

Background: Exposure and phosphorylation of the core protein C-terminal domain (CTD) regulate important viral functions.
Results: Empty capsids contain at least two populations of CTDs with different rates of exposure that are influenced by phosphorylation.
Conclusion: Adding negative charge to CTDs increases capsid stability and decreases CTD exposure.
Significance: Phosphorylation is used to tune HBV core protein function.
Keywords: cryo-electron microscopy; hepatitis B virus (HBV, Hep B); mass spectrometry (MS); phosphorylation; protein dynamics; capsid stability; limited proteolysis
Abstract
Hepatitis B virus core protein has 183 amino acids divided into an assembly domain and an arginine-rich C-terminal domain (CTD) that regulates essential functions including genome packaging, reverse transcription, and intracellular trafficking. Here, we investigated the CTD in empty hepatitis B virus (HBV) T=4 capsids. We examined wild-type core protein (Cp183-WT) and a mutant core protein (Cp183-EEE), in which three CTD serines are replaced with glutamate to mimic phosphorylated protein. We found that Cp183-WT capsids were less stable than Cp183-EEE capsids. When we tested CTD sensitivity to trypsin, we detected two different populations of CTDs differentiated by their rate of trypsin cleavage. Interestingly, CTDs from Cp183-EEE capsids exhibited a much slower rate of proteolytic cleavage when compared with CTDs of Cp183-WT capsids. Cryo-electron microscopy studies of trypsin-digested capsids show that CTDs at five-fold symmetry vertices are most protected. We hypothesize that electrostatic interactions between glutamates and arginines in Cp183-EEE, particularly at five-fold, increase capsid stability and reduce CTD exposure. Our studies show that quasi-equivalent CTDs exhibit different rates of exposure and thus might perform distinct functions during the hepatitis B virus lifecycle. Our results demonstrate a structural role for CTD phosphorylation and indicate crosstalk between CTDs within a capsid particle.
Introduction
Hepatitis B virus (HBV)2 is an enveloped double-stranded DNA virus that causes chronic infection in more than 240 million people and can lead to liver failure, cirrhosis, and hepatocellular carcinoma (1, 2). HBV has an unusual life cycle that is characterized by packaging of viral pregenomic RNA and, within the virus particle, reverse transcription of the RNA genome into a circular, partially double-stranded DNA (3).
The HBV capsids are formed by 120 or 90 copies of the core protein that assembles into a T=4 or T=3 sized particle, respectively, with T=4 capsids being the predominant form in vivo (4). The core protein has 183 residues, which can be divided into an assembly domain (amino acids 1–149) and a C-terminal domain (CTD) (amino acids 150–183) (Fig. 1). The CTD plays an essential role in regulating multiple steps during the HBV life cycle such as packaging of the RNA genome, reverse transcription, and intracellular trafficking (5, 6). The CTD contains 16 arginine residues and a total of 7 serine residues (Fig. 1). Three of these serine residues, Ser-155, Ser-162, and Ser-170, have been shown to be important phosphorylation sites in regulating reverse transcription (7–9).
FIGURE 1.

Schematic of the HBV core protein showing the assembly domain (residues 1–149) and CTD (residues 150–183). The CTD contains a total of 16 arginines, 6 serines, and 1 threonine in 34 residues. The serines mutated to make the phosphorylation mimic Cp183-EEE are bolded and underlined. The amino acid sequence numbers are indicated above the sequence. Shown below the sequence is the predicted likelihood of trypsin cleavage for specific arginine residues calculated by the ExPASy peptide cutter software. Arginine residues that are surrounded by other arginine residues are cleaved with a likelihood of 31%.
In capsids, CTD are clustered around five-fold and quasi-six-fold vertices on the capsid. Studies have suggested that the CTDs can be transiently exposed to the capsid exterior (10–14). It is speculated that exposure of the CTDs is regulated by the phosphorylation state of the core protein and/or the level of genome maturation (10, 15). Indeed, cryo-electron microscopy (cryo-EM) studies of capsid from wild-type core protein and a phosphorylation mimic, Cp183-EEE, carrying three serine to glutamate mutations (S155E, S162E, and S170E), have shown that phosphorylation influences the organization of the CTDs (16). Large stalactite-like structures pointing to the capsid center of empty and RNA-filled capsids were observed in cryo-EM structures of Cp183-EEE capsids. CTDs of Cp183-WT capsids, however, were more disordered (16).
Here we investigated the stability of capsids as well as the exposure of the CTDs based on the phosphorylation state of the core protein. We found that Cp183-EEE capsids are more stable than Cp183-WT capsids. We also found that the EEE mutation affected the rate and distribution of rates for CTD proteolysis. We hypothesize that the difference in exposure can be attributed to CTDs at the five-fold symmetry axes. This is supported by correlation of cryo-EM structures with trypsin digestion. We hypothesize that changes in capsid reactivity are mediated by interactions between CTD arginines and glutamates (or by extension phosphates).
Experimental Procedures
Capsid Preparation
Cp183 was expressed and purified as described previously by Porterfield et al. (17). Briefly, the 183 amino acid HBV core protein was expressed in Escherichia coli using a pet11-based vector. The cells were lysed by sonication, and cell debris was removed by centrifugation for 1 h at 9000 × g. Capsids were pelleted into a sucrose cushion at 200,000 × g for 1 h and further purified by size exclusion chromatography using a 1-liter Sepharose® CL-4B column equilibrated in 20 mm Tris, pH 7.5, 5% Sucrose, 1 mm EDTA, 2 mm DTT at 4 °C. Capsids were concentrated by ammonium sulfate precipitation and further purified by size exclusion chromatography using a 1.5-liter Sephacryl® S-300 column equilibrated in 20 mm Tris, pH 7.5, 5% sucrose, 1 mm EDTA, 2 mm DTT at 4 °C. RNA-filled capsids were concentrated to 2–3 mg/ml and stored at −80 °C.
To obtain empty Cp183-WT and EEE T=4 capsids, RNA-filled capsids were disassembled by dialysis overnight into disassembly buffer (20 mm Tris-HCl, pH 7.5, 1.5 m guanidine HCl, 0.5 m LiCl, 10 mm DTT) at 4 °C. The RNA was pelleted by centrifugation for 1 min at 14,000 × g. Cp183 dimers were purified by size exclusion chromatography using a 250-ml Sephacryl® S-300 column equilibrated in disassembly buffer and concentrated to 1–1.5 mg/ml. Cp183 dimers were reassembled by dialysis overnight into assembly buffer (20 mm Tris-HCl, pH 7.5, 450 mm NaCl, 10 mm DTT) at 4 °C. Capsids were purified by size exclusion chromatography using a 250-ml Sephacryl® S-300 column equilibrated in assembly buffer and concentrated to 1–1.5 mg/ml. 2 mg of capsids was loaded onto a 10–40% continuous sucrose gradient in 50 mm HEPES, pH 7.5, 300 mm NaCl. The T=4 band was extracted, and capsids were dialyzed into assembly buffer at 4 °C and concentrated to 0.7 mg/ml.
Trypsin Preparation
Sequencing grade modified porcine trypsin (Promega) was dissolved in resuspension dilution buffer (0.1 mm HCl) to a final concentration of 3–4 μm, aliquoted into 15-μl aliquots, and stored at −80 °C.
Turbidity Measurements
Purified Cp183-WT and Cp183-EEE T=4 capsids were dialyzed into buffers containing 20 mm Tris, pH 7.5, 10 mm DTT with varying concentrations of NaCl (50–450 mm) for 16 h at 4 °C. After 16 h, the absorbance at 280 nm was recorded using a UV-visible spectrometer and a quartz cuvette with a path length of 1 cm.
Differential Scanning Fluorimetry (DSF)
Fluorescence thermal scans of Cp183-WT and EEE capsids were performed in citric acid-disodium hydrogen phosphate buffer containing 450 mm NaCl at pH 7, 5, and 4. Different pH buffers were prepared by adjusting the ratio of citrate (0.1 m) to phosphate (0.2 m) stock: for pH 4 (30.5% citrate/19.2% phosphate), pH 5 (24.25% citrate/25.5% phosphate), and pH 7 (8.8% citrate/41.2% phosphate). Capsid samples were diluted into citrate phosphate buffer to a final concentration of 1 μm capsid. To each sample, 2.5 μl of 1% SYPRO® Orange dye (Invitrogen, S6651) was added to a final volume of 25 μl. DSF experiments were performed in a quantitative PCR instrument (Corbett Research, RG-3000) with temperature elevating from 25 to 99 °C, increasing 1°/min. Lysozyme was run as positive control at a final concentration of 0.2 mg/ml. The first derivative (dF/dT, the rate of change in fluorescence over temperature) from the fluorescence signal was used to calculate the melting temperature of each capsid. For further details on DSF of viruses, see Ref. 18.
LC-MS
Trypsin digestion of Cp183-WT and EEE capsids was monitored using an Agilent 1290 UPLC coupled to a micro-TOF spectrometer (Bruker Daltonics). Reverse-phase chromatography relied on an Agilent PLRP-S 100 Å, 3-μm column (50 × 1.0 mm) with the following gradient: 1 min, 5% B; 1–12 min, 5–50% B; 12–12.5 min, 50–95% B; 12.5–14 min, 95% B; 14–14.5 min, 5% B. Solvent A consisted of 0.1% formic acid in water, and solvent B consisted of 0.1% formic acid in acetonitrile at a flow rate of 600 μl/min. The buffers, column, and auto sampler compartment were maintained at 25 °C. Electrospray conditions were: drying gas, 6.5 liter/min; drying temperature, 180 °C; capillary exit, 150 V. For each reaction, a final ratio of 10 μm capsid (measured as dimer concentration) and 0.12 μm trypsin was used. Reactions were carried out in the autosampler for 1 h with 10 μl of reaction mixture injected every 15 min. Data processing and deconvolution analysis were performed with the help of Bruker Data Analysis package version 4.0. The maximum entropy deconvolution algorithm was used to determine the capsid mass before and after digestion. The spectral range (900 m/z to 1700 m/z) was selected for deconvolution, which covers the majority of subunit charge envelope.
Dynamic Light Scattering
Dynamic light scattering experiments were performed using a Zetasizer Nano-ZS from Malvern with a 3-mm quartz cuvette with a volume of 45 μl. 0.12 μm trypsin were added to 10 μm Cp183-WT or EEE capsids (measured as dimer concentration), and size measurements were performed after 7.5, 15, 22.5, 30, 60, and 120 min at 23 °C. For each time point, three independent experiments were performed, each based on 15 measurements with a duration of 10 s each.
Tris-Tricine SDS-PAGE
For analysis by Tris-Tricine SDS-PAGE, we used a 6 × 6-cm acrylamide gel consisting of a 16% resolving gel (∼4 cm) overlaid with a 10% resolving and a 4% stacking gel (∼1 cm each). Gels were cast using a freshly prepared acrylamide solution containing 49.5% T and 3% C, based on the protocol described in Schägger et al. (19). The gel was run in a Bio-Rad protean XL gel box with 100 mm Tris-HCl, pH 8.9, as anode buffer and 100 mm Tris-HCl, 100 mm Tricine, pH 8.25, as cathode buffer as described by Schägger et al. (19). The gel was run for a total of 25 h at a constant voltage of 90 V overnight (∼14 h) and at 200 V for an additional 10 h during the next day.
For staining, the gel was placed in fixing solution (10% acetic acid, 50% methanol) overnight and incubated with fluorescent gel stain SYPRO® Ruby for 3 h. The gel was washed for 30 min in 7% acetic acid and 10% methanol again and washed three times with water before imaging. Gels were imaged using Gel DocTM XR station (Bio-Rad). Bands were quantified using the Image LabTM software (Bio-Rad).
Curve Fitting
Curve fitting was performed using the curve fitting tool in OriginPro using a function describing two or three first order exponential decays with the format
For the Cp183-WT, the data were fit to a function with two exponential decays with values for A1 and A2, which were held constant at 25 and 75%. A good fit was achieved with decay constants of k1 = 0.024 min−1 and k2 = 0.276 min−1.
The fitted function for the WT data was
For the Cp183-EEE data, a similar function keeping A1 and A2 constant at 25 and 75% did not result in a good fit. We tried fitting the data to three exponential decay functions with A1 = 25%, A2 = 25%, and A3 = 50%. This function resulted in a good fit with almost identical values for k2 and k3. Thus, we proceeded to fit the data to a function containing two exponential decays with: k1 = 0.0077 min−1 and k2 = 0.456 min−1.
The fitted function for the WT data were
Cryo-EM
To halt trypsin digestions, samples were treated with 0.2 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF). Samples of undigested or digested Cp183-WT and EEE capsids were concentrated to 8–10 mg/ml, and frozen-hydrated cryo-EM specimens were prepared as described previously (20). Briefly, a drop containing 4 μl of HBV sample solution was applied on a glow-discharged continuous carbon-coated grid or Quantifoil® holey carbon grid. The grid was then plunged into liquid ethane using an FEI Vitrobot® Mk III. The grid was transferred into a Gatan 626 cryo-holder and kept at the liquid nitrogen temperature (−177.8 °C) for the subsequent processing. Cryo-EM micrographs were recorded under low-dose conditions (< 25 electrons/Å2) using a Gatan UltraScan® US4000 4k × 4k CCD camera at a nominal magnification of 120,000× on a JEOL JEM-3200FS electron microscopy operated at 300 kV. The image was acquired using the in-column energy filter with a slit width of 20 eV. The resulting pixel is 1.1 Å at the specimen.
Image Processing
The program e2boxer.py (21) was used to select 6706 particles from 95 cryo-EM micrographs for Cp183-EEE, 4797 particles from 100 cryo-EM micrographs for trypsin-digested Cp183-EEE (15 min), 8856 particles from 280 cryo-EM micrographs for trypsin-digested Cp183-EEE (2 h), and 6420 particles from 99 cryo-EM micrographs for trypsin-digested Cp183-WT (15 min). The defocus level was estimated using CTFFIND3 (22). The three-dimensional reconstruction process was computed using images that were corrected for phase reversals. The starting model for each data set was built de novo using the random model method of AUTO3DEM (23, 24). The initial origins and orientations search for each particle was carried out iteratively using the parallel polar Fourier transformation (PPFT) algorithm and refined by the parallel origin and orientation refinement (PO2R) algorithm. The three-dimensional reconstructions were computed using parallel three-dimensional reconstruction (P3DR), imposing 532 icosahedral symmetry. To estimate the resolution of the three-dimensional reconstruction, at the final refinement step, the dataset was evenly divided to compute two three-dimensional reconstructions. The resolution was estimated using Fourier shell correlation by assessing the agreement between these two reconstructions in the Fourier space at a coefficient value of 0.5. The estimated resolutions for Cp183-EEE, trypsin-digested Cp183-EEE (15 min), trypsin-digested Cp183-EEE (2 h), and trypsin-digested Cp183-WT (15 min) were 8.4, 8.6, 8.1, and 8.4 Å, respectively. To build an electron density map for Cp149, the atomic coordinate for a full HBV capsid (Protein Data Bank (PDB) entry 1QGT) was downloaded from VIPERdb (25). The density map was computed using e2pdb2mrc.py (21) and low-pass-filtered to 9 Å. The three-dimensional reconstructions were rendered and visualized using Robem (23) and UCSF Chimera (26). The contour level for Cp183-EEE was chosen using a 100% mass of estimated particle volume. The contour level for the trypsin-digested HBV capsid was rendered at the contour where the atomic model of HBV (PDB entry 1QGT) was fully covered by the electron density map.
Results
Cp183-EEE Capsids Are More Stable than Cp183-WT Capsids
Capsid assembly and stability are highly dependent on ionic strength as shown by assembly experiments using the truncated version of the core protein Cp149 (27). For the full-length protein, Cp183, high ionic strength also plays an important role in shielding repulsive positive charges from arginine residues of the CTD (17).
To investigate the effects of CTD phosphorylation, we utilized a mutant core protein Cp183-EEE, bearing three serine to glutamate mutations (S155E, S162E, and S172E), mimicking phosphorylation. It is highly unlikely that glutamate mutations will be found during natural infections with HBV; however, glutamate mutations have been widely used to study constitutive phosphorylation of the core protein CTD (9, 17, 28, 29).
We first assessed the stability of Cp183-EEE and Cp183-WT capsids as a function of ionic strength using purified in vitro assembled empty T=4 particles. Transmission electron microscopy showed that Cp183-WT and Cp183-EEE capsids were uniform and morphologically indistinguishable under negative stain. To monitor capsid stability, we performed disassembly experiments based on the propensity of free Cp183 dimers to aggregate in buffers that do not contain denaturants such as urea or guanidine-HCl (17). To modulate capsid stability, we dialyzed 10 μm empty Cp183-WT and Cp183-EEE T=4 capsids into buffers of decreasing ionic strength from 450 to 50 mm NaCl at 4 °C for 16 h. We note that all capsid concentrations in this study are displayed in terms of concentration of dimer. Our result showed that Cp183-WT capsids started aggregating at an ionic strength of 250 mm (Fig. 2A). Cp183-EEE capsids, however, remained intact and soluble down to an ionic strength of about 100 mm NaCl, indicating that Cp183-EEE capsids are more stable when compared with Cp183-WT capsids under low ionic strength.
FIGURE 2.
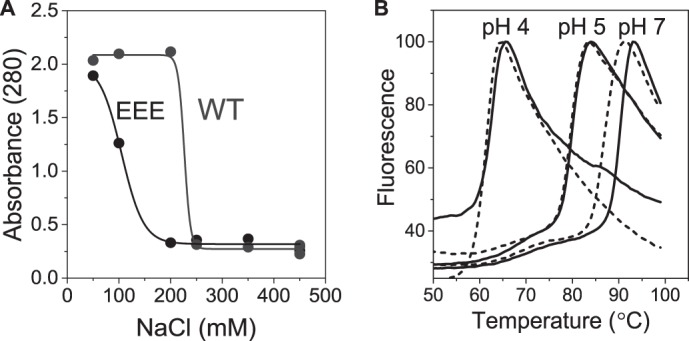
Cp183-WT capsids are less stable than Cp183-EEE. A, representative series of turbidity measurements of Cp183-WT and Cp183-EEE dialyzed for 16 h into 20 mm Tris, pH 7.5, containing 50–450 mm NaCl. An increase in turbidity indicates aggregation of capsid protein released by capsid disassembly. Sigmoidal black and gray lines were plotted to guide the eye. B, differential scanning fluorescence monitoring the thermal dissociation/unfolding of Cp183-EEE (solid lines) and Cp183-WT (dashed lines) capsids through binding of SYPRO® Orange. The melting temperatures are 61.1 ± 0.6 and 63 ± 0.5°C at pH 4, 80 ± 0.2 and 80.5 ± 0.5 °C at pH 5, and 86.8 ± 0.5 and 89.8 ± 0.1°C at pH 7 for Cp183-WT and Cp183-EEE, respectively. DSF data are the normalized average of three independent experiments.
To confirm these results, we assessed the thermal stability of Cp183-WT and Cp183-EEE capsids using DSF. In DSF, dissociation of the subunits and unfolding of the protein can be correlated to an increase in fluorescence of a dye binding in hydrophobic pockets that become accessible during heating. The dissociation/unfolding of 1 μm Cp183-WT and Cp183-EEE capsids in 450 mm NaCl were monitored at three different pHs.
Despite a small difference in the observed values, Cp183-EEE capsids consistently exhibited higher melting temperatures when compared with Cp183-WT capsids (Fig. 2B). The melting temperatures were ∼2 °C higher for Cp183-EEE capsids than for Cp183-WT capsids at pH 7. Both capsids became progressively less stable at lower pH, presumably due to an increase in amino acid protonation.
CTDs Are More Readily Exposed in Cp183-WT than in Cp183-EEE Capsid
We hypothesized that the stability of Cp183-EEE capsids could be attributed to increased interactions of the CTDs inside the capsid particle (16). This also suggests a difference in CTD exposure between Cp183-EEE and Cp183-WT capsids.
To investigate CTD dynamics, we assessed the exposure of the CTDs via limited proteolysis using trypsin. If exposed, the arginine-rich CTDs are predicted to be an excellent substrate for trypsin (Fig. 1). The introduced glutamate residues Glu-155, Glu-162, and Glu-170 are in the P3 and P4′ positions (3 residues N-terminal or 4 residues C-terminal) with respect to the nearest arginine peptide bond. These positions are not likely to have a significant impact on the intrinsic rate of cleavage given their distance from the peptide bond and the position where glutamate has been inserted (30–32). Conversely, with dimensions of ∼43 × 39 × 30 Å, a trypsin molecule is unlikely to enter the capsid shell whose pores have diameters of 10–27 Å (33). Thus, by conducting the experiments using an excess concentration of trypsin, the rate of CTD cleavage can directly be related to the rate of CTD exposure (30, 31).
We incubated 10 μm empty Cp183-EEE and Cp183-WT T=4 capsids with 0.12 μm trypsin for 2 h. Reactions were stopped by rapid heat denaturation, and the cleavage products were identified by LC-MS (Fig. 3) (original LC-MS data in supplemental Fig. 1). In these experiments, we used capsids that had disulfide-linked dimers. Because each dimer subunit contains two CTDs, two cuts per dimer subunit were possible. This allowed us to test for cooperativity of cleavage (Fig. 3).
FIGURE 3.

LC-MS shows a difference in cleavage pattern between Cp183-WT and Cp183-EEE capsids. A and B, LC-MS of trypsin cleavage of 10 μm Cp183-WT (A) and Cp83-EEE (B) capsids with 0.12 μm trypsin. Panels show the normalized deconvoluted masses in Da before and after digestion of capsids for 15, 30, 45, and 60 min. The y axis denotes normalized intensity of the deconvoluted mass. A single asterisk indicates cleavage of a monomer within a dimer, while a double asterisk indicates cleavage of both monomers within a dimer. The numbers in brackets denote the site N-terminal of trypsin cleavage.
Most importantly, the overall pattern of cleavage could be clearly mapped. The masses for a full-length uncut dimer (1–183) and a dimer fragment that was cut symmetrically on both monomer subunits at position 157 (1–157**) could be assigned. The double asterisk indicates that both subunits are cleaved; a single asterisk indicates only one chain is cut. The data clearly showed the depletion of the full-length Cp183-WT and Cp183-EEE dimer and the accumulation of 1–157** over time (Fig. 3, A and B). For Cp183-EEE, at later time points, a dimer subunit that was cut asymmetrically, having only one cleavage at position 150 (1–150*), became visible at low abundance (Fig. 3B).
In Cp183-WT capsids, full-length intact dimers were absent within 30 min of digestion, whereas in Cp183-EEE capsids, some full-length dimers persisted for at least 60 min (Fig. 3). This difference in digestion rate does not arise from Cp183-WT capsids falling apart, however. Dynamic light scattering experiments on Cp183-WT and Cp183-EEE T=4 capsids showed that both capsid particles stayed intact with no measurable quantity of aggregate formed during a 2-h digestion with trypsin (Fig. 4).
FIGURE 4.

Capsid particles stay intact during the 2-h-long trypsin digestion. A and B, dynamic light scattering measurements of the Z-average diameter (A) and the light scattering intensity (derived count rate) (B) for a sample of 10 μm Cp183-WT (black dots) and EEE (gray dots) that was digested with 0.12 μm trypsin. Each data point represents one measurement that is the average of 10 runs.
The observed proteolytic fragments were consistent with predictions based on preferred specificity of trypsin (Fig. 1), and only cleavages with a high likelihood were detected by LC-MS analysis (Fig. 3). To complement the LC-MS analysis, we quantitatively compared trypsin digestion of CTDs using reducing SDS-PAGE. We incubated 10 μm purified Cp183-WT and Cp183-EEE T=4 capsids with 0.12 μm trypsin and followed the reaction for 2 h. At the appropriate time, samples for SDS-PAGE were immediately heated to 80 °C to stop trypsin digestion. To separate cleavage products that ranged from 18 to 21 kDa and only differed by 1–5 amino acids, we used Tris-Tricine SDS-PAGE (19).
Cp183-WT and Cp183-EEE capsid cleavage products were similar but had distinctive kinetics (Fig. 5A), consistent with LC-MS analysis. Cp183-WT capsids were digested faster than Cp183-EEE capsids, indicating that the CTDs of Cp183-WT capsids were more readily exposed (Fig. 5A). Because the gel was run under reducing conditions, each band represents a monomer. Degradation of full-length Cp183 monomer was accompanied by the accumulation of smaller cleavage fragments. Based on the migration on SDS-PAGE and results from LC-MS, putative C termini of the protein fragments were assigned to each gel band (Fig. 5A). However, only masses for the full-length Cp183-monomer and the 157-amino acid-long fragment could be unambiguously assigned. The Cp157 monomer accumulated over time (Fig. 5A). After incubation for 60–120 min with trypsin, a smaller fragment of 150 amino acids became apparent.
FIGURE 5.

SDS-PAGE and data analysis show that Cp183-WT capsids are digested faster than Cp183-EEE capsids and identify two populations of CTDs. A, Tris-Tricine SDS-PAGE of trypsin-cleaved Cp183-WT and Cp183-EEE T=4 capsids, stained with SYPRO® Ruby fluorescent protein stain. Trypsin digestion of Cp183 capsids was initiated, and aliquots were taken after 1–120 min and rapidly heated to 80 °C to stop digestion. An undigested capsid control (indicated by c) was loaded as a standard. Numbers on the right indicate putative cleavage products based on LC-MS results. B, SDS-PAGE results are plotted as percentage of Cp183-monomer versus time. The Cp183-WT data were fit to a function containing two first order decays: Equation 2 with a root mean square difference of 3.5%, showing that 25 and 75% of Cp183-WT CTDs were digested with a half-life of 28.8 min 2.5 min, respectively. The Cp183-EEE data were also fit to a function containing two first order decays: Equation 3 with a root mean square difference of 4.4%, showing that 50% of EEE C termini are digested with a half-life of 89.6 and 1.5 min, respectively. Each data point represents the average of two independent experiments, and the error bars correspond to the range observed.
There Are Two Populations of CTDs with Different Rates of Exposure
To quantify the differences in CTD exposure, we plotted the fluorescence intensities of the SYPRO® Ruby-stained Cp183-monomer bands as a function of the incubation time with trypsin (Fig. 5A). A clear difference in the amount of Cp183 could be observed between Cp183-WT and Cp183-EEE (Fig. 5B). To determine the rate of trypsin cleavage, we fit each data set to a function containing multiple first order decays. Hypothetically, each CTD belonging to a different quasi-equivalent monomer subunit could display a different rate of exposure. For example, in the case of a T=4 capsid, a different rate of exposure could be observed for the 25% of CTDs corresponding to the A, B, C, or D subunits. We tried fitting the data to a function with the fewest number of decays. We found no indication that cleavage had no cooperativity extending across the capsid.
The best fit for Cp183-WT capsids was achieved by fitting the data to a function containing two first order exponential decays, dividing the CTDs into two populations (Fig. 5B). For Cp183-WT capsids, 25% of CTDs had a slow half-life of 28.8 min, whereas 75% of CTDs were digested more quickly, having a half-life of 2.5 min.
We also tried fitting the data of Cp183-EEE capsids to a similar function with two first order exponential decays using pre-exponential coefficients of 25 and 75%. A reasonable fit could not be generated. We were able to fit the data to a function containing two equal populations of CTDs with different decay rates. For Cp183-EEE capsids, 50% of CTDs were cut with a slow half-life of 89.6 min and 50% with a fast half-life of 1.5 min (Fig. 5B). We note that the 89.6-min half-life, although long when compared with the 120-min experiment, is constrained by the fast component and the 25% increment for the pre-exponential term. However, the slow component could in fact have two different, but long half-lives.
Taken together, both capsids exhibited two populations of CTDs corresponding to a fast and a slow rate proteolytic cleavage. For Cp183-EEE, the slow component is larger and, on average, slower.
Cryo-EM Indicates That CTDs at Five-fold Symmetry Axes Are Partially Protected from Trypsin
To identify the sites of cleavage and assess the effects of trypsin cleavage on Cp183 capsids, we investigated trypsin-digested capsids by cryo-EM. Samples were examined at the 15-min time point where we expected the greatest difference between Cp183-WT and Cp183-EEE. Cp183-EEE capsids were also examined at 2 h when we expected that the fast component would be digested but much of the slow component would still be present. To stop capsid cleavage without disturbing structure by pH or temperature shift, we treated cleavage reactions (10 μm purified Cp183-WT and Cp183-EEE T=4 capsids with 0.12 μm trypsin) with 0.2 mm AEBSF, an irreversible protease inhibitor. The efficacy of the inhibitor was confirmed by SDS-PAGE (Fig. 6)
FIGURE 6.

AEBSF inhibits trypsin digestion after 15 min. Shown is a Tris-Tricine SDS-PAGE of Cp183-EEE of Cp183-WT capsids digested for 15 min and 3 h with trypsin. After 15 min, either the sample was boiled (−) or 0.2 mm AEBSF was added (+). The samples with added inhibitor were incubated for 3 h. Control samples had no AEBSF added prior to a 3-h incubation. All samples were analyzed by SDS-PAGE. Note that treatment with AEBSF essentially blocked trypsin cleavage. This allowed trypsin cleavage to be halted at specific time points so that samples could then be studied by cryo-EM.
Differences are clear when comparing central sections of control undigested Cp183-EEE T=4 capsids with Cp183-WT and Cp183-EEE T=4 capsids that were digested for 15 min and 2 h. Although CTDs are largely disordered, clear differences in the visible CTD density could be observed between each sample. Undigested Cp183-EEE exhibited strong CTD density, which was reduced by 2 h of digestion with trypsin. As predicted, after 15 min of trypsin digestion, Cp183-WT capsids had much lower CTD density than the Cp183-EEE capsids (black arrowheads point to the CTD density, Fig. 7A). Also consistent with prediction, CTD density of Cp183-WT capsids after 15 min of digestion is very similar to Cp183-EEE capsids after 2 h. Despite the overall decrease in density, visible density remained at five-fold symmetry axes in cross-sections of digested Cp183-EEE and Cp183-WT capsids (white arrowheads, Fig. 7A).
FIGURE 7.

Cryo-EM shows that CTD density underneath the five-fold symmetry axes is retained during trypsin digestion of Cp183-WT and Cp183-EEE capsids. Selected symmetry axes (icosahedral two-fold, three-fold, and five-fold) are marked as oval, triangle, and pentagon, respectively. A, central sections of the cryo-EM density maps of undigested and digested Cp183-EEE and Cp183-WT capsids are shown in gray scale. The central section of a Cp149 capsid, calculated from x-ray model (PDB entry 1QGT), to 9 Å resolution, is shown as a reference. Black arrowheads indicate quasi-six-fold associated CTD density, while white arrowheads indicate density underneath the five-fold symmetry axes. B, surface-shaded interior views of cryo-EM three-dimensional reconstructions of Cp183-EEE and Cp183-WT capsids were radially color-coded to highlight CTD density. The assembly domain is gray, and the CTD is orange-red. A model of a Cp149 capsid is shown as a reference. The front half of the particles were computationally removed to show the interior. The CTD density under the five-fold axis shows a petal-like organization (black arrows). CTD density under quasi-six-fold (two-fold) and quasi-three-fold axes disappeared with trypsin treatment.
Surface shaded views of these reconstructions emphasize the effects of trypsin digestion (Fig. 7b). Cp183-EEE capsids that were digested for 15 min showed strong CTD density at the five-fold and reduced densities at the icosahedral two-fold (quasi-six-fold) symmetry axes. Cp183-WT capsids that were digested for 15 min show very little density at the quasi-six-fold, quasi-three-fold, and three-fold axes and slightly stronger density at the five-fold symmetry axes (Fig. 7B). The CTDs underneath the five-fold symmetry axes form a petal-like conformation (black arrows Fig. 7B) very similar to those observed in previous cryo-EM reconstructions of Cp183-EEE capsids (16). Small differences in the petal-like conformations between the current and the previous reconstructions were noted due to differences in the experimental settings and the contour levels that used to render the structures. To prevent aggregation, capsids from the current reconstruction were assembled in higher ionic strength (450 mm NaCl) when compared with the previous reconstruction (250 mm NaCl) (16). A ring of density around the stalactite cluster is likely caused by the CTD dynamics, which is more obvious at lower resolution. As in the central sections, the overall CTD density for 15-min digested Cp183-WT capsids was very similar to that of 2-h digested Cp183-EEE capsids. These results are in agreement with cleavage kinetics (Fig. 5B). For Cp183-EEE capsids, we calculated that 45% of CTDs should remain after 15 min of digestion. After 2 h of digestion, we expect 20% of the Cp183-EEE CTDs to remain, very similar to the 19% of Cp183-WT seen at 15 min.
Discussion
Our results strongly suggest that HBV CTD phosphorylation can contribute to capsid stability and alter CTD accessibility. Results from turbidity and DSF experiments showed that Cp183-EEE capsids are more stable than Cp183-WT capsids (Fig. 2)
Although cryo-EM studies show that the CTDs are on the capsid interior (Fig. 7) (20), LC-MS and SDS-PAGE experiments of trypsin-treated capsids have shown that the majority of CTDs were exposed to the capsid exterior (Figs. 3 and 5). Most CTDs were cleaved to residue 157, indicating that at least 8 residues of the CTD had greatly reduced accessibility to trypsin cleavage (Fig. 3). Only after incubation with trypsin for 2 h did another cleavage site, at position 150, begin to appear. The very slow rate of cleavage to Cp150 may be due to rare extrusion of the C terminus or could be due to a slow breathing mode of the capsid, which had been observed in proteolysis experiments with the HBV assembly domain (30).
Our results are in good agreement with previous studies of trypsin-cleaved DNA-filled and RNA-filled capsids that resulted in a cleavage fragment of about 16.5 kDa, suggesting cleavage of about 30 residues from the CTD (10). In these studies, mature DNA-filled capsids were uniformly cleaved at a point similar to what we observed in empty capsids; immature capsids remained partially protected from cleavage (10). Together with the observation that only empty and mature capsids are enveloped for export, these data further support the hypothesis that empty and mature capsids share the same maturation signal (34).
Based on LC-MS and SDS-PAGE analysis, we observed that, overall, CTDs in Cp183-WT capsids were digested more rapidly than those of Cp183-EEE capsids, indicating an increased CTD exposure in Cp183-WT capsids (Figs. 3 and 5). These data indicated that there were two distinct populations of CTDs for both Cp183-EEE and Cp183-WT capsids (Fig. 5B). One population exhibited a fast rate of exposure that was similar for both Cp183-EEE and Cp183-WT capsids (Fig. 5B). The similarity of the two fast rates supports our assumption that the intrinsic cleavage of the Cp183-WT and Cp183-EEE CTDs are about the same so that differences in rate correlate with CTD exposure. Another population of CTDs exhibited slow exposure and was about three times slower for Cp183-EEE than for Cp183-WT capsids (Fig. 5B). Furthermore, only 25% of CTDs in Cp183-WT capsids had slow cleavage kinetics when compared with 50% in Cp183-EEE capsids.
We hypothesize that the 25% of CTD with slow exposure belong to the quasi-equivalent A subunits that are located at the five-fold symmetry axes. Supporting this hypothesis are cryo-EM studies of trypsin-digested capsids showing that CTDs at the five-fold symmetry axes remained partially protected, whereas density corresponding to CTDs at the quasi-six-fold and quasi-three-fold and three-fold symmetry axes disappeared (Fig. 7). This hypothesis is also supported by previous cryo-EM studies that observed that the CTD-binding protein serine arginine protein kinase (SRPK) occupied only quasi-six-fold symmetry vertices in empty Cp183-WT capsids, indicating that the CTDs of the five-fold symmetry axes were not readily exposed (14).
We hypothesize that the decreased CTD exposure of Cp183-EEE capsids is the result of electrostatic interactions between glutamate and arginine residues of the CTD inside the capsid particle. Increased electrostatic interactions at the five-fold symmetry axes also likely explain the 3-fold reduced cleavage/exposure rate of CTDs in Cp183-EEE capsids. Cryo-EM structures of empty and RNA-filled Cp183-EEE capsids suggested that the five CTDs under the five-fold vertex twist into a bundle (16). Arginine-glutamate salt bridges likely also contributed to the increased stability of Cp183-EEE particles.
The cryo-EM data presented here cannot explain why the remaining 25% of CTDs in Cp183-EEE capsids exhibited a slow rate of cleavage/exposure. It is possible that CTDs of subunits distant from the five-fold symmetry axes interact with CTDs of the A subunits and thus are also identified as density at the five-fold symmetry axis during cryo-EM analysis. However, it is not clear which quasi-equivalent subunit these CTDs would belong to. LC-MS data do not show the persistence of Cp183-Cp183 dimer, indicating that AB dimers were not specifically protected.
Based on our results, we can speculate that CTDs at different quasi-equivalent positions play distinct roles during the HBV life cycle. It has been hypothesized that the CTD acts as a nucleic acid chaperone, actively participating in reverse transcription of pregenomic RNA (9, 35). Our cryo-EM reconstruction in combination with solution studies show that CTDs underneath the five-fold vertices are less often exposed. This might indicate that the A subunits play a special role in capsid stability. In turn, subunits more readily exposed could be important for interacting with host factors such as importins α and β and nucleoporin 153 (Nup153) that help to arrest capsid particles in the nuclear basket (15, 36).
The region from 150 to 157 includes three high probability cleavage sites (Fig. 1), which were largely protected from proteolysis. Residues 150–153 have been suggested to be an important nuclear localization signal that acts co-dependently with another nuclear localization signal located at position 164–167 (37). Our studies suggest, however, that this region is rarely exposed to the capsid exterior. In comparison, two C-terminal nuclear export signals, at positions 157–159 and 172–175, which were found to be important players in cytoplasmic localization of expressed HBV core protein, are readily cleaved by trypsin, indicating that they are readily exposed (37).
Studies in permeabilized cells suggested that only mature capsids, containing dsDNA, disassemble and release their genome into the nucleus (11). It is hypothesized that disassembly of the capsid particle is aided by destabilization of the capsid particle due to increased pressure during reverse transcription of pregenomic RNA to the more rigid dsDNA (34, 38, 39). Changes in capsid integrity could also change accessibility of CTDs in unexpected ways.
Author Contributions
L. S. and A. Z. designed the experiments, and L. S. purified the capsid samples. L. S., R. K., and J. C. Y. W. performed the experiments. L. S., R. K., J. C.Y. W., A. Z., and B. B. analyzed the data, and L. S. wrote the manuscript. All authors received and edited the manuscript.
Supplementary Material
Acknowledgments
We thank Michael Boersma and Dr. Malcom Winkler for their experimental contributions and Jonathan Hilmer for assistance with mass spectrometry instrumentation. The mass spectrometry facility at Montana State University receives funding from the Murdock Charitable Trust and National Institutes of Health 5P20RR02437 of the CoBRE program.
This work was supported by a National Institutes of Health Grant R56-AI077688 (to A. Z.) A. Z. is a co-founder of Assembly BioSciences, which is focused on HBV antiviral development. Other co-authors declare that they have no conflicts of interest.

This article contains supplemental Fig. 1.
- HBV
- hepatitis B virus
- cryo-EM
- cryo electron-microscopy
- CTD
- C-terminal domain
- DSF
- differential scanning fluorimetry
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- AEBSF
- 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride.
References
- 1.Lavanchy D. (2005) Worldwide epidemiology of HBV infection, disease burden, and vaccine prevention. J. Clin. Virol. 34, Suppl. 1, S1–S3 [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization (WHO) (2014) Hepatitis B, Fact Sheet Number 204, WHO, Geneva, Switzerland [Google Scholar]
- 3.Nassal M. (2008) Hepatitis B viruses: reverse transcription a different way. Virus Res. 134, 235–249 [DOI] [PubMed] [Google Scholar]
- 4.Stannard L. M., and Hodgkiss M. (1979) Morphological irregularities in Dane particle cores. J. Gen. Virol. 45, 509–514 [DOI] [PubMed] [Google Scholar]
- 5.Birnbaum F., and Nassal M. (1990) Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J. Virol. 64, 3319–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nassal M. (1992) The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 66, 4107–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao W., and Ou J. H. (1995) Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J. Virol. 69, 1025–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gazina E. V., Fielding J. E., Lin B., and Anderson D. A. (2000) Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J. Virol. 74, 4721–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewellyn E. B., and Loeb D. D. (2011) The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. Journal of virology 85, 1298–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabe B., Vlachou A., Panté N., Helenius A., and Kann M. (2003) Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. U.S.A. 100, 9849–9854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rabe B., Delaleau M., Bischof A., Foss M., Sominskaya I., Pumpens P., Cazenave C., Castroviejo M., and Kann M. (2009) Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog. 5, e1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu X., Jin L., Jih J., Shih C., and Zhou Z. H. (2013) 3.5Å cryoEM structure of hepatitis B virus core assembled from full-length core protein. PLoS ONE 8, e69729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wittkop L., Schwarz A., Cassany A., Grün-Bernhard S., Delaleau M., Rabe B., Cazenave C., Gerlich W., Glebe D., and Kann M. (2010) Inhibition of protein kinase C phosphorylation of hepatitis B virus capsids inhibits virion formation and causes intracellular capsid accumulation. Cell Microbiol. 12, 962–975 [DOI] [PubMed] [Google Scholar]
- 14.Chen C., Wang J. C. Y., and Zlotnick A. (2011) A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics in vitro. PLoS Pathog. 7, e1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kann M., Sodeik B., Vlachou A., Gerlich W. H., and Helenius A. (1999) Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J. Cell Biol. 145, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J. C., Dhason M. S., and Zlotnick A. (2012) Structural organization of pregenomic RNA and the carboxy-terminal domain of the capsid protein of hepatitis B virus. PLoS pathogens 8, e1002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porterfield J. Z., Dhason M. S., Loeb D. D., Nassal M., Stray S. J., and Zlotnick A. (2010) Full-length hepatitis B virus core protein packages viral and heterologous RNA with similarly high levels of cooperativity. Journal of virology 84, 7174–7184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rayaprolu V., Kruse S., Kant R., Movahed N., Brooke D., and Bothner B. (2014) Fluorometric estimation of viral thermal stability. Bio-protocol 4, e1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schägger H. (2006) Tricine-SDS-PAGE. Nat. Protocols 1, 16–22 [DOI] [PubMed] [Google Scholar]
- 20.Wang J. C., Dhason M. S., and Zlotnick A. (2012) Structural organization of pregenomic RNA and the carboxy-terminal domain of the capsid protein of hepatitis B virus. PLoS Pathog. 8, e1002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., and Ludtke S. J. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 [DOI] [PubMed] [Google Scholar]
- 22.Mindell J. A., and Grigorieff N. (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 [DOI] [PubMed] [Google Scholar]
- 23.Yan X., Sinkovits R. S., and Baker T. S. (2007) AUTO3DEM: an automated and high throughput program for image reconstruction of icosahedral particles. J. Struct. Biol. 157, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan X., Dryden K. A., Tang J., and Baker T. S. (2007) Ab initio random model method facilitates 3D reconstruction of icosahedral particles. J. Struct. Biol. 157, 211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shepherd C. M., Borelli I. A., Lander G., Natarajan P., Siddavanahalli V., Bajaj C., Johnson J. E., Brooks C. L. 3rd, and Reddy V. S. (2006) VIPERdb: a relational database for structural virology. Nucleic Acids Res. 34, D386–D389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 27.Ceres P., and Zlotnick A. (2002) Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 41, 11525–11531 [DOI] [PubMed] [Google Scholar]
- 28.Köck J., Nassal M., Deres K., Blum H. E., and von Weizsäcker F. (2004) Hepatitis B virus nucleocapsids formed by carboxy-terminally mutated core proteins contain spliced viral genomes but lack full-size DNA. J. Virol. 78, 13812–13818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lan Y. T., Li J., Liao W., and Ou J. (1999) Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259, 342–348 [DOI] [PubMed] [Google Scholar]
- 30.Hilmer J. K., Zlotnick A., and Bothner B. (2008) Conformational equilibria and rates of localized motion within hepatitis B virus capsids. J. Mol. Biol. 375, 581–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park C., and Marqusee S. (2004) Probing the high energy states in proteins by proteolysis. J. Mol. Biol. 343, 1467–1476 [DOI] [PubMed] [Google Scholar]
- 32.Hubbard S. J., Eisenmenger F., and Thornton J. M. (1994) Modeling studies of the change in conformation required for cleavage of limited proteolytic sites. Protein Sci. 3, 757–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zlotnick A., Cheng N., Conway J. F., Booy F. P., Steven A. C., Stahl S. J., and Wingfield P. T. (1996) Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry 35, 7412–7421 [DOI] [PubMed] [Google Scholar]
- 34.Cui X., Ludgate L., Ning X., and Hu J. (2013) Maturation-associated destabilization of hepatitis B virus nucleocapsid. J. Virol. 87, 11494–11503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu T. H., Liou A. T., Su P. Y., Wu H. N., and Shih C. (2014) Nucleic acid chaperone activity associated with the arginine-rich domain of human hepatitis B virus core protein. Journal of virology 88, 2530–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmitz A., Schwarz A., Foss M., Zhou L., Rabe B., Hoellenriegel J., Stoeber M., Panté N., and Kann M. (2010) Nucleoporin 153 arrests the nuclear import of hepatitis B virus capsids in the nuclear basket. PLoS Pathog. 6, e1000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H. C., Huang E. Y., Su P. Y., Wu S. Y., Yang C. C., Lin Y. S., Chang W. C., and Shih C. (2010) Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS pathogens 6, e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dhason M. S., Wang J. C., Hagan M. F., and Zlotnick A. (2012) Differential assembly of Hepatitis B Virus core protein on single- and double-stranded nucleic acid suggest the dsDNA-filled core is spring-loaded. Virology 430, 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo H., Mao R., Block T. M., and Guo J. T. (2010) Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J. Virol. 84, 387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.