

Abstract
Survivin, a member of the inhibitor of apoptosis family of proteins (IAPs) that controls cell division, apoptosis, metastasis and angiogenesis, is overexpressed in essentially all human cancers. As a consequence, the gene/protein is considered an attractive target for cancer treatment. Here, we discuss recent findings related to the regulation of survivin expression and its role in angiogenesis, particularly in the context of hypoxia. We propose a novel role for survivin in cancer, whereby expression of the protein in tumor cells promotes VEGF synthesis, secretion and angiogenesis. Mechanistically, we propose the existence of a positive feed-back loop involving PI3-kinase/Akt activation and enhanced β-Catenin-TCF/LEF-dependent VEGF expression followed by secretion. Finally, we elaborate on the possibility that this mechanism operating in cancer cells may contribute to enhanced tumor vascularization by vasculogenic mimicry together with conventional angiogenesis.
Keywords: Survivin, Hypoxia, Cell survival, ROS, VEGF, Angiogenesis, Oxidative stress, Cancer
Background
Survivin (BIRC5) is a member of the IAP family that participates in cell division, apoptosis inhibition and angiogenesis [1–3]. In humans, survivin is widely expressed in development but generally not present in adult tissues [4]. However, upon malignant transformation the protein survivin is commonly re-expressed [5] leading to the promotion of cell survival, proliferation and metastasis. For these reasons, survivin is considered a potentially interesting target for cancer therapy [2]. In tumors, the expression of survivin and VEGF are closely linked during tumor growth and angiogenesis, and are detected in several types of cancer [6–9]. In this review, we will focus predominantly on the role of survivin in angiogenesis. Recent findings from our group identified a novel pathway by which survivin promotes VEGF (vascular endothelial growth factor) expression in cancer cells, hence promoting angiogenesis, a rate-limiting step in tumor growth. Also, we will elaborate on the relevance of this pathway in the context of hypoxia, reactive oxygen species (ROS) formation and β-Catenin signaling. Finally, we will consider the possibility that survivin may also be relevant to the process of vasculogenic mimicry suggested to occur in regions of tumors that lack endothelial cell-mediated vascularization.
Survivin and cell survival in hypoxia: hypoxia-induced factors
Hypoxia
Under normal physiological conditions, cells are supplied with oxygen at concentrations ranging from 1-13 % O2 depending on the tissue [10, 11]. However, under pathological conditions, as is the case in cancer, tissue oxygenation is severely impaired due to insufficient vascularization resulting in a condition known as hypoxia [10, 12]. In patients, hypoxia has been reported in most solid tumors, including prostate [13], pancreas [14], head and neck [15], breast [16], kidney [17] and liver cancer [18]. Beyond the immediate responses linked to lack of oxygen and nutrients, including the induction of Hypoxia-Induced Factors (HIFs), autophagy and the unfolded protein response (UPR), tumor hypoxia is also associated with resistance to chemotherapy, metastasis and reduced patient survival [19–23]. Unfortunately, these adaptive mechanisms enable cancer cells to adjust to low oxygen concentrations, proliferate and ultimately disseminate to distant sites. In this review, we will center the discussion mainly on HIF-related adaptive responses.
In normal tissues, blood vessels are characterized by a well-defined hierarchical organization and reside in close proximity of target cells in order to ensure a constant oxygen and nutrient supply. However, in tumors an imbalance between oxygen consumption/delivery due to excessive proliferation results in hypoxia [24], a highly dynamic process which involves both periods of chronic and cyclic hypoxia [25]. Several factors contribute to and exacerbate the hypoxic microenvironment. Typically, the tumor blood vessels are disorganized, aberrantly branched and frequently distant from the tumor cells. Also, the blood vessels are more leaky leading to blood deviation and increased compensatory blood flow bypassing tumor blood vessels, thereby reducing oxygen levels even further. In addition, tumor blood vessels are characterized by fragile capillary walls and retarded blood flow, which again accentuates hypoxia [24, 26]. Lower vascular density, inherent limitations in oxygen diffusion, erythrocyte hardening and increased blood viscosity all contribute to reducing the blood flow, and thus generating an extravascular hypoxic niche [27].
Cellular adaptation to hypoxia by HIFs
As previously indicated, cellular responses to hypoxia are complex, and dependent on the severity of hypoxia and the duration of the stimulus. Under conditions of hypoxia (O2 0.1 %-5 %), cells adapt by the activation of the HIF transcription factors that are responsible for metabolic adaptation, pH control and the neovascularization process [28]. HIFs are heterodimeric transcription factors of the bHLH family, comprised of an oxygen-dependent (α) and a constitutive subunit β [29]. To date, three oxygen-sensitive isoforms have been described (1α, 2α and 3α) [29–31], of which HIF1α is the best-described isoform whose expression is more ubiquitous in comparison to HIF2α or HIF3α [32–34].
In normoxia, prolyl-hydroxylase enzymes (PHDs) hydroxylate HIFα on key proline residues that are recognized by the Von Hippel Lindau (VHL) factor, an E3 ubiquitin ligase that promotes proteasomal HIFα degradation [35–37]. Reduced oxygen levels in hypoxia prevent HIFα hydroxylation by PHDs thereby stabilizing HIFα, which then translocates to the nucleus to form an heterodimer with HIF1β and bind to the hypoxia-responsiveness elements (HRE, core sequence 5´-(A/G) CGTG-3´) present in the promoter sequence of a large number of target genes [38–41]. Although both HIF1α and HIF2α bind to the same HRE promoter sequences, the increase in expression of specific target genes depends on HIF interaction with other factors. Thus, HIF target genes are classified according to their HIF subunit dependence as HIF1α-, HIF2α- or HIF1α/HIF2α-dependent [42]. Recently, the interactions between HIF1α/STAT3 and HIF2α/USF2 have been reported to activate the expression of a unique subset of target genes [43–46]. In addition, the activation of HIF-associated Factor (HAF), a HIF1α-specific target gene, permits the transition from HIF1α- to HIF2α-dependent adaptation processes during prolonged hypoxia [12, 47]. In general terms, the HIF1α response appears to be important to trigger adaptive alterations in cellular metabolism [46, 48], while HIF2α-dependent responses are essential to induce cell proliferation in hypoxia, in a manner dependent on c-myc [49, 50]. In summary, exposure to hypoxia triggers a highly complex array of responses that is mediated by different HIFs and the interaction with additional factors.
HIF1α stabilization due to ROS production
While proline hydroxylases (PHD) have been widely regarded as the primary oxygen sensors mediating cellular responses to hypoxia, available evidence indicates that mitochondria also respond to low oxygen tension, generating ROS, which then activate intracellular pathways to control the expression of several pro-survival genes [51]. Indeed, chronic treatment of Hep3B cells with ethidium bromide was shown to generate respiration-deficient p0 cells lacking functional mitochondria and, using this model, the authors then showed that mitochondria-dependent signaling processes involving ROS are required in hypoxia to promote HIF1α stabilization. In addition, mitochondrial ROS were found to be necessary for both HIF1α-DNA binding and the induction of HIF1α-mediated expression of Erythropoietin (EPO), VEGF, as well as glycolytic enzymes [52]. Furthermore, mitochondria-derived ROS are not only required, but are also sufficient to initiate HIF1α stabilization during hypoxia, and this effect requires an active mitochondrial complex III [38, 53, 54]. Importantly, ROS released into the cytosol from mitochondria stabilize not only HIF1α, but also HIF2α [53]. Thus, mitochondrial ROS plays an important role in HIFα stabilization and target gene expression in hypoxia.
In addition, HIF1α may also be stabilized in normoxia due to increased ROS production [38]. Importantly, ROS formation, ROS-dependent HIF1α stabilization and increased VEGF levels have been observed under normoxic conditions in hepatoma, lung carcinoma and osteosarcoma cell lines. Moreover, reduced activity of mitochondrial complex II (succinate-ubiquinone oxidoreductase, Sdh) due to diminished activity of the B subunit (ShdB) in such cells, favors tumor cell growth in a HIF1α-dependent manner [55]. Although it is broadly accepted that ROS can modulate HIF1α activity, often the source(s) of ROS and/or the mechanism(s) leading to ROS generation remain controversial. However, it has become increasingly clear that ROS modulate the rate of HIF1α hydroxylation [56]. A possible mechanism to explain these observations includes direct inhibition of PHD or effects of ROS on the levels of ascorbate, Fe(II) or Krebs cycle (TCA) intermediates [57, 58]. Indeed, ROS are not the only modulators of PHD activity. Multiple mitochondrial products, including TCA cycle intermediates, have been shown to favor HIF1α stabilization and cellular responses similar to those observed upon O2 depletion [37, 59] For instance, an increase in succinate levels in the absence of ROS in HEK cells leads to HIF1α stabilization and increased VEGF mRNA levels by PHD inhibition in normoxia [60]. Also, Pollard et al provided in vivo evidence to support the notion that increased succinate and/or fumarate stabilize HIF1α, possibly through the inhibition of PHD [61].
As tumors develop, tumor cells become increasingly oxygen deprived and need to reprogram their metabolism to adapt. This is achieved initially by decreasing the aerobic respiration rate and increasing glycolytic activity. In this case, increased ROS levels, generated by mitochondrial complex III, stabilize HIF1α via oxidation/inactivation of PHD [62]. Beyond mediating the initial steps in tumor cell adaptation to the hypoxic environment of a growing tumor, ROS formation has also been linked to tumor cell aggressiveness. For instance, Ishikawa et al evaluated the contribution of mutations in mitochondrial DNA to metastasis. Using the cybrid technology, they replaced mitochondrial DNA from a cell line with low–metastatic potential by donor mitochondrial DNA from a highly metastatic mouse cell line. Mitochondrial DNA from metastatic cell lines contains mutations, which result in a Complex I-deficient cell with increased ROS production. This exchange of mitochondrial DNA was sufficient to induce a metastatic phenotype in recipient cells, via ROS formation with elevated HIF1α and VEGF production [63]. Taken together, these observations point towards the existence of an intricate connection between mitochondrial function/ROS levels and HIF1α activation in cancer.
Moreover, it is well established that hypoxia is associated with resistance to chemotherapy. Recently, hypoxia-driven ROS were shown to trigger a biphasic, redox-dependent response that protects cells against etoposide-induced apoptosis. In this case, both mitochondrial- (mtROS) and NADPH oxidase-derived ROS (noxROS) cooperate in HIF1α stabilization, VEGF expression and cell survival. Furthermore, the authors proposed the existence of a VEGF-dependent autocrine loop that results in redox-mediated, prolonged stabilization of HIF1α [64].
Mitochondrial ROS seem to play a dual role in hypoxia signaling related to malignancy in tumors. Hypoxia (5 % O2) generates mROS, which activate both NF-κB through c-Src-mediated phosphorylation of IκB-α on tyrosine residues, and stabilizes HIF1α - and increases VEGF expression. These events promote carcinogenesis by the induction of survival pathways that protect cells in the face of DNA damage and permit tumor progression [58, 65]. Moreover, recently the combined treatment using S13, a Src-specific tyrosine kinase inhibitor, together with paclitaxel, dramatically reduced prostate cancer tumor growth. This effect was attributed to a reduction in ROS production, HIF1α stabilization and de novo formation of blood vessels [66]. These findings indicate that ROS favor tumor progression by activating HIF1α and increasing VEGF expression.
ROS and angiogenesis
Angiogenesis is characterized by the sprouting of new blood vessels from the pre-existing vasculature and is triggered by pro-angiogenic factors, such as fibroblast growth factor (FGF), platelet derived growth factor (PDGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF), Angiopoietins (Ang1, Ang2), TIE1 and TIE2, Ephrins, Neuropeptide Y and the previously mentioned VEGF. This latter family represents the best-characterized group of endothelial growth factors to date [67].
A close relationship exists between angiogenesis and oxidative stress in both physiological and pathological settings [68]. ROS are key mediators of this process that may be produced as a side product of the mitochondrial electron transport reaction, the activation of NADPH oxidases or upon exposure to cytotoxic drugs. ROS are commonly employed in many physiological processes in the cell and thus cannot be considered toxic a priori; however, when produced in excess, the oxidative stress generated in cells can contribute to pathological development. Generation of intracellular ROS is associated with VEGF-dependent signaling in endothelial cells [69]. Importantly, tumor growth is strongly dependent on angiogenesis and in the tumor microenvironment, ROS generated by NADPH oxidases increase VEGF secretion in a HIF1α-dependent manner [70]. Also, inflammatory mechanisms are strongly linked to ROS production and angiogenesis. In the wound healing process, neutrophils and macrophages release ROS, which in turn promote VEGF release [71]. Interestingly, in vitro stimulation of angiogenesis has been observed in bovine thoracic aorta exposed to hydrogen peroxide to promote mild oxidative stress [72]. Moreover, it has been suggested that tumor cells promote angiogenesis by releasing large amounts of hydrogen peroxide [73]. The hypothesis that oxidative stress is an important inducer of angiogenesis is further supported by evidence showing that anti-oxidants inhibit angiogenesis. For instance, leptin, a circulating hormone secreted principally by adipocytes, promotes angiogenesis by enhancing VEGF production, while N-acetylcysteine (NAC) blocks leptin-induced VEGF transcription in microvascular endothelial cells [74]. Likewise, diphenyliodonium and apocinin (a NADPH oxidase inhibitor), mannitol and catalase and other radical scavengers, have all been shown to inhibit angiogenesis [75–77]. Furthermore, vascularization in melanomas is inhibited by over-expressing extracellular superoxide dismutase (SOD) [78]. On the other hand, mice lacking NADPH oxidase 2 display impaired VEGF-induced angiogenesis and neovascularization following hind limb ischemia [79].
The VEGF pathway is modulated by ROS and oxidative stress stimulates VEGF production in several cell types, including endothelial cells, smooth muscle cells and macrophages [68]. ROS enhance angiogenesis by increasing HIF1α, as well as the expression and activity of VEGF receptor-2 (VEGFR2) [69, 70, 80]. This ROS-VEGF connection becomes even more complex when considering that VEGF promotes cell migration and proliferation by increasing intracellular levels of ROS [81]. However, it should be noted that oxidative stress also induces angiogenesis in a VEGF-independent manner by phospholipid oxidization, generating metabolites that act either as ligands or by inducing post-translational modifications (eg. ω-carboxyalkylpirrole: CAP) of proteins within angiogenic signaling pathways. Relevant examples include the Toll like receptor (TLR)2/MyD88 [82] and NFκB activation [83] pathways (Fig. 1).
Fig. 1.
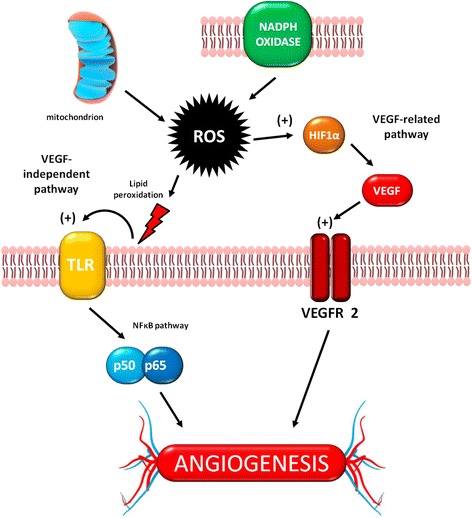
Oxidative stress and angiogenesis. The two main sources of oxidative stress, mitochondria and NADPH oxidases, generate ROS that trigger angiogenesis in two different ways, either by VEGF-related or VEGF-independent pathways – see main text for details. TLR: Toll Like Receptor
In summary, the discussion so far of a considerable body of evidence has revealed the existence of an intricate and complex connection between oxidative stress and angiogenesis. The following sections, will focus on highlighting how survivin fits into this already complex picture.
Survivin
In endothelial cells, the principal targets of HIF-dependent, pro-angiogenic responses are VEGF-A and Survivin [2, 84, 85]. Survivin, also called baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5), is a member of the inhibitor of apoptosis (IAP) family whose expression greatly favors tumor cell survival through activation of multiple pathways (see Lladser et al [2]). HIF activation in hypoxia triggers different strategies to aid in promoting tumor cell survival. While, VEGF-A is controlled by both HIF1α and HIF2α, survivin expression is solely dependent on HIF1α activation [45, 86–88].
Survivin is widely expressed in fetal development, but generally then becomes undetectable in normal adult tissues [4], although there are notable exceptions, as is the case for the gastric mucosa [89]. Importantly, however, survivin is commonly re-expressed in human tumors and is required for cancer cell survival [2, 5]. Suppression of apoptosis is a hallmark of the cancer cell that typically becomes genetically unstable, highly proliferative, and resistant to therapy [90]. Survivin, has emerged as a central player in this context due its roles in proliferation, inhibition of apoptosis, metastasis and angiogenesis [1–3, 91]. In hypoxia, HIF1α-targeting reduces survivin expression, thereby compromising cell viability. For instance, inhibition of HIF1α by Echinomycin reduces survivin expression and sensitizes multiple myeloma cells to melphalan-induced apoptosis [92]. In addition, miRNA-mediated HIF1α knockdown reduces survivin expression and induces cell death, while survivin overexpression prevents apoptosis in A549 lung cancer cells [93]. Furthermore, shRNA-mediated targeting of HIF1α reduces survivin mRNA and protein expression in the SW480 colon cancer cell line, thereby increasing the apoptotic index and reducing in vivo tumor growth [94]. Finally, in the gastric cancer cell lines SGC7901 and BGC823, survivin is upregulated in an AKT/HIF1α dependent manner, and promotes resistance to cisplatin [95]. Taken together, these findings strongly suggest that survivin expression is a downstream target of HIF1α and importantly, that survivin function is required to maintain cell viability in hypoxia. Thus, HIF1α-dependent transcription of survivin may mediate cell survival under the low oxygen conditions commonly associated with tumor growth. In addition, we envisage that increased survivin expression may contribute to VEGF synthesis in hypoxia in a manner dependent on VEGF-A up-regulation by HIF1α and HIF2α. Specifically, as indicated in the schematic (see Fig. 2), recent studies show that the production of VEGF in tumor cells is connected to survivin expression via PI3K/Akt-dependent activation of β-catenin/Tcf-Lef-mediated VEGF transcription [3], as is described below.
Fig. 2.

The Survivin/VEGF connection in angiogenesis. Tumor cells overexpressing Survivin induce VEGF synthesis/release in a β-catenin signaling-dependent manner. Liberated VEGF may act on endothelial cells promoting angiogenesis together with endothelial cell secreted VEGF. Alternatively, in tumors with few endothelial cells, survivin-induced VEGF synthesis/release may promote vasculogenic-mimicry. For details, see main text
Survivin and oxidative stress
Survivin overexpression in human cancers is also associated with drug resistance and, interestingly, in some cases resistance to oxidative stress. As referred to previously, ROS can induce HIF1α stabilization and increase VEGF expression; however, there are no reports linking survivin overexpression to ROS-dependent HIF1α stabilization. Alternatively, evidence for a negative correlation between ROS, VEGF production and survivin expression is available. For instance, reduced tumor cell survival has been observed using treatments with 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (a synthetic triterpenoid and PPARγ ligand) and its methyl ester [96, 97] together with pro-oxidant concentrations of ascorbic acid (5 mM) [98]. This combination was shown to induce the formation of ROS that, via miRNA induction, suppress Sp-transcription factors leading to a reduction in survivin and VEGF expression. This may be taken to indicate that Survivin/VEGF induction by ROS is strongly dependent on the amount of ROS generated. An excess in ROS production may lead to cell death by reduction of Survivin/VEGF expression. Alternatively, oxidative stress induced by phototherapy can lead to a rapid upregulation of inducible nitric oxide synthase (iNOS), which, in turn, promotes a notable increase in survivin expression as part of a protective response in breast cancer cells [99]. On the other hand, oxidative stress also triggers anti-tumor effects by the down-regulation of anti-apoptotic proteins, such as survivin. Resistance to oxidative stress greatly favors tumor cell survival given that tumors are known to produce large amounts of ROS [73], which contribute to tumor progression by enhancing genetic instability [100]. For instance human hepatoma cells undergo apoptosis In a ROS-dependent manner, when treated with the NF-κB inhibitor dehydroxymethyl-epoxyquinomicin (DHMEQ), due to down-regulation of BCL2, Mcl-1 and survivin [101]. Furthermore, a number of studies correlate pro-oxidant cytotoxic effects of compounds with survivin down-regulation, or, alternatively protective effects against oxidative stress with increased levels of survivin, supporting the idea that survivin contributes significantly to protection against pro-apoptotic oxidative stress [102–104]. On the other hand, oxidative stress may induce apoptosis through survivin down-regulation. As an example, Zinc oxide nanoparticles have been shown to induce oxidative stress in human alveolar adenocarcinoma, and this is linked to the down-regulation of survivin and anti-apoptotic proteins [105].
Helicobacter pylori (Hp), a pathogen associated with the development of gastric cancer is known to generate oxidative stress upon infection. Recently, Hp-induced gastritis and damage to the gastric epithelium was linked to loss of survivin expression in the gastric mucosa. Importantly, both loss of survivin and gastric cell line viability was shown to involve enhanced protein degradation via a ROS/Fe-dependent pathway [89, 106]. Taken together, these observations favor the notion that survivin acts as a resistance factor to oxidative stress-induced apoptosis. Furthermore, loss of survivin renders both normal and tumor cells vulnerable to cell death promoting signals.
Thus, although many details remain to be defined, our current understanding of the connections between ROS, HIF1α, VEGF and Survivin point towards the latter is a key point of convergence and a crucial component in determining tumor growth, progression, metastasis and drug resistance.
The up-regulation of VEGF and survivin in cancer
VEGF is required for neo-vascularization under physiological conditions and is fundamental during tumor formation, proliferation and metastasis [85, 107, 108]. The co-expression of VEGF and survivin has been reported in many types of cancer, including small-cell lung [6], bladder [7], thyroid [8] and nasopharyngeal [9] carcinomas. Accordingly, drugs that antagonize VEGFR function reduce angiogenesis and tumor growth, as well as sensitize cells to apoptosis [109], and therefore hold great therapeutic promise for cancer treatment [110]. Interestingly, the anti-apoptotic effects of VEGF are directly associated with the activation of pro-survival signaling pathways [111]. For instance, anti-apoptotic genes, such as survivin and bcl-2, are upregulated in endothelial cells via β-catenin/Tcf-Lef activation following VEGF treatment in vitro [112]. Furthermore, MAPK/ERK activation by VEGF protects endothelial cells against ceramide-induced death [113]. While hypoxia and subsequently HIF1α stabilization are main factors in VEGF production, other signaling pathways also induce this pro-angiogenic protein. For instance, ischemic pre-conditioning leads to cardioprotection through VEGF, survivin and bcl-2 by activating the β-catenin/Tcf-Lef signaling pathway [114, 115].
The apparent correlation between VEGF and survivin expression in cancer can be explained by the fact that VEGF induces survivin transcription. Survivin expression is controlled at the transcriptional and post-transcriptional level [116, 117], through the PI3K [118–120], mTOR [121], Ras [122] (79), AMPK [123] and Bcl-2/ERK [124] pathways. Extracellular stimuli that activate these pathways include VEGF, EGF, and cytokines [125]. Accordingly, the control of survivin can be attributed to the regulation of a large variety of transcription factors, including p53 [126], STAT3 [127–129], PTEN [130], NF-κB [131, 132], KLF4 [133], KLF5 [134], EGR-1 [135], E2F-1 [136], SP-1 and SP-3 [137], FOXO1 [138], HIF1α [87] and β-catenin/Tcf-Lef [139].
As eluded to before, survivin protects cancer cells in the face of pro-apoptotic stimuli. Moreover, down-regulation of survivin correlates with lower levels of VEGF [140] and reduced angiogenesis [107, 141] in cancer cells. Furthermore, in vivo studies in the zebrafish have confirmed that loss of survivin expression impairs angiogenesis, leading to developmental complications. Interestingly, in this model the authors showed that this defective phenotype could be rescued by VEGF treatment [142, 143], demonstrating thereby in vivo the relevance of this link between survivin, VEGF and angiogenesis. Consistent with this interpretation, survivin overexpression augments the secretion of pro-angiogenic molecules, such as VEGF and bFGF, and promotes angiogenesis in glioma cells in vitro and in vivo [144] or in skin flaps [145]. The molecular mechanisms implicated in this process are discussed below.
In transcriptional regulation, survivin overexpression increases the phosphorylation state and activation of proteins, including the transcriptional factors Sp1 and c-Myc [146]. Additionally, it has been demonstrated that survivin overexpression activates PI3K/AKT signaling and subsequent β-catenin/Tcf-Lef-dependent transcription, which increases the expression of VEGF, among other transcriptional target genes [3]. Importantly, survivin-targeting by a shRNA or inhibition of PI3K/Akt reduces β-Catenin/TCF-Lef transcriptional activation, indicating that survivin modulates β-Catenin/TCF-Lef activity via a PI3K/Akt-dependent pathway. In vivo, down-regulation of survivin was shown to reduce the microvessel density and VEGF expression in B16F10 tumors. Taken together, the data suggest that survivin overexpression in tumor cells promotes angiogenesis by PI3K/Akt-mediated activation of β-Catenin/TCF-Lef-dependent VEGF transcription.
Survivin has also been reported to regulate protein expression at the post-transcriptional level via its ability to reduce caspase activity. As an example, may it suffice to say that survivin enhances p53 degradation by inhibiting the caspase-dependent cleavage of Mdm2 and thereby modulating a cell-cycle checkpoint [147]. During the process of mitosis, survivin enhances the activity of the Aurora B kinase by stabilizing the chromosomal passenger protein Aurora B [146, 148]. The deregulation of the Aurora complex may lead to unequal distribution of genetic information and thus contribute to the aneuploidy observed in cancer cells. Beyond modulating caspase activity, a study evaluating survivin binding partners revealed that 18 % of the estimated interactions occurred with kinases [116], and particularly the down-regulation or inhibition of Aurora B kinase was directly associated with reduced PI3K/AKT phosphorylation [149, 150]. This observation raises the specter that survivin may activate PI3K/Akt-β catenin signaling by stabilizing Aurora B. Furthermore, other reports directly link PI3K/Akt to β-catenin signaling by GSK3β phosphorylation/inhibition and β-catenin stabilization [151–154]. Additionally, a positive feedback loop between β-catenin/Tcf-Lef target genes was also observed for COX-2, where PGE2 regulated survivin expression in hepatocellular and colon carcinoma cells through the EP receptors via the EGFR/PI3K and Gs-axin/β-catenin signaling pathways, respectively [155–157].
These observations can be taken to suggest that survivin-mediated Aurora B stabilization combined with a subsequent positive amplification loop mechanism involving PI3K activation may favor β-catenin TCF/Lef activation and VEGF synthesis. However, further experiments are required to corroborate this intriguing possibility.
A potential role for survivin in vasculogenic mimicry?
While angiogenesis has long been accepted as a necessity for tumor growth, in the last decade there have been observations indicating that tumors can continue to grow with limited vasculature. The mechanism behind this survival is speculated to be the process of vasculogenic mimicry [158, 159]. The phenomenon of vasculogenic mimicry describes the formation of tubular structures within the tumor that are of cancer cell origin and thus independent of endothelial cells. As with angiogenesis, an underlying mechanism of induction of vasculogenic mimicry seems to be hypoxia. Unsurprisingly, given the similarities with angiogenesis, genes implemented in vasculogenic mimicry are those previously associated with vascular (VE-cadherin), embryonic (Nodal, Notch4), and hypoxia-related (hypoxia-inducible factor, Twist1) signaling pathways [160]. VEGF and its receptor VEGFR type 2 (also called KDR, Flk-1), have been implicated in vasculogenic mimicry [161, 162]. Expression of the ανβ5 integrin also correlated with vasculogenic mimicry and highly aggressive melanoma [163]. Ovarian tumors exhibiting vasculogenic mimicry demonstrated higher expression of β-catenin and VEGF [164]. In hepatocellular carcinoma cells, VEGF-induced vasculogenic mimicry is also reported to involve Myocyte Enhancer Factor 2C (MEF2C) together with β-catenin via the p38 MAPK and PKC signaling pathways [165]. The search for the exact mechanisms and the unique pathways involved in the process is still very much in its infancy [166]; however, as survivin is overexpressed in almost all human cancers [2] it remains to be determined whether survivin participates in tumor cell mediated vasculogenic mimicry. Indeed, some available evidence suggests that survivin could play a role in this process. First, vasculogenic mimicry is known to be associated with higher β-catenin and VEGF expression [164]. Second, tumor hypoxia accelerates the vasculogenic mimicry process [167] and both Survivin and VEGF expression are upregulated by HIFs in hypoxia [6, 45, 87, 88]. Furthermore, in ovarian cancer, hypoxia has been shown to promote vasculogenic mimicry formation by inducing epithelial-mesenchymal transition (EMT) [167]. The relationship between vasculogenic mimicry and EMT has been reported in numerous cancer types including glioma, liver, head and neck, and stomach cancer [168–172]. Further suggesting a role for survivin, the upregulation of survivin and the induction of EMT has been widely reported in both cellular physiology and cancer, as shown in human retinal pigment epithelial cells and in glioblastoma, among many other cell models [173, 174].
Survivin is also reported to be involved in the interplay between CD31 and VE-Cadherin, both implicated in vasculogenic mimicry. In esophageal carcinoma cells, knock-down of HIF1α inhibited vasculogenic mimicry and HIF1α was shown to upregulate VE-cadherin expression [175].
Evidence is also present connecting CD31, VE-Cadherin, β-catenin and survivin in physiological processes. The endothelial cells of CD31 knock-out mice possess reduced VE-cadherin expression with a corresponding increase in levels of survivin [176]. In accordance, confluence and VE-cadherin and β-catenin are reported to negatively regulate the synthesis of survivin in endothelial cells. Using β-catenin null and positive isogenic endothelial cell lines this down-regulation of survivin has been shown to require β-catenin [177]. Moreover, survivin promotes VEGF synthesis/secretion by tumor cells, thereby favoring angiogenesis [3]. Bearing in mind these observations, one may speculate that in poorly vascularized tumor regions, survivin-mediated VEGF synthesis and/or HIF1α mediated survivin and VEGF expression could promote vasculogenic mimicry and thus favor tumor survival (see Fig. 2).
Survivin expression in human cancers
As eluded to, Survivin plays multiple pleiotropic roles that are important for cancer development and progression. Survivin participates in the cell division process [178–180], protects against cell death via prevention of SMAC/DIABLO release [2] and promotes angiogenesis [3]. Also, survivin participates in the maintenance stemness and promotes cell motility, as well as metastasis [181]. In conjunction, these survivin functions strongly contribute to tumor development, progression and metastasis. This is particularly relevant given that survivin is considered a specific Tumor-Associated-Antigen (TAA) because the protein is overexpressed in most human cancers, but essentially absent in the respective normal tissues, although exceptions do exist [2, 89].
In a meta-analysis including 2703 patients with non-small cell lung cancer (NSCLC), survivin expression was identified as a factor indicative of poorer prognosis in advanced stages of NSCLC (stages III-IV) rather than early stages (I-II) [182]. In a meta-analysis involving 1365 gastric cancer patients, survivin expression was associated with worse overall survival. Specifically, cytoplasmic, but not nuclear, survivin expression was linked to a poorer prognosis for those patients. Hence, not only expression per se, but also the subcellular localization of survivin appears to be important in gastric cancer survival [183].
However, survivin expression is not necessarily always bad. In a study with 60 ovarian cancer patients at advanced stages (stages IIIC, IV FIGO classification) of disease, survivin and p53 expression were analyzed before and after neoadjuvant chemotherapy [184]. Nuclear survivin expression was detected in almost 60 % of patients before treatment, and after neoadjuvant chemotherapy, nuclear survivin expression was reduced. Furthermore, elevated nuclear survivin expression was identified as a favorable prognostic marker in patients treated with neoadjuvant chemotherapy. The median overall survival for p53 positive patients with higher expression of nuclear survivin was 34.6 months, compared to 22.2 months for those patients with lower nuclear survivin expression [184]. These observations implicate nuclear survivin expression as a favorable prognostic marker for chemotherapy in patients with advanced ovarian cancer [184]. This will become important subsequently due to the relevance of angiogenesis in ovarian cancer progression [185, 186], and the increase in vasculogenic mimicry detected in ovarian cancer patients [1, 105].
For the reasons mentioned, there has been great interest in developing approaches that seek to reduce survivin expression in order to limit cancer cell growth, as will be eluded to in the subsequent section. However, as the previous paragraph indicates, survivin expression, particularly in the nucleus, can also be beneficial to patients, thus complicating the expected outcome of such treatments.
Survivin as a target in cancer therapy
Survivin is a member of the IAP family, of which several members are deregulated in human cancers, including solid tumors and hematological malignancies [90, 187–189]. Consistent with these observations, targeting other IAPs in combination with cytotoxic drugs has been suggested as a treatment for hematological malignancies [188]. However, strategies focusing on survivin are generally favored over the targeting of other IAPs because survivin expression is fairly specific, although not exclusive to tumor cells and because survivin displays characteristics of a nodal protein by participating in a great variety of pathways and processes that favor tumor cell development [90]. For precisely these reasons, survivin has been widely exploited as a pharmacological target in cancer (Table 1). Multiple strategies are currently being evaluated in clinical trials, including the use of survivin inhibitors and the development of survivin-based vaccines (Table 1).
Table 1.
Clinical trials targeting survivin in cancer
| Strategy | Pathology | Phase | Clinical trials identifier |
|---|---|---|---|
| Survivin inhibitors | |||
| YM155 (survivin suppressor) together with Paclitaxel and carboplatin | Solid tumors and advanced non-small cell lung carcinoma | I/II | NCT01100931 |
| Terameprocol (EM1421), inhibitor of survivin and cdc2 (cyclin-dependent kinase-1) in continuous intravenous infusion | Refractory Solid tumors | I | NCT00664586 |
| EZN-3042, a locked nucleic acid antisense oligonucleotide | Acute Lymphoblastic Leukemia | I | NCT01186328 |
| Terameprocol (EM-1421), inhibitor of survivin and cdc2 (cyclin-dependent kinase-1) | Leukemia | I | NCT00664677 |
| EZN-3042, a survivin-targeted mRNA antagonist, alone or in combination with standard chemotherapy | Acute Lymphoblastic Leukemia | I | NCT01186328 |
| LY2181308, an antisense oligonucleotid, targeted against survivin mRNA in combination with idarubicin and cytarabine | Acute Myeloid Leukemia | II | NCT00620321 |
| Survivin-based Cellular Therapy | |||
| Dendritic cell vaccine (mRNA from PSA, PAP, survivin and hTERT) plus docetaxel or docetaxel alone | Prostate Cancer (castration resistant and metastatic) | II | NCT01446731 |
| Dendritic cells - transfected with hTERT-, survivin- and tumor cell derived mRNA + ex vivo T cell expansion and reinfusion | Melanoma | I/II | NCT00961844 |
| Drug: Temozolomide | |||
| Procure®, denditric cells loaded with Survivin-peptide and Telomerase mRNA | Ovarian Cancer | I | NCT01456065 |
| Dendritic cell loaded with amplified ovarian cancer stem cell mRNA, hTERT/survivin mRNA | Ovarian Cancer | I/II | NCT01334047 |
| Cell therapy based on dendritic cells transfected with Survivin, hTERT and p53 mRNA | Metastatic breast cancer | I | NCT00978913 |
| Malignant melanoma | |||
| TAA-SPECIFIC CTLs targeting survivin, PRAME, NY-ESO-1, MAGEA4 and SSX | Solid Tumors (TACTASOM) | I | NCT02239861 |
| Treatment with autologous dendritic cells transfected with Survivin, MelanA and MAGE-A3 mRNA or loaded with MAGE-A3, MelanA and Survivin | Melanoma | I/II | NCT00074230 |
| Cell therapy with cytotoxic T lymphocytes exposed to tumor associated antigens: NY-ESO-1, MAGEA4, PRAME, Survivin and SSX. | Hodgkin or Non-Hodgkin Lymphoma | I | NCT01333046 |
| TAA-CTLs may be generated from donors or recipients and will be tested for specificity against 4 tumor antigens commonly found in hematological malignancies (WT1, PRAME, SURVIVIN, and MAGE-A3). | I | NCT02203903 | |
| Hematological | |||
| Malignancies | |||
| Dendritic cell vaccine (MUC-1 and survivin) in combination with cytokine-induced killer cells | Soft Tissue Sarcoma | I/II | NCT01898663 |
| Dendritic cell vaccine (MUC-1 and survivin) in combination with cytokine-induced killer cells | Renal Cell Carcinoma | I/II | NCT01924156 |
| Autologous dendritic cell vaccine (peptides from survivin and telomerase) | Renal Cell Carcinoma | I/II | NCT00197860 |
| Multiple antigen specific cellular therapy: autologous T cytotoxic cells induced by dendritic cells (loaded with p53, her2, survivin and a total of 17 antigens) | Hepatocellular Carcinoma | I/II | NCT02026362 |
| Vaccine therapy (p53, survivin and telomerase) with autologous dendritic cells in combination with adjuvant cytokines | Advanced Melanoma | I/II | NCT00197912 |
| Multiple tumor-associated antigen (TAA)-specific T cells (against WT1, PRAME and survivin) from donors | Acute Lymphoblastic Leukemia | I | NCT02475707 |
| Multiple tumor-associated antigen (TAA)-specific T cells (against WT1, NY-ESO-1, PRAME and survivin) from donors | Acute Lymphoblastic Leukemia/Myelodisplasic Syndrome | I | NCT02494167 |
| Multiple tumor-associated antigen (TAA)-specific cytotoxic T cells (against NY-ESO-1, IMAGEA4, PRAME, SSX and survivin) from donors | Multiple Myeloma | I | NCT02291848 |
| Survivin-vaccines | |||
| Survivin peptide vaccination in combination with sargramostin | Malignant Glioma | I | NCT01250470 |
| Vaccination with DPX-Survivin in combination with low doses of cyclophosphamide (The peptide antigens targeting survivin) | Advanced Stage Ovarian, Fallopian or Peritoneal Cancer | I/II) | NCT01416038 |
| hTERT/survivin/CMV multipeptide vaccine | Breast Cancer | Not provided | NCT01660529 |
| Multipeptide vaccination including survivin-peptide | Multiple Myeloma | I/II | NCT00499577 |
| SurVaxM Vaccine (survivin-peptide vaccine) in combination with temozolamide | Glioblastoma | II | NCT02455557 |
| Peptide vaccine (IDO/survivine peptide) as enhancer of temozolomide chemotherapy | Metastatic Melanoma | II | NCT01543464 |
| Vaccine therapy (MART1 analog, gp100 and survivin) and GM-CSF with or without Aldesleukin | Melanoma | I | NCT00470015 |
| Vaccine therapy (survivin) in patients receiving lenalidomide | Multiple Myeloma | I | NCT02334865 |
| hTERT tumor vaccine (peptides from telomerase, survivin and cytopeptide) in combination with autologous T cell infusion | Multiple Myeloma | I/II | NCT00834665 |
| Immunotherapeutic vaccine DPX-Survivac (targeting survivin) in combination with cyclophosphamide | Diffuse Large B-Cell Lymphoma | II | NCT02323230 |
| Survivin and telomerase peptide vaccination in combination with Daclizumab and Prevnar | Advanced Breast cancer | I | NCT00573495 |
| Survivin peptide vaccination | Advanced Melanoma, pancreatic, colon and cervical cancer | I/II | NCT00108875 |
Unfortunately, studies focusing on pharmacological inhibitors of survivin have shown rather disappointing results. In a phase I clinical trial in patients with acute lymphoblastic leukemia, EZN3042, a locked antisense construct against survivin, was toxic and patients showed poor tolerance to treatment [190]. Subsequently, the application of EZN3042 was suspended for the indicated reasons [190]. In a study in patients with advanced NSCLC solid tumors, YM155, an inhibitor of Sp1-mediated survivin expression, displayed an acceptable safety profile; however, this compound failed to improve responses to chemotherapy treatment [191]. In patients with leukemia, Terameprocol, an inhibitor of survivin and cyclin-dependent kinase-1 was found to be safe in phase I study. In addition, a therapeutic effect (partial response and disease stabilization by terameprocol) was observed in patients treated with this compound [192]. Currently, an additional trial is underway evaluating terameprocol in patients with refractory solid tumors (Clinical trial identifier NCT00664586). Additional studies are required to corroborate the utility of these approaches in cancer treatment.
Multiple survivin-based studies involving cell therapy are currently underway. In this context, the use of dendritic cells loaded with survivin-peptide (Clinical trial identifier NCT01456065) or survivin mRNA (Clinical trial identifier NCT01334047, NCT00978913) in association with telomerase and p53 mRNAs are being evaluated in clinical trials in patients with ovarian cancer, metastatic breast cancer and malignant melanoma. Furthermore, the efficacy of cytotoxic T lymphocytes exposed to a mixture of TAAs, including survivin, is currently being tested in the treatment of hematological malignancies (clinical trial identifier NCT01333046, NCT02203903, NCT02475707). Thus, cellular therapy represents an intense area of contemporary research, although clear benefits of such treatments remain to be established.
In addition to the approaches mentioned, considerable effort is being placed on the development of survivin-based vaccines (survivin mRNA and peptide) for the treatment of several different types of cancer, including breast cancer, kidney cancer, advanced melanomas and ovarian cancer (Table 1). Promising results were obtained in renal carcinoma patients where survivin-vaccination lead to disease stabilization [193] and a response in 35 % of the patients without adverse toxicity effects [194]. The most successful results have been obtained in ovarian cancer, where DPX-Survivac, a vaccine based on the use of survivin peptides in conjunction with a DepoVax™ adjuvant, administrated in a treatment together with cyclophosphamide, yielded favorable results in a phase I clinical trial. This treatment was found to be safe, well-tolerated by patients and yielded strong immune responses against tumors [195]. Recently, DPX-Survivac was designated by the FDA as an orphan drug for maintenance therapy in ovarian cancer patients with no measurable disease after standard treatments (surgery/chemotherapy). These promising results obtained with DPX-Survivac open up a wide array of possibilities and further studies are required to determine the efficacy of this vaccine in phase II trials.
In a phase II trial in patients with metastatic melanoma, a treatment involving vaccination with autologous dendritic cells previously pulsed with survivin, hTERT and p53-derived peptides together with cyclophosphamide and celecoxib (COX-2 inhibitor) was evaluated (clinical trial identifier NCT00197912) [196]. This treatment was shown to be safe and tolerable and an increase in the immune response was detected. Also, for almost 60 % of patients the disease was stabilized for four or more months (clinical trial identifier NCT00197912) [196]. In summary, many clinical trials are currently underway to determine whether targeting survivin represents an effective approach to limit tumor development. Although some trials have unfortunately met with limited success, others, such as those targeting ovarian cancer, have yielded highly promising results. Here, it should be noted that ovarian cancer is precisely a case where angiogenesis represents a highly prevalent “hallmark” trait, underscoring thereby the importance of survivin in this context, as has been discussed throughout this review.
Conclusions
Survivin plays an important role in processes that favor tumor growth and angiogenesis. HIF1α stabilization under low oxygen conditions and/or via ROS production promotes survivin and VEGF expression and favors angiogenesis. In addition to the well-established role of survivin in endothelial cells, survivin in tumor cells enhances β-Catenin Tcf/Lef-dependent VEGF transcription, synthesis and release, thereby promoting angiogenesis of endothelial cells. More recently, cancer cells have also been shown to form vascular-like structures in the absence of endothelial cells in a process known as vasculogenic mimicry. In a poorly vascularized tumor microenvironment, we posit that hypoxia-enhanced survivin levels may increase VEGF production and EMT, thus promoting the process of vasculogenic mimicry. While highly intriguing, this possibility remains to date largely speculative; however, this should represent a fruitful area for research in the future, both in the perspective of developing a better understanding of the underlying mechanisms, as well as how such insight might be harnessed to treat tumors more effectively.
Acknowledgements
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) Postdoctoral Fellowships 3140516, 3140446 (CS, JC); FONDECYT Regular grants 1090071, 1130250 (AFGQ) and 1140970 (GIO); BMRC 13CTI-21526-P6 (GIO), CORFO 13IDL2-18608 (GIO); CONICYT/FONDAP 15010006 and 15130011 (AFGQ).
Footnotes
Competing interests
The authors declare that there are no conflicts of interest.
Authors’ contributions
Review outline (CS, AQ), manuscript preparation (CS, SW, JC, MV, GO, AQ), figures and table (CS, SW). All authors read and approved the final manuscript.
References
- 1.Altieri DC. New wirings in the survivin networks. Oncogene. 2008;27:6276–84. doi: 10.1038/onc.2008.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lladser A, Sanhueza C, Kiessling R, Quest AF. Is survivin the potential Achilles’ heel of cancer? Adv Cancer Res. 2011;111:1–37. doi: 10.1016/B978-0-12-385524-4.00001-5. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez JG, Rodriguez DA, Valenzuela M, Calderon C, Urzua U, Munroe D, et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced beta-catenin/Tcf-Lef dependent transcription. Mol Cancer. 2014;13:209. doi: 10.1186/1476-4598-13-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto H, Ngan CY, Monden M. Cancer cells survive with survivin. Cancer Sci. 2008;99:1709–14. doi: 10.1111/j.1349-7006.2008.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen P, Zhu J, Liu DY, Li HY, Xu N, Hou M. Over-expression of survivin and VEGF in small-cell lung cancer may predict the poorer prognosis. Med Oncol. 2014;31:775. doi: 10.1007/s12032-013-0775-5. [DOI] [PubMed] [Google Scholar]
- 7.Sun YW, Xuan Q, Shu QA, Wu SS, Chen H, Xiao J, et al. Correlation of tumor relapse and elevated expression of survivin and vascular endothelial growth factor in superficial bladder transitional cell carcinoma. Genet Mol Res. 2013;12:1045–53. doi: 10.4238/2013.April.2.21. [DOI] [PubMed] [Google Scholar]
- 8.Zhang HY, Meng X, Du ZX, Fang CQ, Liu GL, Wang HQ, et al. Significance of survivin, caspase-3, and VEGF expression in thyroid carcinoma. Clin Exp Med. 2009;9:207–13. doi: 10.1007/s10238-009-0031-7. [DOI] [PubMed] [Google Scholar]
- 9.Li YH, Hu CF, Shao Q, Huang MY, Hou JH, Xie D, et al. Elevated expressions of survivin and VEGF protein are strong independent predictors of survival in advanced nasopharyngeal carcinoma. J Transl Med. 2008;6:1. doi: 10.1186/1479-5876-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15:1239–53. doi: 10.1111/j.1582-4934.2011.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrera S, Senra J, Acosta MI, Althubiti M, Hammond EM, de Verdier PJ, et al. The role of the HIF-1alpha transcription factor in increased cell division at physiological oxygen tensions. PLoS One. 2014;9:e97938. doi: 10.1371/journal.pone.0097938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37:364–72. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Movsas B, Chapman JD, Hanlon AL, Horwitz EM, Pinover WH, Greenberg RE, et al. Hypoxia in human prostate carcinoma: an Eppendorf PO2 study. Am J Clin Oncol. 2001;24:458–61. doi: 10.1097/00000421-200110000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48:919–22. doi: 10.1016/S0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 15.Becker A, Hansgen G, Bloching M, Weigel C, Lautenschlager C, Dunst J. Oxygenation of squamous cell carcinoma of the head and neck: comparison of primary tumors, neck node metastases, and normal tissue. Int J Radiat Oncol Biol Phys. 1998;42:35–41. doi: 10.1016/S0360-3016(98)00182-5. [DOI] [PubMed] [Google Scholar]
- 16.Vaupel P, Schlenger K, Knoop C, Hockel M. Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res. 1991;51:3316–22. [PubMed] [Google Scholar]
- 17.Lawrentschuk N, Poon AM, Foo SS, Putra LG, Murone C, Davis ID, et al. Assessing regional hypoxia in human renal tumours using 18 F-fluoromisonidazole positron emission tomography. BJU Int. 2005;96:540–6. doi: 10.1111/j.1464-410X.2005.05681.x. [DOI] [PubMed] [Google Scholar]
- 18.Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9:1221–35. doi: 10.1089/ars.2007.1628. [DOI] [PubMed] [Google Scholar]
- 19.Denko NC, Fontana LA, Hudson KM, Sutphin PD, Raychaudhuri S, Altman R, et al. Investigating hypoxic tumor physiology through gene expression patterns. Oncogene. 2003;22:5907–14. doi: 10.1038/sj.onc.1206703. [DOI] [PubMed] [Google Scholar]
- 20.Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist. 2004;9(Suppl 5):4–9. doi: 10.1634/theoncologist.9-90005-4. [DOI] [PubMed] [Google Scholar]
- 21.Vaupel P. Hypoxia and aggressive tumor phenotype: implications for therapy and prognosis. Oncologist. 2008;13(Suppl 3):21–6. doi: 10.1634/theoncologist.13-S3-21. [DOI] [PubMed] [Google Scholar]
- 22.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 24.Pries AR, Hopfner M, le Noble F, Dewhirst MW, Secomb TW. The shunt problem: control of functional shunting in normal and tumour vasculature. Nat Rev Cancer. 2010;10:587–93. doi: 10.1038/nrc2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yasui H, Matsumoto S, Devasahayam N, Munasinghe JP, Choudhuri R, Saito K, et al. Low-field magnetic resonance imaging to visualize chronic and cycling hypoxia in tumor-bearing mice. Cancer Res. 2010;70:6427–36. doi: 10.1158/0008-5472.CAN-10-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 27.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425–37. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365:537–47. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 29.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:4273–8. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makino Y, Kanopka A, Wilson WJ, Tanaka H, Poellinger L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha locus. J Biol Chem. 2002;277:32405–8. doi: 10.1074/jbc.C200328200. [DOI] [PubMed] [Google Scholar]
- 32.Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C, et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003;17:271–3. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- 33.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–85. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka A, Jin Y, Lee SJ, Zhang M, Kim HP, Stolz DB, et al. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol. 2012;46:507–14. doi: 10.1165/rcmb.2009-0415OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–92. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sutter CH, Laughner E, Semenza GL. Hypoxia-inducible factor 1alpha protein expression is controlled by oxygen-regulated ubiquitination that is disrupted by deletions and missense mutations. Proc Natl Acad Sci U S A. 2000;97:4748–53. doi: 10.1073/pnas.080072497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fandrey J, Gorr TA, Gassmann M. Regulating cellular oxygen sensing by hydroxylation. Cardiovasc Res. 2006;71:642–51. doi: 10.1016/j.cardiores.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 39.Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2002;283:L922–31. doi: 10.1152/ajplung.00014.2002. [DOI] [PubMed] [Google Scholar]
- 40.Paul SA, Simons JW, Mabjeesh NJ. HIF at the crossroads between ischemia and carcinogenesis. J Cell Physiol. 2004;200:20–30. doi: 10.1002/jcp.10479. [DOI] [PubMed] [Google Scholar]
- 41.Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–6. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 42.Pawlus MR, Hu CJ. Enhanceosomes as integrators of hypoxia inducible factor (HIF) and other transcription factors in the hypoxic transcriptional response. Cell Signal. 2013;25:1895–903. doi: 10.1016/j.cellsig.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pawlus MR, Wang L, Ware K, Hu CJ. Upstream stimulatory factor 2 and hypoxia-inducible factor 2alpha (HIF2alpha) cooperatively activate HIF2 target genes during hypoxia. Mol Cell Biol. 2012;32:4595–610. doi: 10.1128/MCB.00724-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pawlus MR, Wang L, Murakami A, Dai G, Hu CJ. STAT3 or USF2 contributes to HIF target gene specificity. PLoS One. 2013;8:e72358. doi: 10.1371/journal.pone.0072358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pawlus MR, Wang L, Hu CJ. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene. 2014;33:1670–9. doi: 10.1038/onc.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brahimi-Horn MC, Pouyssegur J. HIF at a glance. J Cell Sci. 2009;122:1055–7. doi: 10.1242/jcs.035022. [DOI] [PubMed] [Google Scholar]
- 47.Koh MY, Lemos R, Jr, Liu X, Powis G. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011;71:4015–27. doi: 10.1158/0008-5472.CAN-10-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J. 2012;31:2448–60. doi: 10.1038/emboj.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franovic A, Holterman CE, Payette J, Lee S. Human cancers converge at the HIF-2alpha oncogenic axis. Proc Natl Acad Sci U S A. 2009;106:21306–11. doi: 10.1073/pnas.0906432106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hielscher A, Gerecht S. Hypoxia and free radicals: role in tumor progression and the use of engineering-based platforms to address these relationships. Free Radic Biol Med. 2015;79:281–91. doi: 10.1016/j.freeradbiomed.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–20. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 54.Bell EL, Klimova TA, Eisenbart J, Schumacker PT, Chandel NS. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol. 2007;27:5737–45. doi: 10.1128/MCB.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–31. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Snyder CM, Chandel NS. Mitochondrial regulation of cell survival and death during low-oxygen conditions. Antioxid Redox Signal. 2009;11:2673–83. doi: 10.1089/ars.2009.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 58.Lluis JM, Buricchi F, Chiarugi P, Morales A, Fernandez-Checa JC. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-{kappa}B via c-SRC and oxidant-dependent cell death. Cancer Res. 2007;67:7368–77. doi: 10.1158/0008-5472.CAN-07-0515. [DOI] [PubMed] [Google Scholar]
- 59.Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27:912–25. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 61.Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–9. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 62.Catalano V, Turdo A, Di Franco S, Dieli F, Todaro M, Stassi G. Tumor and its microenvironment: a synergistic interplay. Semin Cancer Biol. 2013;23:522–32. doi: 10.1016/j.semcancer.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 63.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–4. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 64.Calvani M, Comito G, Giannoni E, Chiarugi P. Time-dependent stabilization of hypoxia inducible factor-1alpha by different intracellular sources of reactive oxygen species. PLoS One. 2012;7:e38388. doi: 10.1371/journal.pone.0038388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lluis JM, Llacuna L, von Montfort C, Barcena C, Enrich C, Morales A, et al. GD3 synthase overexpression sensitizes hepatocarcinoma cells to hypoxia and reduces tumor growth by suppressing the cSrc/NF-kappaB survival pathway. PLoS One. 2009;4(e8059). [DOI] [PMC free article] [PubMed]
- 66.Delle Monache S, Sanita P, Calgani A, Schenone S, Botta L, Angelucci A. Src inhibition potentiates antitumoral effect of paclitaxel by blocking tumor-induced angiogenesis. Exp Cell Res. 2014;328:20–31. doi: 10.1016/j.yexcr.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 67.Bouis D, Kusumanto Y, Meijer C, Mulder NH, Hospers GA. A review on pro- and anti-angiogenic factors as targets of clinical intervention. Pharmacol Res. 2006;53:89–103. doi: 10.1016/j.phrs.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 68.Kim YW, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood. 2014;123:625–31. doi: 10.1182/blood-2013-09-512749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Colavitti R, Pani G, Bedogni B, Anzevino R, Borrello S, Waltenberger J, et al. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J Biol Chem. 2002;277:3101–8. doi: 10.1074/jbc.M107711200. [DOI] [PubMed] [Google Scholar]
- 70.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67:10823–30. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 71.auf dem Keller U, Kumin A, Braun S, Werner S. Reactive oxygen species and their detoxification in healing skin wounds. J Investig Dermatol Symp Proc. 2006;11:106–11. doi: 10.1038/sj.jidsymp.5650001. [DOI] [PubMed] [Google Scholar]
- 72.Yasuda M, Ohzeki Y, Shimizu S, Naito S, Ohtsuru A, Yamamoto T, et al. Stimulation of in vitro angiogenesis by hydrogen peroxide and the relation with ETS-1 in endothelial cells. Life Sci. 1999;64:249–58. doi: 10.1016/S0024-3205(98)00560-8. [DOI] [PubMed] [Google Scholar]
- 73.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8. [PubMed] [Google Scholar]
- 74.Yamagishi S, Amano S, Inagaki Y, Okamoto T, Takeuchi M, Inoue H. Pigment epithelium-derived factor inhibits leptin-induced angiogenesis by suppressing vascular endothelial growth factor gene expression through anti-oxidative properties. Microvasc Res. 2003;65:186–90. doi: 10.1016/S0026-2862(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 75.Al-Shabrawey M, Bartoli M, El-Remessy AB, Platt DH, Matragoon S, Behzadian MA, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koch AE, Cho M, Burrows JC, Polverini PJ, Leibovich SJ. Inhibition of production of monocyte/macrophage-derived angiogenic activity by oxygen free-radical scavengers. Cell Biol Int Rep. 1992;16:415–25. doi: 10.1016/S0309-1651(06)80061-5. [DOI] [PubMed] [Google Scholar]
- 77.Cai T, Fassina G, Morini M, Aluigi MG, Masiello L, Fontanini G, et al. N-acetylcysteine inhibits endothelial cell invasion and angiogenesis. Lab Invest. 1999;79:1151–9. [PubMed] [Google Scholar]
- 78.Wheeler MD, Smutney OM, Samulski RJ. Secretion of extracellular superoxide dismutase from muscle transduced with recombinant adenovirus inhibits the growth of B16 melanomas in mice. Mol Cancer Res. 2003;1:871–81. [PubMed] [Google Scholar]
- 79.Tojo T, Ushio-Fukai M, Yamaoka-Tojo M, Ikeda S, Patrushev N, Alexander RW. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation. 2005;111:2347–55. doi: 10.1161/01.CIR.0000164261.62586.14. [DOI] [PubMed] [Google Scholar]
- 80.Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci U S A. 2002;99:715–20. doi: 10.1073/pnas.022630199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y, Zang QS, Liu Z, Wu Q, Maass D, Dulan G, et al. Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. Am J Physiol Cell Physiol. 2011;301:C695–704. doi: 10.1152/ajpcell.00322.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–6. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mutoh A, Ueda S. Peroxidized unsaturated fatty acids stimulate Toll-like receptor 4 signaling in endothelial cells. Life Sci. 2013;92:984–92. doi: 10.1016/j.lfs.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 84.Fox SB, Gatter KC, Harris AL. Tumour angiogenesis. J Pathol. 1996;179:232–7. doi: 10.1002/(SICI)1096-9896(199607)179:3<232::AID-PATH505>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 85.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–27. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 86.Chen YQ, Zhao CL, Li W. Effect of hypoxia-inducible factor-1alpha on transcription of survivin in non-small cell lung cancer. J Exp Clin Cancer Res. 2009;28:29. doi: 10.1186/1756-9966-28-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peng XH, Karna P, Cao Z, Jiang BH, Zhou M, Yang L. Cross-talk between epidermal growth factor receptor and hypoxia-inducible factor-1alpha signal pathways increases resistance to apoptosis by up-regulating survivin gene expression. J Biol Chem. 2006;281:25903–14. doi: 10.1074/jbc.M603414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang L, Cao Z, Li F, Post DE, Van Meir EG, Zhong H, et al. Tumor-specific gene expression using the survivin promoter is further increased by hypoxia. Gene Ther. 2004;11:1215–23. doi: 10.1038/sj.gt.3302280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Valenzuela M, Perez-Perez G, Corvalan AH, Carrasco G, Urra H, Bravo D, et al. Helicobacter pylori-induced loss of the inhibitor-of-apoptosis protein survivin is linked to gastritis and death of human gastric cells. J Infect Dis. 2010;202:1021–30. doi: 10.1086/656143. [DOI] [PubMed] [Google Scholar]
- 90.Mobahat M, Narendran A, Riabowol K. Survivin as a preferential target for cancer therapy. Int J Mol Sci. 2014;15:2494–516. doi: 10.3390/ijms15022494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53–64. doi: 10.1016/j.ccr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu Y, Kirito K, Yoshida K, Mitsumori T, Nakajima K, Nozaki Y, et al. Inhibition of hypoxia-inducible factor-1 function enhances the sensitivity of multiple myeloma cells to melphalan. Mol Cancer Ther. 2009;8:2329–38. doi: 10.1158/1535-7163.MCT-09-0150. [DOI] [PubMed] [Google Scholar]
- 93.Li W, Chen YQ, Shen YB, Shu HM, Wang XJ, Zhao CL, et al. HIF-1alpha knockdown by miRNA decreases survivin expression and inhibits A549 cell growth in vitro and in vivo. Int J Mol Med. 2013;32:271–80. doi: 10.3892/ijmm.2013.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu XY, Fu ZX, Wang XH. Effect of hypoxia-inducible factor 1-alpha on Survivin in colorectal cancer. Mol Med Rep. 2010;3:409–15. doi: 10.3892/mmr_00000273. [DOI] [PubMed] [Google Scholar]
- 95.Sun XP, Dong X, Lin L, Jiang X, Wei Z, Zhai B, et al. Up-regulation of survivin by AKT and hypoxia-inducible factor 1alpha contributes to cisplatin resistance in gastric cancer. FEBS J. 2014;281:115–28. doi: 10.1111/febs.12577. [DOI] [PubMed] [Google Scholar]
- 96.Jutooru I, Chadalapaka G, Abdelrahim M, Basha MR, Samudio I, Konopleva M, et al. Methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription factors and inhibits pancreatic tumor growth: role of microRNA-27a. Mol Pharmacol. 2010;78:226–36. doi: 10.1124/mol.110.064451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pathi SS, Jutooru I, Chadalapaka G, Sreevalsan S, Anand S, Thatcher GR, et al. GT-094, a NO-NSAID, inhibits colon cancer cell growth by activation of a reactive oxygen species-microRNA-27a: ZBTB10-specificity protein pathway. Mol Cancer Res. 2011;9:195–202. doi: 10.1158/1541-7786.MCR-10-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pathi SS, Lei P, Sreevalsan S, Chadalapaka G, Jutooru I, Safe S. Pharmacologic doses of ascorbic acid repress specificity protein (Sp) transcription factors and Sp-regulated genes in colon cancer cells. Nutr Cancer. 2011;63:1133–42. doi: 10.1080/01635581.2011.605984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bhowmick R, Girotti AW. Cytoprotective signaling associated with nitric oxide upregulation in tumor cells subjected to photodynamic therapy-like oxidative stress. Free Radic Biol Med. 2013;57:39–48. doi: 10.1016/j.freeradbiomed.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–44. doi: 10.1016/S1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 101.Lampiasi N, Azzolina A, Umezawa K, Montalto G, McCubrey JA, Cervello M. The novel NF-kappaB inhibitor DHMEQ synergizes with celecoxib to exert antitumor effects on human liver cancer cells by a ROS-dependent mechanism. Cancer Lett. 2012;322:35–44. doi: 10.1016/j.canlet.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 102.Yang X, Li X, An L, Bai B, Chen J. Silibinin induced the apoptosis of Hep-2 cells via oxidative stress and down-regulating survivin expression. Eur Arch Otorhinolaryngol. 2013;270:2289–97. doi: 10.1007/s00405-013-2444-x. [DOI] [PubMed] [Google Scholar]
- 103.Zheng WX, Wang F, Cao XL, Pan HY, Liu XY, Hu XM, et al. Baicalin protects PC-12 cells from oxidative stress induced by hydrogen peroxide via anti-apoptotic effects. Brain Inj. 2014;28:227–34. doi: 10.3109/02699052.2013.860469. [DOI] [PubMed] [Google Scholar]
- 104.Zhu W, Cromie MM, Cai Q, Lv T, Singh K, Gao W. Curcumin and vitamin E protect against adverse effects of benzo[a]pyrene in lung epithelial cells. PLoS One. 2014;9:e92992. doi: 10.1371/journal.pone.0092992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ahamed M, Akhtar MJ, Raja M, Ahmad I, Siddiqui MK, AlSalhi MS, et al. ZnO nanorod-induced apoptosis in human alveolar adenocarcinoma cells via p53, survivin and bax/bcl-2 pathways: role of oxidative stress. Nanomedicine. 2011;7:904–13. doi: 10.1016/j.nano.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 106.Valenzuela M, Bravo D, Canales J, Sanhueza C, Diaz N, Almarza O, et al. Helicobacter pylori-induced loss of survivin and gastric cell viability is attributable to secreted bacterial gamma-glutamyl transpeptidase activity. J Infect Dis. 2013;208:1131–41. doi: 10.1093/infdis/jit286. [DOI] [PubMed] [Google Scholar]
- 107.Xu GC, Zhang P, Leng F, Pan L, Li ZY, Yu DD, et al. Inhibition of lymphatic metastases by a survivin dominant-negative mutant. Oncol Res. 2012;20:579–87. doi: 10.3727/096504013X13775486749416. [DOI] [PubMed] [Google Scholar]
- 108.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 109.Taeger J, Moser C, Hellerbrand C, Mycielska ME, Glockzin G, Schlitt HJ, et al. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol Cancer Ther. 2011;10:2157–67. doi: 10.1158/1535-7163.MCT-11-0312. [DOI] [PubMed] [Google Scholar]
- 110.Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Samuel S, Fan F, Dang LH, Xia L, Gaur P, Ellis LM. Intracrine vascular endothelial growth factor signaling in survival and chemoresistance of human colorectal cancer cells. Oncogene. 2011;30:1205–12. doi: 10.1038/onc.2010.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Goteri G, Lucarini G, Pieramici T, Filosa A, Pugnaloni A, Montik N, et al. Endothelial cell survivin is involved in the growth of ovarian endometriotic cysts. Anticancer Res. 2005;25:4313–8. [PubMed] [Google Scholar]
- 113.Gupta K, Kshirsagar S, Li W, Gui L, Ramakrishnan S, Gupta P, et al. VEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signaling. Exp Cell Res. 1999;247:495–504. doi: 10.1006/excr.1998.4359. [DOI] [PubMed] [Google Scholar]
- 114.Thirunavukkarasu M, Han Z, Zhan L, Penumathsa SV, Menon VP, Maulik N. Adeno-sh-beta-catenin abolishes ischemic preconditioning-mediated cardioprotection by downregulation of its target genes VEGF, Bcl-2, and survivin in ischemic rat myocardium. Antioxid Redox Signal. 2008;10:1475–84. doi: 10.1089/ars.2008.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaga S, Zhan L, Altaf E, Maulik N. Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. J Mol Cell Cardiol. 2006;40:138–47. doi: 10.1016/j.yjmcc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 116.Nogueira-Ferreira R, Vitorino R, Ferreira-Pinto MJ, Ferreira R, Henriques-Coelho T. Exploring the role of post-translational modifications on protein-protein interactions with survivin. Arch Biochem Biophys. 2013;538:64–70. doi: 10.1016/j.abb.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 117.Boidot R, Vegran F, Lizard-Nacol S. Transcriptional regulation of the survivin gene. Mol Biol Rep. 2014;41:233–40. doi: 10.1007/s11033-013-2856-0. [DOI] [PubMed] [Google Scholar]
- 118.Aoki Y, Feldman GM, Tosato G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood. 2003;101:1535–42. doi: 10.1182/blood-2002-07-2130. [DOI] [PubMed] [Google Scholar]
- 119.Asanuma H, Torigoe T, Kamiguchi K, Hirohashi Y, Ohmura T, Hirata K, et al. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65:11018–25. doi: 10.1158/0008-5472.CAN-05-0491. [DOI] [PubMed] [Google Scholar]
- 120.Ohashi H, Takagi H, Oh H, Suzuma K, Suzuma I, Miyamoto N, et al. Phosphatidylinositol 3-kinase/Akt regulates angiotensin II-induced inhibition of apoptosis in microvascular endothelial cells by governing survivin expression and suppression of caspase-3 activity. Circ Res. 2004;94:785–93. doi: 10.1161/01.RES.0000121103.03275.EC. [DOI] [PubMed] [Google Scholar]
- 121.Kim YS, Jin HO, Seo SK, Woo SH, Choe TB, An S, et al. Sorafenib induces apoptotic cell death in human non-small cell lung cancer cells by down-regulating mammalian target of rapamycin (mTOR)-dependent survivin expression. Biochem Pharmacol. 2011;82:216–26. doi: 10.1016/j.bcp.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 122.Fukuda S, Pelus LM. Activated H-Ras regulates hematopoietic cell survival by modulating Survivin. Biochem Biophys Res Commun. 2004;323:636–44. doi: 10.1016/j.bbrc.2004.08.149. [DOI] [PubMed] [Google Scholar]
- 123.Liu C, Liang B, Wang Q, Wu J, Zou MH. Activation of AMP-activated protein kinase alpha1 alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin. J Biol Chem. 2010;285:15346–55. doi: 10.1074/jbc.M110.102491. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Kumar P, Coltas IK, Kumar B, Chepeha DB, Bradford CR, Polverini PJ. Bcl-2 protects endothelial cells against gamma-radiation via a Raf-MEK-ERK-survivin signaling pathway that is independent of cytochrome c release. Cancer Res. 2007;67:1193–202. doi: 10.1158/0008-5472.CAN-06-2265. [DOI] [PubMed] [Google Scholar]
- 125.Kanwar JR, Kamalapuram SK, Kanwar RK. Targeting survivin in cancer: the cell-signalling perspective. Drug Discov Today. 2011;16:485–94. doi: 10.1016/j.drudis.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 126.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–57. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 127.Fossey SL, Liao AT, McCleese JK, Bear MD, Lin J, Li PK, et al. Characterization of STAT3 activation and expression in canine and human osteosarcoma. BMC Cancer. 2009;9:81. doi: 10.1186/1471-2407-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Weerasinghe P, Garcia GE, Zhu Q, Yuan P, Feng L, Mao L, et al. Inhibition of Stat3 activation and tumor growth suppression of non-small cell lung cancer by G-quartet oligonucleotides. Int J Oncol. 2007;31:129–36. [PubMed] [Google Scholar]
- 129.Kim KW, Mutter RW, Cao C, Albert JM, Shinohara ET, Sekhar KR, et al. Inhibition of signal transducer and activator of transcription 3 activity results in down-regulation of Survivin following irradiation. Mol Cancer Ther. 2006;5:2659–65. doi: 10.1158/1535-7163.MCT-06-0261. [DOI] [PubMed] [Google Scholar]
- 130.Wu ZX, Song TB, Li DM, Zhang XT, Wu XL. Overexpression of PTEN suppresses growth and induces apoptosis by inhibiting the expression of survivin in bladder cancer cells. Tumour Biol. 2007;28:9–15. doi: 10.1159/000097041. [DOI] [PubMed] [Google Scholar]
- 131.Li W, Wang H, Kuang CY, Zhu JK, Yu Y, Qin ZX, et al. An essential role for the Id1/PI3K/Akt/NFkB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol Cell Biochem. 2012;363:135–45. doi: 10.1007/s11010-011-1166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Takada Y, Khuri FR, Aggarwal BB. Protein farnesyltransferase inhibitor (SCH 66336) abolishes NF-kappaB activation induced by various carcinogens and inflammatory stimuli leading to suppression of NF-kappaB-regulated gene expression and up-regulation of apoptosis. J Biol Chem. 2004;279:26287–99. doi: 10.1074/jbc.M400963200. [DOI] [PubMed] [Google Scholar]
- 133.Zhang G, Zhu H, Wang Y, Yang S, Liu M, Zhang W, et al. Kruppel-like factor 4 represses transcription of the survivin gene in esophageal cancer cell lines. Biol Chem. 2009;390:463–9. doi: 10.1515/BC.2009.060. [DOI] [PubMed] [Google Scholar]
- 134.Zhu N, Gu L, Findley HW, Chen C, Dong JT, Yang L, et al. KLF5 Interacts with p53 in regulating survivin expression in acute lymphoblastic leukemia. J Biol Chem. 2006;281:14711–8. doi: 10.1074/jbc.M513810200. [DOI] [PubMed] [Google Scholar]
- 135.Wagner M, Schmelz K, Dorken B, Tamm I. Transcriptional regulation of human survivin by early growth response (Egr)-1 transcription factor. Int J Cancer. 2008;122:1278–87. doi: 10.1002/ijc.23183. [DOI] [PubMed] [Google Scholar]
- 136.Huang CL, Liu D, Nakano J, Yokomise H, Ueno M, Kadota K, et al. E2F1 overexpression correlates with thymidylate synthase and survivin gene expressions and tumor proliferation in non small-cell lung cancer. Clin Cancer Res. 2007;13:6938–46. doi: 10.1158/1078-0432.CCR-07-1539. [DOI] [PubMed] [Google Scholar]
- 137.Xu R, Zhang P, Huang J, Ge S, Lu J, Qian G. Sp1 and Sp3 regulate basal transcription of the survivin gene. Biochem Biophys Res Commun. 2007;356:286–92. doi: 10.1016/j.bbrc.2007.02.140. [DOI] [PubMed] [Google Scholar]
- 138.Zhong Q, Zhou Y, Ye W, Cai T, Zhang X, Deng DY. Hypoxia-inducible factor 1-alpha-AA-modified bone marrow stem cells protect PC12 cells from hypoxia-induced apoptosis, partially through VEGF/PI3K/Akt/FoxO1 pathway. Stem Cells Dev. 2012;21:2703–17. doi: 10.1089/scd.2011.0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619–31. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- 140.Hossain MM, Banik NL, Ray SK. Survivin knockdown increased anti-cancer effects of (-)-epigallocatechin-3-gallate in human malignant neuroblastoma SK-N-BE2 and SH-SY5Y cells. Exp Cell Res. 2012;318:1597–610. doi: 10.1016/j.yexcr.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shen J, Sun H, Meng Q, Yin Q, Zhang Z, Yu H, et al. Simultaneous inhibition of tumor growth and angiogenesis for resistant hepatocellular carcinoma by co-delivery of sorafenib and survivin small hairpin RNA. Mol Pharm. 2014;11:3342–51. doi: 10.1021/mp4006408. [DOI] [PubMed] [Google Scholar]
- 142.Ma A, Lin R, Chan PK, Leung JC, Chan LY, Meng A, et al. The role of survivin in angiogenesis during zebrafish embryonic development. BMC Dev Biol. 2007;7:50. doi: 10.1186/1471-213X-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Delvaeye M, De Vriese A, Zwerts F, Betz I, Moons M, Autiero M, et al. Role of the 2 zebrafish survivin genes in vasculo-angiogenesis, neurogenesis, cardiogenesis and hematopoiesis. BMC Dev Biol. 2009;9:25. doi: 10.1186/1471-213X-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang P, Zhen H, Zhang J, Zhang W, Zhang R, Cheng X, et al. Survivin promotes glioma angiogenesis through vascular endothelial growth factor and basic fibroblast growth factor in vitro and in vivo. Mol Carcinog. 2012;51:586–95. doi: 10.1002/mc.20829. [DOI] [PubMed] [Google Scholar]
- 145.Shu MG, Guo XT, Zhen HN, Han Y, Chen FL, Li LW, et al. Enhancing skin flap survival by a cell-permeable wild-type survivin. Med Hypotheses. 2007;69:888–91. doi: 10.1016/j.mehy.2006.11.055. [DOI] [PubMed] [Google Scholar]
- 146.Furuya M, Tsuji N, Kobayashi D, Watanabe N. Interaction between survivin and aurora-B kinase plays an important role in survivin-mediated up-regulation of human telomerase reverse transcriptase expression. Int J Oncol. 2009;34:1061–8. doi: 10.3892/ijo_00000232. [DOI] [PubMed] [Google Scholar]
- 147.Wang Z, Fukuda S, Pelus LM. Survivin regulates the p53 tumor suppressor gene family. Oncogene. 2004;23:8146–53. doi: 10.1038/sj.onc.1207992. [DOI] [PubMed] [Google Scholar]
- 148.Bolton MA, Lan W, Powers SE, McCleland ML, Kuang J, Stukenberg PT. Aurora B kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Mol Biol Cell. 2002;13:3064–77. doi: 10.1091/mbc.E02-02-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhu LB, Jiang J, Zhu XP, Wang TF, Chen XY, Luo QF, et al. Knockdown of Aurora-B inhibits osteosarcoma cell invasion and migration via modulating PI3K/Akt/NF-kappaB signaling pathway. Int J Clin Exp Pathol. 2014;7:3984–91. [PMC free article] [PubMed] [Google Scholar]
- 150.Shan RF, Zhou YF, Peng AF, Jie ZG. Inhibition of Aurora-B suppresses HepG2 cell invasion and migration via the PI3K/Akt/NF-kappaB signaling pathway. Exp Ther Med. 2014;8:1005–9. doi: 10.3892/etm.2014.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mottet D, Dumont V, Deccache Y, Demazy C, Ninane N, Raes M, et al. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem. 2003;278:31277–85. doi: 10.1074/jbc.M300763200. [DOI] [PubMed] [Google Scholar]
- 152.Sharma M, Chuang WW, Sun Z. Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J Biol Chem. 2002;277:30935–41. doi: 10.1074/jbc.M201919200. [DOI] [PubMed] [Google Scholar]
- 153.Adluri RS, Thirunavukkarasu M, Zhan L, Akita Y, Samuel SM, Otani H, et al. Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: a study using thioredoxin 1 transgenic mice. J Mol Cell Cardiol. 2011;50:239–47. doi: 10.1016/j.yjmcc.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–9. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bai XM, Jiang H, Ding JX, Peng T, Ma J, Wang YH, et al. Prostaglandin E2 upregulates survivin expression via the EP1 receptor in hepatocellular carcinoma cells. Life Sci. 2010;86:214–23. doi: 10.1016/j.lfs.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 156.Krysan K, Dalwadi H, Sharma S, Pold M, Dubinett S. Cyclooxygenase 2-dependent expression of survivin is critical for apoptosis resistance in non-small cell lung cancer. Cancer Res. 2004;64:6359–62. doi: 10.1158/0008-5472.CAN-04-1681. [DOI] [PubMed] [Google Scholar]
- 157.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–10. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 158.Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–52. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Folberg R, Hendrix MJ, Maniotis AJ. Vasculogenic mimicry and tumor angiogenesis. Am J Pathol. 2000;156:361–81. doi: 10.1016/S0002-9440(10)64739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18:2726–32. doi: 10.1158/1078-0432.CCR-11-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Francescone R, Scully S, Bentley B, Yan W, Taylor SL, Oh D, et al. Glioblastoma-derived tumor cells induce vasculogenic mimicry through Flk-1 protein activation. J Biol Chem. 2012;287:24821–31. doi: 10.1074/jbc.M111.334540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yao X, Ping Y, Liu Y, Chen K, Yoshimura T, Liu M, et al. Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by Glioma stem-like cells. PLoS One. 2013;8:e57188. doi: 10.1371/journal.pone.0057188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dunleavey JM, Xiao L, Thompson J, Kim MM, Shields JM, Shelton SE, et al. Vascular channels formed by subpopulations of PECAM1+ melanoma cells. Nat Commun. 2014;5:5200. doi: 10.1038/ncomms6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gao Y, Zhao XL, Gu Q, Wang JY, Zhang SW, Zhang DF, et al. Correlation of vasculogenic mimicry with clinicopathologic features and prognosis of ovarian carcinoma. Zhonghua Bing Li Xue Za Zhi. 2009;38:585–9. [PubMed] [Google Scholar]
- 165.Bai XL, Zhang Q, Ye LY, Liang F, Sun X, Chen Y, et al. Myocyte enhancer factor 2C regulation of hepatocellular carcinoma via vascular endothelial growth factor and Wnt/beta-catenin signaling. Oncogene. 2015;34:4089–97. [DOI] [PubMed]
- 166.Clapp C, Martinez de la Escalera G. Aquaporin-1: a novel promoter of tumor angiogenesis. Trends Endocrinol Metab. 2006;17:1–2. doi: 10.1016/j.tem.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 167.Du J, Sun B, Zhao X, Gu Q, Dong X, Mo J, et al. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol Oncol. 2014;133:575–83. doi: 10.1016/j.ygyno.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 168.Fan YL, Zheng M, Tang YL, Liang XH. A new perspective of vasculogenic mimicry: EMT and cancer stem cells (Review) Oncol Lett. 2013;6:1174–80. doi: 10.3892/ol.2013.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang X, Song Q, Wei C, Qu J. LRIG1 inhibits hypoxia-induced vasculogenic mimicry formation via suppression of the EGFR/PI3K/AKT pathway and epithelial-to-mesenchymal transition in human glioma SHG-44 cells. Cell Stress Chaperones. 2015;20:631–41. doi: 10.1007/s12192-015-0587-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yang Z, Sun B, Li Y, Zhao X, Gu Q, An J, et al. ZEB2 promotes vasculogenic mimicry by TGF-beta1 induced epithelial-to-mesenchymal transition in hepatocellular carcinoma. Exp Mol Pathol. 2015;98:352–9. doi: 10.1016/j.yexmp.2015.03.030. [DOI] [PubMed] [Google Scholar]
- 171.Wang L, Lin L, Chen X, Sun L, Liao Y, Huang N, et al. Metastasis-associated in colon cancer-1 promotes vasculogenic mimicry in gastric cancer by upregulating TWIST1/2. Oncotarget. 2015;6:11492–506. doi: 10.18632/oncotarget.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang W, Lin P, Sun B, Zhang S, Cai W, Han C, et al. Epithelial-mesenchymal transition regulated by EphA2 contributes to vasculogenic mimicry formation of head and neck squamous cell carcinoma. Biomed Res Int. 2014;2014:803914. doi: 10.1155/2014/803914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lee J, Choi JH, Joo CK. TGF-beta1 regulates cell fate during epithelial-mesenchymal transition by upregulating survivin. Cell Death Dis. 2013;4:e714. doi: 10.1038/cddis.2013.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liao A, Shi R, Jiang Y, Tian S, Li P, Song F, et al. SDF-1/CXCR4 Axis regulates cell cycle progression and epithelial-mesenchymal transition via up-regulation of survivin in glioblastoma. Mol Neurobiol. 2014. doi:10.1007/s12035-014-9006-0 [DOI] [PubMed]
- 175.Tang NN, Zhu H, Zhang HJ, Zhang WF, Jin HL, Wang L, et al. HIF-1alpha induces VE-cadherin expression and modulates vasculogenic mimicry in esophageal carcinoma cells. World J Gastroenterol. 2014;20:17894–904. doi: 10.3748/wjg.v20.i47.17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tsuneki M, Madri JA. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J Biol Chem. 2014;289:5357–70. doi: 10.1074/jbc.M113.529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Iurlaro M, Demontis F, Corada M, Zanetta L, Drake C, Gariboldi M, et al. VE-cadherin expression and clustering maintain low levels of survivin in endothelial cells. Am J Pathol. 2004;165:181–9. doi: 10.1016/S0002-9440(10)63287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 179.Deveraux QL, Reed JC. IAP family proteins--suppressors of apoptosis. Genes Dev. 1999;13:239–52. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 180.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–4. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 181.Altieri DC. Survivin - The inconvenient IAP. Semin Cell Dev Biol. 2015;39:91–6. doi: 10.1016/j.semcdb.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang LQ, Wang J, Jiang F, Xu L, Liu FY, Yin R. Prognostic value of survivin in patients with non-small cell lung carcinoma: a systematic review with meta-analysis. PLoS One. 2012;7:e34100. doi: 10.1371/journal.pone.0034100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Liu JL, Gao W, Kang QM, Zhang XJ, Yang SG. Prognostic value of survivin in patients with gastric cancer: a systematic review with meta-analysis. PLoS One. 2013;8:e71930. doi: 10.1371/journal.pone.0071930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gasowska-Bodnar A, Bodnar L, Dabek A, Cichowicz M, Jerzak M, Cierniak S, et al. Survivin expression as a prognostic factor in patients with epithelial ovarian cancer or primary peritoneal cancer treated with neoadjuvant chemotherapy. Int J Gynecol Cancer. 2014;24:687–96. doi: 10.1097/IGC.0000000000000108. [DOI] [PubMed] [Google Scholar]
- 185.Siamakpour-Reihani S, Owzar K, Jiang C, Turner T, Deng Y, Bean SM, et al. Prognostic significance of differential expression of angiogenic genes in women with high-grade serous ovarian carcinoma. Gynecol Oncol. 2015;139:23–9. doi: 10.1016/j.ygyno.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Li J, Li S, Chen R, Yu H, Lu X. The prognostic significance of anti-angiogenesis therapy in ovarian cancer: a meta-analysis. J Ovarian Res. 2015;8:54. doi: 10.1186/s13048-015-0181-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6:1796–803. [PubMed] [Google Scholar]
- 188.Fulda S. Inhibitor of Apoptosis (IAP) proteins in hematological malignancies: molecular mechanisms and therapeutic opportunities. Leukemia. 2014;28:1414–22. doi: 10.1038/leu.2014.56. [DOI] [PubMed] [Google Scholar]
- 189.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–24. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 190.Raetz EA, Morrison D, Romanos-Sirakis E, Gaynon P, Sposto R, Bhojwani D, et al. A phase I study of EZN-3042, a novel survivin messenger ribonucleic acid (mRNA) antagonist, administered in combination with chemotherapy in children with relapsed acute lymphoblastic leukemia (ALL): a report from the therapeutic advances in childhood leukemia and lymphoma (TACL) consortium. J Pediatr Hematol Oncol. 2014;36:458–63. doi: 10.1097/MPH.0b013e3182a8f58f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kelly RJ, Thomas A, Rajan A, Chun G, Lopez-Chavez A, Szabo E, et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann Oncol. 2013;24:2601–6. doi: 10.1093/annonc/mdt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tibes R, McDonagh KT, Lekakis L, Bogenberger JM, Kim S, Frazer N, et al. Phase I study of the novel Cdc2/CDK1 and AKT inhibitor terameprocol in patients with advanced leukemias. Invest New Drugs. 2015;33:389–96. doi: 10.1007/s10637-014-0198-y. [DOI] [PubMed] [Google Scholar]
- 193.Berntsen A, Trepiakas R, Wenandy L, Geertsen PF, thor Straten P, Andersen MH, et al. Therapeutic dendritic cell vaccination of patients with metastatic renal cell carcinoma: a clinical phase 1/2 trial. J Immunother. 2008;31:771–80. doi: 10.1097/CJI.0b013e3181833818. [DOI] [PubMed] [Google Scholar]
- 194.Wang D, Zhang B, Gao H, Ding G, Wu Q, Zhang J, et al. Clinical research of genetically modified dendritic cells in combination with cytokine-induced killer cell treatment in advanced renal cancer. BMC Cancer. 2014;14:251. doi: 10.1186/1471-2407-14-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Berinstein NL, Karkada M, Oza AM, Odunsi K, Villella JA, Nemunaitis JJ, et al. Survivin-targeted immunotherapy drives robust polyfunctional T cell generation and differentiation in advanced ovarian cancer patients. Oncoimmunology. 2015;4:e1026529. doi: 10.1080/2162402X.2015.1026529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ellebaek E, Engell-Noerregaard L, Iversen TZ, Froesig TM, Munir S, Hadrup SR, et al. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: results from a phase II trial. Cancer Immunol Immunother. 2012;61:1791–804. doi: 10.1007/s00262-012-1242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]