-catenin siRNA and then immune co-labeled with BrdU/β-tubulin. Scale bar, 100 μm. Values are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus control. E, the cognitive ability (learning and memory) of the control and ETH-, Aβ-, and Aβ + ETH-treated rats was measured following assessment of two-way conditioned avoidance behavior. ETH significantly reversed the Aβ-induced deficits in learning and memory as compared with control rats. Values are expressed as mean ± S.E. (n = 6). *, p < 0.05 versus control.
ETH Reverses Aβ-mediated Reduced Hippocampal Neurogenesis and Learning and Memory in the Rat Model of AD-like Phenotypes
Prior studies have suggested that impaired neurogenesis in the hippocampus plays an important role in the pathogenesis of AD (2, 9, 10). Hippocampal neurogenesis is functionally involved in learning and memory processes throughout life (2). We observed that ETH stimulates hippocampal neurogenesis via activation of the PI3K/Akt/Wnt/β-catenin pathways. Further, we found that intrahippocampal stereotaxic injection of Aβ significantly inhibited NSC proliferation (Fig. 4, A and B) and neuronal differentiation (Fig. 4, C and D). Because neurogenesis in the hippocampus regulates learning and memory, we assessed the cognitive functions in the rat model of AD-like phenotypes following ETH treatment (Fig. 10E). Whereas Aβ alone caused significantly decreased learning and memory abilities, treatment with ETH resulted in reversal of Aβ-induced cognitive dysfunction in the rats (Fig. 10E). These observations suggested that ETH improves Aβ-mediated reduced neurogenesis and cognitive deficits.
In Silico Prediction of Molecular Targets of ETH; ETH Interacts with Akt, Dkk-1, and GSK-3β
Our experimental studies suggested that ETH up-regulates mRNA expression of genes involved in Akt-Wnt-β-catenin pathway. To understand the molecular mechanism underlying activation of these pathways by ETH, we performed detailed computational molecular docking studies with various key regulatory enzymes in the Akt-Wnt-β-catenin pathway. PharmMapper analysis identified potential target candidates for the given ligand ETH using a pharmacophore mapping approach. Different model receptors from the Protein Data Bank were extracted based on the fixed score matrix to measure its score level among all of the scores of the pharmacophore. Of 300 potential human targets, molecular targets of ETH involved in the Wnt signaling pathways were Akt, GSK-3β, and Dkk-1 with Protein Data Bank codes 3CQW, 1Q5K, and 3S8V, respectively (Fig. 11). We also found plausible binding modes of ETH in the active sites of these molecular targets (Fig. 11). However, no interaction of ETH was observed in the functional sites of Axin, β-catenin, and ICAT (data not shown).
FIGURE 11.

In silico prediction of ETH targets in the Akt/Wnt/β-catenin pathways. In silico molecular docking studies suggest several targets of ETH in the Akt/Wnt/β-catenin pathways. A–C are as described in the key.
Akt regulates many processes, including metabolism, proliferation, cell survival, growth, and neurogenesis. In Akt and ETH interaction, a binding sphere with a radius of ∼14.5 Å was defined around Akt1 molecule binding site from the three-dimensional coordinate file obtained from the Protein Data Bank. We observed the interaction of ETH with Leu-156 amino acid residue, which is involved in the hydrogen bond formation. The best binding pose of Akt1 and ETH complex has CDOCKER energy of −14.17 kcal/mol and CDOCKER interaction energy of −20.51 kcal/mol. Fig. 9 shows ETH in the functional site of Akt1.
Next, we studied the binding efficiency of ETH with GSK-3β. We defined a binding sphere with radius ∼15 Å around the lipid-binding pocket of GSK-3β. ETH showed binding affinity with GSK-3β with CDOCKER energy of −20.20 kcal/mol and CDOCKER energy of −20.55 kcal/mol in the GSK-3β binding cavity. Fig. 11 shows ETH interaction in the functional site of GSK-3β. In the best binding pose, selected on the basis of CDOCKER energy, hydrogen bonds were formed between ETH and Val-135.
Dkk-1 is a secreted glycoprotein that binds with LRP6 and inhibits the Wnt signaling by blocking Wnt-mediated Frizzled·LRP complex formation. In our study, we found up-regulation of Wnt, Frizzled, and LRP-5/6 mRNA expression following ETH treatment. These findings motivated us to investigate whether the up-regulation of the Wnt signaling cascade is mediated by inhibition of Dkk-1 activity resulting from binding of ETH to its LRP-5/6 binding domain. A binding sphere with a radius of ∼12.8 Å was defined around the Dkk-1 and LRP6 binding site. We observed the interaction of ETH with the Thr-221 and Arg-236 amino acid residues of Dkk-1, which are also involved in the hydrogen bond formation with ETH (Fig. 11). The CDOCKER energy and CDOCKER interaction energy for the ETH interaction pose were −10.9408 and −17.2298 kcal/mol, respectively. Taken together, these observations may suggest that ETH interacts with Akt, Dkk-1, and GSK-3β and thereby alters PI3K/Akt/Wnt/β-catenin signaling pathway.
On the basis of these experimental and in silico studies, we propose a schematic representation illustrating the possible mechanism(s) of ETH-induced alterations of NSC proliferation and differentiation through the PI3K/Akt/Wnt/β-catenin signaling pathway (Fig. 12).
FIGURE 12.
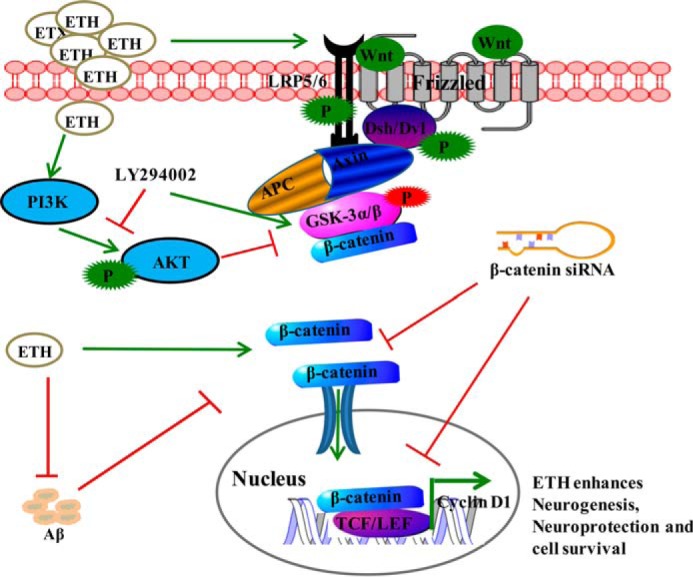
Proposed schematic representation showing the mechanism of ETH and its effect on neurogenesis. On the basis of our experimental and in silico studies, we found that ETH may induce the PI3K/Akt/Wnt/β-catenin signaling. Binding of Wnt ligands with Frizzled receptor and phosphorylated co-receptor low density lipoprotein (LRP-5/6) leads to activation of cytoplasmic dishevelled (Dvl) protein. Activated dishevelled then binds with destruction complex Axin·APC·GSK-3β, inhibits GSK-3β by its phosphorylation, and activates the levels of β-catenin. Inhibition of GSK-3β leads to accumulation of cytoplasmic β-catenin and its translocation into the nucleus. In the nucleus, β-catenin interacts with the Tcf·Lef promoter complex, leading to activation of their target genes, which play an important role in NSC proliferation and differentiation. ETH activates PI3K·Akt, which in turn phosphorylates and inactivates GSK-3β and activates β-catenin. ETH enhances expression of Wnt pathway genes. The blockage of the Wnt·β-catenin signaling (β-catenin siRNA and Dkk-1) and PI3K·Akt pathway (LY294002) results in inhibition of ETH-induced cell proliferation and neuronal differentiation. ETH also blocks Aβ-mediated inhibition of neurogenesis through activation of PI3K/Akt/Wnt/β-catenin signaling.
Discussion
ETH is an AED that is selectively used in the treatment of absence epilepsy (51). It is of potential therapeutic value in other neuropsychiatric disorders (25, 52). In the present study, we demonstrated that ETH promotes adult hippocampal neurogenesis both in vitro and in vivo. Further, we found that it enhances NSC pool and neuronal differentiation and ameliorates cognitive deficits in an Aβ-induced rat model of AD-like phenotypes. NSCs possess the abilities of self-renewal and potential for differentiation toward neurons, astrocytes, and oligodendrocytes (53). We found that ETH induced neurogenesis mainly due to increased NSC proliferation and neuronal differentiation. Several studies have previously suggested that multiple intricate cell signaling cascades, including PI3K·Akt (54–57), Wnt·β-catenin (58), and Notch (59), are involved in NSC proliferation, differentiation, and survival of (60). Herein, we demonstrated that ETH induces NSC proliferation and differentiation through recruitment of the PI3K/Akt/Wnt/β-catenin signaling pathway. Inhibition of these signaling pathways resulted in attenuation of the action of ETH on proliferation and differentiation of the hippocampus-derived NSC. This suggested involvement of the PI3K/Akt/Wnt/β-catenin pathway activation in ETH-induced neurogenesis.
We performed in vitro studies to understand the effect of ETH on rat hippocampal NSC proliferation and differentiation. We found that therapeutic concentrations of ETH (≤100 μm) are non-cytotoxic, whereas higher concentrations adversely affect NSC viability. The neurosphere growth kinetics assay suggested that a non-cytotoxic concentration of ETH enhances the number and size of neurospheres. Neurospheres are considered as free floating bunches of NSC; thus, the increased number and size of neurospheres following ETH treatment suggested that the drug enhances the pool of NSCs. Similarly, the number of nestin/BrdU+ cells was increased after ETH treatment, suggesting an increased NSC pool. Whereas the drug enhanced the number of β-tubulin+ cells, it was not found to alter the number of GFAP+ cells and their morphology. Cumulatively, these results showed that ETH induces proliferation and neuronal differentiation of NSCs in vitro.
We further examined the effects of chronic ETH treatment (125 mg/kg of body weight intraperitoneally) on NSC proliferation and differentiation in the DG region of the hippocampus and in SVZ. The ETH therapeutic plasma concentration range in humans is 25–120 mg/liter (61–64). The dose of 125 mg/kg body weight in the present study was selected on the basis of therapeutic or pharmacologically relevant levels of ETH as described earlier (61–65) and the dose used in rodent studies (25, 66–68). An earlier pharmacokinetic study in rats at a dose of 50 and 200 mg/kg of body weight intraperitoneally resulted in maximum plasma concentration of 33.3 and 121.5 μg/ml, respectively (66). Therefore, it is evident that an in vivo dose of 125 mg/kg of body weight intraperitoneally in rats would yield an in vivo concentration greater than the in vitro effective concentration of ETH (100 μm or 14.1 μg/ml). Keeping in mind the free concentration differences in vitro and in vivo systems, the dose selected in our study is optimal to achieve the effective in vivo ETH concentration after intraperitoneal injection. It was found that ETH induces NSC proliferation in both the DG and SVZ. NSC proliferation was assessed by the differential change in the total number of BrdU+ cells in the DG and SVZ after ETH treatment. This temporal profile of the action of ETH on NSC proliferation is similar to that reported previously for different classes of antidepressant drugs (69, 70). Lamotrigine, an AED with mood-stabilizing and antidepressant potential, has also been found to increase the number of newborn neurons in the rat hippocampus (71). Our observation that the expression of nestin, an NSC marker, is up-regulated in ETH-treated animals suggested that the drug enhances NSC proliferation in the hippocampus and SVZ of the rat brain.
We carried out further analysis, using cell type-specific markers for neurons, astrocytes, oligodendrocytes, and NSC to identify which cells are specifically affected by ETH. We found that ETH increased neuronal differentiation and NSC pool (increased nestin and SOX-2+ cells) in the hippocampus. On the other hand, the glial cell (S100-β and GFAP+ cells) and oligodendrocyte (CNPase+ cells) populations were not altered by ETH treatment. Interestingly, we observed that ETH had no effect on GFAP+ cells but did influence nestin-labeled cells. GFAP also labels adult NSCs; thus, the discrepancy between the observed increase in nestin-positive cells and no change in GFAP+ cells could be explained on the basis of an earlier study (72). In the brain, two distinct subpopulations (type I and type II) of nestin-positive cells exist. Type I nestin-positive cells and their radial processes are GFAP-positive; on the other hand, type II nestin-positive cells are polysialylated neural cell adhesion molecule-positive (72). Thus, it is possible that ETH specifically increased the type II population, which resulted in increased nestin-positive cells and no alteration in GFAP-positive cells. However, ETH had no effect on S100-β-positive cells, suggesting no alteration of the astrocyte population.
In our study, the majority of newly proliferated cells in the DG region following ETH treatment differentiated into neurons, as represented by BrdU/NeuN+- and BrdU/DCX+-co-labeled cells. The expression of genes like β-tubulin and DCX was also up-regulated following ETH treatment. Thus, ETH also causes neuronal differentiation in the hippocampal region of the rat brain. It has previously been shown that established AEDs, such as topiramate, valproate, and carbamazepine, enhance neurogenesis in the adult rodent hippocampus (27, 28, 73–75). Similarly, commonly used new generation AEDs like lamotrigine and topiramate have been found to promote the survival of newborn neurons in the hippocampal region in experimental temporal lobe epilepsy (74). Recently, it was demonstrated that valproate is capable of inducing neurogenesis in the rat forebrain stem cells (76). It enhances neuronal differentiation of NSC and increases neurite outgrowth and the number of GABAergic neurons (76). Further, valproate has been shown to suppress the inhibitory effect of dexamethasone on the proliferation of adult DG-derived NPCs via the GSK-3β and β-catenin pathway (28). Mood-stabilizing drugs enhance the self-renewal capacity of mouse NSC in vitro and the NSC pool in the adult brain through the activation of Notch signaling (59). Similar to the action of various drugs reported in the above studies, we found that ETH also enhances NSC proliferation, neuronal differentiation, and survival in the hippocampal region.
In addition, ETH enhanced the NSC pool and neuronal differentiation in the hippocampus of an Aβ induced phenotypic model of AD and reversed the learning and memory impairment. However, this model has a caveat that Aβ alone only mimics some of the phenotypes in AD, and indeed this limitation has hampered effective therapy development for AD. In addition, local injection of Aβ peptide causes acute neurodegeneration, unlike the stepwise process of neurodegeneration involving plaque and tangle formation in AD. In the future, these studies need to be carried out in transgenic animals, which more closely resemble AD in humans. In an Aβ-induced rat model of AD-like phenotypes, we found a decreased number of BrdU+ cells and neuronal differentiation in the DG region, which was reversed by ETH treatment. Thus, ETH maintained the NSC pool in the DG by increasing their number and reversed the learning and memory deficits induced by Aβ treatment. In order to assess whether the increased NSC proliferation and neuronal differentiation in the ETH-treated group was ETH-specific, we treated rats with an inactive structural analog of ETH (succinimide). Interestingly, we found that succinimide did not induce NSC proliferation and neuronal differentiation in the hippocampus, suggesting a specific role of ETH in neurogenesis. Similarly, we also assessed the effect of scrambled Aβ peptide on hippocampal neurogenesis; this is of particular importance because the short term effects of Aβ injection are considered. Injection of scrambled Aβ peptide showed no significant effect on hippocampal neurogenesis.
Next, we investigated the cellular signaling mechanism underlying the action of ETH on NSC proliferation and differentiation. Activation of the PI3K·Akt and Wnt·β-catenin pathways was examined by assessing phosphorylation of PI3K, Akt, GSK-3β, and β-catenin in the hippocampal NSC culture after ETH treatment. We found increased phosphorylation of PI3K, Akt, and GSK-3β and decreased phosphorylation of β-catenin. From these results, we hypothesized that PI3K·Akt and Wnt·β-catenin signaling pathways may mediate ETH-induced NSC proliferation and neuronal differentiation. Involvement of these pathways was confirmed using inhibitors and activators of PI3K·Akt and Wnt·β-catenin. Our results indicated that inhibition of these pathways attenuated NSC proliferation and neuronal differentiation induced by the drug. PI3K·Akt and Wnt·β-catenin transduce intracellular signals that control adult hippocampal NSC proliferation and differentiation (77, 78). Our results are consistent with the previous finding that valproate enhances NPC proliferation in DG via inhibiting GSK-3β by its phosphorylation and increasing the levels of β-catenin (28).
Hippocampal neurogenesis plays a major role in neuronal plasticity and maintenance of cognitive function (2, 3). In various neurodegenerative disorders, including AD, NSC proliferation and neuronal differentiation are impaired in neurogenic brain regions, leading to learning and memory deficits (8, 9, 31). Importantly, we found that ETH induced hippocampal neurogenesis, reduced neuronal degeneration, and reversed Aβ-induced cognitive deficits in a rat model of AD-like phenotypes via activation of the PI3K/Akt/Wnt/β-catenin pathway. It is notable that two other AEDs, levetiracetam and lamotrigine, have previously been found to ameliorate synaptic and cognitive deficits in a transgenic mouse model of AD, human amyloid precursor protein, and also in presenilin-1 mice (17, 18).
Finally, in our in silico analysis, we found that ETH interacts with different molecules of PI3K/Akt/Wnt/β-catenin signaling pathways that regulate neurogenesis. Thus, it is possible that its interaction with Akt, Dkk-1, and GSK-3β leads to the observed activation of these pathways.
Conclusion
Our results demonstrate that ETH induces proliferation and neuronal differentiation in vitro of NSCs derived from the rat hippocampus, promotes hippocampal neurogenesis in adult rats, and reverses the loss of hippocampal NSCs and the learning and memory deficits characterizing the Aβ rat model of AD-like phenotypes. Evidence provided here suggests that ETH possibly acts through the PI3K/AKT/Wnt/β-catenin signaling pathway to induce neurogenesis. Given these findings, it is tempting to speculate that ETH may promote regeneration of the NSC pool, induce neurogenesis, and provide therapeutic benefits in neurodegenerative disorders.
Author Contributions
S. K. T., A. S., and R. K. C. conceived and coordinated the study, performed experiments, analyzed data, and wrote the paper. S. K. T., B. S., S. A., A. Y., and V. C. designed, performed, and analyzed the experiments shown in Figs. 1–10. S. K. T., S. K. G., and M. K. designed, performed, and analyzed the experiments shown in Fig. 11. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Prof. Alok Dhawan (Director, Council of Scientific and Industrial Research-Indian Institute of Toxicology Research) for constant support during this study.
This work was supported by Council of Scientific and Industrial Research (CSIR) Network Grant UNDO BSC0103 and by a Lady Tata Memorial Young Scientist Grant (to R. K. C.) and PUT513 grant from Estonian Research Council (to V. C.). This work has CSIR-Indian Institute of Toxicology Research (IITR) Manuscript Communication number 3308. The authors declare that they have no conflicts of interest with the contents of this article.
- NSC
- neural stem cell
- PND
- postnatal day
- DG
- dentate gyrus
- SVZ
- subventricular zone
- AD
- Alzheimer disease
- Aβ
- amyloid β
- AED
- antiepileptic drug
- ETH
- ethosuximide
- NPC
- neural progenitor cell
- GFAP
- glial fibriliary acidic protein
- NeuN
- neuronal nuclei
- DCX
- doublecortin
- GCL
- granular cell layer.
References
- 1.Wennstrom M., Hellsten J., Ekstrand J., Lindgren H., and Tingstrom A. (2006) Corticosterone-induced inhibition of gliogenesis in rat hippocampus is counteracted by electroconvulsive seizures. Biol. Psychiatry 59, 178–186 [DOI] [PubMed] [Google Scholar]
- 2.Zhao C., Deng W., and Gage F. H. (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132, 645–660 [DOI] [PubMed] [Google Scholar]
- 3.Ming G. L., and Song H. (2005) Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 28, 223–250 [DOI] [PubMed] [Google Scholar]
- 4.van Praag H., Schinder A. F., Christie B. R., Toni N., Palmer T. D., and Gage F. H. (2002) Functional neurogenesis in the adult hippocampus. Nature 415, 1030–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clelland C. D., Choi M., Romberg C., Clemenson G. D. Jr., Fragniere A., Tyers P., Jessberger S., Saksida L. M., Barker R. A., Gage F. H., and Bussey T. J. (2009) A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 325, 210–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marxreiter F., Regensburger M., and Winkler J. (2013) Adult neurogenesis in Parkinson's disease. Cell Mol. Life Sci. 70, 459–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazarov O., Mattson M. P., Peterson D. A., Pimplikar S. W., and van Praag H. (2010) When neurogenesis encounters aging and disease. Trends Neurosci. 33, 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winner B., Kohl Z., and Gage F. H. (2011) Neurodegenerative disease and adult neurogenesis. Eur. J. Neurosci. 33, 1139–1151 [DOI] [PubMed] [Google Scholar]
- 9.Mu Y., and Gage F. H. (2011) Adult hippocampal neurogenesis and its role in Alzheimer's disease. Mol. Neurodegener. 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steiner B., Wolf S., and Kempermann G. (2006) Adult neurogenesis and neurodegenerative disease. Regen. Med. 1, 15–28 [DOI] [PubMed] [Google Scholar]
- 11.Snyder J. S., Soumier A., Brewer M., Pickel J., and Cameron H. A. (2011) Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 476, 458–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drew M. R., and Hen R. (2007) Adult hippocampal neurogenesis as target for the treatment of depression. CNS Neurol. Disord. Drug Targets 6, 205–218 [DOI] [PubMed] [Google Scholar]
- 13.Mathern G. W., Leiphart J. L., De Vera A., Adelson P. D., Seki T., Neder L., and Leite J. P. (2002) Seizures decrease postnatal neurogenesis and granule cell development in the human fascia dentata. Epilepsia 43, Suppl. 5, 68–73 [DOI] [PubMed] [Google Scholar]
- 14.DeCarolis N. A., and Eisch A. J. (2010) Hippocampal neurogenesis as a target for the treatment of mental illness: a critical evaluation. Neuropharmacology 58, 884–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bakker A., Krauss G. L., Albert M. S., Speck C. L., Jones L. R., Stark C. E., Yassa M. A., Bassett S. S., Shelton A. L., and Gallagher M. (2012) Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tse M. T. (2012) Neurodegenerative diseases: anti-epileptic drug shows benefit in AD mouse model. Nat. Rev. Drug Discov. 11, 748–749 [DOI] [PubMed] [Google Scholar]
- 17.Sanchez P. E., Zhu L., Verret L., Vossel K. A., Orr A. G., Cirrito J. R., Devidze N., Ho K., Yu G. Q., Palop J. J., and Mucke L. (2012) Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc. Natl. Acad. Sci. U.S.A. 109, E2895–E2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang M. Y., Zheng C. Y., Zou M. M., Zhu J. W., Zhang Y., Wang J., Liu C. F., Li Q. F., Xiao Z. C., Li S., Ma Q. H., and Xu R. X. (2014) Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol. Aging 35, 2713–2725 [DOI] [PubMed] [Google Scholar]
- 19.Hwang H., Kim H., Kim S. H., Kim S. H., Lim B. C., Chae J. H., Choi J. E., Kim K. J., and Hwang Y. S. (2012) Long-term effectiveness of ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. Brain Dev. 34, 344–348 [DOI] [PubMed] [Google Scholar]
- 20.Coulter D. A., Huguenard J. R., and Prince D. A. (1989) Specific petit mal anticonvulsants reduce calcium currents in thalamic neurons. Neurosci. Lett. 98, 74–78 [DOI] [PubMed] [Google Scholar]
- 21.Coulter D. A., Huguenard J. R., and Prince D. A. (1989) Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann. Neurol. 25, 582–593 [DOI] [PubMed] [Google Scholar]
- 22.Kostyuk P. G., Molokanova E. A., Pronchuk N. F., Savchenko A. N., and Verkhratsky A. N. (1992) Different action of ethosuximide on low- and high-threshold calcium currents in rat sensory neurons. Neuroscience 51, 755–758 [DOI] [PubMed] [Google Scholar]
- 23.Collins J. J., Evason K., Pickett C. L., Schneider D. L., and Kornfeld K. (2008) The anticonvulsant ethosuximide disrupts sensory function to extend C. elegans lifespan. PLoS Genet. 4, e1000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tauffenberger A., Julien C., and Parker J. A. (2013) Evaluation of longevity enhancing compounds against transactive response DNA-binding protein-43 neuronal toxicity. Neurobiol. Aging 34, 2175–2182 [DOI] [PubMed] [Google Scholar]
- 25.Williams A. J., Bautista C. C., Chen R. W., Dave J. R., Lu X. C., Tortella F. C., and Hartings J. A. (2006) Evaluation of gabapentin and ethosuximide for treatment of acute nonconvulsive seizures following ischemic brain injury in rats. J. Pharmacol. Exp. Ther. 318, 947–955 [DOI] [PubMed] [Google Scholar]
- 26.Kang M. L., Kwon J. S., and Kim M. S. (2013) Induction of neuronal differentiation of rat muscle-derived stem cells in vitro using basic fibroblast growth factor and ethosuximide. Int. J. Mol. Sci. 14, 6614–6623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boku S., Nakagawa S., Masuda T., Nishikawa H., Kato A., Toda H., Song N., Kitaichi Y., Inoue T., and Koyama T. (2011) Effects of mood stabilizers on adult dentate gyrus-derived neural precursor cells. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 111–117 [DOI] [PubMed] [Google Scholar]
- 28.Boku S., Nakagawa S., Masuda T., Nishikawa H., Kato A., Takamura N., Omiya Y., Kitaichi Y., Inoue T., and Kusumi I. (2014) Valproate recovers the inhibitory effect of dexamethasone on the proliferation of the adult dentate gyrus-derived neural precursor cells via GSK-3β and β-catenin pathway. Eur. J. Pharmacol. 723, 425–430 [DOI] [PubMed] [Google Scholar]
- 29.Ponnusamy R., and Pradhan N. (2006) The effects of chronic administration of ethosuximide on learning and memory: a behavioral and biochemical study on nonepileptic rats. Behav. Pharmacol. 17, 573–580 [DOI] [PubMed] [Google Scholar]
- 30.Polack P. O., and Charpier S. (2009) Ethosuximide converts ictogenic neurons initiating absence seizures into normal neurons in a genetic model. Epilepsia 50, 1816–1820 [DOI] [PubMed] [Google Scholar]
- 31.Tiwari S. K., Agarwal S., Seth B., Yadav A., Nair S., Bhatnagar P., Karmakar M., Kumari M., Chauhan L. K., Patel D. K., Srivastava V., Singh D., Gupta S. K., Tripathi A., Chaturvedi R. K., and Gupta K. C. (2014) Curcumin-loaded nanoparticles potently induce adult neurogenesis and reverse cognitive deficits in Alzheimer's disease model via canonical Wnt/β-catenin pathway. ACS Nano 8, 76–103 [DOI] [PubMed] [Google Scholar]
- 32.Mishra D., Tiwari S. K., Agarwal S., Sharma V. P., and Chaturvedi R. K. (2012) Prenatal carbofuran exposure inhibits hippocampal neurogenesis and causes learning and memory deficits in offspring. Toxicol. Sci. 127, 84–100 [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Canabal A., Akers K. G., Josselyn S. A., and Frankland P. W. (2013) Age-dependent effects of hippocampal neurogenesis suppression on spatial learning. Hippocampus 23, 66–74 [DOI] [PubMed] [Google Scholar]
- 34.Chaturvedi R. K., Hennessey T., Johri A., Tiwari S. K., Mishra D., Agarwal S., Kim Y. S., and Beal M. F. (2012) Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntington's disease. Hum. Mol. Genet. 21, 3474–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaturvedi R. K., Calingasan N. Y., Yang L., Hennessey T., Johri A., and Beal M. F. (2010) Impairment of PGC-1α expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington's disease following chronic energy deprivation. Hum. Mol. Genet. 19, 3190–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaturvedi R. K., Adhihetty P., Shukla S., Hennessy T., Calingasan N., Yang L., Starkov A., Kiaei M., Cannella M., Sassone J., Ciammola A., Squitieri F., and Beal M. F. (2009) Impaired PGC-1α function in muscle in Huntington's disease. Hum. Mol. Genet. 18, 3048–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tiwari S. K., Agarwal S., Chauhan L. K., Mishra V. N., and Chaturvedi R. K. (2015) Bisphenol-A impairs myelination potential during development in the hippocampus of the rat brain. Mol. Neurobiol. 51, 1395–1416 [DOI] [PubMed] [Google Scholar]
- 38.Sinha C., Seth K., Islam F., Chaturvedi R. K., Shukla S., Mathur N., Srivastava N., and Agrawal A. K. (2006) Behavioral and neurochemical effects induced by pyrethroid-based mosquito repellent exposure in rat offsprings during prenatal and early postnatal period. Neurotoxicol. Teratol. 28, 472–481 [DOI] [PubMed] [Google Scholar]
- 39.Park D., Xiang A. P., Mao F. F., Zhang L., Di C. G., Liu X. M., Shao Y., Ma B. F., Lee J. H., Ha K. S., Walton N., and Lahn B. T. (2010) Nestin is required for the proper self-renewal of neural stem cells. Stem Cells 28, 2162–2171 [DOI] [PubMed] [Google Scholar]
- 40.Sawicka A., and Seiser C. (2012) Histone H3 phosphorylation: a versatile chromatin modification for different occasions. Biochimie 94, 2193–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng M., Liu J., Ruan Z., Tian S., Ma Y., Zhu J., and Li G. (2013) Intrahippocampal injection of Aβ1–42 inhibits neurogenesis and down-regulates IFN-γ and NF-κB expression in hippocampus of adult mouse brain. Amyloid 20, 13–20 [DOI] [PubMed] [Google Scholar]
- 42.Lie D. C., Colamarino S. A., Song H. J., Désiré L., Mira H., Consiglio A., Lein E. S., Jessberger S., Lansford H., Dearie A. R., and Gage F. H. (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437, 1370–1375 [DOI] [PubMed] [Google Scholar]
- 43.Kuwabara T., Hsieh J., Muotri A., Yeo G., Warashina M., Lie D. C., Moore L., Nakashima K., Asashima M., and Gage F. H. (2009) Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 12, 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalani M. Y., Cheshier S. H., Cord B. J., Bababeygy S. R., Vogel H., Weissman I. L., Palmer T. D., and Nusse R. (2008) Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 105, 16970–16975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahn Y. J., Park S. J., Woo H., Lee H. E., Kim H. J., Kwon G., Gao Q., Jang D. S., and Ryu J. H. (2014) Effects of allantoin on cognitive function and hippocampal neurogenesis. Food Chem. Toxicol. 64, 210–216 [DOI] [PubMed] [Google Scholar]
- 46.Dong C., Rovnaghi C. R., and Anand K. J. (2014) Ketamine affects the neurogenesis of rat fetal neural stem progenitor cells via the PI3K/Akt-p27 signaling pathway. Birth Defects Res. B Dev. Reprod. Toxicol. 101, 355–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruel-Jungerman E., Veyrac A., Dufour F., Horwood J., Laroche S., and Davis S. (2009) Inhibition of PI3K-Akt signaling blocks exercise-mediated enhancement of adult neurogenesis and synaptic plasticity in the dentate gyrus. PLoS One 4, e7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Endo H., Nito C., Kamada H., Nishi T., and Chan P. H. (2006) Activation of the Akt/GSK3β signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 26, 1479–1489 [DOI] [PubMed] [Google Scholar]
- 49.Otero J. J., Fu W., Kan L., Cuadra A. E., and Kessler J. A. (2004) β-Catenin signaling is required for neural differentiation of embryonic stem cells. Development 131, 3545–3557 [DOI] [PubMed] [Google Scholar]
- 50.Gulacsi A. A., and Anderson S. A. (2008) β-Catenin-mediated Wnt signaling regulates neurogenesis in the ventral telencephalon. Nat. Neurosci. 11, 1383–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gören M. Z., and Onat F. (2007) Ethosuximide: from bench to bedside. CNS Drug Rev. 13, 224–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaufman K. R. (2011) Antiepileptic drugs in the treatment of psychiatric disorders. Epilepsy Behav. 21, 1–11 [DOI] [PubMed] [Google Scholar]
- 53.Gobbel G. T., Choi S. J., Beier S., and Niranjan A. (2003) Long-term cultivation of multipotential neural stem cells from adult rat subependyma. Brain Res. 980, 221–232 [DOI] [PubMed] [Google Scholar]
- 54.Fournier N. M., Lee B., Banasr M., Elsayed M., and Duman R. S. (2012) Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK- and PI3K/Akt-dependent signaling. Neuropharmacology 63, 642–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Q., Liu G., Wu Y., Sha H., Zhang P., and Jia J. (2011) BDNF promotes EGF-induced proliferation and migration of human fetal neural stem/progenitor cells via the PI3K/Akt pathway. Molecules 16, 10146–10156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le Belle J. E., Orozco N. M., Paucar A. A., Saxe J. P., Mottahedeh J., Pyle A. D., Wu H., and Kornblum H. I. (2011) Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependent manner. Cell Stem Cell 8, 59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng Y., Jiang B. H., Yang P. H., Cao Z., Shi X., Lin M. C., He M. L., and Kung H. F. (2004) Phosphatidylinositol 3-kinase signaling is involved in neurogenesis during Xenopus embryonic development. J. Biol. Chem. 279, 28509–28514 [DOI] [PubMed] [Google Scholar]
- 58.Ortiz-Matamoros A., Salcedo-Tello P., Avila-Muñoz E., Zepeda A., and Arias C. (2013) Role of wnt signaling in the control of adult hippocampal functioning in health and disease: therapeutic implications. Curr. Neuropharmacol. 11, 465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Higashi M., Maruta N., Bernstein A., Ikenaka K., and Hitoshi S. (2008) Mood stabilizing drugs expand the neural stem cell pool in the adult brain through activation of Notch signaling. Stem Cells 26, 1758–1767 [DOI] [PubMed] [Google Scholar]
- 60.Shioda N., Han F., and Fukunaga K. (2009) Role of Akt and ERK signaling in the neurogenesis following brain ischemia. Int. Rev. Neurobiol. 85, 375–387 [DOI] [PubMed] [Google Scholar]
- 61.Sherwin A. L., Robb J. P., and Lechter M. (1973) Improved control of epilepsy by monitoring plasma ethosuximide. Arch. Neurol. 28, 178–181 [DOI] [PubMed] [Google Scholar]
- 62.Serrano P. A., Hava-Kuri I., Chávez-Lara B., Cervantes-Osorio L., and Zaldivar H. M. (1978) [Hypotension, mental disorders and changes in catecholamine excretion observed during the administration of disulfiram in a case of pheochromocytoma]. Arch. Inst. Cardiol. Mex. 48, 437–443 [PubMed] [Google Scholar]
- 63.Browne T. R., Dreifuss F. E., Dyken P. R., Goode D. J., Penry J. K., Porter R. J., White B. G., and White P. T. (1975) Ethosuximide in the treatment of absence (petit mal) seizures. Neurology 25, 515–524 [DOI] [PubMed] [Google Scholar]
- 64.Bentué-Ferrer D., Tribut O., and Verdier M. C. (2012) [Therapeutic drug monitoring of ethosuximide]. Therapie 67, 391–396 [DOI] [PubMed] [Google Scholar]
- 65.Callaghan N., O'Hare J., O'Driscoll D., O'Neill B., and Daly M. (1982) Comparative study of ethosuximide and sodium valproate in the treatment of typical absence seizures (petit mal). Dev. Med. Child Neurol. 24, 830–836 [DOI] [PubMed] [Google Scholar]
- 66.Mifsud J., Collier P. S., and Millership J. S. (2001) The pharmacokinetics of ethosuximide enantiomers in the rat. Biopharm. Drug Dispos. 22, 83–89 [DOI] [PubMed] [Google Scholar]
- 67.Mares P. (1998) Ontogeny of ethosuximide action against two seizure models in rats is different. Life Sci. 63, PL51–PL57 [DOI] [PubMed] [Google Scholar]
- 68.Mares P., Kubová H., and Mocková M. (1997) Two models of epileptic spike-and-wave rhythm in rats are differently influenced by ethosuximide. Physiol. Res. 46, 397–402 [PubMed] [Google Scholar]
- 69.Santarelli L., Saxe M., Gross C., Surget A., Battaglia F., Dulawa S., Weisstaub N., Lee J., Duman R., Arancio O., Belzung C., and Hen R. (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809 [DOI] [PubMed] [Google Scholar]
- 70.Nakagawa S., Kim J. E., Lee R., Malberg J. E., Chen J., Steffen C., Zhang Y. J., Nestler E. J., and Duman R. S. (2002) Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J. Neurosci. 22, 3673–3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kondziella D., Strandberg J., Lindquist C., and Asztely F. (2011) Lamotrigine increases the number of BrdU-labeled cells in the rat hippocampus. Neuroreport 22, 97–100 [DOI] [PubMed] [Google Scholar]
- 72.Fukuda S., Kato F., Tozuka Y., Yamaguchi M., Miyamoto Y., and Hisatsune T. (2003) Two distinct subpopulations of nestin-positive cells in adult mouse dentate gyrus. J. Neurosci. 23, 9357–9366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dozawa M., Kono H., Sato Y., Ito Y., Tanaka H., and Ohshima T. (2014) Valproic acid, a histone deacetylase inhibitor, regulates cell proliferation in the adult zebrafish optic tectum. Dev. Dyn. 243, 1401–1415 [DOI] [PubMed] [Google Scholar]
- 74.Chen J., Quan Q. Y., Yang F., Wang Y., Wang J. C., Zhao G., and Jiang W. (2010) Effects of lamotrigine and topiramate on hippocampal neurogenesis in experimental temporal-lobe epilepsy. Brain Res. 1313, 270–282 [DOI] [PubMed] [Google Scholar]
- 75.Shankaran M., King C., Lee J., Busch R., Wolff M., and Hellerstein M. K. (2006) Discovery of novel hippocampal neurogenic agents by using an in vivo stable isotope labeling technique. J. Pharmacol. Exp. Ther. 319, 1172–1181 [DOI] [PubMed] [Google Scholar]
- 76.Laeng P., Pitts R. L., Lemire A. L., Drabik C. E., Weiner A., Tang H., Thyagarajan R., Mallon B. S., and Altar C. A. (2004) The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J. Neurochem. 91, 238–251 [DOI] [PubMed] [Google Scholar]
- 77.Wang L., Zhang Z. G., Zhang R. L., Jiao Z. X., Wang Y., Pourabdollah-Nejad D. S., LeTourneau Y., Gregg S. R., and Chopp M. (2006) Neurogenin 1 mediates erythropoietin enhanced differentiation of adult neural progenitor cells. J. Cereb. Blood Flow Metab. 26, 556–564 [DOI] [PubMed] [Google Scholar]
- 78.Peltier J., O'Neill A., and Schaffer D. V. (2007) PI3K·Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 67, 1348–1361 [DOI] [PubMed] [Google Scholar]