

Abstract
Usher syndrome (USH) is the leading cause of inherited deaf-blindness, with type 2 (USH2) being the most common clinical form. Studies suggest that proteins encoded by USH2 causative genes assemble into the ankle link complex (ALC) at the hair cell stereociliary bundle; however, little is known about the in vivo assembly and function of this complex. Using various USH2 mutant mice, we showed by immunofluorescence that USH2 proteins play different roles in cochlear ALC assembly, with G protein-coupled receptor 98 being the most important protein. Complex assembly likely occurs at the stereociliary bundle but not along the protein transport route in the cell body. Stereociliary morphological defects in USH2 mutant mice suggest roles for the ALC in regulating inner hair cell stereociliary growth and differentiation as well as outer hair cell stereociliary rigidity and organization during development. These roles are unique from the bundle cohesion role of Usher syndrome type 1 protein complexes. Loss of individual USH2 gene expressions leads to variable morphological and functional consequences, correlating with the severity of ALC disruption. This finding suggests a potential genotype–phenotype correlation in USH2 patients. In summary, this study provides novel insights into the molecular mechanism underlying cochlear stereociliary bundle development and hearing loss pathogenesis of various USH2 subtypes. Our thorough phenotypical characterization of USH2 mouse models is essential for future use of these animal models in therapeutic development.
Introduction
The stereociliary bundle of cochlear hair cells is a highly specialized structure critical for transducing mechanical sound stimuli into electrical signals. This structure consists of actin-based stereocilia arranged into three rows of increasing length and a microtubule-based kinocilium that exists only during early development. In the stereociliary bundle, orderly stereociliary organization and appropriate stereociliary biophysical properties are essential for high-fidelity mechanotransduction and eventual sound perception (1–3). During development, the stereocilia of hair cell bundles differentiate from microvilli and grow differentially to reach their final lengths, thicknesses and rigidity. Simultaneously, the growing stereocilia and the kinocilium move from the center to the periphery on the hair cell apex to establish bundle polarity (4–8). Throughout this process, various fibrous links develop among the kinocilium and stereocilia (9), and it is hypothesized that these links assist in maintaining stereociliary cohesion. Previous studies have discovered that genes associated with Usher syndrome (USH), an incurable genetic disease, encode protein components of some of these fibrous links (10–12).
USH is the major cause of combined hearing impairment and retinal degeneration (13–15). The predominant clinical form of USH is type 2 (USH2). Currently, ADGRV1 (adhesion G protein-coupled receptor V1, also known as GPR98, VLGR1 or MASS1, MIM *602851) (16), USH2A (MIM *608400) (17) and DFNB31 (also known as Whrn in mice, MIM *607928) (18) have been identified as causative genes, and PDZD7 (PDZ domain-containing 7, MIM *612971) as a modifier gene in USH2 patients (19). G protein-coupled receptor 98 (GPR98) protein encoded by ADGRV1 is a major component of ankle links (20,21), which connect stereocilia at their bases and exist transiently in developing mammalian cochlear hair cells (9). Usherin, whirlin and PDZD7 proteins are the products of USH2A, DFNB31 and PDZD7 genes, respectively. They colocalize with GPR98 at the ankle link region of stereociliary bundles in hair cells (21–24). Recent in vitro biochemical studies demonstrated that whirlin and PDZD7 can heterodimerize with each other and that both proteins are required to link usherin and GPR98 in a dynamic quaternary protein complex (25). These findings suggest that the three USH2 proteins (GPR98, usherin and whirlin) and the PDZD7 protein assemble into a multiprotein complex, the ankle link complex (ALC), in hair cell stereociliary bundles. To support this, mutations in the USH2 and PDZD7 orthologous genes in mice have been shown to disrupt the distribution of some of the USH2 and PDZD7 proteins in cochlear hair cells (21,24). However, previous studies of the ALC in mice were not systematic or comprehensive; they focused on one specific mutant mouse line, one specific type of cochlear hair cell, such as the inner hair cell (IHC), or only some of the USH2 and PDZD7 proteins. Accordingly, a complete picture is missing on how the ALC is assembled at the stereociliary base in cochlear hair cells.
Up to now, eight mouse lines carrying mutations in the USH2 and PDZD7 gene orthologs have been reported (20–22,24,26–29). These mouse models share similar inner ear phenotypes, including stereociliary disorganization and degeneration, as well as hearing loss (20–22,24,26–29). Considering that the ALC exists only during stereociliary bundle development from postnatal day 2 (P2) to P12 (9), these observed phenotypes indicate strongly that the ALC plays an essential role during stereociliary bundle development. However, most of the phenotypical characterizations in the reported mouse models, except the Pdzd7 knockout mouse (24), were conducted four to five days after the emergence of the ALC or in adult animals. Thus, the observed phenotypes in these previous reports may represent a combination of primary and secondary defects caused by disruption of the ALC. In some of these studies, cochlear morphology was examined using low-magnification electron microscopy or phalloidin fluorescence staining, and thus details of the stereociliary bundle defects were not revealed. Furthermore, the hearing loss in Ush2a knockout mice was characterized as late-onset, mild and only at high frequency (28), which differs significantly from the early-onset hearing loss throughout all frequencies tested in other USH2 mutant mouse lines (20,22,24,26,27,30). Therefore, it is currently still unclear how the ALC keeps stereocilia well-organized during stereociliary bundle development and how the individual components contribute to the function of the whole complex.
Here, we investigated thoroughly the ALC using a series of USH2 mutant mouse lines, all of which are on a similar genetic background. We addressed the assembly and function of the ALC at an early postnatal time point, P4, when the complex and its associated ankle links become robust (9,20,21,23,24,31). Using high-resolution immunofluorescence, we examined systematically the roles of each USH2 protein in the normal localizations of ALC components, which is a prerequisite for complex assembly. We also studied the functions of individual USH2 proteins during cochlear stereociliary bundle development and described their involvement in regulating stereociliary thickness, rigidity and organization, roles which are different from the stereociliary cohesion function of Usher syndrome type 1 (USH1) proteins. Our study reveals different contributions of individual USH2 proteins to the assembly and function of the ALC and indicates a potential genotype–phenotype correlation in USH2 patients. Our findings are significant for understanding hair cell development and USH2 pathogenesis and will have a potential impact on USH2 diagnosis, prognosis and treatments.
Results
GPR98 is required for the assembly of the ALC in cochlear hair cells
Usherin, whirlin and PDZD7 distribution was examined in cochlear hair cells of Adgrv1−/− mice at P4. These mutant mice were verified to express no GPR98 protein by immunostaining of the cochlea and immunoblotting of the retinal lysate using antibodies against GPR98 N- and C-terminal regions (Fig. 1A and data not shown). Usherin immunofluorescence was found at the stereociliary base, the location of the ALC, in both IHCs and outer hair cells (OHCs) of wild-type cochleas (Fig. 1B), whereas it was completely absent in IHC and OHC stereociliary bundles of Adgrv1−/− mice (Fig. 2A). Whirlin was detected at both the tip and ALC of stereocilia by an antibody against both whirlin long and short isoforms in wild-type cochlear IHCs and OHCs (Fig. 1C), in agreement with previous publications (24,31,32). In contrast, whirlin immunoreactivity was undetectable at ALCs but was present at the tips of Adgrv1−/− cochlear stereociliary bundles (Fig. 2B). The observed localization of usherin and whirlin in P4 Adgrv1−/− cochlear hair cells are consistent with an earlier report of usherin and whirlin localization in cochlear hair cells of another Adgrv1 knockout mouse line, Adgrv1tm1Msat, at P6 (21). Although we demonstrated previously that PDZD7 is mislocalized in Adgrv1−/− cochlear stereociliary bundles (24), image quality in that study prevented a detailed analysis. The improved technique applied in this study revealed that the PDZD7 signal was mislocalized from stereociliary bases (shown for the wild-type, Fig. 1D) to almost the entire stereociliary length except for the tips in Adgrv1−/− cochlear hair cells (Fig. 2C). Note that these changes of usherin, whirlin and PDZD7 distribution in Adgrv1−/− mice were observed throughout the entire cochlea from the basal to the apical turn (data not shown). Because hair cells along the cochlear longitudinal axis develop progressively, with the developmental stage at the cochlear base 2 days ahead of that at the cochlear apex (33), the similar distribution patterns of the ALC components across the entire cochlea in the absence of GPR98 indicate that the abnormal distribution of ALC components starts earlier than P4. In summary, GPR98 was found to be indispensable for normal localization of other ALC components (usherin, whirlin and PDZD7) in cochlear hair cells, and thus plays a critical role in the assembly of the ALC. These data also suggest that transport of whirlin and PDZD7 from the cell body to the stereociliary bundle may not require expression of Adgrv1 and that an unknown mechanism appears to exclude PDZD7 from the stereociliary tip where whirlin is localized.
Figure 1.

Localization of USH2 and PDZD7 proteins at the ALC of developing cochlear hair cells. Double immunofluorescence demonstrates that GPR98 (A), usherin (B), whirlin (C) and PDZD7 (D) are localized at the ALC, i.e. the stereociliary base, while whirlin (C) is also present at the stereociliary tip in P4 wild-type (WT) mouse IHCs and OHCs. Absence of USH2 and PDZD7 immunoreactivities in their corresponding mutant cochlear hair cells verifies loss of protein expression and the specificity of our antibodies. For each panel, two WT IHC bundles, viewed from the medial side of hair cells (front) at slightly different angles, show USH2 or PDZD7 signals in either one or multiple rows of stereocilia. Amplified views of several IHC stereocilia are shown on the left of original images. The Adgrv1−/−, Ush2a−/− and Pdzd7−/− OHC bundles are viewed from the back side (the lateral side of hair cells), while other stereociliary bundles are shown from either front or side views. Arrows point to stereociliary bases. Magenta signals in stereociliary bundles are specific signals labeled from USH2 and PDZD7 antibodies. Magenta signals outside stereociliary bundles are non-specific. Green signals are from phalloidin staining. Scale bars, 1 μm.
Figure 2.
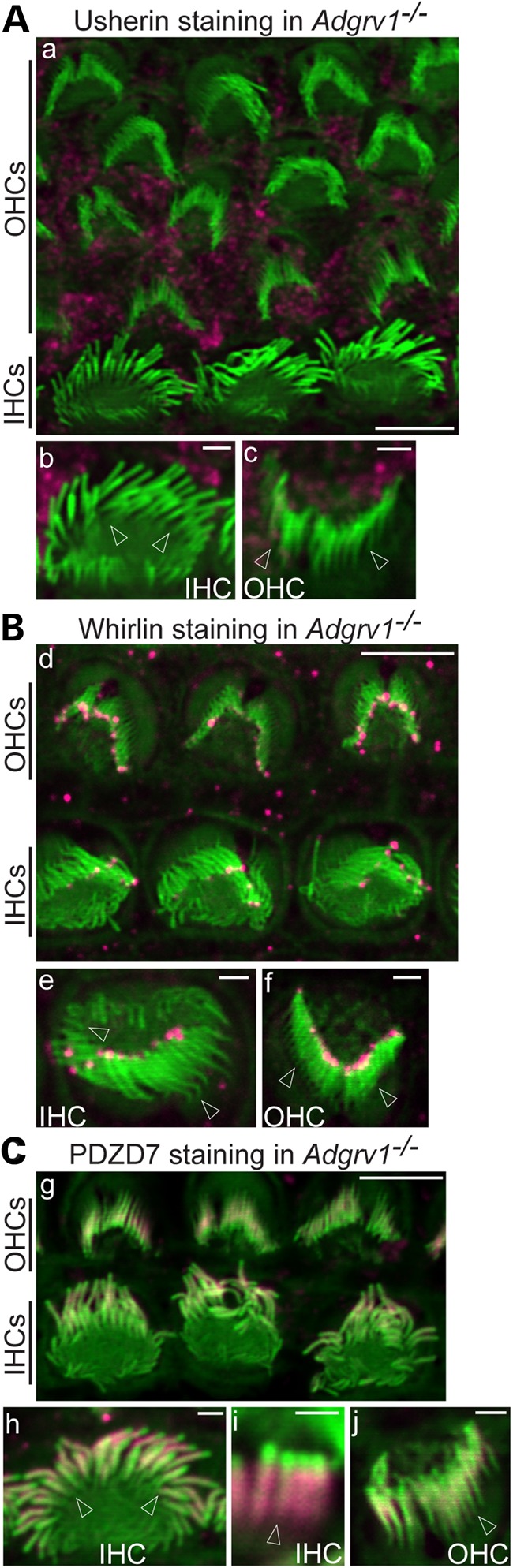
GPR98 is required for localization of usherin, whirlin and PDZD7 at the ALC of cochlear hair cells. (A) Usherin immunoreactivity is undetectable in the stereociliary bundle of P4 Adgrv1−/− IHCs and OHCs. (B) Whirlin immunofluorescence is detectable only at stereociliary tips but not at ALCs in P4 Adgrv1−/− cochlear hair cells. (C) PDZD7 immunoreactivity is distributed along the entire stereocilia, except the tips, in P4 Adgrv1−/− IHCs and OHCs. Images (a), (d) and (g) are low-magnification views of Adgrv1−/− IHC and OHC stereociliary bundles; (b) (front view), (e) (back view) and (h) (front view) are single Adgrv1−/− IHC stereociliary bundles; (c), (f) and (j) are back views of single Adgrv1−/− OHC stereociliary bundles; (i) is an amplified view of several stereocilia within an Adgrv1−/− IHC bundle. Arrows point to stereociliary bases. Magenta signals in stereociliary bundles are from staining of USH2 and PDZD7 proteins, and magenta signals outside stereociliary bundles are non-specific. Phalloidin signals are green. Scale bars, 5 μm (a, d and g) and 1 μm (b and c, e and f, h and j).
Because GPR98 is a major component of ankle links in cochlear hair cells (20,21), we next investigated whether disruption of ankle links could disturb the assembled ALC. Subtilisin proteolysis was shown to disrupt ankle links and GPR98 immunofluorescence in cochlear stereociliary bundles (9,21). We therefore examined whether disruption of ankle links and degradation of GPR98 by subtilisin alters the localization of usherin, whirlin and PDZD7 at the ALC. We found that the treatment of wild-type cochleas with subtilisin for 15 min eliminated immunofluorescence from an antibody against the GPR98 N-terminal extracellular region (Fig. 3A) but not from an antibody against the GPR98 C-terminal intracellular region (Fig. 3B), indicating that subtilisin treatment under our condition degraded only the GPR98 extracellular region, i.e. the ankle links. We then examined the effect of the subtilisin-mediated loss of the GPR98 extracellular region on the distribution of usherin, whirlin and PDZD7 in cochlear stereociliary bundles. Note that usherin is a second transmembrane protein of the ALC, and our usherin antibody detected the usherin cytoplasmic region. Compared with the phosphate-buffered saline (PBS)-treated control group, localization of usherin, whirlin and PDZD7 at the ALC did not change significantly along the entire cochlea after subtilisin treatment (Fig. 3C–E). These findings demonstrate that, at least for a short-time period, GPR98 is anchored by its intracellular region and that this region of GPR98 is sufficient for maintenance of the ALC in cochlear hair cells.
Figure 3.

Subtilisin treatment disrupts ankle links but not the ALC in cochlear stereociliary bundles. Subtilisin treatment eliminates ankle links in P4 cochlear hair cells, shown by a lack of immunoreactivity with an antibody directed against the GPR98 extracellular region (A). However, this treatment does not affect distribution of the GPR98 intracellular fragment at the P4 stereociliary base, shown by immunofluorescence using an antibody against the GPR98 cytoplasmic region (B). Localization of usherin (C), PDZD7 (D) and whirlin (E) at P4 stereociliary bases appears normal after subtilisin treatment (right column) compared with PBS-treated negative controls (left column). Stereociliary bundles are viewed from the front and side except the PBS-treated IHC bundle in (B), which is a back view. Arrows point to stereociliary bases. Green signals are from phalloidin staining. Magenta signals in stereociliary bundles are specific signals of USH2 and PDZD7 proteins, while magenta signals outside stereociliary bundles are non-specific. Scale bars, 1 µm.
Usherin is essential for recruiting whirlin and PDZD7 to the ALC in cochlear hair cells
We examined whether usherin plays a role in the localization of GPR98, whirlin and PDZD7 at the ALC by immunostaining P4 Ush2a−/− cochlear hair cells and observing the immunofluorescence along the entire cochlea. GPR98, detected by the antibody against the GPR98 N-terminal intracellular region, was localized at ankle links of the stereociliary bundle in wild-type IHCs and OHCs (Fig. 1A), as shown in other reports (20,21,23,24). Elimination of Ush2a expression resulted in significant changes in the GPR98 signal pattern (Fig. 4A). Among 44 IHCs examined from more than three litters of Ush2a−/− pups, GPR98 signals were found to be mislocalized (30% at both the tip and base, 27% mainly at the tip, 16% mainly at the base, 11% others; Fig. 4Aa and c). Occasionally, normal GPR98 distribution at the ALC was clearly observed (16%, Fig. 4Ab). Among 76 OHCs examined from the same pool of animals, ∼63% showed random GPR98 punctate signals along the entire stereocilium (Fig. 4Ad), 15% had GPR98 signals at the stereociliary tip or both the stereociliary tip and base and 22% appeared to have normal GPR98 signal at the stereociliary base. In Ush2a−/− mice, whirlin immunofluorescent signal was missing at the base of stereocilia, while it appeared normal at the stereociliary tip in almost all IHCs and OHCs examined (Fig. 4B). Furthermore, PDZD7 protein was mislocalized in a pattern similar to that found in Adgrv1−/− cochlear hair cells, which was along the entire stereocilium except the tips (Fig. 4C). Therefore, usherin is indispensable for normal whirlin and PDZD7 localization and plays a partial role in GPR98 localization at the ALC in cochlear hair cells. A small amount of GPR98 can be localized at ankle links independent of usherin, whirlin and PDZD7. These findings also imply that GPR98, whirlin and PDZD7 can be transported to the stereociliary bundle without assistance of usherin.
Figure 4.

Usherin plays differential roles in localization of GPR98, whirlin and PDZD7 at the cochlear ALC. (A) GPR98 (magenta) distribution at the ALC is altered in most IHCs and OHCs of P4 Ush2a−/− mice. GPR98 punctate signals are localized predominantly at both the tip and base of Ush2a−/− IHC stereocilia (a–c). (a) Back view of four Ush2a−/− IHC stereociliary bundles showing abnormal GPR98 distribution; (b) front view of an Ush2a−/− IHC bundle showing normal GPR98 distribution; (c) side view of an Ush2a−/− IHC bundle with abnormal GPR98 signal (left) and front view of another Ush2a−/− IHC bundle with near-normal GPR98 signal (right); (d) GPR98 punctate signals are randomly distributed along Ush2a−/− OHC stereocilia. (B) Whirlin (magenta) is completely absent at the stereociliary base but detectable at the stereociliary tip in both P4 Ush2a−/− IHCs and OHCs. (e and f) Front and back views of Ush2a−/− IHC bundles, respectively; (g) back view of an Ush2a−/− OHC bundle. (C) PDZD7 (magenta) distribution in Ush2a−/− IHC (h and i) and OHC (j and k) stereociliary bundles is similar to that in Adgrv1−/− IHC and OHC bundles at P4. Insets (h′ and i′) are amplified views and shown in original images for context. Images (h), (j) and (k): back view; (i): side view. Arrows point to stereociliary bases. Green signals are from phalloidin. Magenta signals outside stereociliary bundles are non-specific. Scale bars, 1 μm.
Whirlin plays a minor role in recruiting ALC components for complex assembly in cochlear hair cells
Immunofluorescent staining of cochleas from two different Dfnb31 mutant mouse lines was performed at P4 for GPR98, usherin and PDZD7. Dfnb31neo/neo mice have a deletion at the 3′ region of Dfnb31 exon 1 (22), and Dfnb31wi/wi mice have a spontaneous deletion between Dfnb31 exons 6 and 9 (29). In both Dfnb31 mutant mouse lines, no whirlin protein was present at the ALC in IHC and OHC stereociliary bundles (Fig. 1C and data not shown). Signal for GPR98 was present at the stereociliary tip as well as the ALC in a small percentage of Dfnb31 mutant IHCs (Dfnb31neo/neo: 18.6%, 8 of 43 IHCs examined; Dfnb31wi/wi: 4.3%, 2 of 46 IHCs examined) with the rest of the IHCs having normal GPR98 distribution (Figs 5A and 6A). Note that the short IHC stereocilia in Dfnb31wi/wi mice made it difficult to distinguish the weak GPR98 signal at the stereociliary tip from the GPR98 signal at the stereociliary base; thus, the percentage of IHCs with abnormal GPR98 signal may be underestimated in Dfnb31wi/wi mice. An abnormal GPR98 signal pattern similar to that in IHCs was also found in approximately one-third of Dfnb31 mutant OHCs (Dfnb31neo/neo: 34.5%, 39 of 113 OHCs examined; Dfnb31wi/wi: 33.6%, 40 of 119 OHCs examined), while GPR98 immunostaining signal appeared normal at the ALC in the remaining mutant OHCs (Figs 5A and 6A). Compared with wild-type IHCs and OHCs (Fig. 1B), all mutant IHCs (Dfnb31neo/neo: 100%, 48 IHCs; Dfnb31wi/wi: 100%, 28 IHCs) and most mutant OHCs (Dfnb31neo/neo: 96%, 141 of 147 OHCs; Dfnb31wi/wi: 96.7%, 88 of 91 OHCs) showed intact usherin signal distribution at the ALC (Figs 5B and 6B). Note that our observations of GPR98 and usherin in P4 Dfnb31wi/wi IHCs differ from those of a previous report showing mislocalization of GPR98 and the absence of usherin in P6 Dfnb31wi/wi IHCs (21). The reason for this discrepancy is unclear, but the observations made at P6 could include defects secondary to the Dfnb31wi/wi mutation. Previously, we showed that PDZD7 localization was relatively normal in P4 Dfnb31neo/neo and Dfnb31wi/wi cochlear hair cells (24). In this study, we confirmed this finding (Fig. 6C) and further observed that PDZD7 remained at the ALC in both Dfnb31neo/neo IHCs and OHCs at P8, a later developmental age (Fig. 5C). No difference in the distribution of ALC components was found along the entire longitudinal axis of the Dfnb31neo/neo and Dfnb31wi/wi cochleas (data not shown). Therefore, disruption of whirlin localization at the ALC resulted in mislocalization of GPR98 in only a small fraction of IHCs and OHCs, and rarely altered the localization of usherin and PDZD7 in cochlear hair cells. Accordingly, we concluded that whirlin plays a minor role in recruiting the known ALC components to the correct location within stereocilia for complex assembly and is dispensable for transport of these components from the cell body to the stereociliary bundle.
Figure 5.

Minor mislocalization of GPR98 and normal localization of usherin and PDZD7 in Dfnb31neo/neo cochlear hair cells. (A) GPR98 (magenta) is localized normally at stereociliary bases of most P4 Dfnb31neo/neo IHC (a, b, back and front views, respectively) and OHC (d) bundles, while GPR98 is partially mislocalized to stereociliary tips in a small number of Dfnb31neo/neo IHCs (c) and OHCs (e). (f) Single magenta channel image of (e). (B) Usherin signals (magenta) remain at stereociliary bases of P4 Dfnb31neo/neo cochlear hair cells. (g and h) Back and front views of Dfnb31neo/neo IHC bundles, respectively; (i and j) back and front views of Dfnb31neo/neo OHC bundles, respectively. (C) PDZD7 (magenta) localization appears normal in P8 Dfnb31neo/neo IHC (m) and OHC (n) relative to age-matched wild-type (WT) IHC (k) and OHC (l). Arrows point to stereociliary bases. Green signals are from phalloidin labeling. Magenta signals outside stereociliary bundles are non-specific. Scale bars, 1 μm.
Figure 6.
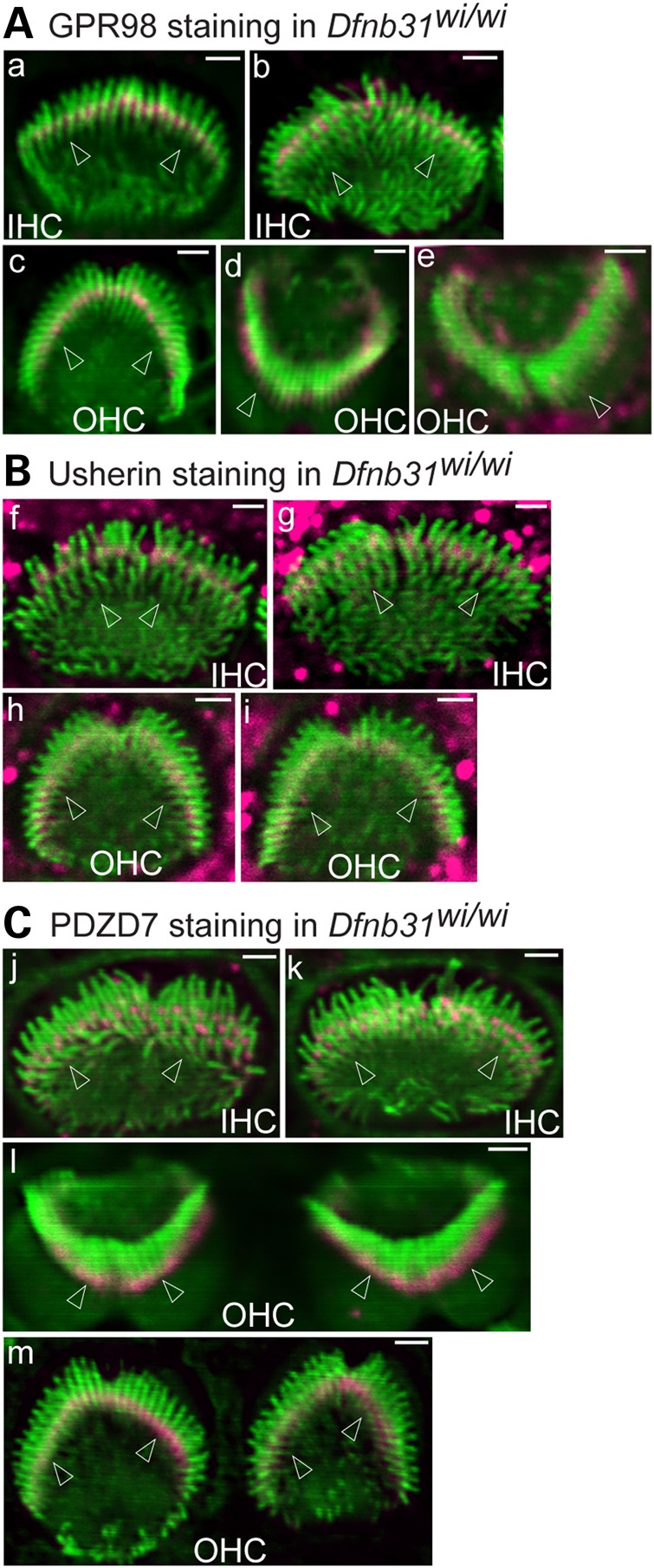
Minor mislocalization of GPR98 and normal localization of usherin and PDZD7 in Dfnb31wi/wi cochlear hair cells. (A) GPR98 (magenta) is localized at the stereociliary base in most P4 Dfnb31wi/wi IHCs (a and b, front views) and OHCs (c, front view). GPR98 is sometimes present at both the stereociliary tip and base in P4 Dfnb31wi/wi OHC stereociliary bundles (d and e). (B) Usherin (magenta) distribution is normal in P4 Dfnb31wi/wi IHC (f and g) and OHC (h and i) stereociliary bundles. (C) PDZD7 (magenta) is localized normally at stereociliary bases in Dfnb31wi/wi IHCs (j and k) and OHCs (l and m) at P4. Arrows point to stereociliary bases. Green signals are from phalloidin staining. Magenta signals outside stereociliary bundles are non-specific. Scale bars, 1 µm.
USH2 proteins contribute differently to the function of the ALC in stereociliary bundle morphogenesis
To study the function of the ALC and contributions of each USH2 protein to this function, we performed scanning electron microscopy (SEM) to examine stereociliary bundle morphology in the middle turn of cochleas of various USH2 mutant mice, except Dfnb31wi/wi mice, which have already been well-characterized (34,35). To reveal the stereociliary bundle defects caused primarily, but not secondarily, by disruption of the ALC, P4 was chosen as the time point in this study. Additionally, our Adgrv1−/−, Ush2a−/−, Dfnb31neo/neo and wild-type control mice were on a similar genetic background (mixed C57BL6/129sv), allowing direct comparison of their phenotypes.
Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo OHC bundles all exhibited obvious morphological defects, while wild-type OHC bundles displayed a sharp V-shaped array of stereocilia organized in three rows of different lengths (Fig. 7A). Adgrv1−/− mice showed stereocilia tilted at their bases, blocking the view of shorter stereociliary rows in many OHC bundles, although these stereocilia still remained cohesive as a bundle with apparently normal length (Fig. 7A and Bf and g). The tilting of Adgrv1−/− OHC stereocilia was not due to an improper imaging angle, because IHCs in the same cochlear region showed standing stereocilia (Fig. 7Ab). Although we could not measure directly the stereociliary rigidity due to lack of appropriate experimental settings, this phenotype implied that the stereociliary rigidity was reduced at the stereociliary base in the absence of GPR98. This ‘fallen’ stereociliary phenotype has also been reported in P14 BUB/BnJ mice, which carry the same Adgrv1 mutant allele (26). Missing stereocilia in the shortest row of Adgrv1−/− OHC bundles were also observed on rare occasions when stereocilia did not tilt (Fig. 7Bh and i). In Ush2a−/− OHCs (Fig. 7Bj–l), distorted bundle shapes and mislocalized kinocilia were common, although stereocilia were connected along their entire length and usually did not tilt. Sometimes, only two rows of stereocilia were observed in the bundle (data not shown). These phenotypes were more severe than those of adult Ush2a−/− mice reported previously (28) (see next section for the possible explanation). Dfnb31neo/neo mice showed a less severe OHC bundle morphological phenotype, with various U-shapes (Fig. 7Bm–o). Considering that immature stereociliary bundles usually exhibit thin and short stereocilia with less obvious increases of stereociliary length between rows (33), the above observed morphological defects in USH2 mutant mice are likely not due to developmental delay.
Figure 7.

Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo mice exhibit various morphological defects in cochlear stereociliary bundles. (A) Low-magnification SEM images showing abnormal hair cell bundles in Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo mice at P4. (B) SEM images of individual Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo OHC bundles. Compared with the WT OHC bundle (e), Adgrv1−/− OHC bundles show stereocilia tilted at stereociliary bases (f and g) and missing stereocilia in the shortest stereociliary row (h and i, arrows); Ush2a−/− OHC bundles have distorted shapes (j–l) and mislocalized kinocilia (k and l, arrows); and Dfnb31neo/neo OHC bundles usually have a blunt U- rather than a sharp V-shape (m and o). (C) SEM images of individual Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo IHC bundles. Compared with WT IHCs (p), Adgrv1−/− IHCs have thick stereocilia (q and r) in the bundle and ectopic stereocilia at the neural edge outside the bundle (r, arrows); many Ush2a−/− IHCs have morphological defects similar to those observed in Adgrv1−/− IHCs (arrows, ectopic stereocilia, s and t) but with normal stereociliary thickness; Dfnb31neo/neo IHC bundles have thick stereocilia (u and v) and sometimes more than three rows of stereocilia (u). Scale bars, 5 µm (A) and 1 μm (B and C).
IHC stereociliary bundle phenotypes in USH2 mutant mice were less evident than those of OHCs at low magnification (Fig. 7A). At high magnification, Adgrv1−/− mice showed thick stereocilia in bundles and stereocilia-like microvilli at the neural (modiolar) edge of the IHC apex outside bundles (Fig. 7Cq and r), indicating abnormalities in stereociliary thickness and differentiation from microvilli. Ush2a−/− mice also displayed stereocilia-like microvilli at the neural side of the IHC apex (Fig. 7Cs and t). This ectopic stereociliary phenotype was not apparent in Dfnb31neo/neo mice (Fig. 7Cu and v); rather, Dfnb31neo/neo mice sometimes exhibited more than three rows of stereocilia within one bundle. To quantify the stereociliary thickness of Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo IHC bundles, stereocilia in the tallest row of bundles at the cochlear middle turn were measured and compared (Fig. 8). Adgrv1−/− and Dfnb31neo/neo mice had thicker IHC stereocilia than wild-type mice, and the Adgrv1−/− IHC stereocilia were the thickest. The thick stereocilia phenotype found in Dfnb31neo/neo mice is consistent with previous studies of P60 Dfnb31neo/neo mice and P20 Dfnb31wi/wi mice (32,35). Although SEM was not performed on Dfnb31wi/wi cochlear hair cells, phalloidin staining of Dfnb31wi/wi cochleas revealed occasionally ectopic stereocilia outside the IHC bundle (Figs 1C and 6Aa and Ck) and multiple stereociliary rows in one IHC bundle (Fig. 6Ab and Bf and g), in addition to the previously reported phenotype of widespread short stereocilia (34,36). Again, the IHC stereociliary bundle defects observed in USH2 mutant mice are likely not due to developmental delay, because these phenotypes have not been described in wild-type IHCs at an early development stage (37).
Figure 8.

Quantification of stereociliary thickness in Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo IHCs. Stereociliary thickness was measured using ImageJ on high-magnification SEM images as described in the Materials and Methods section. Student's t-tests were conducted to analyze the significance of differences among genotype groups. Numbers of cells and animals examined in each genotype group are indicated before and after the comma, respectively. *P < 0.05, **P < 0.01; error bars, standard error of the mean.
In summary, Adgrv1−/− and Dfnb31neo/neo mice show the most and least severe stereociliary bundle defects in cochlear hair cells, respectively, with Ush2a−/− mice having an intermediate phenotype. Dfnb31wi/wi mice were not included in the comparison, because their phenotypes probably result from lack of WHRN at both the ALC and stereociliary tip. Considering the role of GPR98 in ALC assembly as described previously in this study, the abnormal phenotypes in Adgrv1−/− mice are highly likely caused by complete disruption of the ALC. Therefore, the observed stereociliary defects in Adgrv1−/− mice reveal the comprehensive functions of the ALC, which are the regulation of stereociliary base rigidity and V-shaped three-row staircase arrangement in OHCs as well as regulation of stereociliary thickness and differentiation in IHCs.
Loss of individual USH2 proteins at the ALC results in different severities of hearing impairment
The different cochlear stereociliary morphological defects observed in USH2 mutant mice prompted us to evaluate the severity of hearing impairment in these mice by auditory brainstem response (ABR) tests. ABR tests usually give consistent and reliable results when the mouse cochlea reaches maturation. Therefore, we conducted ABR tests on Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo mice at P30. We found that all mutant mice had significant ABR threshold elevations in the frequency range of 4–45.2 kHz relative to wild-type control mice, with Adgrv1−/− mice being affected the most (Fig. 9). Adgrv1−/− response thresholds exceeded the measurement ceiling at all tested frequencies except 8.0 kHz, at which an increase of 61 dB sound pressure level (SPL) was shown. Dfnb31neo/neo mice had the mildest hearing loss with the largest threshold increase of about 42 dB SPL at frequencies of 11.322.6 kHz. Ush2a−/− mice had an intermediate degree of hearing loss similar to that of Pdzd7−/− mice (24). The gradient of hearing impairment in the three USH2 mutant mice correlated with the degrees of ALC disruption (Figs 2, 4, 5 and 6) and stereociliary bundle defects (Fig. 7) found in these mice.
Figure 9.
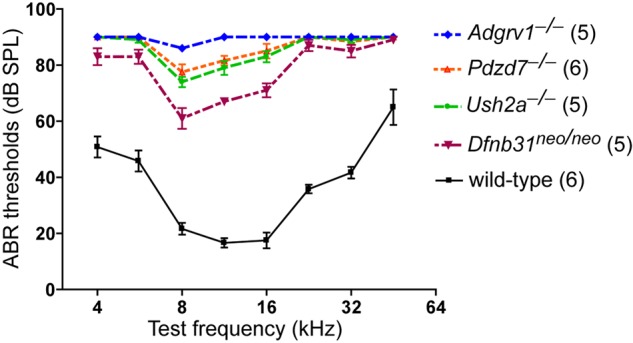
Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo mice have different degrees of hearing loss. Hearing function of 1-month-old Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo mice was assessed by ABR tests at frequencies of 4, 5.6, 8.0, 11.3, 16.0, 22.6, 32.0 and 45.2 kHz. Compared with wild-type controls, these mutant mice have significantly elevated ABR thresholds across all the frequencies tested (P < 0.01, 2-way ANOVA with post hoc tests). Threshold differences between Adgrv1−/− and Ush2a−/−, Adgrv1−/− and Dfnb31neo/neo, and Ush2a−/− and Dfnb31neo/neo mice are statistically significant (P < 0.01) at 5.6, 8.0, 11.3 and 16.0 kHz. Adgrv1−/− and Dfnb31neo/neo mice have the most and least severe hearing loss, respectively. For comparison, ABR results on Pdzd7−/− mice at P21–P28 are also shown here (24). Threshold differences between Ush2a−/− and Pdzd7−/− mice are not statistically significant at any tested frequency. Numbers in parentheses indicate numbers of animals tested. Error bars, standard error of the mean.
Interestingly, the hearing loss found in the Ush2a−/− mice was much more severe than what was previously reported in Ush2a−/− mice using distortion product otoacoustic emission (DPOAE) tests (28). Our Ush2a−/− mice and those used in the previous study carried exactly the same Ush2a mutant allele and were originally from the same colony, which was on a mixed genetic background of C57BL/6 and 129sv. The published Ush2a−/− mice was crossed with CBA/Caj mice for two generations to eliminate the Cdh23ahl allele. These Ush2a−/− mice showed moderate DPOAE threshold increases at frequencies higher than 20 kHz and normal DPOAE thresholds at frequencies lower than 20 kHz at 4 months of age (28). The phenotypical difference between our Ush2a−/− mice and those in the previous study could be due to the use of different hearing test techniques and/or different genetic backgrounds of the Ush2a−/− mice. Therefore, three male littermates from the original Ush2a−/− mouse colony generated in the previous study (28) were tested using both ABR and DPOAE at P30 (Fig. 10). We found that these three Ush2a−/− littermates had very large variations in their ABR and DPOAE thresholds at frequencies lower than 20 kHz, indicating that some modifier gene(s) may exist in the original Ush2a−/− mouse colony. Our Ush2a−/− mice had been crossed with our Dfnb31neo/neo mice for more than five generations and carried no Cdh23ahl allele as verified by PCR and deoxyribonucleic acid (DNA) sequencing. Further, no large ABR (Fig. 9) threshold variation was found in our Ush2a−/− mice. Therefore, the modifier genes in the original Ush2a−/− mouse colony, which could rescue the Ush2a−/− inner ear phenotypes, are highly likely to be eliminated in our Ush2a−/− mice.
Figure 10.

Original Ush2a−/− mouse colony has highly variable ABR and DPOAE responses. Three male littermates from the original Ush2a−/− colony were tested by ABR (left) and DPOAE (right) at 1 month of age. Responses from individual mice are shown. These mice have consistently high thresholds at high frequencies but variable thresholds at low frequencies in both ABR and DPOAE tests.
Discussion
This study is the first to systematically investigate the in vivo protein assembly and function of the ALC during cochlear stereociliary bundle development. We showed that USH2 proteins play distinct roles in ALC assembly by localizing the complex components, but are dispensable for complex component transport to the stereociliary bundle post-biosynthesis. GPR98 and usherin, but not whirlin, were found to be essential for ALC assembly. Our morphological study revealed that, unlike the roles of USH1 protein complexes in stereociliary bundle cohesion and orientation, the ALC and the ankle link itself are essential for stereociliary diameter and differentiation in IHCs and for stereociliary rigidity and V-shaped three-row organization in OHCs. The loss of individual USH2 gene expression led to different severities of cochlear stereociliary bundle defects and hearing loss, indicating a potential correlation between genotype and phenotype in USH2 patients.
As shown in Figure 11A, the three USH2 proteins participated differentially in the localization of ALC components in cochlear hair cells. GPR98 determined the localization of all other components (usherin, whirlin and PDZD7) at the ALC. Vezatin, a candidate ALC component, has also been shown to require GPR98 for its localization at the ALC (21). Thus, GPR98 appears to be indispensable for anchoring the ALC. Similarly, usherin was also crucial for retaining the ALC by controlling the normal localization of whirlin, PDZD7 and a fraction of GPR98. The partial role of usherin in GPR98 localization is probably due to lack of direct interaction between usherin and GPR98 (25) and the presence of relatively less usherin than GPR98 in the ALC, as suggested by a 20:1 molecular ratio of GPR98 to usherin reported in chicken vestibular stereociliary bundles (23). In contrast, whirlin only partially localized GPR98 and had no effect on usherin and PDZD7 localization. The trivial role of whirlin in recruiting other components to the ALC is probably compensated to some extent by its close homolog protein, PDZD7, which was shown to be involved in localizing GPR98, whirlin and a small fraction of usherin in cochlear hair cells (Fig. 11A) (24). The distinct contributions of USH2 proteins to the assembly of the hair cell ALC differ from their roles in assembly of the photoreceptor periciliary membrane complex, where each USH2 protein is responsible for recruiting the entire pools of other USH2 proteins for complex assembly and PDZD7 is dispensable (22,24).
Figure 11.

Roles of individual USH2 and PDZD7 proteins in the assembly and function of the ALC in cochlear hair cells. This summary includes data from Pdzd7−/− mice (24). (A) Interdependence of USH2 and PDZD7 proteins for their normal localization in cochlear stereociliary bundles. Proteins pointed by arrows are those whose localization is determined by the proteins on the other end of lines. Continuous and dashed lines indicate complete and partial dependence, respectively. (B) Hypothesis on the assembly of the ALC in hair cells. Arrows represent the recruitment directions of proteins. Dashed arrow, indirect recruitment. G, GPR98; U, usherin; P, PDZD7; and W, whirlin. (C and D) USH2 and PDZD7 protein distribution and stereociliary bundle morphological defects in various mutant IHCs (C) and OHCs (D). Dashed circles denote the normal or disrupted ALCALC. Symbol color matches the color of USH2 and PDZD7 proteins shown in (A). Straight line arrangement of symbols along stereociliary diagrams indicates even distribution of their represented proteins. Random distribution of symbols in the stereocilia indicates random punctate distribution of the represented proteins.
Our findings strongly support the notion that GPR98, usherin, whirlin and PDZD7 proteins assemble into the ALC in vivo. The assembly of this complex probably occurs in the stereociliary bundle, because the four component proteins of the complex appear to be transported to the stereociliary bundle without any assistance from each other. Interestingly, mislocalization patterns of ALC components in stereociliary bundles varied among the different complex components and genotypes. For example, different GPR98 and PDZD7 distributions were found in Ush2a−/− OHC bundles, and different GPR98 distributions were observed in Ush2a−/− and Dfnb31neo/neo OHC bundles. These results indicate that the ALC components are probably tethered at the stereociliary base by different mechanisms. Our earlier in vitro study showed that the four ALC components interact to form a dynamic quaternary protein complex when mixed together (25): GPR98 prefers to bind to PDZD7 rather than whirlin; usherin prefers to bind to whirlin rather than PDZD7; and GPR98 binds to usherin through the dimerization of PDZD7 and whirlin. Therefore, based on our previous and current studies, we propose that interlocking relationships between the four proteins occur during ALC assembly in cochlear hair cells (Fig. 11B). Upon being delivered at the stereociliary bundle from the cell body, GPR98 and PDZD7 are kept at the stereociliary base, probably through their direct interaction and associations with other local extra- and intracellular structures; some GPR98 is anchored to the stereociliary base independently of any USH2 proteins; usherin is maintained at the stereociliary base by PDZD7 as well as a so far unidentified mechanism that may indirectly depend on GPR98; both GPR98 and usherin recruit the two scaffold proteins, PDZD7 and whirlin, to their proximity with distinct preferences; and, finally, when the four USH proteins are all colocalized at the stereociliary base, they assemble into the ALC.
We believe that the cochlear stereociliary bundle defects and hearing impairment of USH2 mutant mice observed in this study result primarily from the disruption of the ALC, rather than the loss of individual USH2 protein functions before ALC formation or dysfunction of whirlin at the stereociliary tip. GPR98, usherin and whirlin protein expression in the cochlea starts at embryonic day (E) 17 (21), E18 (24) and E20 (31), respectively; however, the cochlear hair cell phenotypes of Adgrv1−/−, Ush2a−/−, and Dfnb31neo/neo mice are evident only at or after P2 when the ankle links emerge (20–22,26–29), suggesting that the three USH2 proteins have minor roles before their involvement in the ALC. Furthermore, the correlation between the severity of ALC disruption and stereociliary bundle defects in USH2 mutant mice at P4 (Fig. 11C and D) suggests that no other dysfunction of USH2 proteins except the ALC disruption contributes to the stereociliary bundle phenotypes observed in this study. Additionally, after the disappearance of ankle links in cochlear hair cells at P12, GPR98 (21,23) and whirlin (31) proteins are no longer present at the stereociliary base and thus probably function insignificantly, although whirlin at the stereociliary tip still functions in stereociliary elongation (31). In Dfnb31neo/neo mice, the function of whirlin at the ALC but not the stereociliary tip is affected (38). Therefore, we conclude that the hearing impairment observed in Adgrv1−/−, Ush2a−/− and Dfnb31neo/neo mice at P30 is caused mainly by ALC disruption.
The correlation of molecular and morphological phenotypes in USH2 mutant mice allows us to infer the functions of the ALC in vivo. Stereocilia of USH2 mutant bundles remain connected with each other without splitting into multiple tufts, suggesting that the ALC has a unique role from USH1 protein complexes in stereociliary bundle cohesion (39). Immunostaining for the GPR98 extracellular region, a major component of ankle links, showed that ankle links are missing in Adgrv1−/− but not in Dfnb31neo/neo or Dfnb31wi/wi cochlear hair cells, in accordance with previous SEM findings in two other Adgrv1 mutant (Adgrv1tm1Pwh and Adgrv1tm1Msat) (20,21) and Dfnb31wi/wi (23) mice. Our immunostaining study also revealed that a small amount of extracellular GPR98, presumably representing some ankle link fibers, exist in Ush2a−/− cochlear hair cells. Therefore, the ALC is important for establishing and maintaining ankle links in cochlear hair cells. In IHCs, Adgrv1−/− and Ush2a−/− but not Dfnb31neo/neo mice exhibited abnormal stereociliary differentiation with ectopic stereocilia outside the bundle on the neural side. Adgrv1−/− mice had the thickest IHC stereocilia. In OHCs, the bundles of Adgrv1−/−, Pdzd7−/− (24) and Ush2a−/− but not Dfnb31neo/neo mice were severely distorted in shape. Tilted stereocilia were frequently displayed in Adgrv1−/− and Pdzd7−/− (24) but not Ush2a−/− or Dfnb31neo/neo OHCs. Missing stereocilia were found in the shortest stereociliary row of Adgrv1−/− and Ush2a−/− OHC bundles (Pdzd7−/− were not examined). Therefore, these data suggest that the ALC participates in cochlear hair bundle morphogenesis, with slightly different roles in IHC and OHC bundles, i.e., regulation of stereociliary thickness and differentiation in IHCs versus stereociliary base rigidity and V-shaped three-row organization in OHCs. The regulation of stereociliary thickness by the ALC is probably mediated by the recently discovered interaction between whirlin and espin proteins (32). Further, the ALC probably plays an essential role in strengthening the stereociliary base before maturation of the stereociliary rootlet (at P14), a dense cytoskeleton bundle originating from the stereociliary base and extending into the cuticular plate, which provides a resilient mechanic support to stereocilia in mature hair cells (40,41). However, the molecular basis of ALC function during stereociliary bundle development needs to be further elucidated.
Correlated with the severities of ankle link/ALC disruption and cochlear stereociliary bundle defects, ABR tests revealed a gradient of hearing impairment in USH2 and Pdzd7 mutant mice, with Adgrv1−/− and Dfnb31neo/neo mice having the most and least severe impairment, respectively. This gradient of hearing impairment is consistent with previous findings from in vitro single-cell recordings that showed abnormal mechanotransduction responses in Adgrv1 and Pdzd7 knockout (20,21,24) but not Dfnb31wi/wi (42) cochlear hair cells. In support of this notion, ADGRV1 patients tend to have more severe hearing loss than USH2A patients (statistically insignificant due to small patient numbers examined) (43), and few USH2 patients have been found to carry DFNB31 mutations (44). USH2 genes are known to have multiple splice variants. Thus, specific mutations in the same USH2 genes may affect their alternatively-spliced isoforms differently and lead to variable symptoms. The molecular and morphological defects found in our USH2 mutant mice are shared by other USH2 mutant mice, such as another Adgrv1 mutant mouse line, Adgrv1tm1Msat (21,27). Further, our two Dfnb31 mutant mouse lines with different mutant alleles demonstrated consistent molecular defects in cochlear stereociliary bundles (Figs 5 and 6). Therefore, the mutations in our USH2 mutant mice are probably loss-of-function mutations at the ALC. Our study indicates a potential genotype–phenotype correlation in USH2 patients carrying loss-of-function mutations of USH2 genes. In summary, this study sheds novel light on the molecular mechanism underlying stereociliary bundle development and the disease mechanism underlying hearing loss in USH2.
Materials and Methods
Animals
Dfnb31 targeted mutant (Dfnb31neo/neo also known as Dfnb31tm1Tili), whirler (Dfnb31wi/wi), Ush2a knockout (Ush2a−/− also known as Ush2atm1Tili) and Pdzd7 knockout (Pdzd7−/−) mice have already been described (22,24,28,29). Adgrv1 mutant mice (Adgrv1−/− also known as Adgrv1frings) were obtained by crossing BUB/BnJ mice (Jax stock # 000653) and wild-type mice with a mixed genetic background of C57BL/6 and 129sv to eliminate the Pderd1 mutation. Both Ush2a−/− and Adgrv1−/− mice were crossed with Dfnb31neo/neo mice for approximately five generations and thus their genetic backgrounds are close to each other, which are a mixture of C57BL/6 and 129sv. Ush2a−/− mice are free of the Cdh23ahl allele, determined by PCR amplification and DNA sequencing of the allele region. All experiments involving animals were approved by the Institutional Animal Care and Use Committee at the University of Utah. For terminal experiments, mice were euthanized by CO2 inhalation, consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association.
Antibodies
Recombinant polyhistidine (His)-tagged usherin (5053–5193 amino acids, NP_067383), GPR98 (1212–2211 amino acids, NP_473394) and whirlin (375–800 amino acids, NP_082916) protein fragments were expressed in BL21-CodonPlus (DE3)-arginine, -isoleucine, -proline, -leucine cells (Agilent Technologies, Santa Clara, CA, USA) using their pET28 constructs, and purified by chromatography using Ni2+-charged His•Bind resin columns (EMD Millipore, Billerica, MA, USA). The obtained recombinant proteins were used to immunize rabbits. Antibodies (except the whirlin antibody) were purified by flowing the sera of immunized rabbits through a column cross-linked with a His-tag to clear antibodies against His-tag, and a second column cross-linked with the cognate antigen. The antibody directed against both whirlin long and short isoforms was affinity-purified against a glutathione S-transferase-tagged whirlin fragment (721–907 amino acids, NP_082916). Specificity of each antibody was confirmed by lack of immunoreactivity in corresponding mutant cochlear hair cells (Fig. 1). The antibody against the GPR98 C-terminal region was described previously (22). Alexa fluorochrome-conjugated phalloidin and secondary antibodies were purchased from Life Technologies (Carlsbad, CA, USA).
Immunofluorescence and subtilisin treatment
Mouse cochleas at P4–P8 were dissected, fixed in 4% formaldehyde/PBS from 30 min to overnight (depending on the antibody to be used) and permeabilized by 0.5% Triton X-100/PBS for 15–20 min. For usherin and whirlin antibodies, subsequent incubations of cochleas in 50 mm NH4Cl for 15 min and in tween Tris-buffered saline (TTBS) (20 mm Tris pH 7.5, 150 mm NaCl, 0.1% Tween 20) for 10 min were performed. Whole-mount cochleas were then blocked in 5% goat serum/PBS for 1 h, incubated with primary antibodies in 5% goat serum/PBS at 1:2000 at 4°C overnight, washed several times with PBS, and then incubated with Alexa Fluor® 594-conjugated secondary antibodies and Alexa Fluor® 488-conjugated phalloidin in 5% goat serum/PBS for 1–2 h. The cochleas were then viewed and photographed using a confocal laser scanning microscope (Olympus Fluoview 1000, Tokyo, Japan). For each antibody and genotype, more than three experiments were performed by two independent researchers using pups from at least three litters.
Subtilisin treatment was performed twice by incubating exposed stereociliary bundles of wild-type mouse cochleas in PBS containing 50 μg/ml subtilisin (protease type XXIV, Sigma-Aldrich) for 15 min at room temperature. Incubation in PBS under the same condition was conducted simultaneously as a negative control. After subtilisin treatment, the above immunofluorescence procedure was followed to localize USH2 and PDZD7 proteins.
SEM and stereociliary thickness measurement
Mouse cochleas at P4 were isolated, fixed in 4% formaldehyde/PBS for 1 h, dissected to remove the tectorial membrane and further fixed in 2.5% glutaraldehyde/PBS overnight. Cochleas were then rinsed in PBS and post-fixed by alternative incubations (alternative treatments of osmium tetroxide and thiocarbohydrizide (OTOTO)) in 1% osmium tetroxide (O) for 1 h, water 1 min six times and 0.3% thiocarbohydrizide (T) for 20 min. The cochlear tissues were then washed in water 15 min twice, dehydrated by passing through a graded ethanol series (30, 50, 70, 90, 95 and 100% for 5 min each), dried by incubation with hexamethyldisilazane (50% in 100% ethanol for 5 min and 100% for 5 min twice) and kept in a desiccator overnight. Images were taken on a Hitachi S-4800 scanning electron microscope. For each genotype, several pups per litter were examined from at least three litters.
Stereociliary thickness was measured on high-magnification SEM images (∼11 000×) taken from the cochlear middle turn using ImageJ (NIH). For each IHC, two stereocilia with a clear top view in the tallest row of stereociliary bundles were measured, and the average of the two measurements was taken to represent the stereociliary thickness of the cell. In the case that the top profile of stereocilia was an oval-like shape, the largest diameter was measured. Measurements were conducted from IHCs of more than four pups for each genotype (Fig. 8). Data were normalized using wild-type cochleas in the same sample processing batches. Student's t-tests were performed to analyze the significance of differences among different genotype groups.
ABR and DPOAE tests
ABR and DPOAE tests were conducted in a double-walled sound chamber (IAC) as described previously (24). Briefly, mice were anesthetized with a combination drug of ketamine (100 mg/kg) and xylazine (10 mg/kg) administered by intraperitoneal injection. For ABR tests, an electrostatic speaker (EC-1, Tucker-Davis Technology, Alachua, FL, USA) fitted with a 1.5-cm long polyethylene tube was placed abutting the ear canal. Needle electrodes were placed subcutaneously at the mastoid of the tested side and vertex, with a remote ground placed in the rump area. ABR signals were amplified with a TDT RA4 pre-amplifier, bandpass (100–3000 Hz) filtered, digitized and averaged with a RA16BA processor controlled by TDT BioSigRP software. Stimuli were generated digitally in SigGenRP, processed by a RX6 real-time processor and passed through a PA5 attenuator prior to delivery to the speaker. Responses to 1024 sweeps were averaged for a series of responses to tone pips ranging from 4 to 45.2 kHz (5 ms with 0.5 ms cos2 rise and fall) using 5 or 10 dB intensity steps, over a 15–90 dB SPL range. ABR thresholds were determined as the lowest intensity at which the response was clearly discernible. DPOAEs were measured using digitally generated stimuli of two primary tones f1 and f2 (f2/f1 = 1.2) with f2 = f1 − 10 dB. Primary tones (f1) were stepped from 30 to 80 dB SPL in 10 dB increments and swept from 8 to 32 kHz in ½ octave steps. The ear canal sound pressure was preamplified and digitized. A fast Fourier transformation was computed. The sound pressures at f1, f2 and 2f1 − f2 were extracted after spectral averaging from 50 serial waveform traces. Student's t-tests and two-way analysis of variance (ANOVA) post hoc tests were performed to analyze the significance of differences among genotypes at various sound frequencies.
Funding
This work was supported by the National Institutes of Health (EY020853 to J.Y., EY014800 to the Department of Ophthalmology &Visual Sciences, University of Utah); Foundation Fighting Blindness (to J.Y.); E. Matilda Ziegler Foundation for the Blind, Inc. (to J.Y.); Research to Prevent Blindness, Inc. (to J.Y. and the Department of Ophthalmology & Visual Sciences, University of Utah); Hearing Health Foundation (to J.Z.); National Organization for Hearing Research Foundation (to J.Z.); and a startup package from the Moran Eye Center, University of Utah (to J.Y.).
Acknowledgements
The authors thank Dr Tiansen Li (National Eye Institute) for shipping mice from the original Ush2a−/− mouse colony and Dr Jeanne M. Frederick (University of Utah) for critical reading of our manuscript. The authors also thank Drs Qing Yin Zheng (Case Western Reserve University) and Jeremy S. Duncan (University of Utah) for helping us to establish mouse inner ear immunofluorescence and SEM protocols.
Conflict of Interest statement. None declared.
References
- 1.Assad J.A., Shepherd G.M., Corey D.P. (1991) Tip-link integrity and mechanical transduction in vertebrate hair cells. Neuron, 7, 985–994. [DOI] [PubMed] [Google Scholar]
- 2.Hudspeth A.J. (1985) Models for mechanoelectrical transduction by hair cells. Prog. Clin. Biol. Res., 176, 193–205. [PubMed] [Google Scholar]
- 3.Fettiplace R., Kim K.X. (2014) The physiology of mechanoelectrical transduction channels in hearing. Physiol. Rev., 94, 951–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frolenkov G.I., Belyantseva I.A., Friedman T.B., Griffith A.J. (2004) Genetic insights into the morphogenesis of inner ear hair cells. Nat. Rev. Genet., 5, 489–498. [DOI] [PubMed] [Google Scholar]
- 5.Nayak G.D., Ratnayaka H.S., Goodyear R.J., Richardson G.P. (2007) Development of the hair bundle and mechanotransduction. Int. J. Dev. Biol., 51, 597–608. [DOI] [PubMed] [Google Scholar]
- 6.Brown S.D., Hardisty-Hughes R.E., Mburu P. (2008) Quiet as a mouse: dissecting the molecular and genetic basis of hearing. Nat. Rev. Genet., 9, 277–290. [DOI] [PubMed] [Google Scholar]
- 7.Richardson G.P., de Monvel J.B., Petit C. (2011) How the genetics of deafness illuminates auditory physiology. Annu. Rev. Physiol., 73, 311–334. [DOI] [PubMed] [Google Scholar]
- 8.Petit C., Richardson G.P. (2009) Linking genes underlying deafness to hair-bundle development and function. Nat. Neurosci., 12, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodyear R.J., Marcotti W., Kros C.J., Richardson G.P. (2005) Development and properties of stereociliary link types in hair cells of the mouse cochlea. J. Comp. Neurol., 485, 75–85. [DOI] [PubMed] [Google Scholar]
- 10.Bonnet C., El-Amraoui A. (2012) Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr. Opin. Neurol., 25, 42–49. [DOI] [PubMed] [Google Scholar]
- 11.Cosgrove D., Zallocchi M. (2013) Usher protein functions in hair cells and photoreceptors. Int. J. Biochem. Cell Biol., 46, 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathur P., Yang J. (2015) Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. BBA Mol. Basis Dis., 1852, 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boughman J.A., Vernon M., Shaver K.A. (1983) Usher syndrome: definition and estimate of prevalence from two high-risk populations. J. Chronic Dis., 36, 595–603. [DOI] [PubMed] [Google Scholar]
- 14.Hartong D.T., Berson E.L., Dryja T.P. (2006) Retinitis pigmentosa. Lancet, 368, 1795–1809. [DOI] [PubMed] [Google Scholar]
- 15.Keats B.J., Corey D.P. (1999) The usher syndromes. Am. J. Med. Genet., 89, 158–166. [PubMed] [Google Scholar]
- 16.Weston M.D., Luijendijk M.W., Humphrey K.D., Moller C., Kimberling W.J. (2004) Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet., 74, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eudy J.D., Weston M.D., Yao S., Hoover D.M., Rehm H.L., Ma-Edmonds M., Yan D., Ahmad I., Cheng J.J., Ayuso C. et al. (1998) Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science, 280, 1753–1757. [DOI] [PubMed] [Google Scholar]
- 18.Ebermann I., Scholl H.P., Charbel Issa P., Becirovic E., Lamprecht J., Jurklies B., Millan J.M., Aller E., Mitter D., Bolz H. (2007) A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum. Genet., 121, 203–211. [DOI] [PubMed] [Google Scholar]
- 19.Ebermann I., Phillips J.B., Liebau M.C., Koenekoop R.K., Schermer B., Lopez I., Schafer E., Roux A.F., Dafinger C., Bernd A. et al. (2010) PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Invest., 120, 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGee J., Goodyear R.J., McMillan D.R., Stauffer E.A., Holt J.R., Locke K.G., Birch D.G., Legan P.K., White P.C., Walsh E.J. et al. (2006) The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci., 26, 6543–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michalski N., Michel V., Bahloul A., Lefevre G., Barral J., Yagi H., Chardenoux S., Weil D., Martin P., Hardelin J.P. et al. (2007) Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci., 27, 6478–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J., Liu X., Zhao Y., Adamian M., Pawlyk B., Sun X., McMillan D.R., Liberman M.C., Li T. (2010) Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet., 6, e1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grati M., Shin J.B., Weston M.D., Green J., Bhat M.A., Gillespie P.G., Kachar B. (2012) Localization of PDZD7 to the stereocilia ankle-link associates this scaffolding protein with the Usher syndrome protein network. J. Neurosci., 32, 14288–14293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou J., Zheng T., Ren C., Askew C., Liu X.P., Pan B., Holt J.R., Wang Y., Yang J. (2014) Deletion of PDZD7 disrupts the Usher syndrome type 2 protein complex in cochlear hair cells and causes hearing loss in mice. Hum. Mol. Genet., 23, 2374–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Q., Zou J., Shen Z., Zhang W., Yang J. (2014) Whirlin and PDZ domain containing 7 (PDZD7) proteins are both required to form the quaternary protein complex associated with usher syndrome type 2. J. Biol. Chem., 289, 36070–36088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson K.R., Zheng Q.Y., Weston M.D., Ptacek L.J., Noben-Trauth K. (2005) The Mass1frings mutation underlies early onset hearing impairment in BUB/BnJ mice, a model for the auditory pathology of Usher syndrome IIC. Genomics, 85, 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yagi H., Tokano H., Maeda M., Takabayashi T., Nagano T., Kiyama H., Fujieda S., Kitamura K., Sato M. (2007) Vlgr1 is required for proper stereocilia maturation of cochlear hair cells. Genes Cells, 12, 235–250. [DOI] [PubMed] [Google Scholar]
- 28.Liu X., Bulgakov O.V., Darrow K.N., Pawlyk B., Adamian M., Liberman M.C., Li T. (2007) Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. U. S. A., 104, 4413–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mburu P., Mustapha M., Varela A., Weil D., El-Amraoui A., Holme R.H., Rump A., Hardisty R.E., Blanchard S., Coimbra R.S. et al. (2003) Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat. Genet., 34, 421–428. [DOI] [PubMed] [Google Scholar]
- 30.Fleming J., Rogers M.J., Brown S.D., Steel K.P. (1994) Linkage analysis of the whirler deafness gene on mouse chromosome 4. Genomics, 21, 42–48. [DOI] [PubMed] [Google Scholar]
- 31.Delprat B., Michel V., Goodyear R., Yamasaki Y., Michalski N., El-Amraoui A., Perfettini I., Legrain P., Richardson G., Hardelin J.P. et al. (2005) Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum. Mol. Genet., 14, 401–410. [DOI] [PubMed] [Google Scholar]
- 32.Wang L., Zou J., Shen Z., Song E., Yang J. (2012) Whirlin interacts with espin and modulates its actin-regulatory function: an insight into the mechanism of Usher syndrome type II. Hum. Mol. Genet., 21, 692–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waguespack J., Salles F.T., Kachar B., Ricci A.J. (2007) Stepwise morphological and functional maturation of mechanotransduction in rat outer hair cells. J. Neurosci., 27, 13890–13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holme R.H., Kiernan B.W., Brown S.D., Steel K.P. (2002) Elongation of hair cell stereocilia is defective in the mouse mutant whirler. J. Comp. Neurol., 450, 94–102. [DOI] [PubMed] [Google Scholar]
- 35.Mogensen M.M., Rzadzinska A., Steel K.P. (2007) The deaf mouse mutant whirler suggests a role for whirlin in actin filament dynamics and stereocilia development. Cell Motil. Cytoskeleton, 64, 496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belyantseva I.A., Boger E.T., Naz S., Frolenkov G.I., Sellers J.R., Ahmed Z.M., Griffith A.J., Friedman T.B. (2005) Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol., 7, 148–156. [DOI] [PubMed] [Google Scholar]
- 37.Chen J., Johnson S.L., Lewis M.A., Hilton J.M., Huma A., Marcotti W., Steel K.P. (2014) A reduction in Ptprq associated with specific features of the deafness phenotype of the miR-96 mutant mouse diminuendo. Eur. J. Neurosci., 39, 744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathur P., Zou J., Zheng T., Almishaal A., Wang Y., Chen Q., Wang L., Vashist D., Brown S., Park A. et al. (2015) Distinct expression and function of whirlin isoforms in the inner ear and retina: an insight into pathogenesis of USH2D and DFNB31. Hum. Mol. Genet., 24, 6213–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lefevre G., Michel V., Weil D., Lepelletier L., Bizard E., Wolfrum U., Hardelin J.P., Petit C. (2008) A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development, 135, 1427–1437. [DOI] [PubMed] [Google Scholar]
- 40.Kitajiri S., Sakamoto T., Belyantseva I.A., Goodyear R.J., Stepanyan R., Fujiwara I., Bird J.E., Riazuddin S., Riazuddin S., Ahmed Z.M. et al. (2010) Actin-bundling protein TRIOBP forms resilient rootlets of hair cell stereocilia essential for hearing. Cell, 141, 786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furness D.N., Mahendrasingam S., Ohashi M., Fettiplace R., Hackney C.M. (2008) The dimensions and composition of stereociliary rootlets in mammalian cochlear hair cells: comparison between high- and low-frequency cells and evidence for a connection to the lateral membrane. J. Neurosci., 28, 6342–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stepanyan R., Belyantseva I.A., Griffith A.J., Friedman T.B., Frolenkov G.I. (2006) Auditory mechanotransduction in the absence of functional myosin-XVa. J. Physiol., 576, 801–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abadie C., Blanchet C., Baux D., Larrieu L., Besnard T., Ravel P., Biboulet R., Hamel C., Malcolm S., Mondain M. et al. (2012) Audiological findings in 100 USH2 patients. Clin. Genet., 82, 433–438. [DOI] [PubMed] [Google Scholar]
- 44.Le Quesne Stabej P., Saihan Z., Rangesh N., Steele-Stallard H.B., Ambrose J., Coffey A., Emmerson J., Haralambous E., Hughes Y., Steel K.P. et al. (2012) Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet., 49, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]