

Background: The proposed model for promoter escape predicts the destabilization of interactions of σ region 4 with RNA polymerase and DNA.
Results: Using a two-component σ factor, we show that YvrI, mimicking the σ region 4, is released, whereas YvrHa, mimicking σ region 2, is retained after promoter escape.
Conclusion: This study validates the proposed mechanism for promoter escape.
Significance: This study suggests the possibility of certain σ-factors to be retained in elongation complex.
Keywords: Bacillus, promoter, RNA polymerase, transcription, transcription initiation factor, YvrI and YvrHa, promoter escape
Abstract
The transition from the formation of the RNA polymerase (RNAP)-promoter open complex step to the productive elongation complex step involves “promoter escape” of RNAP. From the structure of RNAP, a promoter escape model has been proposed that suggests that the interactions between σR4 and RNAP and σR4 and DNA are destabilized upon transition to elongation. This accounts for the reduced affinity of σ to RNAP and stochastic release of σ. However, as the loss of interaction of σR4 with RNAP results in the release of intact σ, assessing this interaction remains challenging to be experimentally verified. Here we study the promoter escape model using a two-component σ factor YvrI and YvrHa from Bacillus subtilis that independently contributes to the functions of σR4 and σR2 in a RNAP-promoter complex. Our results show that YvrI, which mimics σR4, is released gradually as transcription elongation proceeds, whereas YvrHa, which mimics σR2 is retained throughout the elongation complexes. Thus our result validates the proposed model for promoter escape and also suggests that promoter escape involves little or no change in the interaction of σR2 with RNAP.
Introduction
Transcription is the first step in gene regulation that involves initiation, elongation, and termination. For transcription initiation in bacteria, a σ factor must bind to RNA polymerase (RNAP)3 rendering the enzyme capable of promoter recognition and nucleation of DNA prior to the open complex formation (RPo). Upon addition of NTP, RNAP in the open complex begins synthesis of RNA as an initial transcribing complex and releases short RNA transcripts between 2 and 8 nucleotides. Once the length of the nascent RNA becomes greater than 9–11 nucleotides, RNAP is able to escape from the promoter and start RNA synthesis productively as a transcription elongation complex (EC). The step between the open complex formation and elongation complex formation is known as “promoter escape.” Initially, it was proposed that promoter escape is possible only upon the release of σ from RNAP (1–5). The conclusion was based on biochemical assays with Escherichia coli σ70, in which σ70 was observed to be present in the open complex, but absent in the elongation complex. This observation lead to the generalized proposal that promoter escape in bacteria involves “σ-cycle” in which σ binds to RNAP to initiate transcription and is released from RNAP upon transition from transcription initiation to elongation (1–5). This obligatory release of σ70 for promoter escape was subsequently challenged by several observations that suggested that the release of σ70 does not occur immediately upon transition from transcription initiation to elongation, but rather occurs slowly during the course of transcription elongation (6–12). In contrary to the “σ-release model,” it was further reported that a fraction of RNAP of E. coli in stationary phase does not release σ70 throughout the elongation step on some genes (13, 14). When RNAP covalently tethered with σ70 was expressed in E. coli, there was no adverse effect on the cell growth. Because there was no release of σ70 in vivo in these cells, this observation argues that the σ-cycle may not be essential for bacteria.
The principal σ factors of bacterial species contain four conserved regions: (σR1.1, σR2, σR3, and σR4), all of which interact extensively with β and β′ subunits of RNAP (15–17). In addition, σR4 and σR2 are responsible for recognition and interaction with promoter elements −35 and −10, respectively. σR1.1 is located inside the active center cleft in the RNAP holo but is displaced from the cleft once the open complex is formed (15). The structures of RNAP holoenzyme of different bacterial species show that the σR3/σR4 linker occupies the “RNA exit channel” that mediates the egress of nascent RNA. This linker encounters a steric clash with the nascent RNA once it reaches a threshold length of ∼9–11 nucleotides and tries to enter the RNA exit channel (15–17). Thus, the transition from transcription initiation to elongation involves the displacement of the σR3/σR4 linker from the RNA exit channel. From the structure of RNAP, Murakami and Darst (18) proposed a model for the promoter escape, which predicts that displacement of the σR3/σR4 linker could destabilize the interactions between σR4 and β, which in turn, further destabilizes the interactions of σR4 and the −35 element, allowing RNAP to escape from the promoter and start productive elongation. Although the interaction of σR2 with RNAP possibly remains intact at this stage, this transition into elongation is responsible for the reduced affinity of σ for RNAP (19) and results in a stochastic release of σ.
In Bacillus subtilis σ-like factors YvrI and coregulator YvrHa activate transcription from a small set of conserved promoters (PoxdC, PyvrI, and PyvrJ) (20). YvrI includes a σ region 4 domain that interacts with the β subunit of RNAP and is responsible for recognizing the −35 element of specific promoters. On the other hand, YvrHa functions as σR2 and is responsible for recognizing the −10 element and DNA melting of the promoter (20–22). Thus, these two proteins together function as a σ factor in an RNAP-promoter complex and is referred to as a two-component σ factor. This two-component σ factor could posit an ideal system to study the proposed promoter escape model that predicts that the interaction between RNAP and YvrI (which mimics σR4) may break once nascent RNA reaches a length of 9∼11 nucleotides, whereas interaction between YvrHa (which mimics σR2) and RNAP may remain intact. Here, we observed that YvrI was released as RNAP proceeds through elongation, whereas YvrHa was fully retained in the elongation complex. Thus, our study validates the predictions of the proposed model for promoter escape and establishes that σR2 retains its interaction with RNAP in the elongation phase.
Materials and Methods
Cloning Strategies
The genes encoding YvrI and YvrHa were subcloned from plasmids pSM11 and pSM17, respectively (a kind gift from Dr. Shawn Maclellan), into pET32a with NcoI and BamHI, for the incorporation of the C-terminal histidine tag. DNA fragments containing oxdC and yvrI promoters were amplified from B. subtilis genomic DNA (strain Bs168) using primers (Table 1). The portions of the rpoD gene encoding regions 2/3.1 and 3.2/4 of σ70 were amplified from plasmids pGEMD-396Cys and pGEMD-578Cys (7), respectively (a kind gift from Dr. Richard H. Ebright), using primers (Table 1) and cloned into pET32a with NcoI-EcoRI and NcoI-BamHI, respectively.
TABLE 1.
Oligonucleotide primers used in this study
| Sequence | |
|---|---|
| yvrI fw (NcoI) | ccatggatgaagcatcccatcgtgaagc |
| yvrI rv (BamHI) | ggatccttacttttccccctttttttgcgc |
| yvrHa fw (NcoI) | ccatgggtggacggcc agtttgaacaa |
| yvrHa rv (BamHI) | ggatccttatgagttctcatctaccatgtacaaaataaac |
| yvrI S88C fw | ggcgggagatcggctttcttgcgaaacagg |
| yvrI S88C rv | cctgtttcgcaagaaagccgatctcccgcc |
| yvrI A142C fw | gccacgatgcaggagattgcatgttcattagg |
| YvrI A142C rv | cctaatgaacatgcaatctcctgcatcgtggc |
| YvrI S25C fw | gttcaaaaacgtaatggaatgccctaacgaaaaag |
| YvrI S25C Rv | ctttttcgttagggcattccattacgtttttgaac |
| yvrI S59C fw | gattcgcatcttttgtattgattttgataagcggg |
| yvrI S59C rv | cccgcttatcaaaatcaatacaaaagatgcgaatc |
| yvrI L168C fw | cagaaagcagttgccggcgcaaaaaaaggggg |
| yvrI L168C rv | ccccctttttttgcgccggcaactgctttctg |
| yvrHa C24S fw | gagcacctgattgcaagcttttcaccgatgatc |
| yvrHa C24S Rv | gatcatcggtgaaaagcttgcaatcaggtgctc |
| yvrHa C61S fw | ggctgatatgcttttaagtcaggatgtaccgggg |
| yvrHa C61S Rv | ccccggtacatcctgacttaaaagcatatcagcc |
| yvrHa S37C fw | gaaaaaaactcagcaatacgtgctatcaagaaagagaag |
| yvrHa S37C Rv | cttctctttcttgatagcacgtattgctgagtttttttc |
| yvrHa T15C fw | gaaacaaaaagacgagtgttatgacattgagcacct |
| yvrHa T15C Rv | caggtgctcaatgtcataacactcgtctttttgtttc |
| yvrHa I50C fw | gagcaagagctgaagtgcaaaatgtttgaaaaggctg |
| yvrHa I50C rv | cagccttttcaaacattttgcacttcagctcttgctc |
| PoxdC fw biotin | biotin-gacagaactgaaagatcatgc |
| PoxdC 98rv | atggtaccctgcggaatgtcattttgttttttc |
| PoxdC 54rv | catttaaatgtgatagcgggcggcggcgg |
| PyvrI fw biotin | biotin-gcaaagtattttggaagacactaggcgg |
| PyvrI 98rv | gcttcacgatgggatgcttcattatctcc |
| PyvrI 54rv | ctccttacattttgaccgtaaaagactttg |
| T7A1 fw biotin | biotin-cggaattcggatccagatcccg |
| T7A1 rv | cgagtagcttcttcgattcgattcgcc |
| M13 fw biotin | biotin-gtaaaacgacggccagtgaattcgagc |
| lacUV5 rv | atggatcctgtgtgaaattgttatccgc |
| σ70-R2 fw | atccatggttcaatgctccgttgctgaatatcc |
| σ70-R2 rv | atgaattccgatcttcagcactttgcgg |
| σ70-R4 fw | atccatggccaaagagccaatctcgatggaaac |
| σ70-R4 rv | atggatccatcgtccaggaagctacgcagc |
Preparation of RNAP Core of B. subtilis
For the purification of B. subtilis (Bs) RNAP core, plasmids pNG545 (containing α and β) and pNG540 (containing β′ and ω) (a kind gift from Dr. Xiao Yang and Peter J. Lewis) were transformed into E. coli B384 (DE3) cells (23), and grown in 4 liters of LB (with 0.1% dextrose, 100 μg/ml of ampicillin, and 35 μg/ml of chloramphenicol) at 37 °C until the OD reached 0.5. Protein expression was induced by adding 0.5 mm isopropyl 1-thio-β-d-galactopyranoside and further grown at 16 °C until the OD reached 1.2. The cells were harvested, and RNAP was purified essentially as described by Mukhopadhyay et al. (7), except that the pellet obtained from the polyminP precipitation was washed with buffer containing 0.4 m NaCl instead of 0.5 m NaCl.
Purification of YvrI and YvrHa Proteins of B. subtilis
pET32a-yvrI and pET32a-yvrHa were transformed into E. coli BL21(DE3) cells and grown in 2 liters of 2× YT (16 g of tryptone, 10 g of yeast extract, and 5 g of NaCl per liter) with 0.1% dextrose and 100 μg/ml of ampicillin at 37 °C until the OD reached 0.4. Protein expressions were induced with addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside and the cells were grown for another 3 h. The cells were lysed with TGB (50 mm Tris-HCl, 5% glycerol, 2 mm β-mercaptoethanol) and 0.2 m NaCl, 0.25% deoxycholate, 1 mm PMSF, followed by sonication and centrifugation at 14000 rpm for 20 min at 4 °C. The pellets were washed with TGB, 0.2 m NaCl, and 0.5% Triton X-100 followed by centrifugation at 8000 rpm for 20 min at 4 °C. The pellets were solubilized in buffer A (TGB + 8 m urea) and incubated for 1 h, followed by centrifugation at 14000 rpm at room temperature. The supernatant containing the solubilized protein was passed through 5-ml nickel-nitrilotriacetic acid-agarose, pre-equilibrated with buffer A, washed with 25 ml of buffer A, and eluted with 5 ml each of buffer A containing 10, 20, 40, 80, and 160 mm imidazole. Both proteins were eluted at 40 mm or higher concentration of imidazole. The purified YvrI sample was then dialyzed in 4 liters of TGB buffer for 24 h with two changes at 8-h intervals and the renatured protein was stored with 50% glycerol at −80 °C.
The 5 ml of YvrHa protein sample was renatured by serial dilution with an equal volume of TGB buffer to make a final volume of 80 ml. The sample was applied to a MonoQ HR 10/10 column in Akta Purifier (GE Healthcare), preequilibrated with TGB buffer. The column was washed with 16 ml of TGB buffer and eluted in 1-ml fractions with a linear gradient 0 to 1 m NaCl in 160 ml of TGB buffer. The protein was eluted at 0.4 m NaCl. The purified protein sample was added with an equal volume of 100% glycerol and stored at −80 °C.
Preparation of E. coli RNAP Core
E. coli RNAP core was purified as in Mukhopadhyay et al. (7).
Purification of E. coli σ70
E. coli σ70 (a derivative with a single-Cys residue at position 578) was purified as in Mukhopadhyay et al. (7).
Purification of E. coli σ70-R2 and σ70-R4
Plasmids pET32a-rpoD-R2 and pET32a-rpoD-R4 containing Cys at positions 396 and 578, respectively, were transformed into E. coli C43 cells and grown in 1 liter of 2× YT containing 100 μg/ml of ampicillin. After growing the cells to OD of 0.4 at 37 °C, the protein induction was initiated at 16 °C with the addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and the cells were further grown for 12 h. The cells were lysed and the proteins that appeared in soluble forms were purified essentially following the same method as for YvrI except that protein samples were passed through a nickel-nitrilotriacetic acid column using TGB + 0.2 m NaCl buffer, and no denaturation/renaturation steps were involved.
Labeling of YvrI and YvrHa mutants of B. subtilis and σ70, σ70-R2, and σ70-R4 of E. coli with TMR-6-maleimide
For labeling of YvrI and YvrHa, we generated single Cys derivatives of each protein. Because YvrI does not contain any cysteine, this residue was incorporated at amino acid position 25 (originally Ser) using a site-directed mutagenesis. YvrHa contains two cysteines at amino acid positions 24 and 61. First these cysteines residues were mutated to serine to generate a no-Cys protein derivative. Then Cys was incorporated at amino acid position 15 (originally Thr) of YvrHa using a site-directed mutagenesis kit (Table 1). Both the single Cys YvrI and YvrHa derivatives were purified as their wild type counterparts.
The purified ammonium sulfate precipitates of YvrI S25C, YvrHa T15C mutants of B. subtilis, and σ70-578C, σ70-R2–396C, and σ70-R4–578C of E. coli (containing single Cys residues) were labeled with TMR-6-maleimide as described by Rudra et al. (24, 25). The labeled protein fractions were confirmed by running on SDS-PAGE and viewed in a Fluorescence Imager. The efficiency of labeling was detected by the ratio of the dye concentration (estimated by absorbance at 555 nm) and the protein concentration (estimated by Bradford assay). The labeling efficiencies of YvrI S25C, YvrHa T15C, σ70-578C, σ70-R2–396C, and σ70-R4–578C were found to be 90, 95, 98, 72, and 60% respectively.
Preparation of 5′-Biotinylated Promoter DNA Fragments
The promoter DNA templates (Table 2) were generated by PCR amplification of either genomic DNA (Bs168) or from synthetic oligonucleotides with their respective 5′-biotinylated forward and reverse primers (Table 1).
TABLE 2.
Promoter DNA templates used in the assays
The bold letter A denotes the transcription start site. Other bold letters denote the first non-template strand T or C nucleotide.
| Sequence | |
|---|---|
| Bs PoxdC EC+11 | 5′-gacagaactgaaagatcatgcaaaaaaataatttttcaatcgaagttgacttttcactggtttttttcacttaacaaaacAgaagggaaaaCgaaaggcctttcaccttctctttctgctatcacatttaaatg taaggaggaaacatttcatgaaaaaacaaaatgacattccgcag-3′ |
| Bs PoxdC EC+20 | 5′-gacagaactgaaagatcatgcaaaaaaataatttttcaatcgaagttgacttttcactggtttttttcacttaacaaaacAgaagggaaaacgaaaggccTttcaccttctctttctgctatcacatttaaatgt aaggaggaaacatttcatgaaaaaacaaaatgacattccgcag-3′ |
| Bs PoxdC EC+39 | 5′-gacagaactgaaagatcatgcaaaaaaataatttttcaatcgaagttgacttttcactggtttttttcacttaacaaaacAgaagggaaaacgaaaggccacacaccaccaccacccgcTatcacatttaaatg-3′ |
| Bs PyvrI EC+10 | 5′-gcaaagtattttggaagacactaggcggaaagtagtagtttacttttttttgctttccttcatttaatttagtAgggaggcggTttgatggactatcaaagtcttttacggtcaaaatgtaaggagataatgaagcatcccatcgtgaagc |
| Bs PyvrI EC+19 | 5′-gcaaagtattttggaagacactaggcggaaagtagtagtttacttttttttgctttccttcatttaatttagtAgggaggaggtttgatggaCtatcaaagtcttttacggtcaaaatgtaaggag-3′ |
| Ec T7A1 EC+23 | 5′-cggaattcggatccagatcccgaaaatttatcaaaaagagtattgacttaaagtctaacctataggatacttacagccAtcgagagggacacgggcgaagcTtggcgaatcgaatcgaagaagctactcg-3′ |
| Ec lacUV5 EC+15 | 5′-taggcaccccaggcttgacactttatgcttcggctcgtataatgtgtggAattgtgaggagaggCggataacaatttcacacaggatccat-3′ |
| Ec lacUV5 EC+25 | taggcaccccaggcttgacactttatgcttcggctcgtataatgtgtggAattgtgaggagaggtggataataaCggataacaatt tcacacagg |
| Ec lacUV5 EC+40 | taggcaccccaggcttgacactttatgcttcggctcgtataatgtgtggAattgtgaggagaggtggataataatttaatataggattaCaggcggataac |
Formation of Open Complexes on Streptavidin Beads
E. coli RNAP holoenzyme was prepared by incubation of 200 nm unlabeled RNAP core with 500 nm labeled σ70 (or σ70-R2 and σ70-R4) for 20 min at 25 °C in transcription buffer (TB: 40 mm Tris, pH 8.0, 150 mm KCl, 10 mm MgCl2, 0.01% Triton-X-100, 2 mm DTT). Open complexes were formed by incubating the E. coli RNAP holoenzymes sample with 100 nm 5′-biotinylated DNA fragments containing either T7A1 or lacUV5 promoter at 37 °C for 20 min.
B. subtilis RNAP holoenzyme was prepared by incubation of 200 nm unlabeled RNAP core and 500 nm each of TMR-labeled YvrI and YvrHa in transcription buffer (18 mm Tris (pH 8.0), 10 mm NaCl, 8 mm β-mercaptoethanol,10 mm MgCl2) for 20 min at room temperature. Open complexes were formed by incubating the RNAP holoenzyme sample with 100 nm 5′-biotinylated DNA fragments containing either PoxdC or PyvrI promoter at 37 °C for 20 min. 10 μl RPo samples were immobilized on 20 μl of streptavidin beads pre-equilibrated with the respective transcription buffer, by incubating the samples at 25 °C for 1 h with regular tapping. The beads were washed 4 times with the respective transcription buffer; each time the samples were centrifuged at 4000 × g for 1 min.
Formation of Stalled Elongation Complexes
For formation of stalled elongation complex EC+n, we used the DNA derivative that contained the first non-template strand CTP or UTP at position n+1, (Table 2). After formation of the open complex as above, transcription reactions were initiated with subset of NTPs and 0.25 μg/μl of heparin as following: EC+11 on PoxdC, 500 μm ApG, GTP, UTP, ATP; EC+20 and EC+39 on PoxdC, 500 μm ApG, GTP, CTP, ATP; EC+10 on PyvrI, 500 μm ApG, GTP, CTP, ATP; EC+19 on PyvrI, 500 μm ApG, GTP, UTP, ATP; EC+23 T7A1, 500 μm ApU, GTP, CTP, ATP; EC+15, EC+25 and EC+40 on lacUV5, 500 μm ApA, GTP, UTP, ATP.
These complexes were washed thoroughly several times with TB to remove the excess unbound proteins, followed by centrifugation at 4000 × g for 1 min each time. A set of reactions containing RPo and EC are run on 10% SDS-PAGE and scanned in the TMR channel (excitation 530 nm, emission 580 nm) of a fluorescence scanner, (Typhoon Trio+, GE Healthcare). The amounts of YvrI, YvrHa, and σ70 in each of the complexes were quantitated from the fluorescence intensities of the corresponding protein bands on the fluorescence scanned gel, and the amount of RNAP was quantitated from the intensity of the ββ′ band on the same gel stained with Coomassie Blue. Previously a standard curve with intensity versus concentration was generated for each protein sample by running various amounts of each protein on a 10% SDS-PAGE and measuring the intensity of each band on the gel either scanned by fluorescence on the TMR channel or stained by Coomassie Blue. The fractional occupancies of YvrI, YvrHa, σ70, σ70-R2, and σ70-R4 with respect to RNAP were calculated from the ratio of the amount of the proteins to RNAP.
Determination of the Transcription Efficiency of RPo
To quantitate the amount of transcripts in stalled EC, an in vitro transcription assay was performed with each RPo as above but with 32P-labeled ATP. The samples were run on 12% urea PAGE and scanned by phosphorimaging (Typhoon Trio+, GE Healthcare). The amounts of transcripts were determined from the intensity of band for each stalled EC as described by Mukhopadhyay et al. (7). The amounts of open complexes formed were determined from the quantity of DNA bound to the beads by a fluorescence based assay with SyBr Gold as per the manufacturer's protocol (Life Technologies). The subpopulation of RPo competent to form EC (f) was determined from the ratio of the amount of transcripts to RPo.
The fractional occupancies of YvrI, YvrHa, σ70, σ70-R2, and σ70-R4 were corrected for the subpopulation of the RPo competent to form EC using the formula: EEC = (ERPo+NTPs − (1-f) ERPo)/f, where f = fraction of molecules competent to undergo the transition to elongation, E = fractional occupancies of YvrI, YvrHa, σ70, σ70-R2, and σ70-R4.
Fluorescence Anisotropy Assay
20 nm TMR-labeled YvrI and unlabeled YvrHa in 60 μl of TB was titrated with an increasing concentration of RNAP at 37 °C and the anisotropy values were monitored with excitation at 540 nm and emission at 580 nm using a PTI Fluorescence master QM400 system fitted with automatic polarizer (25, 26). The anisotropy values remained unchanged on the addition of unlabeled YvrHa to labeled YvrI, in the absence of RNAP (considered as anisotropy value (Ao) of free YvrI. When saturation was reached with the addition of around 50 nm RNAP, PoxdC promoter DNA was added at a concentration equal to the saturating level of RNAP core, to form the open complex. The addition of DNA did not change the anisotropy value (considered as A1 of fully bound YvrI).
Stalled elongation complexes (EC+11, EC+20, and EC+39) were formed by adding appropriate NTP and heparin (as indicated above) and incubating with RPo and the anisotropy values of the samples were monitored. The fractions of bound YvrI in RPo and ECs with anisotropy value A (with both free and bound molecules) were determined from the equation: fA0 +(1 − f) A1 = A, where f is the fraction of free molecules of YvrI.
The subpopulation of RPo competent to form EC was estimated using in vitro transcription assay as above but in solution. The fractional occupancies of YvrI and YvrHa in the stalled elongation complexes were corrected for the subpopulation of RPo competent to form EC using the formula mentioned earlier. A similar set of experiments were repeated with labeled YvrHa (and unlabeled YvrI) to determine the fractional occupancies of this protein in RPo and ECs.
Results
Previously the occupancy of σ relative to RNAP in the context of the open complex and elongation complex had been determined by separating the complexes from the free components either by gel electrophoresis or chromatography and subsequently analyzing their contents by SDS-PAGE and Coomassie staining. In this report, we use a similar technique that involves immobilization of open complex (RPo) on streptavidin beads using biotin-labeled promoter DNA fragments. First RNAP core and TMR-labeled σ or two-component σ factor were incubated to form RNAP holo before further incubation with promoter DNA fragments to form RPo. The beads were washed several times to remove unbound excess RNAP and σ. A part of the open complexes were used to form the stalled elongation complexes (EC+n, where n is the length of RNA) and further washed. To form EC, the promoter DNA fragment derivatives first having UTP or CTP residues at the position n+1 base on the non-template strand of the transcribed region (sequences listed in Table 2) were used to form the RPo and added with ATP, GTP, and CTP or UTP, so that RNAP synthesizes RNA of length n nucleotide and halts. Both RPo and EC+n were resolved on SDS-PAGE gel, scanned with a fluorescence imager at the TMR channel, stained with Coomassie Blue, and scanned further. The amounts of RNAP in the complexes were quantified by comparing the intensity of ββ′ band on the Coomassie-stained gel with the intensity of the same band of RNAP of known quantity run in parallel on the same gel. The amount of labeled YvrI and YvrHa (or σ) in the complexes were quantified in the same way by estimating the fluorescence intensity of the band on the fluorescently scanned gel. Because the size of YvrI and YvrHa is small (much less as compared with ββ′), the protein bands from the above complexes were not visible on the Coomassie-stained gel. Therefore these small proteins were labeled with fluorescent dye for better quantification. The labeled YvrI and YvrHa were active in transcription (results not shown). The fractional occupancy of these factors in RPo and EC were determined from the ratio of the amount of protein to RNAP in the complexes. The fractional occupancy was further corrected for the subpopulation of RPo that was unable to form EC. The amount of RPo was determined from the quantity of DNA bound to the beads as the DNA was the limiting factor in the formation of RPo. The amount of EC was determined from the quantity of radiolabeled transcripts formed with RPo using the in vitro transcription assay identical to the formation of EC as above, but with 32P-labeled NTP.
Study with E. coli σ70
Because it was previously shown that σ70 in E. coli is released upon transition from transcription initiation to elongation, we first tested whether the result was reproducible using the above assay. Two different promoter DNA fragments, lacUV5 and T7A1, were used to form RPo. Subsequently EC+23 and EC+15 were formed with T7A1 and lacUV5 promoter DNA fragments, respectively (Fig. 1A). Six replicates of a set of RPo and EC were formed for each promoter. Fractional occupancy of σ70 in these complexes were estimated for each set. The mean value of fractional occupancy of σ70 was found to be 0.61 for the T7A1 DNA fragment and 0.57 for the lacUV5 DNA fragment (Fig. 1B). Fractions of RPo that were competent to form elongation complexes were estimated for each set and the typical values were 0.55 of RPo in T7A1 and 0.60 in lacUV5 (Fig. 1C). After correcting the subpopulation that were competent to undergo the transition from transcription initiation to elongation, the fractional occupancy of σ70 was estimated to be 0.19 and 0.13 in T7A1 and lacUV5 promoters, respectively (Fig. 1D). Thus, the majority of σ70 was released from the elongation complexes on both promoters in this assay, consistent with previous observation that σ70 was released during transition from transcription initiation to elongation using a similar assay.
FIGURE 1.
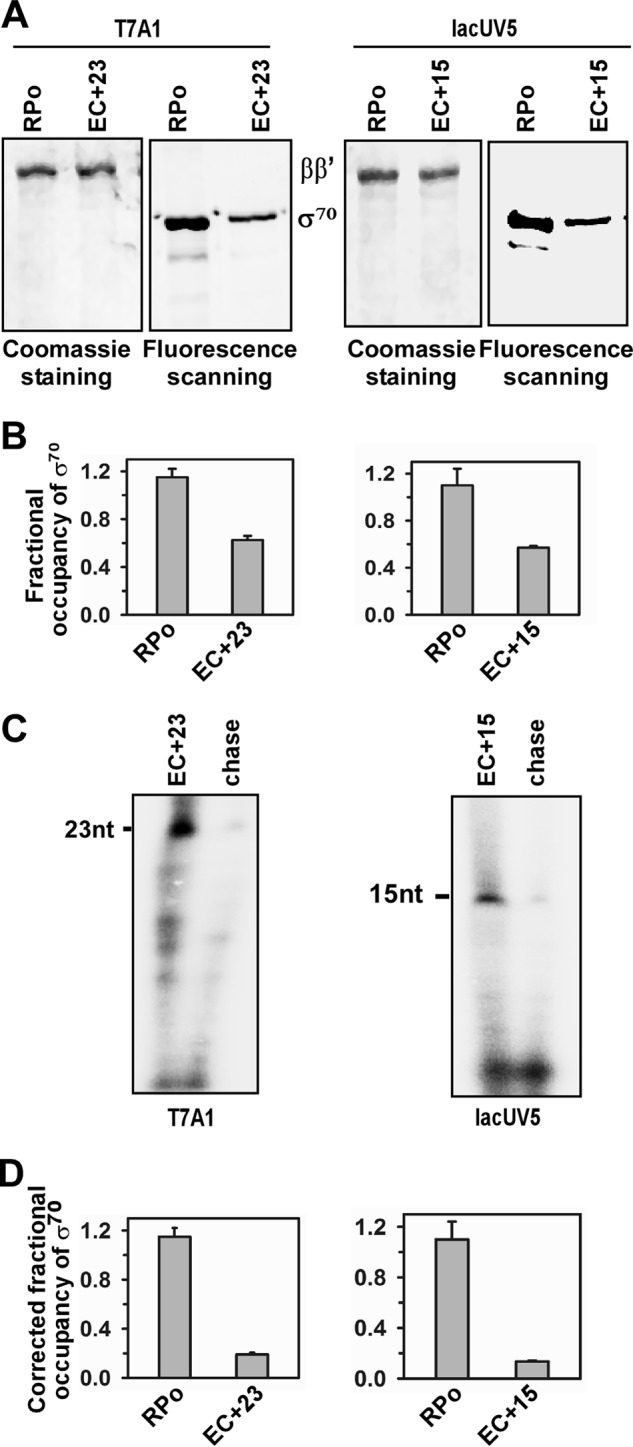
Fractional occupancy of σ70 of E. coli in EC on T7A1 and lacUV5 promoters. A, representative data assessing the components of RPo and EC. RPo were formed by incubating 200 nm Ec RNAP core, 500 nm each of TMR σ70, and 100 nm biotinylated promoter DNA fragment at 37 °C for 20 min, and immobilized on streptavidin beads. ECs were formed by adding NTP (indicated under ”Materials and Methods“). The beads containing RPo and EC were washed before resolving on 10% SDS-PAGE, followed by fluorescence scanning and Coomassie staining. Left panel, data for T7A1 promoter: EC+23 were formed. Right panel, data for lacUV5 promoter: EC+15 were formed. B, fractional occupancies of σ70 with respect to RNAP core: determined by quantifying the amount of each protein from the gel. Data were average of six replicates. Left panel, data for T7A1 promoter: EC+23 were formed. Right panel: data for lacUV5 promoter: EC+15 were formed. C, representative data for in vitro transcription assay with RPo immobilized on streptavidin beads. Stalled elongation complexes were generated using 32P-labeled NTP; chase, all four NTP were added to EC to produce runoff products. The samples were run on 12% urea PAGE and scanned on a phosphorimager. Left panel, data for T7A1 promoter: EC+23 were formed. Right panel, data for the lacUV5 promoter: EC+15 were formed. D, fractional occupancies of σ70 after correction for the subpopulation that are competent to form EC. Data were average of six replicates. Left panel, data for T7A1 promoter: EC+23 were formed. Right panel, data for lacUV5 promoter: EC+15 were formed.
Study with B. subtilis YvrI and YvrHa
We then tested the fate of two-component σ factors YvrI and YvrHa of B. subtilis during the transition from transcription initiation to elongation. RPo were formed on the PoxdC promoter DNA fragments with RNAP core, YvrI and YvrHa. Three PoxdC promoter DNA derivatives were used in the assay that permitted formation of EC+11, EC+20, and EC+39, respectively, using a subset of NTPs (Fig. 2A). The assay was repeated six times and the mean value of the fractional occupancies (Fig. 2B) of YvrI with respect to RNAP were estimated to be 0.85, 0.85, and 0.69 in EC+11, EC+20, and EC+39, respectively. The subpopulation of RPo competent to undergo transition to elongation was estimated to be 0.45 for EC+11, 0.36 for EC+20, and 0.30 for EC+39 (Fig. 2C). After correction, the fractional occupancies of YvrI were estimated to be 0.48 in EC+11, 0.16 in EC+20, and 0.0 in EC+39 (Fig. 2D). On the other hand, the fractional occupancy of YvrHa for all three ECs was estimated to be around 0.99 and 0.97, respectively, before and after the correction. Please note that as the fraction of RPo that was competent to undergo transition to elongation was estimated only from the intensity of stalled EC, not taking into account the intermediate paused EC, and therefore there could be a 10–20% error in the estimation, and these errors could be incorporated in the estimation of fractional occupancies of YvrI and YvrHa. The data indicate a gradual release of YvrI from RNAP as transcription elongation proceeds, whereas there is no release of YvrHa from RNAP for all the ECs.
FIGURE 2.

Fractional occupancies of YvrI and YvrHa of B. subtilis in EC on PoxdC promoter. A, representative data assessing the components of RPo and EC. RPo were formed by incubating 200 nm B, subtilis RNAP core, 500 nm each of TMR-labeled YvrI and YvrHa, and 100 nm biotinylated promoter PoxdC DNA fragment at 37 °C for 20 min, and immobilized on streptavidin beads. ECs were formed by adding NTP (indicated under ”Materials and Methods“). The beads containing RPo and EC were washed before resolving on 10% SDS-PAGE, followed by fluorescence scanning and Coomassie staining. B, fractional occupancy of YvrI and YvrHa with respect to the RNAP core: determined by quantifying the amount of each protein from the gel. Data were average of six replicates. Gray bar, YvrI; black bar, YvrHa. C, representative data for in vitro transcription assay with RPo immobilized on streptavidin beads. Stalled elongation complexes EC+11, EC+20, and EC+39 were generated using 32P-labeled NTP; chase, all four NTP were added to EC to produce runoff products. The samples were run on 12% urea PAGE and scanned on a phosphorimager. D, fractional occupancies of YvrI and YvrHa after correction for the subpopulation that are competent to form EC. Data were average of six replicates. Gray bar, YvrI; black bar, YvrHa.
Identical results were obtained with these factors using another promoter DNA PyvrI. Two elongation complexes, EC+10 and EC+19, were formed using the PyvrI promoter derivatives (Fig. 3A). The fractional occupancy of YvrI at EC+10 and EC+19 was estimated to be 0.79 and 0.72 before correction (Fig. 3C); and 0.45 and 0.11 after correcting for the subpopulation not competent to undergo transition to elongation (fraction of RPo efficient in transcription were 0.39 for EC+10 and 0.30 for EC+19, Fig. 3D). The fractional occupancy of YvrHa for the above ECs was estimated to be around 1 and 1.1, respectively, before and after the correction.
FIGURE 3.

Fractional occupancies of YvrI and YvrHa of B. subtilis in EC on PyvrI promoter. A, representative data assessing the components of RPo and EC. Same as described in the legend to Fig. 2A but with the PyvrI promoter. B, fractional occupancy of YvrI and YvrHa with respect to the RNAP core: determined by quantifying the amount of each protein from gel. Data were the average of six replicates. Gray bar, YvrI; black bar, YvrHa. C, representative data for the in vitro transcription assay with RPo immobilized on streptavidin beads. Stalled elongation complexes EC+10 and EC+19 were generated using 32P-labeled NTP; chase, all four NTP were added to EC to produce runoff products. The samples were run on 12% urea PAGE and scanned on a phosphorimager. D, fractional occupancies of YvrI and YvrHa after correction for the subpopulations that are competent to form EC. Data were average of six replicates. Gray bar, YvrI; black bar, YvrHa.
Study with B. subtilis YvrI and YvrHa Using Anisotropy
In addition to the above method that involved a separation step, we used another complimentary approach involving fluorescence anisotropy that does not involve any separation step for RPo or EC from the free components. As the fluorescence anisotropy value of a free molecule (Ao) is different from the anisotropy value of the molecule bound to another protein (A1), the fraction of free molecules (f) in a mixture of samples with anisotropy value A (both free and bound molecules) can be monitored from the equation: A = fAo + (1 − f)A1. The anisotropy values of TMR-labeled YvrI were monitored in the presence of an equal amount of unlabeled YvrHa (taken as an anisotropy value of free YvrI, Fig. 4A) before and after titration with RNAP core. Once saturation was reached, the PoxdC promoter DNA fragment was added (at a concentration equal to the saturating level of RNAP core) to form the open complex. After addition of DNA, the anisotropy value of the complex did not change and was taken as the anisotropy value of bound YvrI (Fig. 4A). Three sets of RPo and ECs (EC+11, EC+20, and EC+39) were formed, and the anisotropy values were monitored. Each set was repeated six times and the mean values of fractional occupancies of YvrI in ECs were estimated to be 0.75 for EC+11, 0.65 for EC+20, and 0.52 for EC+39 (Fig. 4C). The competent subpopulation of RPo efficient in forming ECs in solution was determined to be 0.42 for EC+11, 0.36 for EC+20, and 0.30 for EC+39 (Fig. 4D), which in turn, determined the corrected fractional occupancy of EC+11, EC+20, and EC+39, respectively, as 0.52, 0.22, and 0 (Fig. 4E). Similar assays were performed with TMR-labeled YvrHa and unlabeled YvrI (Fig. 4B), in which the fractional occupancy of YvrHa remained around 0.98 for all three ECs. Observation of the fluorescence anisotropy assay was consistent with the previous assay.
FIGURE 4.
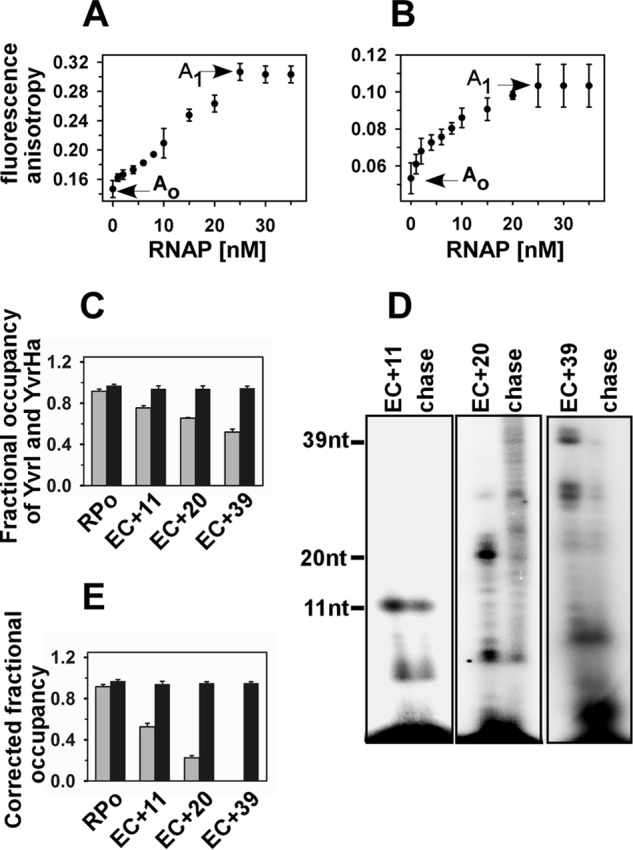
Fractional occupancy of YvrI and YvrHa of B. subtilis in EC:fluorescence anisotropy assay. A, fluorescence anisotropy values of 20 nm TMR-labeled YvrI (and 20 nm unlabeled YvrHa) upon titration with B. subtilis RNAP core. Ao (0.15) and A1 (0.3) were the values of free and fully bound YvrI, respectively. B, fluorescence anisotropy values of 20 nm TMR-labeled YvrHa (and 20 nm unlabeled YvrI) upon titration with B. subtilis RNAP core. Ao (0.051) and A1 (0.104) were the values of free and fully bound YvrHa, respectively. C, open complexes were formed by incubating 50 nm B. subtilis RNAP core, 20 nm each of labeled YvrI and YvrHa with 50 nm PoxdC promoter in solution at 37 ºC. Stalled elongation complexes are generated by adding 500 μm each of ApG, heparin, and other NTPs at +11 (without CTP), +20 (without UTP), and +39 (without UTP). The mean anisotropy values of RPo, EC+11, EC+20, and EC+39 were 0.297, 0.26, 0.24, and 0.22, respectively, for YvrI; 0.104, 0.102, 0.1, and 0.1, respectively, for YvrHa. The fractional occupancies of YvrI and YvrHa were determined by estimating the amount of bound proteins in RPo and EC from the anisotropy values of the protein in the respective complexes. Gray bar, YvrI; black bar, YvrHa. D, representative data for in vitro transcription assay in solution. Stalled elongation complexes EC+11, EC+20, and EC+39 were generated using 32P-labeled NTP; chase, all four NTP were added to EC to produce runoff products. The samples were run on 12% urea PAGE and scanned on a phosphorimager. E, fractional occupancies of YvrI and YvrHa after correction for the subpopulations that are competent to form EC. Data were average of six replicates. Gray bar, YvrI; black bar, YvrHa.
Study with E. coli σ70-R2 and σ70-R4
To test whether the promoter escape study with YvRI and YvrHa is valid for any other two-component σ factor, we prepared two truncated derivatives of σ70, comprising amino acids 130–500 of the σ region 2/3.1 (σ70-R2) and amino acids 501–613 of the σ region 3.2/4 (σ70-R4), respectively. Interestingly, these two truncated derivatives of σ70 are able to initiate transcription from the σ70-dependent promoter. Each protein derivative contained a single Cys residue and was labeled with TMR. The promoter escape study with these two σ70 derivatives was performed essentially as with YvrI and YvrHa of B. subtilis except that the lacUV5 promoter derivatives and E. coli RNAP core were used to form RPo and ECs (Fig. 5A). The mean value of the fractional occupancies of σ70-R4 with respect to RNAP was estimated to be 0.67, 0.56, and 0.49 in EC+15, EC+25, and EC+40, respectively (Fig. 5B). The subpopulations of RPo competent to undergo the transition to elongation were estimated to be 0.60 for EC+15, 0.52 for EC+25, and 0.45 for EC+40 (Fig. 5C). After correction, the fractional occupancies of σ70-R4 were estimated to be 0.45 in EC+15, 0.15 in EC+25, and 0.0 in EC+40 (Fig. 2D). On the other hand, the fractional occupancies of σ70-R2 for all three ECs were estimated to be around 0.99 and 0.98, respectively, before and after the correction.
FIGURE 5.

Fractional occupancies of E. coli σ70-R2 and σ70-R4 in EC on lacUV5 promoter. A, representative data assessing the components of RPo and EC. RPo and ECs were formed as described in the legend to Fig. 2A. The beads containing RPo and EC were washed before resolving on a 4–20% gradient SDS-PAGE, followed by fluorescence scanning and Coomassie staining. B, fractional occupancy of σ70-R2 and σ70-R4 with respect to RNAP core: determined by quantifying the amount of each protein from the gel. Data were the average of six replicates. Gray bar, σ70-R2; black bar, σ70-R4. C, representative data for in vitro transcription assay with RPo immobilized on streptavidin beads. Stalled elongation complexes EC+15, EC+25, and EC+40 were generated using 32P-labeled NTP; chase, all four NTP were added to EC to produce runoff products. The samples were run on 12% urea-PAGE and scanned on phosphorimager. D, fractional occupancies of σ70-R2 and σ70-R4 after correction for the subpopulation that are competent to form EC. Data were average of six replicates. Gray bar, σ70-R2; black bar, σ70-R4.
Discussion
Our results show that the amount of YvrI relative to RNAP is gradually decreased in the elongation complexes as elongation proceeds, whereas the amount of YvrHa relative to RNAP remains constant in the elongation complexes (Fig. 6). Because a certain amount of YvrI is present in the early elongation complexes (fractional occupancy is 0.48 at EC+11), our result is consistent with previous observations that shows a significant fraction of σ70 remains associated with RNAP in the early elongation complex (6, 12). The result (fractional occupancy of YvrI are 0.48, 0.16, and 0, respectively for EC+11, EC+20, and EC+39) is also consistent with the observation that demonstrated the stochastic release of σ in transcription elongation (11). The RNAP structure-based proposed model for promoter escape predicts that, upon transition from transcription initiation to elongation, the interaction of σR4 with RNAP is destabilized upon transition to elongation reducing the overall affinity of σ to RNAP and this accounts for stochastic release of σ. However, as the destabilization of interactions of σR4 with RNAP in EC results in the release of intact σ70, assessing the overall interaction of this σ region with RNAP during the promoter escape is not possible using a simple biochemical assay. In principle, assessing the above interaction could be possible using a FRET assay involving fluorescently labeled RNAP and σ derivatives. However, this assay could be technically challenging because of the difficulty in labeling RNAP at specific sites. Thus validation of the proposed model for promoter escape remains a difficult task. The two-component σ factor YvrI and YvrHa individually contribute to the functions of σR4 and σR2 to an RNAP-promoter complex. Using simple biochemical and biophysical assays we show that YvrI (that mimics σR4) is released from EC due to the loss of its interaction with RNAP, whereas YvrHa (that mimics σR2) is retained in the EC. Similar results were obtained with the truncated σ70 derivatives: σ70-R4 is released, whereas σ70-R2 is retained in the EC. Therefore, our results validate the proposed model for promoter escape in bacteria. As YvrHa is not covalently attached to YvrI, the release of YvrI does not alter the interaction of YvrHa with RNAP in EC. This further suggests that there is no or little change in the interaction between σR2 with RNAP upon transition to elongation. But this interaction is not strong enough to retain the whole σ70 in the elongation complex as the rest of σ regions, σR1.1, σR2, and σR3/4 linker, lose their interaction with RNAP. However, the observation raises the possibility that the σR2-RNAP interaction of a certain σ factor from other bacterial species could be strong enough to retain the σ factor in the EC throughout the elongation phase.
FIGURE 6.
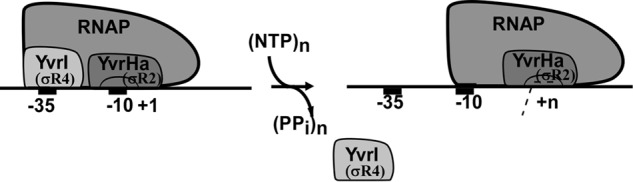
Proposed model for promoter escape: YvrI (σR4) is released from RNAP, whereas YvrHa (σR2) is retained by RNAP upon transition from transcription initiation to elongation.
Author Contributions
J. M. conceived and designed the experiments; S. S. performed the experiments; S. S. preformed PAGE analysis, in vitro transcription assays, protein labeling, and fluorescence anisotropy assays, R. K. P. purified the B. subtilis RNAP; S. S. analyzed the data; and S. S. and J. M. wrote the paper.
Acknowledgment
We thank Dr. Runa Sur (University of Calcutta) for critically reading the manuscript and comments.
This work was supported in part by Research Grant BT/PR 5345/MED/29/648/2012 from the Department of Biotechnology, Ministry of Science and Technology, India. The authors declare that they have no conflicts of interest with the contents of this article.
- RNAP
- RNA polymerase
- EC
- elongation complex
- σR2
- σRegion2
- σR4
- σRegion4.
References
- 1.Burgess R. R., Travers A. A., Dunn J. J., and Bautz E. K. (1969) Factor stimulating transcription by RNA polymerase. Nature 221, 43–46 [DOI] [PubMed] [Google Scholar]
- 2.Hansen U. M., and McClure W. R. (1980) Role of the sigma subunit of Escherichia coli RNA polymerase in initiation: II. release of σ from ternary complexes. J. Biol. Chem. 255, 9564–9570 [PubMed] [Google Scholar]
- 3.Straney D. C., and Crothers D. M. (1985) Intermediates in transcription initiation from the E. coli lac UV5 promoter. Cell 43, 449–459 [DOI] [PubMed] [Google Scholar]
- 4.Krummel B., and Chamberlin M. J. (1989) RNA chain initiation by Escherichia coli RNA polymerase: structural transitions of the enzyme in early ternary complexes. Biochemistry 28, 7829–7842 [DOI] [PubMed] [Google Scholar]
- 5.Metzger W., Schickor P., Meier T., Werel W., and Heumann H. (1993) Nucleation of RNA chain formation by Escherichia coli DNA-dependent RNA polymerase. J. Mol. Biol. 232, 35–49 [DOI] [PubMed] [Google Scholar]
- 6.Mukhopadhyay J., Kapanidis A. N., Mekler V., Kortkhonjia E., Ebright Y. W., and Ebright R. H. (2001) Translocation of σ(70) with RNA polymerase during transcription: fluorescence resonance energy transfer assay for movement relative to DNA. Cell 106, 453–463 [DOI] [PubMed] [Google Scholar]
- 7.Mukhopadhyay J., Mekler V., Kortkhonjia E., Kapanidis A. N., Ebright Y. W., and Ebright R. H. (2003) Fluorescence resonance energy transfer (FRET) in analysis of transcription-complex structure and function. Methods Enzymol. 371, 144–159 [DOI] [PubMed] [Google Scholar]
- 8.Nickels B. E., Mukhopadhyay J., Garrity S. J., Ebright R. H., and Hochschild A. (2004) The σ 70 subunit of RNA polymerase mediates a promoter-proximal pause at the lac promoter. Nat. Struct. Mol. Biol. 11, 544–550 [DOI] [PubMed] [Google Scholar]
- 9.Brodolin K., Zenkin N., Mustaev A., Mamaeva D., and Heumann H. (2004) The σ 70 subunit of RNA polymerase induces lacUV5 promoter-proximal pausing of transcription. Nat. Struct. Mol. Biol. 11, 551–557 [DOI] [PubMed] [Google Scholar]
- 10.Wade J. T., and Struhl K. (2004) Association of RNA polymerase with transcribed regions in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 101, 17777–17782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raffaelle M., Kanin E. I., Vogt J., Burgess R. R., and Ansari A. Z. (2005) Holoenzyme switching and stochastic release of σ factors from RNA polymerase in vivo. Mol. Cell 20, 357–366 [DOI] [PubMed] [Google Scholar]
- 12.Kapanidis A. N., Margeat E., Laurence T. A., Doose S., Ho S. O., Mukhopadhyay J., Kortkhonjia E., Mekler V., Ebright R. H., and Weiss S. (2005) Retention of transcription initiation factor σ70 in transcription elongation: single-molecule analysis. Mol. Cell 20, 347–356 [DOI] [PubMed] [Google Scholar]
- 13.Bar-Nahum G., and Nudler E. (2001) Isolation and characterization of σ70-retaining transcription elongation complexes from Escherichia coli. Cell 106, 443–451 [DOI] [PubMed] [Google Scholar]
- 14.Mooney R. A., and Landick R. (2003) Tethering σ70 to RNA polymerase reveals high in vivo activity of σ factors and σ70-dependent pausing at promoter-distal locations. Genes Dev. 17, 2839–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mekler V., Kortkhonjia E., Mukhopadhyay J., Knight J., Revyakin A., Kapanidis A. N., Niu W., Ebright Y. W., Levy R., and Ebright R. H. (2002) Structural organization of bacterial RNA polymerase holoenzyme and the RNA polymerase-promoter open complex. Cell 108, 599–614 [DOI] [PubMed] [Google Scholar]
- 16.Vassylyev D. G., Sekine S., Laptenko O., Lee J., Vassylyeva M. N., Borukhov S., and Yokoyama S. (2002) Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature 417, 712–719 [DOI] [PubMed] [Google Scholar]
- 17.Murakami K. S., Masuda S., Campbell E. A., Muzzin O., and Darst S. A. (2002) Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science 296, 1285–1290 [DOI] [PubMed] [Google Scholar]
- 18.Murakami K. S., and Darst S. A. (2003) Bacterial RNA polymerases: the wholo story. Curr. Opin. Struct. Biol. 13, 31–39 [DOI] [PubMed] [Google Scholar]
- 19.Gill S. C., Weitzel S. E., and von Hippel P. H. (1991) Escherichia coli σ70 and NusA proteins: I. binding interactions with core RNA polymerase in solution and within the transcription complex. J. Mol. Biol. 220, 307–324 [DOI] [PubMed] [Google Scholar]
- 20.MacLellan S. R., Wecke T., and Helmann J. D. (2008) A previously unidentified σ factor and two accessory proteins regulate oxalate decarboxylase expression in Bacillus subtilis. Mol. Microbiol. 69, 954–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacLellan S. R., Helmann J. D., and Antelmann H. (2009) The YvrI alternative σ factor is essential for acid stress induction of oxalate decarboxylase in Bacillus subtilis. J. Bacteriol. 191, 931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacLellan S. R., Guariglia-Oropeza V., Gaballa A., and Helmann J. D. (2009) A two-subunit bacterial σ-factor activates transcription in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 106, 21323–21328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X., and Lewis P. J. (2008) Overproduction and purification of recombinant Bacillus subtilis RNA polymerase. Protein Expr. Purif. 59, 86–93 [DOI] [PubMed] [Google Scholar]
- 24.Kim Y., Ho S. O., Gassman N. R., Korlann Y., Landorf E. V., Collart F. R., and Weiss S. (2008) Efficient site-specific labeling of proteins via cysteines. Bioconjug. Chem. 19, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudra P., Prajapati R. K., Banerjee R., Sengupta S., and Mukhopadhyay J. (2015) Novel mechanism of gene regulation: the protein Rv1222 of Mycobacterium tuberculosis inhibits transcription by anchoring the RNA polymerase onto DNA. Nucleic Acids Res. 43, 5855–5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moerke N. J. (2009) Fluorescence polarization (FP) assays for monitoring peptide-protein or nucleic acid-protein binding. Curr. Protoc. Chem. Biol. 1, 1–15 [DOI] [PubMed] [Google Scholar]