

Abstract
Stem cell-based therapy of retinal degenerative conditions is a promising modality to treat blindness, but requires new strategies to improve the number of functionally integrating cells. Grafting semidifferentiated retinal tissue rather than progenitors allows preservation of tissue structure and connectivity in retinal grafts, mandatory for vision restoration. Using human embryonic stem cells (hESCs), we derived retinal tissue growing in adherent conditions consisting of conjoined neural retina and retinal pigment epithelial (RPE) cells and evaluated cell fate determination and maturation in this tissue. We found that deriving such tissue in adherent conditions robustly induces all eye field genes (RX, PAX6, LHX2, SIX3, SIX6) and produces four layers of pure populations of retinal cells: RPE (expressing NHERF1, EZRIN, RPE65, DCT, TYR, TYRP, MITF, PMEL), early photoreceptors (PRs) (coexpressing CRX and RCVRN), inner nuclear layer neurons (expressing CALB2), and retinal ganglion cells [RGCs, expressing BRN3B and Neurofilament (NF) 200]. Furthermore, we found that retinal progenitors divide at the apical side of the hESC-derived retinal tissue (next to the RPE layer) and then migrate toward the basal side, similar to that found during embryonic retinogenesis. We detected synaptogenesis in hESC-derived retinal tissue, and found neurons containing many synaptophysin-positive boutons within the RGC and PR layers. We also observed long NF200-positive axons projected by RGCs toward the apical side. Whole-cell recordings demonstrated that putative amacrine and/or ganglion cells exhibited electrophysiological responses reminiscent of those in normal retinal neurons. These responses included voltage-gated Na+ and K+ currents, depolarization-induced spiking, and responses to neurotransmitter receptor agonists. Differentiation in adherent conditions allows generation of long and flexible pieces of 3D retinal tissue suitable for isolating transplantable slices of tissue for retinal replacement therapies.
Introduction
Stem cell-based therapy is a promising approach to repair vision loss, but to this day, it has yet to deliver any Food and Drug Administration-approved cell replacement therapies, leaving neuroprotection as the best current alternative [1]. Photoreceptors (PRs) and the retinal pigment epithelium (RPE) are commonly considered the most important targets of stem cell therapies [1–3]. PRs and RPE interact functionally and structurally [2–6], appear to polarize each other [7], and are reported to show improved engraftment when transplanted together [8,9]. After engraftment, stem cell-derived PRs must survive and form functional synaptic connections. Furthermore, complicating cell-based therapies is the complex milieu of degenerative pathologies of retinal tissue. Transplanted dissociated cells may not survive long term in such abnormal conditions [10]. The survival of transplanted PRs is especially challenging if the recipient retina lacks a functional RPE [11]. These considerations lead to the conclusion that grafting retinal tissue (such as embryonic retina) rather than dissociated PRs may be more effective for repairing the retina [12] in patients suffering from advanced retinal degeneration. While fetal tissue challenges efforts at quality control, lot-to-lot uniformity, ethics, and scalability [13], human pluripotent stem cells (hPSCs) are a perpetually renewable source of cells and therefore may be instrumental in developing industrially scalable therapies for early and advanced retinal dystrophies such as Leber congenital amaurosis (LCA) and retinitis pigmentosa (RP), and likely age-related macular degeneration (AMD), diabetic retinopathy (DR), and retinopathy of prematurity (ROP). The optic nerve remains intact in these blinding conditions, which enables restoration of connectivity between the 3D retinal graft and downstream visual nuclei to transmit the PR visual signals to the cortex [14,15].
Blindness is a major burden on society [16–18] with an estimated annual cost of $35.4 billion [19]. There are many unmet clinical needs associated with the challenging task of treating RP, LCA, AMD, ROP, and DR. About 15 million Americans currently suffer from AMD [20,21], the leading cause of vision loss in adults aged over 50 years in developed nations, with the number of cases projected to nearly double by 2030. RP is the third most frequent cause of inherited visual impairment and is estimated to affect up to 100,000 people in the United States and ∼1.5 million people worldwide [22]. DR affects 4.2 million adults in the United States, among them 655,000 have advanced DR with conditions such as clinically significant macular edema and proliferative DR that could lead to blindness [23]. These numbers illustrate the urgent need for new and efficient retinal therapies.
A viable new direction of treating blindness is retinal grafting with tissue derived from human embryonic stem cells (hESCs). Recent reports demonstrated that hESCs and induced pluripotent stem cells (iPSCs) can generate optic vesicle- and optic cup-like structures and produce retinal progenitors that differentiate into RPE, PRs, inner nuclear layer (INL) neurons, and retinal ganglion cells (RGCs) [24–26]. Culturing hPSC-derived retinal spheres in suspension for up to 6 months demonstrated the ability of retinal organoids to form cell layers, including PRs with outer disk-like protrusions and photosensitivity [26], which are challenging to purify in 2D monolayer culture [27]. However, the main advantage of deriving 3D tissue rather than PR progenitors is that the organization of embryonic-like tissue can be preserved. This facilitates subsequent subretinal grafting and likely the survival of PRs. Retinal repair with human fetal grafts and vision improvements have been achieved in animals [14,28] and in patients with advanced retinal degeneration [9,29–31]. Self-organization of 3D retinal tissue is especially efficient if the transplant includes the RPE [8,9,30,32]. It has been observed that stem cell-derived 3D retinas support lamination and outer segment (OS) outgrowth demonstrates the tissue's potential to perform visual function after grafting. However, the retinal tissue cannot be too differentiated to survive the surgical procedure [33]. In addition, the structural rigidity of retinospheres (cultured in suspension) makes it difficult to isolate a transplantable slice of hESC-derived retina [34]. In this study, we derived immature, long, and flexible 3D retinal tissue from hESCs in adherent conditions. This tissue containing layers of RPE cells, PRs, INL cells, and RGCs is capable of forming synapses and exhibiting a range of electrophysiological responses. The ability of hESC-derived retinal tissue to form synapses is especially important as this increases the likelihood of establishing functional connections with the recipient retinal neurons in subretinal grafts [14,15]. The results will lay the groundwork for transitioning this stem cell technology to clinical trials.
Materials and Methods
Pluripotent hESC culture
The hESC line, WA01 (formerly H1) [35], was obtained from WiCell at passage (P-23) (mTeSRT1/MatrigelT Platform) and cultured in feeder-free conditions using mTeSR1 protocol and basic fibroblast growth factor (Sigma-Aldrich) [36,37] with the addition of heparin (10 ng/mL) [38] and amphotericin-B/gentamicin (Life Technologies) on 1xES-qualified, growth factor-reduced (GFR) Matrigel-coated (Fisher Scientific) plates. Cells were passaged every 6–7 days (reaching ∼80% confluency by day 7) on GFR-coated 35-mm plates using the enzymatic protocol with Versene/EDTA (at a ratio 1:10) from Lonza Group. RHO-kinase inhibitor (ROCK) [39] 10 μM Y-27632 (Catalog #72302) was used for initial plating of hESCs from cryostorage, and then removed from culture media. Colonies containing clearly visible differentiated cells were marked and mechanically removed before passaging with Versene, as recommended by mTeSR1 protocol [36].
Retinal differentiation
See Supplementary Data and Supplementary Fig. S1 (Supplementary Data are available online at www.liebertpub.com/scd) for detailed protocol.
RNA isolation and quantitative reverse transcription–coupled polymerase chain reaction analysis of gene expression
Total RNA was prepared from (1) undifferentiated hESCs (control) and (2) differentiated cells using the RNeasy Mini kit (Qiagen) according to the manufacturer's protocol and our earlier published procedures [27,40]. One microgram (μg) of RNA was converted to cDNA with the Maxima H Minus First-strand cDNA synthesis kit (Thermo scientific). Quantitative polymerase chain reaction (qPCR) analysis was performed using SsoAdvanced Universal SYBR Green supermix (Bio-Rad) on the CFX384 Touch Real-Time PCR System. Primers were selected using the Universal Probe Library program (Roche Life science, see primer list in Supplementary Table S1). All experiments were performed in triplicates, and the data are expressed as mean ± SD. The comparative Ct method was applied in quantitative real-time PCR assay according to the ddCt method with GAPDH as internal control [41]. The data are represented as fold change over control.
Immunohistochemistry
hESC-derived retinal tissue aggregates growing in adherent condition were fixed in fresh ice-cold paraformaldehyde (4% PFA; Sigma-Aldrich) for 15 minutes (min), rinsed with 1× phosphate-buffered saline (PBS), and washed thrice in ice-cold PBS (5 min each). The aggregates were cryoprotected in 20% sucrose (prepared in PBS, pH 7.8), and then 30% sucrose (until tissue sank), and snap-frozen (dry ice/ethanol bath) in optimum cutting temperature (OCT) embedding material (Tissue-Tek). hESC-derived retinal tissue aggregates were serially sectioned at 12 μm. The sections were first permeabilized with 0.1% Triton X-100/PBS (PBS-T) at room temperature for 30 min, followed by 1 h of incubation in blocking solution [5% preimmune normal goat serum (Jackson Immunoresearch) and 0.1% PBS-T] at room temperature, and then were incubated with primary antibodies (Supplementary Table S2) diluted in blocking solution at 4°C overnight. The following day sections were washed thrice (10–15 min each time) with PBS-T, and then incubated with the corresponding secondary antibodies (Alexa Fluor 568 goat anti-mouse, Alexa Fluor 488 goat anti-rabbit, 1:1,000, or vice versa) at room temperature for 45 min. The slides were washed thrice with 0.1% PBS-T solution, incubated with 4′, 6-diamidino-2-phenylindole (DAPI) solution (1 μg/mL) for 10 min, and then washed again with 0.1% PBS-T solution. As a negative control for primary antibody-specific binding, we stained tissue sections with secondary antibodies only. The specimens were mounted with ProLong Gold Antifade medium (Life Technologies) and examined using a Nikon Eclipse Ni epifluorescent microscope with ZYLA 5.5 sCMOS (ANDOR Technologies) black and white charge-coupled device high-speed camera or Olympus FluoView FV1000 confocal microscope (Olympus). All antibodies are listed in Supplementary Table S2.
Immunoblot
Total lysates of undifferentiated and differentiated hESCs were homogenized by motorized handheld homogenizer (VWR) in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors (Roche complete). At least 40 individual large hESC-derived retinal tissue aggregates were collected for immunoblotting. The protein concentration was measured by using Bradford assay. Equal concentrations (40 μg) of protein samples were resolved in 4%–20% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to the Immobilon-FL membrane (Bio-Rad). Membranes were blocked with blocking buffer (BLOTTO) for 30 min at room temperature. After blocking, membranes were incubated with primary antibodies. See Supplementary Data for details.
Electrophysiological recording
hESC-derived retinal tissue aggregates grown in 35-mm cell culture dishes were positioned on a fixed-stage upright microscope (Eclipse FN1; Nikon Instruments) and superfused by Ames' medium gassed with 95% O2 5% CO2. The superfusate was delivered to the dish at 2–3 mL/min and maintained at 32°C using a temperature controller (Warner Instruments). Retinal tissue aggregates were visualized using infrared transillumination and NIS Elements D imaging software (Nikon Instruments). Whole-cell recordings were obtained from medium to large round somas located on the interior of the hESC-derived retinal tissue using either a MultiClamp 700A or 700B amplifier (Molecular Devices). Glass micropipettes with tip resistances 6–8 MΩ were pulled from thick-walled borosilicate tubings on a Narishige PC-10 puller and filled with either a K+- or Cs+-based intracellular solution (described in Supplementary data). PCLAMP 9 or PCLAMP 10 software (Molecular Devices) was used for data acquisition. See the “Electrophysiological recording - chemicals and solutions” section in Supplementary Data for further methodological details.
Results
Evaluating retinal development by quantitative reverse transcription–coupled polymerase chain reaction identifies markers of all retinal lineages in 3D retina-like tissue
WA01 hESC cultures maintained pluripotency while growing in feeder-free conditions (Supplementary Fig. S2). hESC-derived retinal tissue aggregates started to appear by about week 4 after initiation of a differentiation protocol and rapidly increased in size by 6 weeks (Supplementary Fig. S1). We did not observe further growth between weeks 7 and 12, and maintaining cells on the plates at later time points (around weeks 10–12) was challenging due to the visible degradation of Matrigel. The growth of retinal tissue aggregates in cultures was not synchronous, producing various shapes and sizes, and the number of such aggregates varied between 2 and 15 or more per 35-mm plate. Large and elongated aggregates (Supplementary Fig. S1) were chosen for further study, which we reasoned may be most suitable for isolating long and flexible pieces of hESC-derived retinal tissue for grafting. We analyzed differentiating cells by quantitative reverse transcription–coupled polymerase chain reaction (qRT-PCR) at 2, 3, 4, and 12 weeks (2D monolayer culture), and also in hESC-derived retinal tissue at 6 weeks.
Early neuroectoderm (NE), anterior neuroectoderm (ANE), early neuronal (EN), and the eye field (EYF) markers were robustly upregulated between 2 and 6 weeks in our neural cultures, indicating efficient induction of NE and early retinal cell fate (Fig. 1 and Supplementary Table S3), while astroglial (ASTR), oligodendroglial (OLIG), neural crest (NCR), mesodermal, and endodermal markers were undetectable. We verified the acquisition of NE fate in differentiating hESC-derived retinal tissue by tracing the expression of the earliest pan-neuroectodermal transcription factor, SOX1 [42,43], [11.2 ± 0.3]. ANE specification in hESC-derived retinal tissue was confirmed by tracing the expression of OTX2 [peaked at 4 weeks (7.5 ± 0.2)] and FOXG1 genes (rising steadily, 218.2 ± 10.7 at 6 weeks). The acquisition of retinal fate in hESC-derived retinal tissue was determined by analyzing the expression of several key eye field genes, [RAX (206.9 ± 31.3), LHX2 (88.4 ± 4.8), SIX3 (9.9 ± 1.4), SIX6 (13.1 ± 1.1), and PAX6 (43.7 ± 1.1)]. Expression of RAX, PAX6, and LHX2 on a protein level was confirmed by immunoblots (Supplementary Fig. S3). Most of the cells in the developing hESC-derived retinal tissue were strongly positive for RAX (retina and anterior neural fold homeobox transcription factor), a key retinal cell fate marker [44–47] (Supplementary Fig. S4). Proneural/retinal markers NEUROD1, responsible for the development of PRs and amacrines [48], and ASCL1 (MASH1) [49], which determine competence-restricted progenitor lineage in the retina [50], demonstrated very high expression reaching a peak at 4 weeks (377.3 ± 5.5 and 134.2 ± 12.5, respectively). The CHX10 marker of multipotential retinal progenitors and bipolar cell fate [51,52] was not expressed between 2 and 4 weeks in retinal cultures, but was sharply upregulated in hESC-derived retinal tissue at 6 weeks [98.7 ± 10.1]. The IKZF1 [IKAROS], which provides temporal competence to early, but not late, retinal progenitors [53], was expressed between 2 and 4 weeks after the initiation of retinal differentiation (3.9 ± 1.3 and 2.3 ± 0.0, respectively). Several PR markers showed high expression in hESC-derived retinal tissue at 6 weeks, specifically CRX [11.2 ± 0.1], RCVRN [49.2 ± 1.7], PDE6B [6.9 ± 0.1], and OPN1SW [48.4 ± 4.3]. Key rod cell fate-determining transcription factors, NRL, NR2E3, and THRB (TRβ2, M-cones) [54], were upregulated 2.0–2.3-fold in hESC-derived retinal tissue (Fig. 1 and Supplementary Table S3). Among the genes important for RGC development, MATH5 [55], BRN3B [56,57], and ISL1 were most upregulated in hESC-derived retinal tissue (48.1 ± 4.2, 119.3 ± 5.8, and 17.2 ± 2.3, respectively). Müller glia markers, GLUL and CCND3, peaked at 4 weeks, but were not upregulated in hESC-derived retinal tissue at 6 weeks. INL markers (PROX1, CALB1–horizontal cells, CALB2–amacrine cells) were upregulated (9.2 ± 0.7, 4.3 ± 0.1 and 2.0 ± 0.0, respectively). Rod bipolar marker, PRKCA (PKCα), was not upregulated in hESC-derived retinal tissue. Last, we observed marked upregulation of key RPE markers [MITF (359.3 ± 22.9), RPE65 (3035.2 ± 267.4), TYR (13676.8 ± 402), TYRP (4446.5 ± 8.4), DCT (11462.7 ± 280.9), and PMEL (6.9 ± 0.2)] in hESC-derived retinal tissue at 6 weeks. (Supplementary Table S3).
FIG. 1.

Quantitative reverse transcription-coupled polymerase chain reaction analysis of human embryonic stem cell (hESC)-derived retinal cells differentiated in 2D adherent monolayer condition (2, 3, 4, 12 weeks) and in 3D retina-like tissue (6 weeks). Expression values of key eye field, photoreceptor (PR), horizontal, amacrine, and ganglion cell markers are presented as fold change over control (pluripotent hESCs, day 0) using the comparative ddCt method [41]. Fold ratio of 1 denotes no change in gene expression. GAPDH was used as internal control. The analysis was done in technical triplicates. The error bars represent standard deviation (SD) ± mean.
Maintaining most 3D retina-like tissue aggregates for 12 weeks on the same plate proved to be challenging due to Matrigel degradation and dislodging of aggregates from the plates. However, we evaluated gene expression of hESC-derived retinal cells growing in 2D culture (12-week time point, Fig. 1 and Supplementary Table S3). We confirmed that the expression levels of some key NE, ANE, and EYF markers continue to increase (compared with that found in 6-week-old hESC-derived retinal tissue), specifically SOX1 [43.1 ± 4.8], FOXG1 [355 ± 31.2], SIX3 [13.5 ± 2.4], LHX2 [214.6 ± 9.5], CHX10 [314.0 ± 12.3], DCX [33.4 ± 1.9], ASCL1 [144.2 ± 6.4], PROX1 [41.4 ± 2.3], CALB1 [35.9 ±2.8], CALB2 [4.8 ± 0.3], and NRL [4.9 ± 0.0]. PRKCA became detectable only in 12-week-old cultures [2.3 ± 0.1]. Importantly, markers of early retinal cell types [58], such as RPE genes, amacrines (LGR5), EYF (RX, PAX6), RGC (BRN3B, ISL1, MATH5), and PR (RCVRN), showed reduced expression compared with hESC-derived retinal tissue at 6 weeks.
Trophic factors
We noted the steady upregulation of selected retina-specific neurotrophic factor genes from 2 to 4 weeks such as ciliary neurotrophic factor (CNTF, 7.3 ± 0.3 at 4 weeks), pigment epithelial-derived factor (PEDF [4] or SERPINF1 8.5 ± 0.6 at 4 weeks), and nerve growth factor (NGF) (4.7 ± 0.3 at 4 weeks). Brain-derived neurotrophic factor (BDNF) and Glial-derived neurotrophic factor (GDNF) maintained a low expression level. The hESC-derived retinal tissue showed high expression of PEDF and NGF at 6 weeks (Supplementary Table S3).
MicroRNA-processing genes (DROSHA, AGO2, PASHA, DICER)
These genes showed similar expression levels compared with hESCs, while LIN28 was downregulated (Supplementary Table S3).
Evidence of subdivision of 3D retina-like tissue into outer and inner neuroblast-like zones
To evaluate the mitotic profiles and the distribution of dividing progenitors in histological sections of hESC-derived retinal tissue, we used anti-Ki67 antibody [59]. Ki67 is a widely used marker of cell proliferation present at all stages of the cell cycle in the dividing cells [60], except for G0 (dormant or resting phase), with peak levels in mitosis [61]. We found about 4.5% ± 0.8% (n = 4) Ki67-positive (Ki67 [+]) cells with strong Ki67 nuclear signal (mitosis) in hESC-derived retinal tissue (Supplementary Fig. S5). The majority of Ki67 [+] cells localized to the apical side (next to the RPE layer). Such distribution is typical for cells in the M-phase undergoing interkinetic nuclear migration in vertebrate retinogenesis [62–67]. It is also characteristic of M-stage dividing progenitors in other types of developing stratified neuroepithelia [68]. Ki67 [+] nuclei were enlarged and had round morphology (mitotic rounding [68]), confirming that these cells were not migratory.
Localization of mitotic cells to the apical side in the large 6-week-old hESC-derived retinal tissue prompted us to search for the presence of cells with the progenitor signature. We used antibodies for the catalytic component of telomerase (TERT) and stem cell marker, LGR5, and observed a strong signal in the apical side (Supplementary Fig. S6). Adult stem cell marker, LGR5, originally selected as a marker of glycinergic amacrine neurons [69], demonstrated very high gene expression level at 2–3 weeks (306.8 ± 6.0 and 280.3 ± 2.8, respectively) and remained very high at 6 weeks in hESC-derived retinal tissue (493.7 ± 21.8) (Supplementary Table S3).
RPE in hESC-derived retinal tissue and 2D retinal monolayer cultures
To better define the similarity between the developing mammalian retina and the hESC-derived retinal tissue, we evaluated the presence of RPE cells with antibodies to human EZRIN [70,71] and EBP50/NHERF [72]. EZRIN, an actin-binding protein, and EBP50/NHERF1, the ERM-binding phosphoprotein 50/sodium–hydrogen exchanger regulatory factor 1, are typical markers of the apical RPE. Immunohistological analysis found a strong signal in the apical side of hESC-derived retinal tissue (Fig. 2 and Supplementary Fig. S7). We confirmed the presence of RPE in hESC-derived retinal tissue by the immunoblot with the antibody to the RPE-specific marker, MITF (Supplementary Fig. S3). We did not detect the typical microvilli staining pattern expected with antibodies to NHERF1 [72] and EZRIN [2,72]. Nevertheless, we did observe visible pigmentation of RPE cells in hESC-derived retinal tissue (apical side) matching the EZRIN/NHERF1 immunostaining (Supplementary Fig. S7). We were unable to do flat-mount analysis of this hESC-derived retinal tissue to observe the typical hexagonal shape of RPE cells because of the small size of each piece of tissue. However, we observed the typical honeycomb shape and pigmentation in 2D adherent monolayer cultures (Supplementary Fig. S7d, d′). Interestingly, we also observed some RPE cells migrating along the hESC-derived retinal tissue and sometimes residing on the basal side (Supplementary Fig. S7c). This possibly explains finding traces of EZRIN/NHERF1 [+] signal on the other side of hESC-derived retinal tissue section.
FIG. 2.
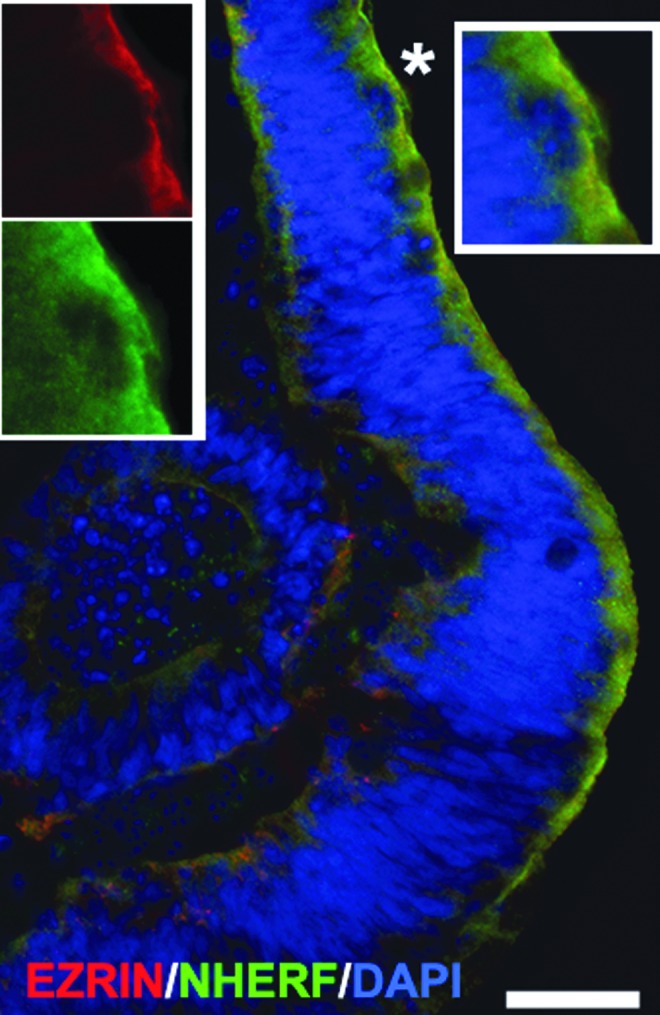
Presence of retinal pigment epithelium (RPE) layer in hESC-derived retinal tissue. Immunostaining of hESC-derived retinal tissue (6 weeks) with two mature RPE markers, EZRIN, an actin-binding protein, and ERM-binding phosphoprotein50/sodium–hydrogen exchanger regulatory factor1 (EBP50/NHERF1), shows strong staining in the apical side. Inset represents the magnification of the area marked with an asterisk. 4′, 6-diamidino-2-phenylindole (DAPI)-stained nucleus. Scale bar: 20 μm.
Finding layers of early retinal cell types, synaptogenesis, and axonogenesis in hESC-derived retinal tissue
Finding an RPE layer prompted us to search for cells with progenitor markers in the apical side of hESC-derived retinal tissue. OTX2, a paired-type homeodomain protein, is important for specification and postmitotic function of PRs [73], bipolar neurons, and RPE [74–77]. OTX2 [+] cells first appear in the developing PRs, and then in the bipolar and RPE cells during mammalian retinogenesis [78]. Six or more enhancers of the OTX2 gene [79] control the timing and activation of OTX2 at different stages of ANE and ocular development [73,80], including PRs [68] and RPE [81]. We detected a strong presence of OTX2 by the immunoblot (Supplementary Fig. S3) and immunohistochemistry (IHC) (Fig. 3a, b, d) in hESC-derived retinal tissue. The pattern of staining resembled that reported in 6–10-week-old human fetal retinal sections [81] (Supplementary Table S4). The majority of OTX2 [+] cells were localized to the apical side of the hESC-derived retinal tissue. Otx2 is expressed at the time of the last mitotic cycle in retinal progenitors [54] and also in early PR precursors committed to the PR cell fate during mammalian retinogenesis [73]. The nuclei of PRs are expected to localize near the apical side of developing mammalian retina [54]. OTX2 [+] cells were also found in the basal side of hESC-derived retinal tissue and could belong to precursors of bipolar cells, which appear in the late stage of retinogenesis [58]. All OTX2 [+] cells had round nuclei, indicating that cells are not migrating. To determine if OTX2 [+] cells in the apical side are mitotic or postmitotic, we did confocal IHC analysis with OTX2 and Ki67 antibodies (Fig. 3b–e′) and found that all OTX2 [+] cells in the apical side are postmitotic. However, we did not find colocalization between MAP2, an early postmitotic cytoplasmic marker present in developing postmitotic RGCs, and OTX2 [+] nuclei (Fig. 3a). We found most of the cells in the ciliary margin-like zone strongly positive for OTX2 (Fig. 3a, arrows). Similar findings have been reported by Sasai laboratory [82] and others [83].
FIG. 3.

OTX2 [+] cells in hESC-derived retinal tissue are postmitotic. (a) Immunolabeling of hESC-derived retinal tissue (6 weeks) with antibodies to OTX2 and postmitotic neuronal marker, MAP2. Insets are magnifications of framed area showing no colocalization between OTX2 and MAP2. Arrows point to the presence of OTX2 [+] cells in the ciliary margin-like zone. Scale bar: 50 and 10 μm. (b–e) Confocal images (single optical sections) showing no colocalization of OTX2 and Ki67 (marker of cell division) in hESC-derived retinal tissue, indicating that OTX2 [+] cells are postmitotic. DAPI-stained nucleus. Scale bar: 100 μm. (d′, e′) Confocal (z-stack) images of the area marked with an asterisk in (b–e). The confocal image was virtually resectioned at the x and y planes to confirm the identity of the double-labeled cell. Scale bar: 50 μm.
Next, we determined the distribution of CRX [+] and PAX6 [+] cells within the hESC-derived retinal tissue stained with anti-CRX and anti-PAX6 antibodies. The majority of cells were positive for PAX6 protein, which is a reliable eye field marker and is also expressed in multipotential retinal progenitors [84] and later in RGCs and amacrine neurons. We found a large number of strongly positive PAX6 [+] cells near the apical side of hESC-derived retinal tissue. The apical side of embryonic mammalian retina contains progenitors [62,63], where they acquire cell fate after the last mitotic cycle and migrate into the retinal layers to undergo maturation. The majority of CRX [+] cells were also localized to the apical side of hESC-derived retinal tissue. We did not colocalize them with mitotic Ki67 [+] cells, but the localization of CRX [+] cells at the apical side of hESC-derived retinal tissue and the round shape of these CRX [+] nuclei (Fig. 4, insets) provide indirect evidence that many CRX [+] cells have exited the mitotic cycle and acquired PR cell fate [85]. CRX is expressed in early postmitotic PR precursors and is regulated by OTX2 [54,73,74,85]. Both markers robustly localized to the apical side of hESC-derived retinal tissue in multiple IHC experiments (Figs. 3 and 4). We identified a small number of CRX [+] cells, which also had residual weak staining for PAX6, likely indicating ongoing loss of PAX6 expression in these cells and acquisition of CRX [+] signal. Similar findings were reported during PR development in mammalian retinogenesis, where PAX6 suppresses premature activation of CRX [86]. We also found a number of CRX [+] PAX6 [−] cells in hESC-derived retinal tissue. Many of these nuclei were elongated (Fig. 4, insets), suggesting the continuing movement of these cells typical for interkinetic nuclear migration [63,68] after cell fate acquisition. We confirmed strong expression of PAX6 and CRX by immunoblots (Supplementary Fig. S3).
FIG. 4.

Distribution of rod–cone marker, CRX, and multipotential retinal progenitor marker, PAX6, in hESC-derived retinal tissue. Immunostaining of hESC-derived retinal tissue (6 weeks) with antibodies to CRX and PAX6 (a–c). (a) Preferential localization of CRX [+] nuclei to the apical side of hESC-derived retinal tissue (arrows). Insets represent the magnification of the area marked with an asterisk (b). The white arrows in the first inset (*) point to a CRX [+] nucleus with a round shape (indicating postmitotic PR progenitor), which is negative for PAX6. The second inset (**) shows several cells, which are CRX [+], but not PAX6 [+], and vice versa. Note CRX [+] cells in the inner neuroblast-like layer are exclusively of elongated shape, indicating the interkinetic nuclear migration process in less mature CRX [+] PR progenitors. Scale bar: 20 and 10 μm, respectively.
Identification of CRX [+] cells near the apical side of hESC-derived retinal tissue (Fig. 4) prompted us to look for rod–cone PR markers, recoverin (RCVRN) and rhodopsin (RHO). RCVRN, a calcium-binding protein, plays a key role in sensory adaptation during phototransduction [87], and RHO is a major rod PR rim OS protein, which belongs to G-protein-coupled receptors and is a primary light-sensing pigment in rods [88]. We found a large number of RCVRN [+] cells in the layer of cells carrying CRX [+] nuclei (Fig. 5a–a″, c). RCVRN [+] cells and CRX [+] cells were colocalized by confocal IHC (Fig. 5b–b″′). Every RCVRN [+] cell had a CRX [+] nucleus, although only about half of cells with CRX [+] nuclei had RCVRN [+] cytoplasm. We also noted a variety of developmental stages in the RCVRN [+] PR population, from more to less mature, likely reflecting the asynchronous nature of PR development in hESC-derived retinal tissue (Fig. 5c, inset and Supplementary Fig. S8). However, preferential localization of RCVRN [+] cells to the apical side was very consistent in retinal sections. RCVRN [+] cells are found in human embryonic retina at about fetal weeks 10–13 [24,69,89,90] (Supplementary Table S4) and in mouse developing retina by embryonic day 18 [85]. No RHO [+] cells were found in several sets of histological sections, although we saw upregulation of the RHO gene expression. In developing mouse retina, RHO could be identified by IHC at postnatal days 2–6 [91], while few cells could be found immediately at birth (P0).
FIG. 5.

Localization of PR and amacrine markers to the apical and basal sides of hESC-derived retinal tissue. (a–a′′) Low-magnification confocal image of hESC-derived retinal tissue section stained with RCVRN and CRX antibodies. Note the identical distribution pattern of CRX [+] and RCVRN [+] cells within the apical side. DAPI-stained nucleus. Scale bar: 100 μm. (b–b′′′) High-magnification confocal (z-stack) images (60×, optical zoom 2) of the area marked with an asterisk in a–a′′. The confocal image was virtually resectioned at the x and y planes to confirm the identity of the double-labeled cell. Note that all cells with RCVRN [+] cytoplasm carry CRX [+] nucleus. However, about half of cells with CRX [+] nuclei have RCVRN [+] cytoplasm. DAPI-stained nucleus. Scale bar: 100 μm. (c) Epifluorescent image of RCVRN [+] PRs with more mature morphology (including long cytoplasmic processes) distributed along the apical side. Left (RCVRN) and right (RCVRN and DAPI) insets represent magnification of the area indicated with an asterisk. DAPI-stained nucleus. (d) Immunostaining of hESC-derived retinal tissue with cone outer segment marker lectin peanut agglutinin (PNA). PNA staining pattern was specific to the apical side of hESC-derived retinal tissue (see epifluorescent image in d and the high-magnification confocal image, left inset). The right inset depicts short PNA [+] protrusions in the apical side (high-magnification confocal image with 60×, zoom 3). DAPI-stained nucleus. Scale bar: 10 μm. (e) Immunostaining of hESC-derived retinal tissue with CRX (rod–cone marker) and CALB2 (Calretinin, amacrine marker) antibodies shows localization of PRs (double arrowheads) and amacrines (arrowheads) to the apical and basal sides, respectively. Inset represents the magnification of the area (*) showing the well-developed CALB2 [+] axons of amacrine neurons. The inset with a purple arrow is the magnification of the area shown with a double asterisk (**) and depicts an elongated CALB2 [+] cell migrating from the apical toward the basal side of hESC-derived retinal tissue. DAPI-stained nucleus. Scale bar: 50 and 10 μm, respectively.
We assayed the presence of cone PRs in hESC-derived retinal tissue by IHC using peanut agglutinin (PNA). PNA lectin binds to terminal galactose-beta (1–3) N-acetyl galactosamine disaccharide residues of glycoconjugates found on the surface of cone (but not rod) outer/inner segment cell membranes and cone synaptic pedicles [54]. We noted a robust punctate PNA staining in the apical side in hESC-derived retinal tissue (Fig. 5d, inset). Under high magnification, PNA staining decorated very short stubby protrusions reminiscent of developing PR OSs [2] (Fig. 5d, left and right insets).
The presence of amacrine neurons in hESC-derived retinal tissue was determined using anti-Calretinin (CALB2) antibody. Calretinin is a member of the calcium-binding protein expressed from about embryonic week 11 in human development (Supplementary Table S4). A large number of Calretinin [+] amacrine neurons with well-developed axons were found in all sections (Fig. 5e, arrowheads and insets). Amacrine neurons localized to the basal side of hESC-derived retinal tissue, where they migrate after cell fate acquisition during retinogenesis. We observed a number of migrating Calretinin [+] cells moving from the apical toward the basal side (Fig. 5e, purple arrow). Colabeling with CRX (Fig. 5e) and CALB2 antibodies showed that the PRs and the amacrines are localized to two different layers.
Next, we examined the presence of RGCs by IHC with the anti-BRN3B antibody. Transcription factor, BRN3B, is a member of the POU domain family of transcription factors critical for RGC differentiation [56,57] and activated by RGC master cell fate control transcription factor, ATOH7 (MATH5) [55]. In agreement with finding the messages for BRN3B and MATH5 sharply upregulated by qRT-PCR, we found the abundant presence of BRN3B [+] nuclei by IHC in the basal side of hESC-derived retinal tissue (Fig. 6 and insets). In developing mammalian retina, BRN3B [+] cells were found starting from embryonic days 13–15 [92]. We found occasional BRN3B [+] cells migrating from the apical to the basal side of hESC-derived retinal tissue (Fig. 6, insets). Interestingly, as with RCVRN [+] cells (Supplementary Fig. S8, double asterisk), we discovered patches of BRN3B [+] cells located in the peripheral part of the hESC-derived retinal tissue (Fig. 6, double asterisk). We confirmed the presence of RGC marker, BRN3A, in hESC-derived retinal tissue by the immunoblot (Supplementary Fig. S3).
FIG. 6.

Localization of BRN3B-positive retinal ganglion cells (RGCs) to the basal side of hESC-derived retinal tissue. Immunostaining of hESC-derived retinal tissue with BRN3B antibody shows the prominent distribution of BRN3B [+] cells (with acquired RGC fate) in the central part of the section. The large inset represents the magnification of the area marked with an asterisk and a white arrow. The top small inset depicts a migrating BRN3B [+] cell with an elongated nucleus (*). The bottom small inset shows a BRN3B [+] cell with a round nucleus (likely mature BRN3B [+] cell) present in the basal side. Small patches of BRN3B [+] cells were also found in the periphery (**). DAPI-stained nucleus. Scale bar: 50 and 10 μm, respectively.
Identification of BRN3B [+] cells in hESC-derived retinal tissue prompted us to search for RGC axonogenesis and neuronal maturation. We used anti-NF200 antibody (NF- Heavy chain, NF-H) specific for mature RGC axons [93]. NF200 marker appears later than BRN3B in RGC development [56,93] and is associated with stabilization of neuronal circuitries in developing mammalian CNS. NF200 cytoplasmic staining was found in the basal side of hESC-derived retinal tissue. We identified numerous NF200 [+] axons traversing the retinal tissue and migrating along the apical side. Synaptophysin IHC identified positive cells in the apical and basal sides of hESC-derived retinal tissue (Fig. 7b, d). Confocal analysis of single optical sections identified numerous synaptic boutons around some (but not all) cells in the RGC and PR layers (Fig. 7e, f, f′). We also observed stretches of synaptic puncta resembling bouton en passant in the PR and RGC layers (data not shown).
FIG. 7.

Axonogenesis and synaptogenesis in hESC-derived retinal tissue. (a, a′) Immunostaining of hESC-derived retinal tissue with antibodies to NF200 (mature axonal marker) and human Synaptophysin (Syn). DAPI-stained nucleus. Scale bar: 20 μm. (b, c, c′) NF200 [+] axons outgrowing from the basal side, traversing the retinal tissue, and running along the apical side of hESC-derived retinal section (purple dots). Scale bar: 50 μm. (d) Magnified area shown in b (*) and shows the presence of Synaptophysin [+] cells in the basal layer. (e, f, f′) Confocal images demonstrating the presence of many Synaptophysin [+] synaptic boutons in the apical side. Double asterisk in e marks a group of Synaptophysin [+] cells depicted in f, magnified image. Scale bar: 20 μm. Arrows point to multiple Synaptophysin [+] boutons. (f) A confocal (z-stack) image of two cells in e. Scale bar: 20 μm. (f′) Confocal (z-stack) image of the area marked with white arrows in f. The stack image was virtually resectioned at the x and y planes to demonstrate the presence of individual synaptic boutons.
To characterize cells located between the RPE/PR and RGC/amacrine layers of hESC-derived retinal tissue, we used anti-DCAMKL1 antibody, a marker of young developing neurons in the retina (reported at embryonic day 14.5) and brain [13]. DCAMKL1 is highly expressed during axonogenesis [40,94]. The presence of this marker identifies commitment to neuronal, but not glial, identity [40]. We found the cytoplasm of most cells positive for DCAMKL1. However, the strongest staining was consistently found in the apical and basal sides (Supplementary Fig. S9).
To confirm the specificity of the IHC signals described herein, we performed staining of hESC-derived retinal tissue with only secondary antibodies (rabbit Alexa-488 and mouse Alexa-568 or vice versa) and routinely found no signal with secondary antibodies alone (Supplementary Fig. S10).
Electrophysiology of inner retinal neurons
To investigate whether neurons in hESC-derived retinal tissue are functional, we whole-cell recorded from them and characterized their intrinsic electrophysiological properties as well as responses to neurotransmitter receptor agonists. We targeted cells with medium to large somas (>8 μm diameter; Fig. 8a) toward the interior region of the hESC-derived retinal tissue. Intracellular dye fills of the recorded cells revealed that they possessed multiple branching processes that often extended more than 50 μm, confirming that they were amacrine and/or ganglion cells (Fig. 8a).
FIG. 8.

Inner retinal neurons in 12-week-old hESC-derived retinal tissue aggregates are electrically excitable. (a) Left: infrared image of a cell being recorded. Right: Lucifer Yellow fill of the same cell. Scale bar is 10 μm. (b–d) Current responses (top traces) of three different cells to a series of voltage steps (bottom traces). The dark traces highlight responses to the −13 mV step. Each cell's responses are plotted at a longer timescale (left traces) to show the complete responses and at a shorter timescale (right traces) to illustrate early components of the responses. (b) Voltage-step responses of a typical 6-week-old neuron. These responses showed no signs of voltage-gated channels. (c) Voltage-step responses of a 12-week-old neuron. The transient inward currents (arrows) induced shortly after the capacitive currents were voltage-gated Na+ currents, whereas the slowly decaying outward currents were voltage-gated K+ currents. (d) Voltage-step responses of another 12-week-old neuron recorded using the Cs+-based intracellular solution containing QX-314. Both voltage-gated Na+ and K+ currents were virtually eliminated. (e) Current step responses of another 12-week-old neuron. The most negative current step induced a sagging response reflecting Ih, whereas most of the depolarizing steps induced one or more spikes. The response highlighted in black exhibited repetitive spiking. (f) Quantitative RT-PCR analysis of hESC-derived retina differentiated for 6 and 12 weeks. Expression values of genes for Na+ and K+ voltage-gated channels, SCN1A, SCN2A, KCNA1, and KCNA6, are presented as fold change over control (pluripotent hESCs, day 0) using the comparative ddCt method [41]. Fold ratio of 1 denotes no change in gene expression. GAPDH was used as internal control. The analysis was done in technical triplicates. RT-PCR, reverse transcription-coupled polymerase chain reation.
We then examined the voltage-step responses of neurons in hESC-derived retinal tissue at either 6 or 12 weeks of age. At the younger age (6 weeks), the majority of cells tested (8 of 9) exhibited nearly ohmic current responses (Fig. 8b), suggesting that most (but not all) cells at this age lacked functional voltage-gated ion channels. At the more mature age, however, the responses of all cells studied (n = 12) displayed pronounced voltage gating. Specifically, all cells exhibited rapidly inactivating inward currents (Fig. 8c, arrows), which were followed by a slower, more sustained outward current (Fig. 8c). Similar voltage-gated currents have been shown to arise from voltage-gated Na+ and K+ channels, respectively [95]. Indeed, when we recorded from five other cells using an intracellular solution containing QX-314 (which blocks voltage-gated Na+ channels) and Cs+ (which blocks K+ channels), both voltage-gated response components were largely eliminated (Fig. 8d). To investigate whether these channels supported the generation of action potentials, we returned to the normal K+-based intracellular solution and studied voltage responses to a series of current steps. All five cells tested showed spiking activity in response to most of the depolarizing current steps (Fig. 8e). Furthermore, in response to the most negative current steps, all cells exhibited hyperpolarization that sagged over time (Fig. 8e), reflecting the hyperpolarization-activated inwardly rectifying current (Ih) [96].
Next, we tested for the presence of functional neurotransmitter receptors in the 12-week-old neurons. All cells tested (n = 7) responded to 5 s puffs of 2 mM l-glutamate and of 1 mM muscimol (a GABAA agonist) plus 1 mM glycine under both voltage clamp (Supplementary Fig. S11a, b) and current clamp (Supplementary Fig. S11c). The voltage-clamp responses to muscimol + glycine were increases in conductance and reversed near −65 mV (Supplementary Fig. S11a, b), reflecting the opening of inhibitory Cl− channels. The glutamate-induced responses were also conductance increases, but they remained inward at the most positive holding potential tested (−33 mV) (Supplementary Fig. S11a, b), consistent with the opening of excitatory cation channels. When cells were recorded under zero current clamp, responses to puffed l-glutamate were depolarizing, whereas responses to muscimol + glycine were hyperpolarizing (Supplementary Fig. S11c).
Last, we examined the change in expression of voltage-gated Na+ and K+ channel genes, SCN1A, SCN2A, KCNA1, and KCNA6, in 6- and 12-week-old hESC-derived retinal tissue by qRT-PCR. Our results support the increase in voltage-gated response in whole-cell recorded cells between 6 weeks (absent to weak) and 12 weeks (robust) (Fig. 8f).
Discussion
The regeneration of normal cellular architecture in the diseased retina through cell-based therapies will require new strategies to repair a very delicate and highly organized retinal tissue. A critical number of PRs need to be functionally reconnected into the retinal circuitry to restore vision. A number of elegant studies demonstrated the ability of some grafted PRs to integrate into the PR layer and synapse onto bipolar neurons [97–99]. However, only a limited number of PR progenitors, harvested at a narrow window of time (postnatal days 3–5 for mouse), showed successful integration [100]. Differentiating PRs in 2D monolayers further reduce the numbers of PR progenitors available for successful transplantation as PR development in 2D cultures is not synchronized. This presents a major challenge for selecting the right cell population of hESC-derived PRs for subretinal grafting. In addition, the degenerating environment in dystrophic retinas may cause integrated PRs to die [101], similar to degeneration of neurons transplanted into the brains of Huntington's patients [102].
The remarkable ability of retinally differentiated hPSCs to self-organize into rudimentary 3D retinas was first described by Sasai laboratory [103]. Compared with PRs differentiated in the 2D monolayer [27], PRs derived from hPSCs develop in a more synchronized manner and are more likely to be transplantable [104]. Sasai's discovery also provided a way to generate immature, but partially laminated, retina-like human tissue rather than PRs, resembling human embryonic retina, and use it for translational applications. Recently, several laboratories have demonstrated the ability of retinospheres to partially recapitulate retinal development [24–26] using this elegant 3D in vitro model.
Nevertheless, efforts of grafting 3D retinal tissue derived from hESCs and iPSCs are still in their infancy. Earlier attempts in fetal retina transplantation have been reported in patients [9,29–31], although it is ethically controversial and quality control and the scalability of the required tissue are limited. Both limitations can be circumvented by the use of hPSC-derived retina. However, retinospheres cultured in suspension are suboptimal for grafting because it is difficult to cut a flat and large piece of tissue from them and position it in the subretinal space (Fig. 9). The extracted tissue would likely have to be crumbled during isolation [34], which will reduce tissue's full developmental potential shown by Seiler and Aramant [105]. We demonstrate that larger pieces of hESC-derived retinal tissue, resembling human retinas during weeks 6–12 of development (Supplementary Table S4), could be derived in adherent conditions. Culturing hESC-derived retinal tissue as attached aggregates allows derivation of long and flat (rather than spherical) pieces of retina. The retinal markers we tested suggest that our hESC-derived retinal tissue roughly corresponds to the first trimester of human retinal development; this developmental stage allows successful survival and differentiation of human fetal retinal grafts [33]. Specifically, we observed the organization of hESC-derived retinal tissue into RPE, PR, and INL/RGC layers. Next, we found many early-born retinal cells (RPE, RGCs, amacrines) and strong upregulation of Opn1sw (S-Cones, 48.4 ± 4.3), while RHO, PKCα, CALB1, and GLUL (late markers) were undetected by IHC, and RHO, NRL, NR2E3, PRKCA, GLUL, and CCND3 expression was low. We also found a number of migrating cells (typical for embryonic retinogenesis), including amacrines, RGCs, and PR progenitors. Our research [27] and that of other laboratories [32,105] indicates that the subretinal niche may provide further developmental cues for immature retinal cells. To advance work on designing retinal replacement strategies, we improved our previously published retinal differentiation protocol [27] to produce hESC-derived retinal tissue in adherent conditions. Unlike culturing in suspension, culturing in adherent monolayers allows the formation of flexible and long stretches of 3D retinal tissue suitable for subretinal transplantation while not compromising the temporospatial dynamics of retinal maturation. The mitotic index in hESC-derived retinal tissue remained low at 6 weeks (Supplementary Fig. S5), suggesting that uncontrollable cell growth is highly unlikely. Our hESC-derived retinal tissue shows striking resemblance to normal embryonic mammalian retina, including emerging synaptogenesis (including boutons en passant), robust axonal growth in the RGC layer, differentiation initiating from the center of the tissue, distribution of postmitotic PRs along the apical side, and the migration of amacrines, RGCs, and putative CRX [+] PR progenitors from the apical to the basal side of the tissue (Figs. 3–7). The distribution of OTX2 [+] cell nuclei, which are typically found in PR precursors [54,85], resembled that in 6–10-week-old human fetal retina [81]. RPE pigmentation together with strong upregulation of several key RPE signature genes [106] (MITF [RPE isoform] [107], DCT, RPE-65, TYR, TYRP, and even PMEL) suggests the formation of an RPE layer around the hESC-derived retinal tissue, providing additional evidence for self-organization and polarization of this tissue. We noted that PMEL (human homolog of the Silver gene in mice), which is responsible for melanocyte granule formation, was upregulated about sevenfold by 6 weeks in hESC-derived retinal tissue. Interestingly, in cases when we found RPE-like cells on the basal side of hESC-derived retinal tissue, we observed some immature recoverin [+] PRs on the basal side as well, in line with our findings that PRs and RPE cells develop in sync during retinogenesis and PRs need close proximity to the RPE to mature and develop OSs [2]. However, we cannot exclude the possibility that these RCVRN [+] cells were cone bipolar neurons [108,109].
FIG. 9.

Retinal repair strategy using hESC-derived retinal tissue. (a) Strategy to isolate a long piece of hESC-derived retinal tissue from the large aggregate for subretinal grafting. (b) Presents our estimates of therapeutically important cell type numbers in 1 mm slices of hESC-derived retinal tissue.
We found PNA lectin signal in the apical side of hESC-derived retinal tissue, which looked like either puncta or small stubby protrusions (Fig. 5d, inset). PNA is very specific to cone, but not rod, PRs [110]. In chick retina, it was detected as early as embryonic day 7, although Müller glia was also noted to be PNA immunopositive [111]. Because we did not detect any Müller glia-specific markers (GLUL, CCND3) by IHC and by qRT-PCR, we conclude that the PNA [+] staining belongs to cones. Our hESC-derived retinal tissue probably only approximates the timing of the developmental processes in mammalian retinogenesis. Therefore, the expression onset of some markers in hESC-derived retinal tissue may not exactly match what happens in developing mammalian retina. However, the PNA signal in the apical side was an encouraging observation as robust PR development in hESC-derived retinal tissue is a prerequisite for vision restoration after retinal grafting.
Our current culturing conditions are unlikely to support the development of PR OSs, which requires a mature RPE with apical microvilli [2]. Nevertheless, our observation that PRs do develop in our hESC-derived retinal tissue suggests that hESC-derived retina might form PR OSs after being grafted subretinally, together with an intact RPE. Our results also provide reasonable expectation that hESC-derived retinal tissue will slowly complete retinal development in subretinal grafts, as has been described for grafted human embryonic retina [105]. The finding of a ciliary margin-like zone in larger hESC-derived retinal tissue (OTX2 [+], CRX [+], and LGR5 [+] cells, Figs. 3, 5, and Supplementary Fig. S6) confirms Sasai's observation [82]. The identification of many LGR5 [+] and TERT [+] cells in addition to Ki67 [+] cells in the apical side of the developing hESC-derived retinal tissue was intriguing. These data support our finding that retinal progenitors are born near the apical side of hESC-derived retinal tissue, similar to that found in mammalian retinogenesis [62,63]. In the most thoroughly characterized intestinal crypt stem cell niche, mTERT labels slow cycling cells [112], while LGR5 [+] marks rapidly cycling cells. However, both cell types appear to be Ki67 negative. In our hESC-derived retinal tissue, we identified a small number of strongly Ki67 [+] (mitotic) cells at the apical side, while more cells were weakly positive, indicating recent mitosis [60]. We used LGR5 as a marker of glycinergic amacrine cells [69], and we were intrigued by the fact that LGR5 expression became highly upregulated after the initiation of hESC-retinal differentiation and remained very strong afterward (Supplementary Table S3), likely indicating its role in retinogenesis. Collectively, it is evident that similar to the apical region of the developing mammalian retina [62,63], the apical side of the hESC-derived retinal tissue contains cells with progenitor characteristics, which divide there, acquire cell fate, and then migrate out (Figs. 3–6).
Developmental gradient in hESC-derived retinal tissue
We discovered patches of BRN3B [+] cells and recoverin [+] cells located in the periphery of the developing hESC-derived retinal tissue (Fig. 6 and Supplementary Fig. S8), while the central region, which was the most developmentally advanced part of the tissue, consistently had high concentration of these cells. RGC development starts at the central region of the developing retina and gradually spreads to the periphery [113]. This maturation pattern also applies to other cell types during mammalian retinogenesis [114,115]. Our 6-week-old hESC-derived retinal tissue followed that pattern.
RGC axons and maturation
Tracing of NF200 [+] axons allowed the identification of cells within the BRN3B [+] layer (purple dots, Fig. 7b, c, c′). In the absence of the optic nerve, these axons traversed the tissue and traveled along the apical side. NF200 appears only in postnatal RGCs in the developing mammalian retina [116,117], although other markers in our hESC-derived retinal tissue approximately matched the developmental stage to 6–10-week-old human fetal retina or embryonic days 14–15 stage in mouse retinogenesis [46,58,118]. These data support our conclusion that not all markers follow precisely their expression time course during retinogenesis. Cell fate acquisition and maturation are regulated by different intrinsic and extrinsic factors during retinogenesis, and the lack of many extrinsic developmental signals likely prevents precise coordination of retinogenesis events in hESC-derived retinal tissue. We compared our data with other reports where human fetal retina or hPSC-derived retinal organoids were studied by IHC (Supplementary Table S4). This comparison validates our finding of early PR marker CRX, RCVRN, OTX2, retinal progenitor/PR/RGC marker PAX6, early RGC marker BRN3B, and amacrine marker Calretinin in 6-week-old hESC-derived retinal tissue and also the absence of RHO and R/G opsins.
Synaptogenesis
Establishing synaptic connectivity between the INL of the graft and the host retina's RGCs is crucial for vision restoration. The robust synaptogenesis by hESC-derived retinal neurons after transplantation into adult rodent CNS and the high frequency of human/rat (chimeric) synapse formation have been demonstrated [40]. Such synapses looked mature and functional as they had Synaptophysin [+] synaptic vesicles and postsynaptic densities. Using the same monoclonal antibody to human Synaptophysin used in this study, we show that hESC-derived retinal tissue can initiate synaptogenesis and form multiple boutons on some cells (Fig. 7e–f′) identical to that found in earlier reports [40,119]. We project from this finding that our hESC-derived retinal tissue would be able to synapse with the RGCs of the recipient retina in subretinal grafts (as shown in Fig. 9) similar to that reported by Seiler et al. [28]. Transplantation of hPSC-derived retinal tissue is a promising approach to repair vision and should be extensively tested in animals with PR degeneration because in vitro studies can reveal only a limited amount of translational information.
The size of the hESC-derived retinal tissue and numbers of therapeutically valuable cell types
Large and elongated hESC-derived retinal tissue aggregates cultured in our conditions were about 150–300 somas in diameter and 8–12 somas in thickness. We estimated that an average midsagittal section carried about 80 PRs, 50 amacrines, and 80 RGCs (Fig. 9). Because our sections were 12 μm thick and assuming that every other section within a 1 mm slice of hESC-derived retinal tissue produces similar cell counts, we estimate that a large 1-mm-thick section of hESC-derived retinal tissue provides about 3,200 PRs, about 2,000 amacrines, and 3,200 RGCs. The size of this 3D retinal slice is comparable with that used for routine fetal retinal grafting (2 mm2) [105]. The transplant size for human patients (reported by Seiler and colleagues [9], also personal communication) was 2–5 mm2. The size of the grafts for rat transplants was about 1–1.5 × 0.6 mm [120,121]. Two or three large pieces of hESC-derived retinal tissue, generated by our approach, could be grafted together to have a graft of about 5 mm length in size.
Electrophysiology
A recent study showed that retinal tissue derived from human iPSCs contained all five classes of neurons (rod/cone PRs, horizontal, bipolar, amacrine, and ganglion) as well as Müller glia and that these cells organized systematically into layers [26]. Furthermore, some PRs responded to light, indicating that they had acquired functional properties in addition to expressing molecular markers of PRs [26]. In the current study, we tested whether inner retinal neurons in hESC-derived retinal tissue have acquired functional characteristics in addition to expressing amacrine and RGC markers. We found that in 6-week-old hESC-derived retinal tissue aggregates, only one of nine recorded cells had significant voltage-gated currents. By contrast, all 12 tested cells from 12-week-old hESC-derived retinal tissue aggregates exhibited both slowly decaying, Cs+-sensitive outward currents and rapidly inactivating, QX-314-sensitive inward currents, indicating the presence of voltage-gated K+ and Na+ channels, respectively (Fig. 8). These more mature neurons also generated spikes in response to depolarizing current injection, suggesting a capacity for long-range signal propagation. However, these responses usually consisted of only one spike (see the responses to the four most positive current steps in Fig. 8e), suggesting that at 12 weeks of age, these neurons are still not fully developed [122]. Finally, all 12-week-old inner retinal neurons responded to l-glutamate, muscimol, and/or glycine with reversal potentials expected for ionotropic glutamate and GABA/glycine receptors (Supplementary Fig. S11), indicating the presence of both excitatory and inhibitory receptors. In conclusion, these more mature neurons should be able to respond to input from presynaptic bipolar and amacrine cells and to signal to postsynaptic neurons through spiking. Thus, this retinal tissue has the potential to perform at least some electrophysiological functions needed for visual processing. It is likely that its full potential could be revealed only in subretinal grafts 6–9 months after grafting [32,105].
Limitations of our method include the inability to keep hESC-derived retinal tissue aggregates for a prolonged period of time on the same Matrigel-coated plates and variable yield (from 2–3 to 15 or more of such aggregates per 35-mm Petri dish). While we solved the second problem by scaling up the cultures, maintaining hESC-derived retinal tissue aggregates on the same Matrigel plates for up to 12 weeks was challenging. Many of them were gradually dislodging from degenerating Matrigel. Introducing novel matrices for longer-term culture of hESC-derived retinal tissue in adherent conditions may provide a significant advancement in process development leading to the industrial-scale manufacture of clinical-grade product. On the other hand, the developmental stage of our hESC-derived retinal tissue at 6 weeks of differentiation (approximately equal to 6–10-week-old human fetal retina, Supplementary Table S4) may be sufficient for successful survival of such grafts in the subretinal space, and culture for longer periods of time may generate more mature retinal tissue, which may not survive retinal grafting [33].
Process and assay development leading to products that can ultimately be utilized in animal preclinical and subsequently human clinical development will likely need to be performed using clinical good manufacturing practice (cGMP)-grade hESCs and potentially cells genetically modified to escape the immune surveillance of the host. Alternatively, patient-specific iPSCs could be utilized, although patient-specific protocols would be expected to add significantly to the cost of care. A key aspect of process development will need to include quality assurance systems that precisely characterized the cellular component of the resulting retinal tissues. The characterization of the cellular components, while not necessarily pluripotent stem cells, could nevertheless cause inappropriate ectopic tissue formation and will therefore need additional study.
In summary, the key observations of our study are the (1) ability to partially reproduce early retinogenesis and histogenesis in hESC-derived retinal tissue, (2) potential to generate longer and flexible pieces of retina suitable for subretinal grafting in adherent conditions, (3) low mitotic index by 6 weeks after initiation of hESC-> retinal differentiation, indicating low danger of tumorigenesis, (4) approximate matching of developmental events in hESC-derived retinal tissue to ∼6–10 week human fetal retina (which is close to developmental time point successfully used for subretinal grafting [105]), and (5) the evidence of emerging synaptogenesis and electrophysiological activity, indicating the potential of such hESC-derived retinal tissue to establish synaptic connectivity in subretinal grafts.
Supplementary Material
Acknowledgments
The authors acknowledge WiCell tech support (for culturing hESCs WA01 in feeder-free conditions); Edwin S. Monuki, MD, PhD, UCI, Connie Cepko, PhD, Harvard, and Tudor Badea, MD, PhD, NEI, for antibodies; Department of Ophthalmology, University of Pittsburgh (startup funding to I.O.N.); Department of Ophthalmology and Visual Sciences, University of Michigan (startup funding to K.Y.W.); Dr. E. Ronald Salvitti (I.O.N., R.K.S.); National Institutes of Health CORE Grant P30 EY008098 (to University of Pittsburgh); National Institutes of Health CORE Grant P30 EY007003 (to University of Michigan); Eye and Ear Foundation of Pittsburgh, PA; Jane Love Family Foundation; Lions PA Foundation; and Unrestricted Grant from Research to Prevent Blindness, New York, NY. The authors thank Jerome Roger, PhD, for critically reading the manuscript and Michael Dyer, PhD, for helpful suggestions.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Kolomeyer AM. and Zarbin MA. (2014). Trophic factors in the pathogenesis and therapy for retinal degenerative diseases. Surv Ophthalmol 59:134–165 [DOI] [PubMed] [Google Scholar]
- 2.Nasonkin IO, Merbs SL, Lazo K, Oliver VF, Brooks M, Patel K, Enke RA, Nellissery J, Jamrich M, et al. (2013). Conditional knockdown of DNA methyltransferase 1 reveals a key role of retinal pigment epithelium integrity in photoreceptor outer segment morphogenesis. Development 140:1330–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Longbottom R, Fruttiger M, Douglas RH, Martinez-Barbera JP, Greenwood J. and Moss SE. (2009). Genetic ablation of retinal pigment epithelial cells reveals the adaptive response of the epithelium and impact on photoreceptors. Proc Natl Acad Sci U S A 106:18728–18733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu D, Deng X, Spee C, Sonoda S, Hsieh CL, Barron E, Pera M. and Hinton DR. (2011). Polarized secretion of PEDF from human embryonic stem cell-derived RPE promotes retinal progenitor cell survival. Invest Ophthalmol Vis Sci 52:1573–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazzoni F, Safa H. and Finnemann SC. (2014). Understanding photoreceptor outer segment phagocytosis: use and utility of RPE cells in culture. Exp Eye Res 126:51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strauss O. (2005). The retinal pigment epithelium in visual function. Physiol Rev 85:845–881 [DOI] [PubMed] [Google Scholar]
- 7.German OL, Buzzi E, Rotstein NP, Rodriguez-Boulan E. and Politi LE. (2008). Retinal pigment epithelial cells promote spatial reorganization and differentiation of retina photoreceptors. J Neurosci Res 86:3503–3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aramant RB, Seiler MJ. and Ball SL. (1999). Successful cotransplantation of intact sheets of fetal retina with retinal pigment epithelium. Invest Ophthalmol Vis Sci 40:1557–1564 [PubMed] [Google Scholar]
- 9.Radtke ND, Aramant RB, Petry HM, Green PT, Pidwell DJ. and Seiler MJ. (2008). Vision improvement in retinal degeneration patients by implantation of retina together with retinal pigment epithelium. Am J Ophthalmol 146:172–182 [DOI] [PubMed] [Google Scholar]
- 10.Yao J, Tucker BA, Zhang X, Checa-Casalengua P, Herrero-Vanrell R. and Young MJ. (2011). Robust cell integration from co-transplantation of biodegradable MMP2-PLGA microspheres with retinal progenitor cells. Biomaterials 32:1041–1050 [DOI] [PubMed] [Google Scholar]
- 11.Seiler MJ. and Aramant RB. (2012). Cell replacement and visual restoration by retinal sheet transplants. Prog Retin Eye Res 31:661–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seiler MJ. and Aramant RB. (2005). Transplantation of neuroblastic progenitor cells as a sheet preserves and restores retinal function. Semin Ophthalmol 20:31–42 [DOI] [PubMed] [Google Scholar]
- 13.Boer GJ. (1994). Ethical guidelines for the use of human embryonic or fetal tissue for experimental and clinical neurotransplantation and research. Network of European CNS Transplantation and Restoration (NECTAR). J Neurol 242:1–13 [DOI] [PubMed] [Google Scholar]
- 14.Seiler MJ, Aramant RB, Thomas BB, Peng Q, Sadda SR. and Keirstead HS. (2010). Visual restoration and transplant connectivity in degenerate rats implanted with retinal progenitor sheets. Eur J Neurosci 31:508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seiler MJ, Sagdullaev BT, Woch G, Thomas BB. and Aramant RB. (2005). Transsynaptic virus tracing from host brain to subretinal transplants. Eur J Neurosci 21:161–172 [DOI] [PubMed] [Google Scholar]
- 16.(2010). Access Economics, prepared for AMD Alliance International: The Global Economic Cost of Visual Impairment. www.icoph.org/resources/146/The-Global-Economic-Cost-of-Visual-Impairment.html
- 17.Frick KD, Kymes SM, Lee PP, Matchar DB, Pezzullo ML, Rein DB, Taylor HR; Vancouver Economic Burden of Vision Loss Group. (2010). The cost of visual impairment: purposes, perspectives, and guidance. Invest Ophthalmol Vis Sci 51:1801–1805 [DOI] [PubMed] [Google Scholar]
- 18.Ferris FL, 3rd, and Tielsch JM. (2004). Blindness and visual impairment: a public health issue for the future as well as today. Arch Ophthalmol 122:451–452 [DOI] [PubMed] [Google Scholar]
- 19.Rein DB, Zhang P, Wirth KE, Lee PP, Hoerger TJ, McCall N, Klein R, Tielsch JM, Vijan S. and Saaddine J. (2006). The economic burden of major adult visual disorders in the United States. Arch Ophthalmol 124:1754–1760 [DOI] [PubMed] [Google Scholar]
- 20.Friedman DS, O'Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J; Eye Diseases Prevalence Research Group. (2004). Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 122:564–572 [DOI] [PubMed] [Google Scholar]
- 21.Jager RD, Mieler WF. and Miller JW. (2008). Age-related macular degeneration. N Engl J Med 358:2606–2617 [DOI] [PubMed] [Google Scholar]
- 22.Hartong DT, Berson EL. and Dryja TP. (2006). Retinitis pigmentosa. Lancet 368:1795–1809 [DOI] [PubMed] [Google Scholar]
- 23.Centers for Disease Control and Prevention. (2014). National Diabetes Statistics Report. www.cdc.gov/diabetes/data/statistics/2014statisticsreport.html
- 24.Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, Saito K, Yonemura S, Eiraku M. and Sasai Y. (2012). Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10:771–785 [DOI] [PubMed] [Google Scholar]
- 25.Meyer JS, Howden SE, Wallace KA, Verhoeven AD, Wright LS, Capowski EE, Pinilla I, Martin JM, Tian S, et al. (2011). Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 29:1206–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao LH, Peters A, Park TS, Zambidis ET, et al. (2014). Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun 5:4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hambright D, Park KY, Brooks M, McKay R, Swaroop A. and Nasonkin IO. (2012). Long-term survival and differentiation of retinal neurons derived from human embryonic stem cell lines in un-immunosuppressed mouse retina. Mol Vis 18:920–936 [PMC free article] [PubMed] [Google Scholar]
- 28.Seiler MJ, Thomas BB, Chen Z, Wu R, Sadda SR. and Aramant RB. (2008). Retinal transplants restore visual responses: trans-synaptic tracing from visually responsive sites labels transplant neurons. Eur J Neurosci 28:208–220 [DOI] [PubMed] [Google Scholar]
- 29.Radtke ND, Aramant RB, Seiler M. and Petry HM. (1999). Preliminary report: indications of improved visual function after retinal sheet transplantation in retinitis pigmentosa patients. Am J Ophthalmol 128:384–387 [DOI] [PubMed] [Google Scholar]
- 30.Radtke ND, Aramant RB, Seiler MJ, Petry HM. and Pidwell D. (2004). Vision change after sheet transplant of fetal retina with retinal pigment epithelium to a patient with retinitis pigmentosa. Arch Ophthalmol 122:1159–1165 [DOI] [PubMed] [Google Scholar]
- 31.Radtke ND, Seiler MJ, Aramant RB, Petry HM. and Pidwell DJ. (2002). Transplantation of intact sheets of fetal neural retina with its retinal pigment epithelium in retinitis pigmentosa patients. Am J Ophthalmol 133:544–550 [DOI] [PubMed] [Google Scholar]
- 32.Aramant RB. and Seiler MJ. (2002). Transplanted sheets of human retina and retinal pigment epithelium develop normally in nude rats. Exp Eye Res 75:115–125 [DOI] [PubMed] [Google Scholar]
- 33.Aramant R, Seiler M. and Turner JE. (1988). Donor age influences on the success of retinal grafts to adult rat retina. Invest Ophthalmol Vis Sci 29:498–503 [PubMed] [Google Scholar]
- 34.Assawachananont J, Mandai M, Okamoto S, Yamada C, Eiraku M, Yonemura S, Sasai Y. and Takahashi M. (2014). Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice. Stem Cell Reports 2:662–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS. and Jones JM. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282:1145–1147 [DOI] [PubMed] [Google Scholar]
- 36.Ludwig TE, Bergendahl V, Levenstein ME, Yu J, Probasco MD. and Thomson JA. (2006). Feeder-independent culture of human embryonic stem cells. Nat Methods 3:637–646 [DOI] [PubMed] [Google Scholar]
- 37.Xu RH, Peck RM, Li DS, Feng X, Ludwig T. and Thomson JA. (2005). Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat Methods 2:185–190 [DOI] [PubMed] [Google Scholar]
- 38.Furue MK, Na J, Jackson JP, Okamoto T, Jones M, Baker D, Hata R, Moore HD, Sato JD. and Andrews PW. (2008). Heparin promotes the growth of human embryonic stem cells in a defined serum-free medium. Proc Natl Acad Sci U S A 105:13409–13414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K. and Sasai Y. (2007). A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25:681–686 [DOI] [PubMed] [Google Scholar]
- 40.Nasonkin I, Mahairaki V, Xu L, Hatfield G, Cummings BJ, Eberhart C, Ryugo DK, Maric D, Bar E. and Koliatsos VE. (2009). Long-term, stable differentiation of human embryonic stem cell-derived neural precursors grafted into the adult mammalian neostriatum. Stem Cells 27:2414–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bustin SA. (2000). Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25:169–193 [DOI] [PubMed] [Google Scholar]
- 42.Pevny LH, Sockanathan S, Placzek M. and Lovell-Badge R. (1998). A role for SOX1 in neural determination. Development 125:1967–1978 [DOI] [PubMed] [Google Scholar]
- 43.Suter DM, Tirefort D, Julien S. and Krause KH. (2009). A Sox1 to Pax6 switch drives neuroectoderm to radial glia progression during differentiation of mouse embryonic stem cells. Stem Cells 27:49–58 [DOI] [PubMed] [Google Scholar]
- 44.Muranishi Y, Terada K. and Furukawa T. (2012). An essential role for Rax in retina and neuroendocrine system development. Dev Growth Differ 54:341–348 [DOI] [PubMed] [Google Scholar]
- 45.Mathers PH, Grinberg A, Mahon KA. and Jamrich M. (1997). The Rx homeobox gene is essential for vertebrate eye development. Nature 387:603–607 [DOI] [PubMed] [Google Scholar]
- 46.Livesey FJ. and Cepko CL. (2001). Vertebrate neural cell-fate determination: lessons from the retina. Nat Rev Neurosci 2:109–118 [DOI] [PubMed] [Google Scholar]
- 47.Furukawa T, Kozak CA. and Cepko CL. (1997). rax, a novel paired-type homeobox gene, shows expression in the anterior neural fold and developing retina. Proc Natl Acad Sci U S A 94:3088–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mao CA, Cho JH, Wang J, Gao Z, Pan P, Tsai WW, Frishman LJ. and Klein WH. (2013). Reprogramming amacrine and photoreceptor progenitors into retinal ganglion cells by replacing Neurod1 with Atoh7. Development 140:541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pollak J, Wilken MS, Ueki Y, Cox KE, Sullivan JM, Taylor RJ, Levine EM. and Reh TA. (2013). ASCL1 reprograms mouse Muller glia into neurogenic retinal progenitors. Development 140:2619–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brzezinski JA, 4th, Kim EJ, Johnson JE. and Reh TA. (2011). Ascl1 expression defines a subpopulation of lineage-restricted progenitors in the mammalian retina. Development 138:3519–3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rowan S. and Cepko CL. (2004). Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev Biol 271:388–402 [DOI] [PubMed] [Google Scholar]
- 52.Rowan S, Chen CM, Young TL, Fisher DE. and Cepko CL. (2004). Transdifferentiation of the retina into pigmented cells in ocular retardation mice defines a new function of the homeodomain gene Chx10. Development 131:5139–5152 [DOI] [PubMed] [Google Scholar]
- 53.Elliott J, Jolicoeur C, Ramamurthy V. and Cayouette M. (2008). Ikaros confers early temporal competence to mouse retinal progenitor cells. Neuron 60:26–39 [DOI] [PubMed] [Google Scholar]
- 54.Swaroop A, Kim D. and Forrest D. (2010). Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci 11:563–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brzezinski JA, 4th, Prasov L. and Glaser T. (2012). Math5 defines the ganglion cell competence state in a subpopulation of retinal progenitor cells exiting the cell cycle. Dev Biol 365:395–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bates CA. and Meyer RL. (1993). The heavy neurofilament protein is expressed in regenerating adult but not embryonic mammalian optic fibers in vitro. Exp Neurol 119:249–257 [DOI] [PubMed] [Google Scholar]
- 57.Tanaka S, Toyonaga T, Ohara Y, Yoshizaki T, Kawara F, Ishida T, Hoshi N, Morita Y. and Azuma T. (2015). Esophageal diverticulum exposed during endoscopic submucosal dissection of superficial cancer. World J Gastroenterol 21:3121–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marquardt T. and Gruss P. (2002). Generating neuronal diversity in the retina: one for nearly all. Trends Neurosci 25:32–38 [DOI] [PubMed] [Google Scholar]
- 59.Gerdes J, Schwab U, Lemke H. and Stein H. (1983). Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer 31:13–20 [DOI] [PubMed] [Google Scholar]
- 60.Scholzen T. and Gerdes J. (2000). The Ki-67 protein: from the known and the unknown. J Cell Physiol 182:311–322 [DOI] [PubMed] [Google Scholar]
- 61.Jonat W. and Arnold N. (2011). Is the Ki-67 labelling index ready for clinical use? Ann Oncol 22:500–502 [DOI] [PubMed] [Google Scholar]
- 62.Baye LM. and Link BA. (2008). Nuclear migration during retinal development. Brain Res 1192:29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Norden C, Young S, Link BA. and Harris WA. (2009). Actomyosin is the main driver of interkinetic nuclear migration in the retina. Cell 138:1195–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cayouette M, Poggi L. and Harris WA. (2006). Lineage in the vertebrate retina. Trends Neurosci 29:563–570 [DOI] [PubMed] [Google Scholar]
- 65.Cayouette M. and Raff M. (2003). The orientation of cell division influences cell-fate choice in the developing mammalian retina. Development 130:2329–2339 [DOI] [PubMed] [Google Scholar]
- 66.Dyer MA. and Cepko CL. (2001). Regulating proliferation during retinal development. Nat Rev Neurosci 2:333–342 [DOI] [PubMed] [Google Scholar]
- 67.Gomes FLAF. and Cayouette M. (2009). Retinal development: cell type specification. In: Encyclopedia of Neuroscience. Squire LR, ed. Academic Press, Oxford, pp. 203–209 [Google Scholar]
- 68.Meyer EJ, Ikmi A. and Gibson MC. (2011). Interkinetic nuclear migration is a broadly conserved feature of cell division in pseudostratified epithelia. Curr Biol 21:485–491 [DOI] [PubMed] [Google Scholar]
- 69.Sukhdeo K, Koch CE, Miller TE, Zhou H, Rivera M, Yan K, Cepko CL, Lathia JD. and Rich JN. (2014). The Lgr5 transgene is expressed specifically in glycinergic amacrine cells in the mouse retina. Exp Eye Res 119:106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonilha VL, Finnemann SC. and Rodriguez-Boulan E. (1999). Ezrin promotes morphogenesis of apical microvilli and basal infoldings in retinal pigment epithelium. J Cell Biol 147:1533–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonilha VL. and Rodriguez-Boulan E. (2001). Polarity and developmental regulation of two PDZ proteins in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 42:3274–3282 [PubMed] [Google Scholar]
- 72.Huang J, Possin DE. and Saari JC. (2009). Localizations of visual cycle components in retinal pigment epithelium. Mol Vis 15:223–234 [PMC free article] [PubMed] [Google Scholar]
- 73.Nishida A, Furukawa A, Koike C, Tano Y, Aizawa S, Matsuo I. and Furukawa T. (2003). Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat Neurosci 6:1255–1263 [DOI] [PubMed] [Google Scholar]
- 74.Koike C, Nishida A, Ueno S, Saito H, Sanuki R, Sato S, Furukawa A, Aizawa S, Matsuo I, et al. (2007). Functional roles of Otx2 transcription factor in postnatal mouse retinal development. Mol Cell Biol 27:8318–8329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martinez-Morales JR, Signore M, Acampora D, Simeone A. and Bovolenta P. (2001). Otx genes are required for tissue specification in the developing eye. Development 128:2019–2030 [DOI] [PubMed] [Google Scholar]
- 76.Beby F, Housset M, Fossat N, Le Greneur C, Flamant F, Godement P. and Lamonerie T. (2010). Otx2 gene deletion in adult mouse retina induces rapid RPE dystrophy and slow photoreceptor degeneration. PLoS One 5:e11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Housset M, Samuel A, Ettaiche M, Bemelmans A, Beby F, Billon N. and Lamonerie T. (2013). Loss of Otx2 in the adult retina disrupts retinal pigment epithelium function, causing photoreceptor degeneration. J Neurosci 33:9890–9904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Glubrecht DD, Kim JH, Russell L, Bamforth JS. and Godbout R. (2009). Differential CRX and OTX2 expression in human retina and retinoblastoma. J Neurochem 111:250–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kurokawa D, Ohmura T, Sakurai Y, Inoue K, Suda Y. and Aizawa S. (2014). Otx2 expression in anterior neuroectoderm and forebrain/midbrain is directed by more than six enhancers. Dev Biol 387:203–213 [DOI] [PubMed] [Google Scholar]
- 80.Spieler D, Baumer N, Stebler J, Koprunner M, Reichman-Fried M, Teichmann U, Raz E, Kessel M. and Wittler L. (2004). Involvement of Pax6 and Otx2 in the forebrain-specific regulation of the vertebrate homeobox gene ANF/Hesx1. Dev Biol 269:567–579 [DOI] [PubMed] [Google Scholar]
- 81.Larsen KB, Lutterodt M, Rath MF. and Moller M. (2009). Expression of the homeobox genes PAX6, OTX2, and OTX1 in the early human fetal retina. Int J Dev Neurosci 27:485–492 [DOI] [PubMed] [Google Scholar]
- 82.Kuwahara A, Ozone C, Nakano T, Saito K, Eiraku M. and Sasai Y. (2015). Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat Commun 6:6286. [DOI] [PubMed] [Google Scholar]
- 83.Liu H, Xu S, Wang Y, Mazerolle C, Thurig S, Coles BL, Ren JC, Taketo MM, van der Kooy D. and Wallace VA. (2007). Ciliary margin transdifferentiation from neural retina is controlled by canonical Wnt signaling. Dev Biol 308:54–67 [DOI] [PubMed] [Google Scholar]
- 84.Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F. and Gruss P. (2001). Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105:43–55 [DOI] [PubMed] [Google Scholar]
- 85.Emerson MM, Surzenko N, Goetz JJ, Trimarchi J. and Cepko CL. (2013). Otx2 and Onecut1 promote the fates of cone photoreceptors and horizontal cells and repress rod photoreceptors. Dev Cell 26:59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oron-Karni V, Farhy C, Elgart M, Marquardt T, Remizova L, Yaron O, Xie Q, Cvekl A. and Ashery-Padan R. (2008). Dual requirement for Pax6 in retinal progenitor cells. Development 135:4037–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dizhoor AM, Ray S, Kumar S, Niemi G, Spencer M, Brolley D, Walsh KA, Philipov PP, Hurley JB. and Stryer L. (1991). Recoverin: a calcium sensitive activator of retinal rod guanylate cyclase. Science 251:915–918 [DOI] [PubMed] [Google Scholar]
- 88.Molday RS. and MacKenzie D. (1983). Monoclonal antibodies to rhodopsin: characterization, cross-reactivity, and application as structural probes. Biochemistry 22:653–660 [DOI] [PubMed] [Google Scholar]
- 89.Engelsberg K. and Ghosh F. (2011). Human retinal development in an in situ whole eye culture system. Dev Neurosci 33:110–117 [DOI] [PubMed] [Google Scholar]
- 90.Yan XX. and Wiechmann AF. (1997). Early expression of recoverin in a unique population of neurons in the human retina. Anat Embryol (Berl) 195:51–63 [DOI] [PubMed] [Google Scholar]
- 91.Nasonkin I, Cogliati T. and Swaroop A. (2010). Photoreceptor Development: early steps/fate. In: The Retina and its Disorders. Besharse J. and Bok D, eds. Academic Press, pp 567–574 [Google Scholar]
- 92.Prasov L. and Glaser T. (2012). Dynamic expression of ganglion cell markers in retinal progenitors during the terminal cell cycle. Mol Cell Neurosci 50:160–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tanaka T, Yokoi T, Tamalu F, Watanabe S, Nishina S. and Azuma N. (2015). Generation of retinal ganglion cells with functional axons from human induced pluripotent stem cells. Sci Rep 5:8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB. and Walsh CA. (2006). Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron 49:41–53 [DOI] [PubMed] [Google Scholar]
- 95.Lipton SA. and Tauck DL. (1987). Voltage-dependent conductances of solitary ganglion cells dissociated from the rat retina. J Physiol 385:361–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pape HC. (1996). Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol 58:299–327 [DOI] [PubMed] [Google Scholar]
- 97.Lamba DA, Gust J. and Reh TA. (2009). Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell 4:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Barber AC, Hippert C, Duran Y, West EL, Bainbridge JW, Warre-Cornish K, Luhmann UF, Lakowski J, Sowden JC, Ali RR. and Pearson RA. (2013). Repair of the degenerate retina by photoreceptor transplantation. Proc Natl Acad Sci U S A 110:354–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pearson RA, Barber AC, West EL, MacLaren RE, Duran Y, Bainbridge JW, Sowden JC. and Ali RR. (2010). Targeted disruption of outer limiting membrane junctional proteins (Crb1 and ZO-1) increases integration of transplanted photoreceptor precursors into the adult wild-type and degenerating retina. Cell Transplant 19:487–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, Swaroop A, Sowden JC. and Ali RR. (2006). Retinal repair by transplantation of photoreceptor precursors. Nature 444:203–207 [DOI] [PubMed] [Google Scholar]
- 101.Yao J, Feathers KL, Khanna H, Thompson D, Tsilfidis C, Hauswirth WW, Heckenlively JR, Swaroop A. and Zacks DN. (2011). XIAP therapy increases survival of transplanted rod precursors in a degenerating host retina. Invest Ophthalmol Vis Sci 52:1567–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cicchetti F, Saporta S, Hauser RA, Parent M, Saint-Pierre M, Sanberg PR, Li XJ, Parker JR, Chu Y, et al. (2009). Neural transplants in patients with Huntington's disease undergo disease-like neuronal degeneration. Proc Natl Acad Sci U S A 106:12483–12488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T. and Sasai Y. (2011). Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472:51–56 [DOI] [PubMed] [Google Scholar]
- 104.Lakowski J, Gonzalez-Cordero A, West EL, Han YT, Welby E, Naeem A, Blackford SJ, Bainbridge JW, Pearson RA, Ali RR. and Sowden JC. (2015). Transplantation of photoreceptor precursors isolated via a cell surface biomarker panel from embryonic stem cell-derived self-forming retina. Stem Cells 33:2469–2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Seiler MJ. and Aramant RB. (1998). Intact sheets of fetal retina transplanted to restore damaged rat retinas. Invest Ophthalmol Vis Sci 39:2121–2131 [PubMed] [Google Scholar]
- 106.Strunnikova NV, Maminishkis A, Barb JJ, Wang F, Zhi C, Sergeev Y, Chen W, Edwards AO, Stambolian D, et al. (2010). Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum Mol Genet 19:2468–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bharti K, Liu W, Csermely T, Bertuzzi S. and Arnheiter H. (2008). Alternative promoter use in eye development: the complex role and regulation of the transcription factor MITF. Development 135:1169–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Milam AH, Dacey DM. and Dizhoor AM. (1993). Recoverin immunoreactivity in mammalian cone bipolar cells. Vis Neurosci 10:1–12 [DOI] [PubMed] [Google Scholar]
- 109.Haverkamp S. and Wassle H. (2000). Immunocytochemical analysis of the mouse retina. J Comp Neurol 424:1–23 [PubMed] [Google Scholar]
- 110.Kawano K, Uehara F, Sameshima M. and Ohba N. (1984). Binding sites of peanut agglutinin in mammalian retina. Jpn J Ophthalmol 28:205–214 [PubMed] [Google Scholar]
- 111.Arregui C, Barra HS. and Landa CA. (1992). Peanut agglutinin binding glycoproteins in the chick retina: their presence in Muller glia cells. J Neurosci Res 31:532–542 [DOI] [PubMed] [Google Scholar]
- 112.Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk ME, Henderson DE, Baffour-Awuah NY, Ambruzs DM, Fogli LK, Algra S. and Breault DT. (2011). Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A 108:179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Furukawa T, Mukherjee S, Bao ZZ, Morrow EM. and Cepko CL. (2000). rax, Hes1, and notch1 promote the formation of Muller glia by postnatal retinal progenitor cells. Neuron 26:383–394 [DOI] [PubMed] [Google Scholar]
- 114.Prada C, Puga J, Perez-Mendez L, Lopez R. and Ramirez G. (1991). Spatial and temporal patterns of neurogenesis in the chick retina. Eur J Neurosci 3:559–569 [DOI] [PubMed] [Google Scholar]
- 115.Prada F, Medina JI, Lopez-Gallardo M, Lopez R, Quesada A, Spira A. and Prada C. (1999). Spatiotemporal gradients of differentiation of chick retina types I and II cholinergic cells: identification of a common postmitotic cell population. J Comp Neurol 410:457–466 [PubMed] [Google Scholar]
- 116.Vergara MN. and Canto-Soler MV. (2012). Rediscovering the chick embryo as a model to study retinal development. Neural Dev 7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vergara MN, de la Rosa EJ. and Canto-Soler MV. (2012). Focus on molecules: proinsulin in the eye: precursor or pioneer? Exp Eye Res 101:109–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bassett EA. and Wallace VA. (2012). Cell fate determination in the vertebrate retina. Trends Neurosci 35:565–573 [DOI] [PubMed] [Google Scholar]
- 119.Xu L, Yan J, Chen D, Welsh AM, Hazel T, Johe K, Hatfield G. and Koliatsos VE. (2006). Human neural stem cell grafts ameliorate motor neuron disease in SOD-1 transgenic rats. Transplantation 82:865–875 [DOI] [PubMed] [Google Scholar]
- 120.Yang PB, Seiler MJ, Aramant RB, Yan F, Mahoney MJ, Kitzes LM. and Keirstead HS. (2010). Trophic factors GDNF and BDNF improve function of retinal sheet transplants. Exp Eye Res 91:727–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seiler MJ, Jones BW, Aramant RB, Yang PB, Keirstead HS. and Marc RE. (2012). Computational molecular phenotyping of retinal sheet transplants to rats with retinal degeneration. Eur J Neurosci 35:1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Skaliora I, Scobey RP. and Chalupa LM. (1993). Prenatal development of excitability in cat retinal ganglion cells: action potentials and sodium currents. J Neurosci 13:313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.