

Abstract
Mutations in subunits or regulators of cohesin cause a spectrum of disorders in humans known as the ‘cohesinopathies’. Cohesinopathies, including the best known example Cornelia de Lange syndrome (CdLS), are characterized by broad spectrum, multifactorial developmental anomalies. Heart defects occur at high frequency and can reach up to 30% in CdLS. The mechanisms by which heart defects occur are enigmatic, but assumed to be developmental in origin. In this study, we depleted cohesin subunit Rad21 by 70–80% in a zebrafish cohesinopathy model. The hearts of Rad21-depleted animals were smaller, often failed to loop, and functioned less efficiently than size-matched controls. Functional deficiency was accompanied by valve defects and reduced ejection fraction. Interestingly, neural crest cells failed to populate the heart and instead exhibited a wandering behavior. Consequently, these cells also failed to condense correctly into pharyngeal arches. Transcriptome analysis revealed that Wnt pathway, chemokine and cadherin genes are dysregulated at the time of cardiac neural crest development. Our results give insight into the etiology of heart defects in the cohesinopathies, and raise the possibility that mild mutations in cohesin genes may be causative of a fraction of congenital heart disease in human populations.
Introduction
Mutations in subunits of the cohesin complex, or regulators of cohesin activity, are associated with a broad spectrum of human disorders known as the ‘cohesinopathies’ (1–6), the best known of which is Cornelia de Lange syndrome (CdLS; MIM #122470, #300590, #610759 and #614701). CdLS is a complex, multisystem developmental syndrome affecting between 1:10 000 and 1:30 000 live births. CdLS is characterized by growth retardation, developmental delay, microcephaly, facial dysmorphism, cognitive impairment, hirsutism and upper limb defects ranging from small hand size to severe limb differences. Developmental problems in CdLS also include gastrointestinal, musculoskeletal and cardiac, as well as hearing loss and urogenital abnormalities (4,7,8).
Most cases of CdLS (up to 80%) are caused by heterozygous mutations in the NIPBL protein that loads cohesin onto DNA (9–11). However, a smaller fraction of atypical cases falling within the CdLS spectrum are caused by mutations in cohesin subunits SMC1A (MIM #300040) (12,13), SMC3 (MIM #606062) (13) and RAD21 (MIM #606462) (14), and the SMC3 lysine deacetylase, HDAC8 (MIM #300269) (15,16). The cohesin complex has essential roles in the cell cycle (17) and in DNA damage repair (18). Therefore, gene mutations that characterize the cohesinopathies involve reduced function of the encoded protein rather than a complete loss of function, which would be incompatible with life. The current view is that reduced function of cohesin/NIPBL alters the transcription of developmental genes, resulting in cohesinopathy phenotypes (1,2,19,20). However, dysregulation of growth pathways and ribosome biogenesis may also be contributory (21).
Congenital heart disease (CHD) is very common in CdLS and related cohesinopathies, affecting from 30% (22) up to 70% (23) of individuals, and leading to significant morbidity and mortality when present (23). The most common abnormalities include (in descending order) ventricular septal defects, atrial septal defect, pulmonic stenosis, tetralogy of Fallot and hypoplastic left heart syndrome (23,24). CHD occurs at more or less the same frequency in CdLS individuals who have cohesin subunit mutations as well as those with NIPBL mutations (23). Of the nine human probands with RAD21 mutations, two have congenital heart defects, one with tetralogy of Fallot (14,24). CHD was also found frequently in SMC3 and SMC1A patients, in whom the anomalies were usually milder than for NIPBL patients (23). Although CHD is prevalent in cohesinopathies, the causes are poorly understood and may be multifactorial.
Congenital heart defects are recapitulated in animal models of CdLS (25–27). In a zebrafish model, reduced Nipbl function caused heart, gut and laterality defects via dysregulated expression of endodermal differentiation and left–right patterning genes (26). Expression of sox32 and foxa2 in Nipbl-deficient embryos rescued some but not all types of cardiac defect, implying that not all heart defects were endoderm-derived (26). Another study showed that stimulation of cell growth via the mTOR pathway rescues growth and some developmental defects in cohesin-deficient zebrafish, but not heart defects (21), implying that cardiac defects caused by cohesin deficiency are not due to a paucity of cells alone. These findings prompted us to seek additional causes for heart anomalies upon cohesin gene deficiency.
Here, we used Rad21-depleted zebrafish to expose developmental phenotypes that result from partial cohesin deficiency. Complete loss of Rad21 function leads to early embryo lethality, so we used low doses of morpholino oligonucleotide (MO) to deplete Rad21 protein by up to 80%. Surprisingly, zebrafish partially depleted of Rad21 developed almost normally, except for the presence of structural abnormalities of the heart. We found that heart defects were at least in part caused by the failure of cardiac neural crest (CNC) cells to reach the heart and were accompanied by dysregulation of genes involved in neural crest development (28,29). Our results are consistent with other studies in which organs and systems derived from neural crest are dysfunctional upon mutation of cohesinopathy genes (27,30). Our results also raise the possibility that CHD of unknown etiology could involve sub-threshold expression or function of cohesinopathy genes not previously appreciated in the general population.
Results
Moderate depletion of Rad21 as a model for human cohesinopathies
In previous research, we demonstrated that zebrafish embryos homozygous mutant for a null allele of rad21 (rad21nz171) are characterized by dysregulated expression of genes involved in development and cell proliferation (31,32). rad21nz171 zygotic mutant embryos are initially indistinguishable from wild-type, but display a delay in development from 22 h post-fertilization (hpf) onwards and do not survive beyond 72 hpf, precluding analysis of later development (31). At 2 days post-fertilization (dpf), homozygous rad21nz171 mutants have a beating, yet tiny heart that is not looped and unable to pump blood. In contrast, heterozygous rad21nz171 mutant zebrafish develop relatively normally (but have a low incidence of heart defects, as described in this study), despite having a ∼40% reduction in rad21 transcript (32).
In CdLS, the amount of functional protein present is only partially compromised, as complete loss of function would disrupt cell division and lead to non-viability. Thus, cohesinopathies result from haploinsufficiency/impaired function of cohesin genes. For example, heterozygous NIPBL mutation results in only 15–30% protein reduction due to up-regulation of the other allele (25), while homozygous loss of ESCO2 (33) is sometimes survivable, and is likely to be compensated by other mechanisms in survivors (34). Human RAD21 mutations include missense point mutations and loss of one allele, while homozygous mutations have not been reported (14,24). This implies that reduced or altered function of RAD21 might be tolerated, while complete loss of function is not. We reasoned that if we reduced the amount of functional Rad21 below a threshold level, we might be able to expose the etiology of specific developmental phenotypes that result from insufficiency of cohesin.
We have thoroughly characterized antisense MOs targeting rad21 and determined that their activity accurately recapitulates the mutant phenotype (31,32,35). To create a zebrafish model that approximates a human cohesinopathy, we used very low doses of the previously characterized MO targeting the rad21 ATG. This established a partial rad21 knock down condition that led to 70–80% depletion of Rad21 protein (Supplementary Material, Fig. S1). The degree of Rad21 depletion was mild enough to allow embryos to grow up to the larval stage and to feed normally. We found that 0.035 pmol of MO led to 60% survival, and 0.07 pmol allowed 20% survival at 12 dpf when grown at 25°C (Fig. 1A).
Figure 1.

Partial depletion of Rad21 produces heart defects in zebrafish larvae that otherwise appear normal. Zebrafish embryos were injected at the one-cell stage with 1 nl phenol red dye, or with various quantities of Rad21 ATG MO, then raised at 25°C. (A) Percentage of Rad21-depleted larvae surviving to swim and eat normally at 12 dpf (n > 20 for each condition). (B) Larval size at 12 dpf. Rad21-depleted larvae were significantly smaller than control embryos (P < 0.05, unpaired t-test). n = 6 for each condition, error bars are ±SEM. (C) Rad21-depleted larva (below) and control (top) at 12 dpf. Dorsal view, anterior to the right. Apart from their slightly smaller size, Rad21-depleted larvae appeared externally normal. Scale bar = 100 µm. (D) Percentage of larvae displaying a heart defect and reduced circulation at 12 dpf. Eighty-three percent of Rad21-depleted larvae (n = 10/12) displayed this phenotype. Heart defects were never observed in control larvae (n = 20/20). (E) Heartbeats per minute (bpm) at 12 dpf. Heartbeat did not differ between Rad21-depleted larvae and controls (P > 0.05, unpaired t-test). n = 9 for each condition, error bars are ±SEM. (F and G) Heart morphology at 12 dpf, visualized using a double-transgenic Tg(gata1:dsred), Tg(β-actin:GFP) zebrafish line. Black arrows indicate the blood-filled atrium, white arrows indicate the ventricle. Ventral views, anterior to the left. Scale bars are 100 µm. In control larvae (F), the orientation of the atrium and ventricle were at right angles, whereas in Rad21-depleted larvae (G), the orientation of the atrium and ventricle were more aligned (see Supplementary Material, Video S1).
Growth retardation is a common feature of the cohesinopathies. We compared the size of 10 Rad21-depleted (0.035 pmol rad21 MO) and 10 control embryos at 12 dpf and found that growth in the Rad21-depleted embryos is slightly but significantly reduced relative to controls (Fig. 1B and C; 4.7 versus 4.2 mm). Other than their smaller size, Rad21-depleted embryos and larvae developed relatively normally, except that most had prominent heart defects (Fig. 1D; F compared with G). Heart defects were partially rescued when Rad21 MO was injected into the p53 mutant background, tp53zdf1 (36) (Supplementary Material, Fig. S2), indicating enhancing cell survival can compensate for Rad21 deficiency. We hypothesized that partial Rad21-deficient zebrafish represent a good model to better understand the etiology of heart defects in cohesinopathy patients (14,24).
We next reasoned that heterozygous embryos from a rad21nz171 incross might be sensitized for heart defects by an even lower dose of the MO (0.02 pmol), well below the threshold for inducing heart defects in wild-type animals. Indeed, 65% of heterozygous, MO-injected embryos from rad21nz171 incrosses presented with heart defects. Moreover, ∼30% of their homozygous wild-type sibs also exhibited heart defects (Supplementary Material, Fig. S3). These results suggest both decreased zygotic expression and a decreased maternal contribution (since heterozygous mothers have lower maternal contribution of Rad21) sensitize embryos to low-dose MO treatment. Importantly, these results argue against the likelihood of off-target effects of the MO, since the same amount of MO does not interfere with heart development in embryos from wild-type parents (Supplementary Material, Fig. S3).
On close inspection, a proportion of uninjected control embryos from rad21nz171 incrosses exhibited transient heart defects that later resolved (Supplementary Material, Fig. S3). These data indicate that even a slight depletion of Rad21, independent of MO treatment, predisposes embryos to abnormal heart development. This result is significant because it raises the possibility that ideopathic embryonic heart defects in humans may arise from partial loss-of-function mutations in cohesin genes.
Rad21 deficiency leads to failure of heart looping in zebrafish embryos
To visualize the heart defects in Rad21-depleted zebrafish larvae, we used double transgenic zebrafish containing Tg(bactin2:GFP)zp5 (37), which labels the myocardium, and Tg(gata1:dsred) (38), which labels erythrocytes. Heart morphology and function was analyzed at 12 dpf following injection of 0.035 pmol rad21 MO at the one-cell stage. Our analysis revealed that 83% (10/12) Rad21-depleted larvae had malformed hearts and reduced circulation (Fig. 1D, Supplementary Material, Video S1). However, the heart rate was unchanged between Rad21-depleted larvae and controls at this developmental stage (Fig. 1E, Supplementary Material, Video S1). To examine the developmental origin of heart malformation in Rad21-depleted embryos, we observed hearts at 2 and 3 dpf, the time when heart looping takes place in zebrafish. To allow for the developmental delay in Rad21-depleted embryos, we grew MO-injected embryos at 28°C and control embryos at 25°C. Under these conditions, both groups reached the same developmental stage at 2 and 3 dpf as confirmed by equivalent embryo size (Fig. 2A–D and J).
Figure 2.

Cardiac phenotypes in Rad21-depleted embryos. Zebrafish embryos were injected at the one-cell stage with 1 nl phenol red dye, or with 0.035 pmol Rad21 ATG MO, then raised as described below. (A and B) Control embryos and (C and D) Rad21-depleted embryos at 2 and 3 dpf. Rad21-depleted embryos incubated at 28.5°C were equivalent in size to controls incubated at 25°C. Apart from cardiac defects, Rad21-depleted embryos displayed no obvious malformations. (E–H) Heart morphology at 2 and 3 dpf, visualized using a double-transgenic Tg(gata1:dsred), Tg(β-actin:GFP) zebrafish line, in which erythrocytes are labeled red, and myocardium is light blue. Scale bar is 50 µm. (F and H) At both 2 and 3 dpf, the hearts of Rad21-depleted embryos failed to loop, and blood cells accumulated under the heart. n > 12 for each condition. (I) Heart rate in beats per minute (bpm) at 2 and 3 dpf. Heart rate was significantly reduced in Rad21-depleted embryos at both 2 and 3 dpf (**P < 0.01 and ****P < 0.0001, respectively, unpaired t-test). Error bars are ±SEM. (J) Embryo size at 2 and 3 dpf. There was no difference in size at either time point between Rad21-depleted and control embryos, when controls were grown at 25°C and Rad21-depleted embryos at 28°C (P > 0.05, unpaired t-test). Three days post-fertilization embryos were significantly larger than 2 dpf (****P < 0.0001, two-way unpaired ANOVA). Error bars are ±SEM. (K) Phenotypic abnormalities observed at 2 and 3 dpf. Rad21-depleted embryos had reduced blood flow and unlooped hearts at 2 and 3 dpf, as well as cardiac edema at 3 dpf (see also Supplementary Material, Video S2).
We observed that the heart failed to loop correctly in 95% (19/20) of Rad21-depleted embryos at 2 and 3 dpf. In contrast, the heart looped correctly in 100% (20/20) of dye-injected control embryos, all of which had established normal circulation at 2 and 3 dpf (Fig. 2E–H and K; Supplementary Material, Video S2). The heart rate was significantly reduced in Rad21-depleted animals compared with controls at this stage (Fig. 2I). Only 10% of Rad21-depleted animals (2/20) had circulation at 2 dpf, but by 3 dpf, circulation had been established in 55% (11/20) (Fig. 2K). This implies that the onset of circulation was delayed in a proportion of Rad21-depleted embryos. Cardiac edema was present in 35% of Rad21-depleted embryos (6/20) compared with 5% (1/20) of controls at 3 dpf (Fig. 2K).
Rad21 deficiency leads to heart valve defects in zebrafish embryos
Defects in heart valve development are very common in cohesinopathy patients (23). To determine whether reduced Rad21 function leads to abnormalities in zebrafish valve development, we used a transgenic zebrafish line, Tg(5xERE:EGFP262), which expresses GFP in the developing heart valves in response to estrogen (39). This line enabled visualization of the developing atrioventricular valve (AV) and the ventriculobulbar valve (VB) at 5 dpf in Rad21-depleted and control larvae. Valve morphology, orientation and function were normal in 10 out of 10 control embryos (Fig. 3A and E, and Supplementary Material, Video S3). In contrast, three out of eight Rad21-depleted larvae did not develop a VB valve, and had a non-functional AV valve that failed to close properly (Fig. 3B and E, and Supplementary Material, Video S3). The remaining five out of eight Rad21-depleted larvae had developed both valves, but they appeared notably smaller than control valves (Fig. 3C and D and Supplementary Material, Video S3). The orientation of the AV/VB valve openings were vertically aligned in Rad21-depleted embryos, as opposed to at right angles as observed for controls. We visualized the ejection fraction, the volumetric fraction of blood pumped out of the ventricle, using Tg(gata1:dsred) Tg(5xERE:EGFP262) fish. The apparent ejection fraction of one contraction was much lower in Rad21-depleted embryos compared with control larvae at 5 dpf (Fig. 3C and D, and Supplementary Material, Video S3).
Figure 3.
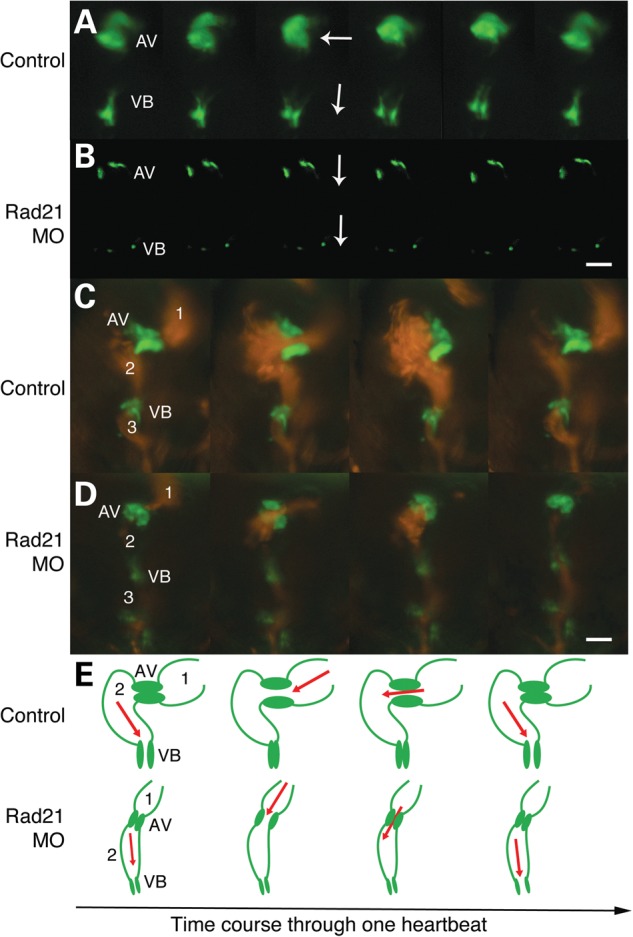
Rad21 depletion causes cardiac valve defects and decreased ejection of blood from the heart. Zebrafish embryos were injected at the one-cell stage with 1 nl Phenol Red dye, or with 0.035 pmol Rad21 ATG MO, then raised as for Figure 2. Scale bars are 20 µm. (A and B) Time series of heart valves at 5 dpf, visualized using a transgenic Tg(5xERE:EGFP262) zebrafish line in which the heart valves are GFP-labeled. (A) Normal valve morphology and function in control embryos. The opening of the VB valve is oriented at right angles to the AV valve (white arrows). The AV and VB valves open and close alternately. Valve defects were never observed in controls (n = 10). (B) Example of the extreme heart valve defects observed in Rad21-depleted embryos. In three out of eight Rad21-depleted embryos analyzed, the VB valve failed to develop and the AV valve failed to close properly. In the remaining five out of eight Rad21-depleted embryos, both AV and VB valves developed, but were notably smaller compared with control valves, as shown in (D). In all Rad21-depleted embryos, the orientation of the VB valve was in line with the AV valve (white arrows), in contrast to the perpendicular orientation of the valves seen in controls. (C and D) Time series of blood pumping through the heart at 5 dpf, visualized using a double-transgenic Tg(gata1:dsred), Tg(5xERE:EGFP262) zebrafish line, in which erythrocytes are labeled red and the heart valves are green. (C) Normal heart function in a control embryo. Blood flows from the atrium (1) into the ventricle (2) in a contralateral direction, and from the ventricle intro the bulbus arteriosus (3) in a cranial direction. (D) Rad21-depleted embryo. The volume of blood pumped into both the ventricle and the atrium is reduced in Rad21-depleted embryos (see also Supplementary Material, Video S3). (E) Pictorial representation of the heart morphologies shown in C and D. AV, atrioventricular valve; VB, ventriculobulbar valve. 1, atrium; 2, ventricle; 3, bulbus arteriosus.
In humans, CNC is involved in heart septum formation and evidence suggests that tetralogy of Fallot is associated with CNC insufficiency (28). Therefore, it is possible that cardiac defects present in RAD21-deficient patients and in other cohesinopathies are associated with dysfunction of CNC. In zebrafish, Li et al. (40) showed that ablating pre-migratory CNC leads to heart looping failure, similar to the phenotype observed in Rad21-depleted embryos. These observations led us to investigate if neural crest insufficiency could underlie the cardiac defects observed in Rad21-depleted zebrafish.
Essentially normal specification and survival of cardiac neural crest in Rad21-depleted zebrafish
First, we asked if CNC is specified and remains viable in Rad21-depleted zebrafish. Expression of sox10 marks pre-migratory neural crest in zebrafish. The spatial expression pattern of sox10 was unchanged between controls and Rad21-depleted embryos at the 10-somite stage (Supplementary Material, Fig. S4), indicating that neural crest is specified normally in Rad21-depleted embryos. However, RNA-seq revealed that total levels of sox10 transcript were reduced in Rad21-depleted embryos, which might affect subsequent neural crest development (see Supplementary Material, Table S1 and text below).
It is well known that neural crest cells are sensitive to genetic and environmental causes of cell death (41–43). Moderate Rad21 depletion could activate cell cycle checkpoints and cause death of neural crest cells, which would in turn lead to abnormal heart development. Whole-mount TUNEL staining revealed only a slight non-significant increase in apoptotic cells in Rad21-depleted embryos (0.07 pmol) compared with controls at the 10-somite stage. At the lower MO concentration (0.035 pmol), there was no significant change in the numbers of apoptotic cells (Supplementary Material, Fig. S5). Apoptotic cells were not particularly concentrated to neural crest locations, indicating that this population of cells appears not to be especially vulnerable to Rad21 depletion. However, heart looping defects caused by Rad21 depletion with 0.07 pmol MO were partially rescued by a zebrafish line homozygous for a missense mutation in p53 (tp53zdf1) (36) (Supplementary Material, Fig. S2), indicating that enhancing cell survival can compensate for developmental heart defects in the embryo. Together, the results argue that a slight deficiency in sox10 mRNA together with a modest increase in cell death leading to lower cell numbers could together contribute to the heart looping phenotypes observed in Rad21-depleted embryos.
Cardiac neural crest cells in Rad21-depleted zebrafish are able to migrate, but fail to populate the heart
CNC is specified normally in Rad21-depleted embryos, and cell survival is not significantly impacted; so next, we asked whether migration and location of CNC is impaired. There is no known CNC marker in zebrafish; therefore, we traced presumptive CNC cells by using caged fluorescein dextran (40). Previously, neural crest just anterior to somite 1 was found to be the primary source of CNC that migrates to the heart by 24 hpf in zebrafish (40,44). Caged fluorescein dextran was injected into control, Rad21-depleted or rad21nz171 mutant embryos at the one-cell stage, and uncaged as previously described (40) (Fig. 4A–A″). By 2 dpf, 87% control embryos showed fluorescent CNC cells in the myocardium (Fig. 4B–B″ and D), indicating that cells had successfully migrated from their origin anterior to somite 1. In contrast, embryos that were Rad21-depleted (0.035 and 0.07 pmol MO) had only 38 and 16% fluorescent cells in the heart, respectively (Fig. 4D). Furthermore, 0 out of five homozygous mutant rad21nz171 embryos showed fluorescent cells in the heart (Fig. 4D).
Figure 4.

Cardiac neural crest cells fail to contribute to the heart in Rad21-depleted embryos. Wild-type, Rad21-depleted or rad21nz171 homozygous mutant zebrafish embryos were injected at the one-cell stage with caged fluorescein dextran. (A–A″) Uncaging of fluorescein dextran in the pre-migratory neural crest at three- to eight-somite stages (10.5–11.5 hpf). (A) Brightfield image of an embryo at the six-somite stage. (A′) Fluorescein dextran was uncaged in the region along the neural tube cranial to somite 1 by exposure to 360 nm UV light. (A″) A merged view shows the position of the uncaged region. Scale bar is 250 µm. (B–B″) In control embryos, fluorescein-labeled cardiac neural crest cells were present in the myocardium by 35 hpf. (B) Brightfield image of the heart (h) of a control embryo. (B′) Darkfield image showing fluorescent cardiac neural crest cells in the heart. (B″) Merged images reveal that the fluorescent cells are part of the myocardium. Scale bar is 10 µm. (C–C″) In rad21nz171 mutants, fluorescein-labeled neural crest cells never reached the heart, but instead, accumulated posterior to the otic vesicle by 35 hpf. (C) Brightfield image of the otic vesicle (ov) and the adjacent posterior region in a rad21 mutant embryo. (C′) Darkfield image showing fluorescent cells accumulating posterior to the otic vesicle. (C″) Merge of (C) and (C′). Scale bar is 100 µm. (D) Percentage of embryos with fluorescent cells in the heart. Rad21-depleted embryos (0.035 pmol MO, n = 16; 0.07 pmol MO, n = 7) displayed fluorescent cells in the heart less frequently than control embryos (n = 23). Fluorescent cells were never observed in the hearts of rad21nz171 mutants (n = 5).
In rad21nz171 mutants, uncaged fluorescent cells of neural crest origin never appeared in the heart at 26, 35 or 48 hpf. Instead, fluorescent cells were observed to accumulate just behind the otic vesicle at 35 hpf (Fig. 4C–C″). This result indicates that CNC cells are able to migrate, but fail to populate the heart in rad21nz171 mutants. Melanocytes are neural crest-derived (45). At 2 dpf, ectopic melanocytes appear at the same location, just behind the otic vesicle, in both Rad21-depleted embryos (Fig. 5B and E; n = 15/20) and rad21nz171 mutants (n = 20/20; Fig. 5C and F, compare with A and D). Some melanocytes in Rad21-depleted embryos were abnormally located far under the epidermis (Fig. 5G–G″, n = 4), which is <4 μm thick at 2 dpf (46). Melanocytes were never observed at this location in control embryos (n = 20). We also observed increased pigmentation along the neural tube in both Rad21-depleted and rad21nz171 mutant embryos (Fig. 5B and C compared with A). We hypothesized that the failure of CNC cells to populate the heart, together with the inappropriate location of melanocytes, reflects a general defect in neural crest delamination and/or migration pathways.
Figure 5.

Rad21 depletion causes mislocalisation of melanocytes. (A–C) Neural tube pigmentation at 3 dpf. In Rad21-depleted (0.035 pmol MO) embryos (B), we observed increased pigmentation along the neural tube compared with controls (A) (black arrowheads). Abnormal pigmentation was observed in all rad21nz171 mutant embryos (C). (D–F) Ectopic melanocytes were present behind the otic vesicle (arrows) in 15 out of 20 Rad21-depleted embryos (0.035 pmol MO) (E), and 20 out of 20 rad21nz171 mutants (F) at 3 dpf. (G–G″) Images of the ectopic melanocyte clusters of Rad21-depleted embryos, taken in different focal planes, revealed that ectopic melanocytes were present far beneath the epidermis. (G) Level of the first layer of skin (0 µm), showing one melanocyte in focus (arrowhead). (G′) Nine micrometers deeper, another melanocyte can be seen in focus (arrowhead). (G″) Twenty-four micrometers deeper, further melanocytes can be seen (arrowhead). At this developmental stage, the epidermis is <4 µm thick. The average depth of the ectopic melanocytes was 42.75 µm (n = 4). Pigmentation was not observed behind the otic vesicles of 20 out of 20 control embryos laid in the same clutch. Anterior is to the left and dorsal is to the top in all panels. Scale bars are 250 µm.
To determine if neural crest delamination and migration is abnormal in Rad21-depleted embryos, we used time-lapse imaging of rad21nz171 homozygous mutant embryos on a Tg(sox10:GFP) background to track cranial neural crest migration to the pharyngeal arches. Time-lapse analysis (n = 4) revealed that neural crest successfully delaminated from the neural tube and was able to reach the developing pharyngeal arches in rad21nz171 homozygous mutant embryos (Supplementary Material, Video S4). However, we found that neural crest failed to condense and to shape the typical pharyngeal arch structures in rad21nz171 homozygous mutant embryos (Fig. 6B–B″ compared with controls in A–A″). Rad21-depleted embryos also exhibited aberrant pharyngeal arch condensation, with misshaped arches appearing in 9 out of 20 embryos at the 0.035 pmol MO dose (data not shown), and 18 out of 18 embryos displaying abnormal arches at the 0.07 pmol dose (Fig. 6C–C″). In rad21nz171 homozygous mutant embryos, it appeared that cells continue to migrate, sometimes in the reverse direction (Supplementary Material, Video S4), indicating that guidance cues driving condensation may be absent.
Figure 6.

Rad21 deficiency causes defects in cranial neural crest condensation into pharyngeal arch structures. Neural crest from wild-type, Rad21-depleted or rad21nz171 homozygous mutant zebrafish embryos were visualized using Tg(sox10:GFP) zebrafish. Scale bars are 250 µm. (A) In 28 hpf wild-type zebrafish embryos, neural crest cells condense and form distinctive structures representing the developing pharyngeal arches (arrows). (A′) By 36 hpf, six pharyngeal arches are clearly visible (arrows). (A″) Higher magnification of (A′) showing the pharyngeal arches in more detail. (B–B″) sox10:GFP-positive cells in rad21nz171homozygotes on the Tg(sox10:GFP) background. (B) At 28 hpf, sox10:GFP-positive neural crest cells had migrated to the anticipated location cranio-ventral to the otic vesicle (ov), but failed to correctly condense to form the characteristic arch structure. (B′) At 36 hpf, the pharyngeal arch structures remained unformed in rad21nz171 mutants. (B″) Higher magnification of (B′) showing disorganization of sox10:GFP-positive cells in the pharyngeal region. (C–C″) sox10:GFP-positive cells in Tg(sox10:GFP) embryos depleted of Rad21 using 0.07 pmol Rad21 MO. (C) Rad21-depleted embryos displayed disorganization of the pharyngeal arches at 28 hpf (18/18) as marked by sox10:GFP-positive cells. In 7 out of 18 instances, the defects recapitulated the mutant phenotype. (C′) At 36 hpf, the pharyngeal arch structures remained unformed or disorganized in Rad21-depleted embryos. (C″) Higher magnification of (C′) showing disorganization of sox10:GFP-positive cells in the pharyngeal region. See also Supplementary Material, Video S4.
Dysregulation of genes involved in neural crest cell function in Rad21-depleted embryos
We previously determined that multiple developmental genes are dysregulated in Rad21-depleted embryos at 24 and 48 hpf (1,32). Therefore, it is possible that failure of neural crest cells to reach the heart field, or to condense into pharyngeal arches, is associated with aberrant developmental gene expression between 8 somites and 24 hpf in Rad21-depleted zebrafish embryos. To investigate this possibility, we analyzed RNA-seq data from Rad21-depleted zebrafish embryos at the 8-somite stage (10 hpf), and found 236 genes were up-regulated and 322 were down-regulated (FDR < 0.05) (Fig. 7A). Many genes involved in cellular and developmental processes were dysregulated (Fig. 7B). Of these, we found dysregulated genes were enriched in pathways known to contribute to neural crest induction, migration and condensation into pharyngeal arches and cardiac outflow tract (29) (Fig. 7C, Supplementary Material, Table S1). Dysregulated genes belonged to the Wnt and BMP signaling pathways, cell adhesion, guidance and migration pathways, and transcription factors involved in neural crest identity. Selected genes were confirmed by quantitative reverse transcriptase–polymerase chain reaction (qRT–PCR) to be dysregulated in rad21nz171 mutants at 24 hpf as well as MO-treated Rad21-depleted embryos (Fig. 7D).
Figure 7.

Genes involved in neural crest function are dysregulated upon Rad21 depletion in eight-somite zebrafish embryos. (A) Scatter plot comparing global gene expression profiles between wild-type and Rad21 ATG MO knockdown (Rad21 MO, 0.5 pmol MO) 10 hpf embryos (8 somites). Differentially expressed genes (false-discovery rate FDR < 0.05, log2 fold change >0.8) are marked in green (up-regulated) and red (down-regulated). (B) GO term enrichment analysis was performed on down-regulated genes in Rad21 MO using the R package ClusterProfiler (52) using a cutoff of 0.05 FDR corrected P-value. Bar lengths represent number of genes in each category, bar color stands for the −log10 of the adjusted P-value of each GO term. No significant GO enrichment was found in the up-regulated gene set. (C) Bar graph of genes involved in neural crest development showing fold change and FDR in bar color. Neural crest genes were those identified by Kirby and Hutson (29). Raw data including direction and degree of regulation can be found in Supplementary Material, Table S1, and information on transcriptome production and analysis can be found in the Supplementary Methods. (D) Confirmation by qRT–PCR of dysregulated expression of candidate neural crest-related genes identified by RNA-seq in 10 hpf Rad21 ATG MO embryos (0.5 pmol MO). Dysregulated expression was similar to that observed in rad21nz171 homozygous mutants at 24 hpf. The vertical axis shows the relative mRNA expression normalized to rpl13a and actb mRNA. Data are means of three independent biological replicates ±SEM; asterisks indicate significant changes in expression, P-value < 0.05.
Discussion
A moderate deficiency of cohesin subunit Rad21 leads to specific cardiac defects in zebrafish
Here, we show that reduced levels of Rad21 in zebrafish result in heart defects characteristic of humans with CdLS-related cohesinopathies. Cohesinopathies are characterized by impaired, but not loss-of-function of the affected genes, owing to the essential nature of the cohesin complex and its regulators. To date, creating animal models that accurately recapitulate the cohesinopathies has been difficult because even modest reductions in the function of cohesinopathy genes can result in drastic developmental consequences. Tissue-specific genetic techniques that completely remove all cohesin function may not accurately recapitulate human disease either, since dosage is likely to be an important consideration in disease etiology (27,47). In previous work, we characterized a homozygous lethal genetic mutation in cohesin subunit rad21, which has defects in gene expression and blood development, but does not survive long enough for subsequent phenotypes to be analyzed (31,32,35). Zebrafish heterozygous for a rad21 null allele have 40% less rad21 mRNA than wild-type animals (32), and they develop normally with a small proportion showing temporary cardiac defects that later resolved (Supplementary Material, Fig. S3). In the present study, we more accurately model human RAD21 mutations by partially knocking down Rad21 protein in zebrafish to ∼20% of wild-type levels using a gene-specific morpholino (Supplementary Material, Figs S1 and S3). Remarkably, zebrafish embryos with only 20% of wild-type Rad21 developed relatively normally and were only slightly smaller than wild-type controls. However, the vast majority of Rad21-depleted zebrafish exhibited some form of cardiac defect (Figs 1–3). The hearts of Rad21-depleted fish were much smaller than wild-type, remained unlooped, had deficiencies or defects in valvular structures and were unable to pump blood efficiently. Our data suggest that heart development is exquisitely sensitive to the available amount of Rad21/cohesin. We found that heart development is compromised even when there is sufficient cohesin present to support relatively normal growth. Accordingly, our results also reflect the clinical findings that CHD are prevalent in CdLS and related cohesinopathies (23,24). However, human cohesinopathy cases generally have a lower incidence of cardiac defects than did the Rad21-depleted zebrafish. This could reflect species-specific differential sensitivity (or plasticity) of cell populations in developing embryos (Fig. 8).
Figure 8.

Model illustrating how a slight decrease in cohesin could sensitize the heart to abnormal development. Only a small proportion of cohesin is needed for sister chromatid cohesion, while the majority of cohesin has other cellular roles such as gene expression and DNA damage repair (1). In the graph, the curved line represents the requirement for cohesin function in a given cell type as development progresses. During a particular developmental window, the requirement for cohesin in (for example) neural crest could be up to maximum wild-type levels. Cells in this window would be sensitized to abnormal development by partial cohesin knockdown, as described by our study, or by genetic insufficiency. This model could account for the specific heart phenotype observed with Rad21 depletion (our study), or the multifactorial developmental abnormalities observed in the cohesinopathies. It could explain why many developmental processes remain untouched, while others are compromised.
Cardiac defects induced by Rad21 deficiency have a neural crest origin
A previous study showed that expression of endodermal differentiation genes sox32, sox17 and foxa2 was altered in Nipbl-depleted, but normal in Rad21-depleted zebrafish embryos (26). Heart looping defects in Nipbl-depleted embryos, which are similar to those we observed in Rad21-depleted embryos, were not rescued by injection of sox32 or gata5 transcript. In contrast, other heart phenotypes in Nipbl-deficient embryos, such as cardia bifida, were rescued by these transcripts (26). We did not observe cardia bifida in Rad21-depleted zebrafish. These results imply that different cohesinopathy mutations cause CHD via divergent pathways, and that defects in heart looping are not due to problems with endoderm differentiation.
Proper development of the vertebrate heart depends on contribution from cardiac neural crest cells as well as from endoderm. Cardiac neural crest cells contribute to valve development, cardiac septation and the outflow tract (28). Highlighting their developmental importance, cardiac neural crest cells are central to the pathogenesis of several human cardiocraniofacial syndromes (28). Cardiac neural crest cells have been identified in zebrafish (44), and their ablation caused a very similar cardiac phenotype to Rad21 depletion (40). We found that cardiac neural crest cells failed to populate the myocardium in Rad21-depleted zebrafish and rad21 homozygous mutants (Fig. 4). Our observation that the VB valve is often severely reduced or absent in Rad21-depleted zebrafish (Fig. 3) is consistent with the idea that cardiac neural crest cells contribute to this structure.
Other studies also indicate neural crest involvement in the cohesinopathies. Deficiency of cohesin subunit Pds5 in mice caused cleft palate, congenital heart defects and delayed migration of enteric neuron precursors (27); neural crest cells contribute to all of these structures. More recently, mutations in SGOL1, a cohesin-related gene, were found to cause chronic atrial and intestinal dysrhythmia in humans (30) with the possibility that the affected structures are neural crest-derived. Together, the results indicate that cardiac neural crest defects may be an important contributory factor to CHD in the cohesinopathies.
Cardiac defects induced by Rad21 deficiency are accompanied by dysregulation of genes important for neural crest function
We found that neural crest is specified normally in Rad21-depleted animals (Supplementary Material, Fig. S4), and we also found that neural crest can migrate from its dorsal origin (Fig. 6, Supplementary Material, Video S4). So why don't Rad21-deficient neural crest cells populate the heart field? Rad21-depleted embryos (0.07 pmol) had slightly higher levels of cell death (Supplementary Material, Fig. S5), and heart defects owing to Rad21 deficiency were compensated in a p53 mutant background (Supplementary Material, Fig. S2). Therefore, it is possible that insufficient numbers of neural crest cells reach the heart field because of cell death or reduced proliferation of neural crest cells, or surrounding cells. However, Xu et al. (21) showed that while stimulation of growth pathways rescued some of the growth and developmental anomalies of Rad21-depleted embryos, cardiac defects were not rescued. Furthermore, we were unable to rescue cardiac defects in rad21−/− zebrafish embryos by overexpressing Myc, which promotes growth, and is normally down-regulated upon cohesin deficiency (32) (data not shown). The results suggest that while cell proliferation and survival downstream of Rad21 deficiency might partially account for the observed heart defects, additional factors are likely to contribute.
Our previous microarray study showed that several developmental genes are dysregulated in 24 and 48 hpf rad21−/− zebrafish embryos (32). In the present study, our RNA-seq analysis of Rad21-depleted embryos at the 8-somite stage revealed that dysregulated genes included those essential for neural crest development and migration (Fig. 7, Supplementary Material, Table S1). Together, the results support the idea that dysregulated gene expression might contribute to the failure of neural crest cells to populate the heart in Rad21-depleted embryos, and to condense into pharyngeal arches in the rad21nz17 mutants (Fig. 6, Supplementary Material, Video S4). Several dysregulated genes belong to guidance molecules and their receptors, chemokines, cell adhesion molecules and the Wnt signaling pathway (Fig. 7). Abnormal function of these pathways could explain the observation that while neural crest cells are able to migrate, they appear to ‘wander’, sometimes reversing their direction, without reaching their destination (Supplementary Material, Video S4). Therefore, it is possible that developmental cues directing neural crest contribution to the heart are compromised in Rad21-depleted embryos, leading to the observed cardiac phenotypes. However, it is not known whether such developmental cues would originate from within neural crest cells themselves, or from surrounding tissue that contributes toward guiding these cells to their destination.
In conclusion, heart development must be precisely timed, and is exquisitely sensitive to changes in multiple developmental pathways. Our results show that abnormal migration of CNC is likely to contribute to CHD in CdLS patients, perhaps depending on which cohesinopathy gene is mutated. We found that lowering the threshold of cohesin function can expose the embryo to cardiac anomalies, while otherwise preserving essentially normal development. A hypothetical model for how cardiac development might be particularly exposed to cohesin depletion is presented in Figure 8. Our results raise the interesting possibility that cardiac defects arising from genetic mutations in the cohesin apparatus could affect the general population, without any other apparent clinical manifestation.
Materials and Methods
Zebrafish
Zebrafish were maintained as described previously (48). Zebrafish lines tp53zdf1 (36), Tg(bactin2:GFP)zp5 (37), Tg(gata1:dsred) (38) and Tg(sox10:GFP) (49) were obtained from the Zebrafish International Resource Center (University of Oregon), or from the University of Auckland with appropriate permissions. The Tg(5xERE:EGFP262) (50) line was a gift from Daniel Gorelick and Marnie Halpern. The University of Otago Animal Ethics Committee approved all zebrafish research.
Microinjection
An MO targeting the ATG codon of the zebrafish rad21 gene (sequence: 5′-AGGACGAAGTGGGCGTAAAACATTG-3′) was obtained from GeneTools LLC and diluted in water. Characterization and validation of this MO was reported in detail previously (31,32). For microinjection, 1 nl containing the stated pmol of MO was injected into the yolk of wild-type zebrafish embryos, or those from various transgenic reporter lines, at the one-cell stage. Control embryos were injected with 1 nl Phenol Red dye solution. Rad21 protein knockdown was confirmed by western blot analysis (Supplementary Material, Fig. S1 and Supplementary Methods). To exclude the possibility of non-specific MO effects, we established an MO dose that caused no defects in wild-type embryos, and demonstrated that this dose was able to cause heart defects in embryos from a rad21nz171/+ background (Supplementary Material, Fig. S2 and Supplementary Methods).
Microscopy and image capture
All imaging used either a Leica M205 FA stereofluorescent microscope and Leica Applications Suite software or a Nikon Eclipse Ni-E microscope and NIS Elements software. Time-lapse movies were acquired using a Nikon Eclipse Ni-E microscope with a DS-U3 camera using water immersion 20× and 40× objectives, and processed using NIS Elements or ImageJ software.
Cell tracing
Caged fluorescein dextran was synthesized as described (51). One-cell stage embryos were injected with 1 nl of RNase-free water containing Rad21 MO or a water control, and caged fluorescein dextran stock diluted 1:5. For mutant analysis, embryos from a rad21nz171 heterozygous incross were injected with caged fluorescein dextran stock diluted 1:5. Embryos were incubated in the dark at 22°C overnight. Subsequently, three- to eight-somite stage embryos were placed in a petri dish containing a bed of 2% agarose and dechorionated using forceps. Embryos were mounted in a drop of 3% methylcellulose (Sigma) on a depression slide with the lateral surface facing upwards. Fluorescein was uncaged in the pre-migratory neural crest region as previously described (40). Briefly, using a 20 µm pinhole on a Nikon Eclipse Ni-E microscope and a 40× water immersion objective, neural crest cells immediately anterior to somite one were exposed to UV light (360 nm for 10 s). The location of fluorescein-labeled cells was examined at 2 dpf using a Nikon Eclipse Ni-E microscope. Mutant rad21nz171 embryos and Rad21 MO-injected embryos were compared with wild-type.
RNA extraction and RNA-seq
Wild-type embryos were collected at the one-cell stage, synchronized and either morpholino injected (0.5 pmol Rad21 ATG MO) or kept as control and allowed to develop to the eight-somite stage at 28°C. Three biological replicates each containing total RNA from 100 pooled embryos were isolated using the NucleoSpin® RNAII Kit (Macherey-Nagel). The quality of the RNA was confirmed using the Agilent 2100 Bioanalyzer; all samples had RIN numbers above 9. Strand-specific libraries were prepared using the TruSeq stranded total RNA-ribozero kit (Illumina) and 100 bp paired-end sequencing was performed to depth of 10 million reads per library on an Illumina HiSeq 2000. See Supplementary Methods for details on RNA-seq analysis.
Quantitative reverse transcriptase–polymerase chain reaction
One microgram of total RNA was used for cDNA synthesis with qScript cDNA Supermix kit (95048-100; Quanta Biosciences). qRT–PCR reactions were performed in duplicates on a Roche LightCycler 480 Instrument II Real-time PCR System with SYBR Green PCR master mix (no. 4309155; Applied Biosystems, Life Technologies). Standard curves were run for all primer pairs to ensure high efficiencies (>97%; R2 > 0.98) and a single shoulder-free peak upon melt curve analysis. Transcript expression was normalized to reference genes rpl13a, actb1, acta1 using qBase Plus (Biogazelle) and results are represented as fold change in expression level relative to control sample (mean ± SEM, n = 3 biological replicates), all primers are listed in Supplementary Material, Table S2. Statistical analysis was performed using qBase Plus (Biogazelle) (unpaired t-test). A P-value of <0.05 was considered to indicate statistical significance and marked by an asterisk.
Statistical analysis
Unless otherwise specified, statistical analysis and graphing were performed using Prism version 6.00 for OSX (Graphpad Software).
Supplementary Material
Funding
This work was supported by the Neurological Foundation of NZ (to J.A.H.) and the Royal Society of NZ Marsden Fund (grant number 11-UOO-027 to J.A.H.). S.B. is supported by National Institutes of Health (grant number R01 GM075119).
Supplementary Material
Acknowledgements
We thank Noel Jhinku for expert management of the Otago Zebrafish Facility and Stephen Robertson for a critical reading of the manuscript. We are grateful to Daniel Gorelick and Marnie Halpern for providing the Tg(5xERE:GFP) zebrafish line.
Conflict of Interest statement. None declared.
References
- 1.Horsfield J.A., Print C.G., Monnich M. (2012) Diverse developmental disorders from the one ring: distinct molecular pathways underlie the cohesinopathies. Front. Genet., 3, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ball A.R. Jr., Chen Y.Y., Yokomori K. (2014) Mechanisms of cohesin-mediated gene regulation and lessons learned from cohesinopathies. Biochim. Biophys. Acta, 1839, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu J., Krantz I.D. (2009) Cornelia de Lange syndrome, cohesin, and beyond. Clin. Genet., 76, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mannini L., Cucco F., Quarantotti V., Krantz I.D., Musio A. (2013) Mutation spectrum and genotype–phenotype correlation in Cornelia de Lange syndrome. Hum. Mutat., 34, 1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bose T., Gerton J.L. (2010) Cohesinopathies, gene expression, and chromatin organization. J. Cell. Biol., 189, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Remeseiro S., Cuadrado A., Losada A. (2013) Cohesin in development and disease. Development, 140, 3715–3718. [DOI] [PubMed] [Google Scholar]
- 7.Liu J., Krantz I.D. (2008) Cohesin and human disease. Annu. Rev. Genomics Hum. Genet., 9, 303–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kline A.D., Krantz I.D., Sommer A., Kliewer M., Jackson L.G., FitzPatrick D.R., Levin A.V., Selicorni A. (2007) Cornelia de Lange syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance. Am. J. Med. Genet. A., 143A, 1287–1296. [DOI] [PubMed] [Google Scholar]
- 9.Krantz I.D., McCallum J., DeScipio C., Kaur M., Gillis L.A., Yaeger D., Jukofsky L., Wasserman N., Bottani A., Morris C.A. et al. (2004) Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet., 36, 631–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tonkin E.T., Wang T.J., Lisgo S., Bamshad M.J., Strachan T. (2004) NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet., 36, 636–641. [DOI] [PubMed] [Google Scholar]
- 11.Liu J., Baynam G. (2010) Cornelia de Lange syndrome. Adv. Exp. Med. Biol., 685, 111–123. [PubMed] [Google Scholar]
- 12.Musio A., Selicorni A., Focarelli M.L., Gervasini C., Milani D., Russo S., Vezzoni P., Larizza L. (2006) X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet., 38, 528–530. [DOI] [PubMed] [Google Scholar]
- 13.Deardorff M.A., Kaur M., Yaeger D., Rampuria A., Korolev S., Pie J., Gil-Rodriguez C., Arnedo M., Loeys B., Kline A.D. et al. (2007) Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet., 80, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deardorff M.A., Wilde J.J., Albrecht M., Dickinson E., Tennstedt S., Braunholz D., Monnich M., Yan Y., Xu W., Gil-Rodriguez M.C. et al. (2012) RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet., 90, 1014–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deardorff M.A., Bando M., Nakato R., Watrin E., Itoh T., Minamino M., Saitoh K., Komata M., Katou Y., Clark D. et al. (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature, 489, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaiser F.J., Ansari M., Braunholz D., Concepcion Gil-Rodriguez M., Decroos C., Wilde J.J., Fincher C.T., Kaur M., Bando M., Amor D.J. et al. (2014) Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet., 23, 2888–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nasmyth K., Haering C.H. (2009) Cohesin: its roles and mechanisms. Annu. Rev. Genet., 43, 525–558. [DOI] [PubMed] [Google Scholar]
- 18.Dorsett D., Strom L. (2012) The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr. Biol., 22, R240–R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorsett D., Merkenschlager M. (2013) Cohesin at active genes: a unifying theme for cohesin and gene expression from model organisms to humans. Curr. Opin. Cell Biol., 25, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuin J., Franke V., van Ijcken W.F., van der Sloot A., Krantz I.D., van der Reijden M.I., Nakato R., Lenhard B., Wendt K.S. (2014) A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS. Genet., 10, e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu B., Sowa N., Cardenas M.E., Gerton J.L. (2014) l-leucine partially rescues translational and developmental defects associated with zebrafish models of Cornelia de Lange syndrome. Hum. Mol. Genet., 24, 1540–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selicorni A., Colli A.M., Passarini A., Milani D., Cereda A., Cerutti M., Maitz S., Alloni V., Salvini L., Galli M.A. et al. (2009) Analysis of congenital heart defects in 87 consecutive patients with Brachmann-de Lange syndrome. Am. J. Med. Genet. A, 149A, 1268–1272. [DOI] [PubMed] [Google Scholar]
- 23.Chatfield K.C., Schrier S.A., Li J., Clark D., Kaur M., Kline A.D., Deardorff M.A., Jackson L.S., Goldmuntz E., Krantz I.D. (2012) Congenital heart disease in Cornelia de Lange syndrome: phenotype and genotype analysis. Am. J. Med. Genet. A, 158A, 2499–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyle M.I., Jespersgaard C., Brondum-Nielsen K., Bisgaard A.M., Tumer Z. (2015) Cornelia de Lange syndrome. Clin. Genet., 88, 1–12. [DOI] [PubMed] [Google Scholar]
- 25.Kawauchi S., Calof A.L., Santos R., Lopez-Burks M.E., Young C.M., Hoang M.P., Chua A., Lao T., Lechner M.S., Daniel J.A. et al. (2009) Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet., 5, e1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muto A., Calof A.L., Lander A.D., Schilling T.F. (2011) Multifactorial origins of heart and gut defects in nipbl-deficient zebrafish, a model of Cornelia de Lange Syndrome. PLoS Biol., 9, e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang B., Chang J., Fu M., Huang J., Kashyap R., Salavaggione E., Jain S., Shashikant K., Deardorff M.A., Uzielli M.L. et al. (2009) Dosage effects of cohesin regulatory factor PDS5 on mammalian development: implications for cohesinopathies. PLoS ONE, 4, e5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keyte A., Hutson M.R. (2012) The neural crest in cardiac congenital anomalies. Differentiation, 84, 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirby M.L., Hutson M.R. (2010) Factors controlling cardiac neural crest cell migration. Cell. Adh. Migr., 4, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chetaille P., Preuss C., Burkhard S., Cote J.M., Houde C., Castilloux J., Piche J., Gosset N., Leclerc S., Wunnemann F. et al. (2014) Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm. Nat. Genet., 46, 1245–1249. [DOI] [PubMed] [Google Scholar]
- 31.Horsfield J.A., Anagnostou S.H., Hu J.K., Cho K.H., Geisler R., Lieschke G., Crosier K.E., Crosier P.S. (2007) Cohesin-dependent regulation of Runx genes. Development, 134, 2639–2649. [DOI] [PubMed] [Google Scholar]
- 32.Rhodes J.M., Bentley F.K., Print C.G., Dorsett D., Misulovin Z., Dickinson E.J., Crosier K.E., Crosier P.S., Horsfield J.A. (2010) Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev. Biol., 344, 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vega H., Waisfisz Q., Gordillo M., Sakai N., Yanagihara I., Yamada M., van Gosliga D., Kayserili H., Xu C., Ozono K. et al. (2005) Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat. Genet., 37, 468–470. [DOI] [PubMed] [Google Scholar]
- 34.Percival S.M., Thomas H.R., Amsterdam A., Carroll A.J., Lees J.A., Yost H.J., Parant J.M. (2015) Variations in sister chromatid cohesion dysfunction in esco2 mutant zebrafish reflects the phenotypic diversity of Roberts Syndrome. Dis. Model. Mech., 8, 941–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marsman J., O'Neill A.C., Kao B.R., Rhodes J.M., Meier M., Antony J., Monnich M., Horsfield J.A. (2014) Cohesin and CTCF differentially regulate spatiotemporal runx1 expression during zebrafish development. Biochim. Biophys. Acta, 1839, 50–61. [DOI] [PubMed] [Google Scholar]
- 36.Berghmans S., Murphey R.D., Wienholds E., Neuberg D., Kutok J.L., Fletcher C.D., Morris J.P., Liu T.X., Schulte-Merker S., Kanki J.P. et al. (2005) tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc. Natl Acad. Sci. USA, 102, 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gillette-Ferguson I., Ferguson D.G., Poss K.D., Moorman S.J. (2003) Changes in gravitational force induce alterations in gene expression that can be monitored in the live, developing zebrafish heart. Adv. Space Res., 32, 1641–1646. [DOI] [PubMed] [Google Scholar]
- 38.Traver D., Paw B.H., Poss K.D., Penberthy W.T., Lin S., Zon L.I. (2003) Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol., 4, 1238–1246. [DOI] [PubMed] [Google Scholar]
- 39.Gorelick D.A., Iwanowicz L.R., Hung A.L., Blazer V.S., Halpern M.E. (2014) Transgenic zebrafish reveal tissue-specific differences in estrogen signaling in response to environmental water samples. Environ. Health Perspect., 122, 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y.X., Zdanowicz M., Young L., Kumiski D., Leatherbury L., Kirby M.L. (2003) Cardiac neural crest in zebrafish embryos contributes to myocardial cell lineage and early heart function. Dev. Dyn., 226, 540–550. [DOI] [PubMed] [Google Scholar]
- 41.Morgan S.C., Relaix F., Sandell L.L., Loeken M.R. (2008) Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res. Part A Clin. Mol. Teratol., 82, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith F., Hu D., Young N.M., Lainoff A.J., Jamniczky H.A., Maltepe E., Hallgrimsson B., Marcucio R.S. (2013) The effect of hypoxia on facial shape variation and disease phenotypes in chicken embryos. Dis. Model. Mech., 6, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trainor P.A. (2010) Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am. J. Med. Genet. A, 152A, 2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato M., Yost H.J. (2003) Cardiac neural crest contributes to cardiomyogenesis in zebrafish. Dev. Biol., 257, 127–139. [DOI] [PubMed] [Google Scholar]
- 45.Trainor P.A. (2005) Specification and patterning of neural crest cells during craniofacial development. Brain. Behav. Evol., 66, 266–280. [DOI] [PubMed] [Google Scholar]
- 46.Guzman A., Ramos-Balderas J.L., Carrillo-Rosas S., Maldonado E. (2013) A stem cell proliferation burst forms new layers of P63 expressing suprabasal cells during zebrafish postembryonic epidermal development. Biol. Open, 2, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorsett D. (2011) Cohesin: genomic insights into controlling gene transcription and development. Curr. Opin. Genet. Dev., 21:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westerfield M. (1995) The Zebrafish Book. A Guide for the Laboratory use of Zebrafish (Brachydanio Rerio). University of Oregon Press, Eugene, Oregon. [Google Scholar]
- 49.Carney T.J., Dutton K.A., Greenhill E., Delfino-Machin M., Dufourcq P., Blader P., Kelsh R.N. (2006) A direct role for Sox10 in specification of neural crest-derived sensory neurons. Development, 133, 4619–4630. [DOI] [PubMed] [Google Scholar]
- 50.Gorelick D.A., Halpern M.E. (2011) Visualization of estrogen receptor transcriptional activation in zebrafish. Endocrinology, 152, 2690–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clanton J.A., Shestopalov I., Chen J.K., Gamse J.T. (2011) Lineage labeling of zebrafish cells with laser uncagable fluorescein dextran. J. Vis. Exp., 50, 2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu G., Wang L.-G., Han Y., He Q.-Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.