

Abstract
The autism spectrum disorders (ASD) comprise a broad group of behaviorally related neurodevelopmental disorders affecting as many as 1 in 68 children. The hallmarks of ASD consist of impaired social and communication interactions, pronounced repetitive behaviors and restricted patterns of interests. Family, twin and epidemiological studies suggest a polygenetic and epistatic susceptibility model involving the interaction of many genes; however, the etiology of ASD is likely to be complex and include both epigenetic and environmental factors. 5-hydroxymethylcytosine (5hmC) is a novel environmentally sensitive DNA modification that is highly enriched in post-mitotic neurons and is associated with active transcription of neuronal genes. Here, we used an established chemical labeling and affinity purification method coupled with high-throughput sequencing technology to generate a genome-wide profile of striatal 5hmC in an autism mouse model (Cntnap2−/− mice) and found that at 9 weeks of age the Cntnap2−/− mice have a genome-wide disruption in 5hmC, primarily in genic regions and repetitive elements. Annotation of differentially hydroxymethylated regions (DhMRs) to genes revealed a significant overlap with known ASD genes (e.g. Nrxn1 and Reln) that carried an enrichment of neuronal ontological functions, including axonogenesis and neuron projection morphogenesis. Finally, sequence motif predictions identified associations with transcription factors that have a high correlation with important genes in neuronal developmental and functional pathways. Together, our data implicate a role for 5hmC-mediated epigenetic modulation in the pathogenesis of autism and represent a critical step toward understanding the genome-wide molecular consequence of the Cntnap2 mutation, which results in an autism-like phenotype.
Introduction
The autism spectrum disorders (ASD) comprise a continuum of neurodevelopmental disorders characterized by core deficits in social behavior and communication, accompanied by restricted interests and repetitive behaviors (1). Other non-core symptoms frequently associated with ASD are epilepsy and hyperactivity, as well as abnormalities in sleep, sensory and gastrointestinal function (2). Genetic studies have revealed extraordinary heterogeneity in the etiology of ASD, predicting hundreds of rare risk genes that cumulatively account for <25% of ASD instances (3–5). Much of these data are from family and twin studies that find the concordance rates ranging from 60 to 90% in monozygotic twins and 0 to 20% in dizygotic twins (6). While a portion of these rates depends on the diagnosis and on the autism subtype, genetic contributions are not sufficient to explain the entirety of ASD etiology. Thus, autism is considered a multifactorial hereditary disorder, resulting from contributions from numerous genes (polygenic heredity) and environmental factors.
Environmentally sensitive epigenetic modifications are emerging as important factors in the long-term biological trajectories leading to psychiatric-related outcomes. Epigenetic mechanisms have been reported in various genetic disorders associated with autism, including maternal 15q11-q13 duplication and several syndromes such as Fragile X, Rett, Down, Turner, Phelan-Mcdermid, Beckwith-Wiedermann, Willians-Beuren, CHARGE, Angelman and Prader-Willi (6). Given the number of disorders associated with epigenetic etiologies comorbid with autism, it can be suggested that epigenetic mechanisms involving gene × environment interactions might be a common pathway for many cases of ASD. DNA methylation is an epigenetic modification with important roles in chromatin remolding, gene silencing, embryonic development, cellular differentiation and the maintenance of cellular identity (7–10). Traditional studies of DNA methylation have focused on the dynamic variation of a methyl group on cytosine (5-methylcytosine; 5mC) that plays a role in many crucial cellular processes and has been linked to neurological disorders as well as psychiatric disorders, including depression, anxiety, post-traumatic stress disorders and schizophrenia (11–14). Recently, it was shown that 5mC can be oxidized to 5-hydroxymethylcytosine (5hmC) by the ten-eleven translocation (TET) methylcytosine dioxygenase family of enzymes. Initial studies found that 5hmC is enriched in neuronal cells and is associated with the regulation of neuronal activity (15). Genomic studies revealed that 5hmC is located within distal cis-regulatory elements and in the gene bodies of synaptic plasticity-related loci, particularly at intron–exon boundaries, suggesting an important role for 5hmC in coordinating transcriptional activity (15). Unlike 5mC, the overall abundance of 5hmC dramatically varies among tissues, with ∼10 times more found in brain tissues like striatal neurons than in other tissues (16–20). This differential distribution points to the possible functional importance of 5hmC in neuronal development and activity and that aberrant 5hmC patterns may lead to neurological and neurodegenerative disease. These findings have prompted investigations into the potential role(s) of 5hmC in disease, where it appears to play a role in neurological disorders (e.g. Fragile X syndrome, Rett syndrome and Autism) (21–23) and neurodegenerative diseases (e.g. Huntington's and Alzheimer's) (24–27).
Human association, linkage, gene expression and imaging data support the role of both common and rare variants of contactin-associated protein-like 2 (CNTNAP2) in ASD (28–33). The Cntnap2 (or Caspr2) gene encodes a neuronal transmembrane protein member of the neurexin superfamily involved in neuron–glia interactions and clustering of potassium channels in myelinated axons (34,35). Recently, it was shown that mice harboring a homozygous knockout of Cntnap2 (Cntnap2−/−) exhibit parallels to the major neuropathological features in ASD, including cortical dysplasia and focal epilepsy. In addition, core deficits were also observed such as defects in neuronal migration of cortical projection neurons and a reduction in the number of striatal GABAergic interneurons (36). Here, we hypothesized that the loss of CNTNAP2 and the resulting ASD-like neuronal and behavioral outcome might also alter striatal 5hmC levels in genes contributing to the autism phenotype. To test this hypothesis, we used an established chemical labeling and affinity purification method coupled with high-throughput sequencing technology to generate an unbiased genome-wide 5hmC profile of the Cntnap2−/− mutant mouse. These profiles provide new insight into epigenetic contributions to the autism phenotype and are intended to serve as a conceptual basis that will facilitate the future study of cellular and brain regional dynamics of 5hmC, especially as it relates to ASD. Here, we provide a genome-wide map of 5hmC in a mouse model of autism, which reveals known and potentially novel genes contributing to the autism phenotype. These findings establish a role for 5hmC in ASD and provide insights into the immediate genome-wide neuromolecular response to the loss of CNTNAP2, which results in an autism-like phenotype.
Results
Disruption of 5hmC in the striatum of an autism mouse model
To determine the genome-wide 5hmC distribution in both an autism mouse model (Cntnap2−/−) and their wild-type (WT) littermates, we employed an established chemical labeling and affinity purification method, coupled with high-throughput sequencing technology (16,20). Three Cntnap2−/− mice and three age-matched WT littermates were sacrificed at 9 weeks of age as independent biological replicates. 5hmC-containing DNA sequences were enriched from striatum total DNA, and high-throughput sequencing resulted in a range of ∼30–50 million uniquely mapped reads from each biological replicate (Supplementary Material, Table S1; Materials and Methods). Read density mapping showed high correlations between all animals, indicating that the differences between the two groups are small and only in short regions of the genome (Supplementary Material, Table S2). Moreover, these data showed no visible differences among the chromosomes, except depletion on chromosomes X and Y, which is consistent with previous observations (Fig 1A) (16). Annotation of 5hmC reads from Cntnap2−/− and WT mice to standard overlapping genomic features obtained from the UCSC Tables for NCBI37v1/mm9 showed that 5hmC-mapped reads were found in all regions with enrichment in genic structures compared with intergenic regions (Fig. 1B). Nonetheless, these distributions demonstrated an overall equal distribution of 5hmC levels in both genotypes on these defined genomic features.
Figure 1.

Distribution of sequence reads. The percent distribution (A and C) or the number of reads per million reads per 1 kb (B; y-axis) of monoclonal reads that passed QC for each chromosome (A), standard genomic structure (B) and repetitive element (C) in Cntnap2 (black) and WT (white) mice are shown. (B) The standard genomic structures are defined as the following: 10 kb upstream of the transcription start site (TSS; TSS-10K); 2 kb upstream of the TSS (TSS-2K); 500 bp flanking the TSS (TSS+/−500 bp; 5′ UTR; first exon; exonic; intronic; 3′ UTR; 10 kb downstream of the TES (TES10K); intragenic; or intergenic regions. Asterisk denotes a binomial P-value = <0.001).
DNA hypermethylation on repetitive elements is believed to play a critical role in maintaining genomic stability (37). To investigate the genome-wide distribution of 5hmC on repetitive elements, we aligned the total 5hmC reads to the RepeatMasker and segmental duplication tracks of NCBI37v1/mm9 and found that 5hmC levels were significantly reduced in Cntnap2−/− mice for all classes of repetitive elements (Fig 1C). These data suggest a global reduction of 5hmC at repeat sequences of Cntnap2−/− mice, which could imply stabilization of 5mC in repetitive elements and appear as hypermethylation, a response previously observed in response to stress (38).
Identification and characterization of differentially hydroxymethylated regions in an autism mouse model
To identify distinct 5hmC distribution patterns in Cntnap2−/− and WT mice, we characterized differentially hydroxymethylated regions (DhMRs) with respect to the Cntnap2 mutant genome. A total of 847 Cntnap2−/−-specific increases in hydroxymethylation (hyper-DhMRs) and 801 Cntnap2−/−-specific decreases in hydroxymethylation (hypo-DhMRs) were found, and these loci were distributed across all chromosomes (Fig. 2; Materials and Methods; Supplementary Material, Dataset S1). As specific regions of the genome are differentially methylated based on the biological functions of the genes contained within the region, we determined whether Cntnap2−/−-specific DhMRs are enriched or depleted in certain chromosomes using a binomial test of all detected 5hmC peaks as the background (Materials and Methods). This analysis revealed that chromosomes 1 and 13 have disproportionately less DhMRs and that chromosomes 6 and X have disproportionately more DhMRs than expected by chance alone (binomial P-value <0.05; Fig. 2C). These data indicate that some chromosomes are more sensitive to DNA hydroxymethylation changes in the absence of the Cntnap2-encoded protein.
Figure 2.

Characterization of DhMRs across standard genomic structures. (A and B) Identification of WT- and Cntnap2-specific DhMRs. 847 and 801 WT- and Cntnap2-specific DhMRs were identified by directly comparing the profile of one to the other (Materials and Methods). (C) Modified Manhattan plot of DhMRs from the striatum of Cntnap2−/− mice reveals DhMRs to be distributed across the genome. Positively correlated DhMRs are displayed with a positive −log10 of the P-value and negatively correlated DhMRs are displayed with a negative −log10 of the P-value. Significant DhMRs are displayed outside the dashed lines (P-value <0.05), whereas all DhMRs alternate between black and gray to indicate each chromosome. (D) The percent distribution (y-axis) of all 5hmC peak data (striped), all DhMRs (gray), hyper-DhMRs (black) and hypo-DhMRs (white) across each genomic region is shown. Asterisk denotes permutation P-value of <0.001.
We proceeded to define the genomic features associated with the Cntnap2−/−-specific DhMRs by annotating both hyper- and hypo-DhMRs to overlapping gene structures or to the intergenic region if >100 kb away from any gene structures. The distribution of these data indicated that the largest fraction of DhMRs (∼85%) is in the intragenic regions of the genome (Fig. 2D). To determine the relative abundance of DhMRs in each annotated region, we compared, via permutation testing, the proportion of DhMRs with the total number of 5hmC peaks detected for each region (Materials and Methods). This analysis found that only the 3′ untranslated region (UTR) had a significant abundance of DhMRs (Fig. 2D; permutation P-value <0.01). When the abundance of the hyper and hypo-DhMRs were analyzed separately, we found that only the hyper-DhMRs, and not the hypo-DhMRs, contributed to the significant abundance of DhMRs in the 3′ UTR (Fig 2D; permutation P-value <0.01). In addition, a significant abundance and depletion of hyper and hypo-DhMRs, respectively, were found in intronic regions of the genome, which also is reflected in the profile of the entire intragenic region (Fig 2D; permutation P-value <0.01). These findings suggest genomic preferences for Cntnap2−/−-specific hyper- and hypo-hydroxymethylation (e.g. the 3′ UTR), with only the intronic sequences containing an inverse correlation in abundance of hyper- and hypo-DhMRs, supporting the hypothesis that the intronic sequences are more dynamic in relation to hydroxymethylation in the absence of Cntnap2. Together, these data indicate that Cntnap2−/−-specific hydroxymethylation primarily resides in genic regions of the genome, suggesting that Cntnap2−/−-specific changes are not randomly distributed throughout the genome.
Annotation of DhMRs to genes reveals known and potentially novel autism genes
Annotation of the DhMRs to genes revealed 1843 and 1399 genes that contain only hyper- and hypo-DhMRs, respectively (Fig. 3A; Supplementary Material, Dataset S2). Five hundred and ninety-three genes were found to have more than one DhMR, and 312 genes were identified with both hyper- and hypo-DhMRs, meaning these genes contained regions with both increases (hyper) and decreases (hypo) in 5hmC in the absence of CNTNAP2 (Fig. 3A; Supplementary Material, Dataset S2). Notably, the majority (>80%) of inversely abundant DhMRs were >100 kb away from each other, suggesting that when found on the same gene hyper- and hypo-DhMRs are not located near each other and may have unique roles in the absence of CNTNAP2. An initial observation of the DhMR-associated genes revealed several loci related to Cntnap2 and autism, such as contactin family members (e.g. Cntnap1 and Cntnap4) as well as Nrxn1, Reln and several glutamate receptors (Fig. 3B, DhMR custom genome browser tracks can be found at http://alischlab.psychiatry.wisc.edu/) (39,40). As a means to examine whether the DhMR-associated genes are enriched for autism-related genes, we compared them with a recently updated list of developmental brain disorder genes, which included all current genes that are strongly linked to autism (N = 233 genes; Materials and Methods; C.L. Martin, personal communication). This comparison revealed that a significant number of known autism genes (N = 68 of 233; χ2 test, P-value <0.01) harbor DhMRs in the absence of CNTNAP2, suggesting that 5hmC has an autism-related molecular role in the absence of CNTNAP2. Moreover, these findings also potentially revealed novel genes contributing to the autism phenotype (i.e. a subset of the non-autism-related genes; N = 3486; Fig. 3C). Notably, the DhMR-associated genes also included several genes known to function in epigenetic pathways, including Tet3, Dnmt3a, Hdac7 and Hdac9. Interestingly, expression of CNTNAP2 in the WT brain was first detected by western blot around embryonic day 14 (36). Together, these data indicate that the loss of CNTNAP2 disrupts known autism-related genes and diverse epigenetic pathways, including Tet3, which mediates the oxidation of 5mC to 5hmC in the fetal brain near the same time that Cntnap2 expression begins.
Figure 3.

Annotation of DhMRs to genes. (A) DhMR-associated genes. Venn diagram showing the proportion and overlap of hyper-DhMR-associated genes (N = 1843) and hypo-DhMR-associated genes (N = 1399). The overlap indicates that some genes have both hyper- and hypo-DhMR-associated genes (N = 312). (B) The relative location of DhMRs annotated to two biologically relevant genes. Each gene structure shown contains the transcription start site (broken arrow) and the relative positions of exons (vertical black bar), introns (line connecting exons) and DhMRs (gray boxes), which are below [WT-specific (hypo), Neurexin1] or above [Cntnap2-specific (hyper), Reelin] the gene diagram. (C) Comparison of known autism-related genes. Venn diagram showing the proportion and overlap of hyper- and hypo-DhMR-associated genes and known autism-related genes (N = 233). (D–E) Top 15 GO BPs associated with the Cntnap2-specific (hyper)- and WT-specific (hypo)-DhMR-associated genes (D and E, respectively, ordered by statistical significance from the top of each figure). X-axis indicates the number of Cntnap2-specific (hyper)- and WT-specific (hypo)-DhMR-associated genes that are in each GO term. The shade of the bars shows the P-value based on the legend, as determined by a Fischer test. The % column indicates the proportion of DhMR-associated genes in the GO term compared with the number of genes forming the GO term.
To further examine the biological significance of finding DhMRs associated with these identified genes, we performed gene ontology (GO) analysis of the hyper- and hypo-DhMR genes (N = 2155 and 1711, respectively) and found a significant enrichment of neuronal ontological terms from both gene sets, including axonogenesis, axon development and neuron projection morphogenesis (Fig. 3D and E; permutation P-value <0.01; Supplementary Material, Datasets S3 and S4; Materials and Methods). However, there is a distinct difference among the top GO terms for the hyper- and hypo-DhMR-associated genes: the top hyper-DhMR-associated terms are predominately related to neuronal cell growth and development, whereas the top hypo-DhMR-associated terms involve transporters and ion channels (Fig. 3D and E). Together, these data suggest an intricate regulation of particular cellular pathways in the absence of CNTNAP2.
Characterization of DhMRs in an autism mouse model
Syndromic forms of autism often involve the disruption of transcription factor function (41,42). Thus, we investigated the potential for the Cntnap2−/−-related DhMRs to alter DNA-binding of transcription factors by testing for enrichments of the known transcription factor sequence motifs among the DhMRs, as compared with the total 5hmC peaks (Materials and Methods). Consistent with the distinct differences among the top GO terms for the hyper- and hypo-DhMR-associated genes, this analysis also revealed that unique transcription factor sequence motifs were enriched in the hyper- and hypo-DhMRs. Notably, many of the transcription factors identified have links to psychiatric-related behaviors and disorders, such as RREB1, KLF and NRF1 (Fig. 4; Supplementary Material, Table S3; see Discussion). Together, these findings suggest that 5hmC may regulate the expression of neuronal genes in the absence of CNTNAP2 by modulating the binding or function of transcription factors.
Figure 4.
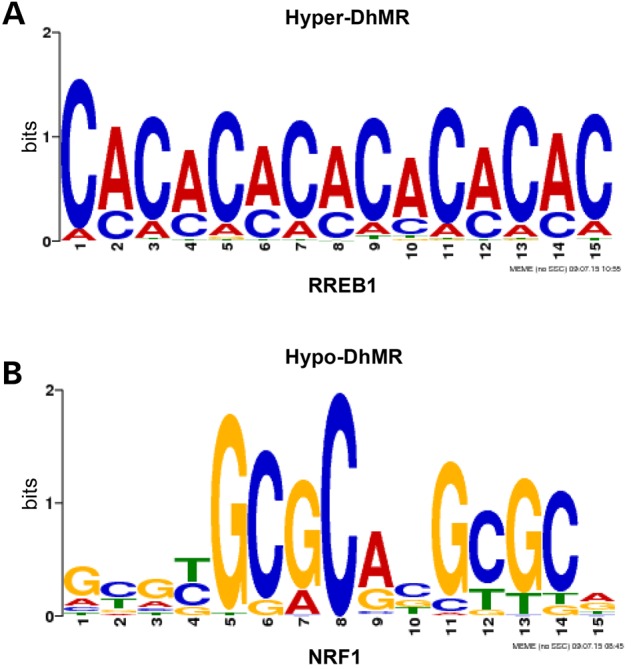
Characterization of the DhMRs in an autism mouse model. (A and B) Identification of DhMR-associated sequence motifs and their putative transcription factors. MEME55 was used to generate adaptive logo plots of RREB1 and NRF1, using the FIMO matches with 5-bp extension on each side as the input (see Materials and Methods).
Finally, the relationship of these data to autism can be summarized through the known synaptic proteins, receptors and signaling pathways associated with ASD (Fig. 5; Supplementary Material, Dataset 5). These pathways contain several known ASD genes with DhMRs (e.g. Nrxn, Nlgn and Reln) as well as a high redundancy of genes with common synaptic and receptor roles containing DhMRs, including genes encoding >20 potassium channels, >10 calcium channels and >10 post-synaptic transmembrane receptors [e.g. gamma-aminobutyric acid (GABA), metabotropic glutamate (mGLU), serotonin, N-methyl-d-aspartate (NMDA), kainate and nicotinic]. Together, these data indicate that the loss of the Cntnap2-encoded protein results in epigenetic disruptions in genes with common roles in ASD pathways, perhaps these genes individually reflect the consequence of losing CNTNAP2 but collectively they produce the autism-like phenotype. Studies focused on the molecular role of 5hmC in these neuroplasticity genes could form the basis of early interventions with the potential of preventing a heightened risk to develop full-blown autism.
Figure 5.

Summary of the DhMRs annotated to genes encoding known synaptic proteins, receptors and signaling pathways linked to ASD. Molecules whose synaptic proteins and receptors are involved in the pathogenesis of ASD. Underline indicates that the genes encoding these proteins and receptors contain DhMRs.
Discussion
Here, we report a genome-wide disruption of 5hmC in the striatum of mice lacking the gene product encoded by Cntnap2, a well-known autism candidate gene. Importantly, these DhMRs significantly reside in known autism-related genes, which serve to validate a role for 5hmC in autism and also reveal potentially novel autism-related genes. While the changes in 5hmC likely represent a complex array of physiological and morphological differences in the striatum of a mouse lacking CNTNAP2, the DhMRs identified here represent a framework that will facilitate the future study of the complex interactions between the genes involved in the pathogenesis and downstream consequences of having autistic-like features, such as those present in the CNTNAP2-deleted mouse model. Moreover, a clearer understanding of the molecular mechanisms regulating the expression of these genes may provide potentially modifiable substrates that ultimately could be targeted to attenuate the progression of autism.
The absence of CNTNAP2 revealed a role for 5hmC in several known autism genes, including Nrxn1, Nlgn, Reln and genes necessary for GABAergic transmission [i.e. Gad1 and Gad2; reviewed in (43)]. As it was previously shown that mice lacking the Cntnap2-encoded protein have a reduction in striatal GABAergic interneurons (36), our data suggest a role for 5hmC in GABAergic interneuron development. In addition, consistent with Cntnap2 being involved in the clustering of potassium channels (34,35), we find DhMRs associated with more than twenty potassium channels. The role of an overwhelming number of DhMRs residing on genes encoding potassium channels is unclear and may result from an unregulated feedback loop in the absence of CNTNAP2. We also observed DhMRs in genes known to function in epigenetic pathways, particularly in Tet3. It is interesting that the expression of Cntnap2 begins at embryonic day 14, which coincides with the timing of Tet3 expression. Further studies are needed to determine the role, if any, of CNTNAP2 on Tet3 expression and 5hmC distribution during this embryonic time period, and beyond. Finally, many of the DhMR-associated genes involve general cell growth processes, including cell adhesion (e.g. cadherin and protocadherin gamma gene families) and cellular developmental processes (e.g. Gdf11 and Pik3r2). Together, these data demonstrate that a remarkable number of known autism-related genes are associated with changes in 5hmC abundance and implicate a role for genes not known to be associated with the autism phenotype. It is of high interest to further examine the molecular contribution of 5hmC in both known and potentially novel autism-related genes.
A descriptive analysis of the hydroxymethylation events found in the striatal tissue revealed that the majority of the hydroxymethylation is in the genic structures (i.e. within the gene body), which is consistent with previous reports of 5hmC (16). However, nearly 20% of the total hydroxymethylation was found in the intergenic region. Despite this somewhat diverse distribution of total 5hmC, the vast majority of DhMRs are found in the genic structures. However, because the role of hydroxymethylation in gene regulation is not fully characterized, these data warrant a deeper investigation of the role of 5hmC on individual gene expression changes caused by the DhMRs in these genomic loci. Notably, we found that the 3′ UTR was enriched for DhMRs. This finding may indicate that 5hmC changes in these regions contribute to direct and/or indirect molecular mechanism(s) that include regulatory sites for the transcription factors that we found associated with the DhMRs. Moreover, noncoding RNAs are highly expressed in the brain and are involved in neurodevelopment and neuroplasticity-related functions; their dysregulation has been described in numerous neurological diseases, including autism (44,45). Hence, future studies should also investigate 5hmC that is not correlated to changes in gene expression, which may reveal that 5hmC mediates the action of a noncoding, but functional, RNA found in the genome.
Several transcription factors that recognize sequence motifs harboring a DhMR have known roles in neurogenesis and neurological activities. For example, the ras-responsive element binding protein 1 (RREB1) is a transcription factor that binds to RAS-responsive elements (RREs), and this binding is disrupted in patients with psychosis (46). The Kruppel-like factor (KLF) family of transcription factors is required for late phase neuronal maturation in the developing dentate gyrus during adult hippocampal neurogenesis (47). While murine studies find associations with Klf-9 and anxiety, human studies link Klf-11 with chronic stress and depressive disorders (47,48). Finally, the nuclear respiratory factor-1 (NRF1) transcription factor mediates neurite outgrowth and was recently shown to regulate several autism-related genes, including Cntnap2 and the fragile X-related 2 gene (49,50). It is notable that the loss of CNTNAP2 resulted in distinct differences between Cntnap2−/−-specific and WT-specific sequence motifs, perhaps suggesting multiple 5hmC roles in transcription factor binding and function. Moreover, Nrf1, and several members of the Klf family, including Klf-9, contain DhMRs, suggesting that 5hmC may also disrupt expression of these transcription factors in the absence of CNTNAP2. Further studies are needed to definitively determine the role of differential hydroxymethylation within transcription factor-binding sites in the autism model.
Annotation of the DhMR-associated genes to gene ontological (GO) functions identified a significant enrichment of neuronal pathways. The biological relevance of this finding was corroborated by the seemingly overabundance of DhMR-associated genes in common pathways related to the ASD neurological phenotype. The Cntnap2 mutants have cortical migration abnormalities and a reduction of GABAergic interneurons (36). As Gad1 and Gad2, as well as GABAergic receptors (e.g. Gabrb1 and Gabbr2), contain DhMRs, our results suggest that 5hmC could be contributing to the altered GABAergic function observed in the Cntnap2 mutants. Substantial dysregulation of Gad mRNA expression, coupled with downregulation of Reln, is observed in schizophrenia and bipolar disorders (51); indeed, Reln also contains a DhMR in the Cntnap2 mutant. In addition, we find DhMRs in several synaptic molecules including neuroligin (52), neurexins (40,53) and Shank (54–56), all of which are linked to autism with mutations resulting in defective transmissions at excitatory and inhibitory synapses, a key mechanism implicated in ASD. In line with this, ASD has been genetically associated with diverse glutamate receptors, and we find DhMRs in many of them, including the kainite receptors (e.g. Grik2) (57), the metabotropic glutamate receptor (e.g. Grm8) and the N-methyl-d-aspartic acid receptor (NMDAR; e.g. Grin2b) (58,59). These data support a role for 5hmC in ASD as well as further implicate known and potentially novel candidate genes contributing to these disorders. Perhaps more importantly, these studies reveal that the loss of CNTNAP2 triggers a cascade of events that includes a genome-wide disruption of 5hmC on molecular substrates involved in neurogenesis, synaptic plasticity, neurite growth and dendritic spine development. Together these findings shed light on possible novel mechanisms for the pathogenesis of autism and may provide modifiable molecular targets for future therapeutic interventions.
Materials and Methods
Mice
Heterozygous male Cntnap2+/− mice were purchased from the Jackson laboratories (Bar Harbor, ME) and maintained on C57BL/6J background. The mice were housed under uniform conditions in a pathogen-free mouse facility with a 12-hour light/dark cycle. Food and water were available ad libitum. All experiments were approved by the University of Wisconsin–Madison Institutional Animal Care and Use Committee (M02529).
Genotyping
Cntnap2 mutants and WT littermates were genotyped using the following primers: Mutant Rev: CGCTTCCTCGTGCTTTACGGTAT, Common: CTGCCAGCCCAGAACTGG, WT Rev 1: GCCTGCTCTCAGAGACATCA. PCR amplification was performed with one cycle of 95°C for 5 min and 31 cycles of 95°C for 30 s, 56°C for 30 s, 68°C for 30 s, 56°C for 30 s and 68°C for 10 min. The mutant allele was obtained with a 350-bp and WT allele with a 197-bp PCR products.
DNA extraction
Seven-week old male Cntnap2−/− mice and their WT littermates (N = 3 per group) were sacrificed (2 h after lights on), and whole brains were extracted and immediately flash-frozen in 2-methylbutane and dry ice. Striatum tissue was excised by micropunch (1.53 to −0.95 mm posterior to bregma), and ∼30 milligrams of tissue was homogenized with glass beads (Sigma) and DNA was extracted using AllPrep DNA/RNA mini kit (Qiagen).
5hmC Enrichment of genomic DNA
Chemical labeling-based 5hmC enrichment was described previously (28). Briefly, a total of 10 µg of striatum DNA was sonicated to 300 bp and incubated for 1 h at 37°C in the following labeling reaction: 1.5 µl of N3-UDPG (2 mm); 1.5 µlβ of -GT (60 µm) and 3 µl of 10× β-GT buffer, in a total of 30 µl. Biotin was added and the reaction was incubated at 37°C for 2 h prior to capture on streptavidin-coupled dynabeads (Invitrogen, 65001). Enriched DNA was released from the beads during a 2-h incubation at room temperature with 100 mm DTT (Invitrogen, 15508013), which was removed using a Bio-Rad column (Bio-Rad, 732-6227). Capture efficiency was ∼5–7% for each sample.
Library preparation and high-throughput sequencing
5hmC-enriched libraries were generated using the NEBNext ChIP-Seq Library Prep Reagent Set for Illumina sequencing, according to the manufacturer's protocol. Briefly, the 5hmC-enriched DNA fragments were purified after the adapter ligation step using AMPure XP beads (Agencourt A63880). An Agilent 2100 BioAnalyzer was used to quantify the amplified library DNA and 20-pM of diluted libraries were used for sequencing. 50-cycle single-end sequencing was performed by Beckman Coulter Genomics. Image processing and sequence extraction were done using the standard Illumina Pipeline.
Analysis of 5hmC data: sequence alignment, fragment length estimation and peak identification
The sequences were mapped to mouse NCBI37v1/ mm9 reference genome using Bowtie 0.12.7 (60), allowing at most two mismatches in the whole read, and only the uniquely mapped reads were kept. The fragment length of each sample were estimated using R package SPP (61). MOSAiCS was used to call peaks from each subject (62). MOSAiCS adjusts for mappability, GC contents and ambiguity score in peak identification, which is very useful when input data are not available. Default parameters were used except for the following: bin size of 300 bp, threshold of ChIP read counts at 95th quantile, FDR level at 0.05, allowing at most five reads with the same starting position, the maximal gap in a peak as 300 bp and the fragment length as estimated by SPP. R package segvis (63) was then used to search for the summit of each peak, and the peaks were redefined as 500 bp on each side of the summit. The peaks for each group were defined as follows using the peaks from all three subjects: the peaks from all subjects in this group were pooled together, the overlapping ones were merged and one such region was called a peak for one group if it overlapped with the peaks from at least two subjects in this group.
Identification of differentially hydroxymethylated regions (DhMRs)
The peaks of both Cntnap2−/− and WT groups were pooled and merged to form the candidate regions, and then Bioconductor package edgeR was used to test whether there was a difference of read counts between the two groups in each candidate region (64). The types of DhMRs (hyper or hypo) were determined by the average log fold change in a normalized read count (logFC) between Cntnap2−/− mutant mice and WT controls.
Annotation of sequence reads, peaks and DhMRs
All the reads were extended to their fragment lengths estimated by SPP. The genomic features and their associated gene symbols were extracted from Bioconductor packages TxDb.Mmusculus.UCSC.mm9.knownGene (65) and org.Mm.eg.db (65). The repetitive elements were downloaded from UCSC genome browser (http://www.repeatmasker.org) (66). A binomial test was used to test the difference in read density over genomic features and repetitive elements, and the over/under-representation of DhMR in each chromosome. For the binomial test for DhMR, the background proportions were calculated from all the peaks in the two groups of animals. Permutation test was used to test the over/under representation of DhMR over genomic features, and the number of subsamples was 105, because one DhMR may overlap with multiple genomic features. In this analysis, the background was also all the peaks from the two groups. Software ngsplot was used to draw the profile plots of the hyper- and hypo-DhMRs and other peaks after slight in-house modification (67).
Enrichment tests of genes and GO analysis
DhMRs were annotated to all genes within 100 kb. To test for an enrichment of autism genes in DhMR-associated genes, we used a chi-square test to compare the DhMR-associated genes to a list of known autism genes (N = 233; C. L. Martin, personal communication). Bioconductor package clusterProfiler was used to test GO biological process (BP) term enrichment with a raw P-value cut-off of 0.05 (68). For these analyses, the gene universe consisted of all the genes associated with 5hmC peaks in both the Cntnap2−/− and WT genomes. To test for an enrichment of neuronal-related GO BP terms among the DhMR-associated GO BP terms, we used a chi-square test and a previously published list of neuronal-related GO BP terms (N = 3046 (69).
Sequence motif analysis
To search for matches between the hyper- and hypo-DhMRs and the known vertebrate transcription factor motifs [N = 205; JASPAR database (70)], we used FIMO (71) and the background peaks with the P-value cut-off of 0.0001. For each genomic feature, a binomial test was used to test the enrichment of each motif in hyper- and hypo-DhMRs overlapping with this genomic feature, where the background proportion were calculated from all the peaks overlapping with this genomic feature. A motif was considered enriched if it satisfied the following criteria: present in at least 20% of DhMRs, enriched at least 1.1-fold compared with the background and satisfied the cut-off false discovery rate (P-value = 0.05). For computational efficiency, only peaks and DhMRs of <2 kb were used in this analysis, which only excluded a small proportion of the regions (∼4%). To generate adaptive logo plots of RREB1 and NRF1, we used MEME and the FIMO matches with 5-bp extension on each side as the input (72). TOMTOM was used to compare these motifs from our data with the one in JASPAR database (73), and the results suggested that the position weight matrices obtained from our data were similar to the counter-parts in JASPAR database (P-value <0.0001), although there was some visual difference in the RREB1 logo plots.
Supplementary Material
Funding
This work was supported in part by the University of Wisconsin-Madison department of Psychiatry (R.S.A.), the University of Wisconsin Neuroscience training grant T32-GM007507 (S.L.), NARSAD Young Investigator Grant from the Brain & Behavioral Research Foundation #22669 (L.P.), and NIH grants HG003747 (S.K.), HG007019 (S.K. and Q.Z.) and U54AI117924 (S.K. and K.C.).
Supplementary Material
Acknowledgements
The authors thank the WISPIC animal facility.
Conflict of Interest statement: None declared.
References
- 1.Association, Psychiatric Association. (2013) Diagnostic and Statistical Manual of Mental Disorders, 5th edn Association, Psychiatric Association, Washington, DC. [Google Scholar]
- 2.Geschwind D.H. (2009) Advances in autism. Annu. Rev. Med., 60, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. et al. (2007) Strong association of de novo copy number mutations with autism. Science, 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature, 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss L.A., Arking D.E., Daly M.J., Chakravarti A. (2009) A genome-wide linkage and association scan reveals novel loci for autism. Nature, 461, 802–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tordjman S., Somogyi E., Coulon N., Kermarrec S., Cohen D., Bronsard G., Bonnot O., Weismann-Arcache C., Botbol M., Lauth B. et al. (2014) Gene x environment interactions in autism spectrum disorders: role of epigenetic mechanisms. Front. Psychiatry, 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li E., Bestor T.H., Jaenisch R. (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell, 69, 915–926. [DOI] [PubMed] [Google Scholar]
- 8.Li E., Beard C., Jaenisch R. (1993) Role for DNA methylation in genomic imprinting. Nature, 366, 362–365. [DOI] [PubMed] [Google Scholar]
- 9.Reik W. (2007) Stability and flexibility of epigenetic gene regulation in mammalian development. Nature, 447, 425–432. [DOI] [PubMed] [Google Scholar]
- 10.Zemach A., McDaniel I.E., Silva P., Zilberman D. (2010) Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science, 328, 916–919. [DOI] [PubMed] [Google Scholar]
- 11.Grayson D.R., Jia X., Chen Y., Sharma R.P., Mitchell C.P., Guidotti A., Costa E. (2005) Reelin promoter hypermethylation in schizophrenia. Proc. Natl Acad. Sci. USA, 102, 9341–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdolmaleky H.M., Cheng K.H., Faraone S.V., Wilcox M., Glatt S.J., Gao F., Smith C.L., Shafa R., Aeali B., Carnevale J. et al. (2006) Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum. Mol. Genet., 15, 3132–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuratomi G., Iwamoto K., Bundo M., Kusumi I., Kato N., Iwata N., Ozaki N., Kato T. (2008) Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol. Psychiatry, 13, 429–441. [DOI] [PubMed] [Google Scholar]
- 14.Poulter M.O., Du L., Weaver I.C., Palkovits M., Faludi G., Merali Z., Szyf M., Anisman H. (2008) GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol. Psychiatry, 64, 645–652. [DOI] [PubMed] [Google Scholar]
- 15.Yao B., Jin P. (2014) Cytosine modifications in neurodevelopment and diseases. Cell. Mol. Life Sci., 71, 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szulwach K.E., Li X., Li Y., Song C.X., Wu H., Dai Q., Irier H., Upadhyay A.K., Gearing M., Levey A.I. et al. (2011) 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci., 14, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szulwach K.E., Li X., Li Y., Song C.X., Han J.W., Kim S., Namburi S., Hermetz K., Kim J.J., Rudd M.K. et al. (2011) Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet., 7, e1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kriaucionis S., Heintz N. (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science, 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munzel M., Globisch D., Bruckl T., Wagner M., Welzmiller V., Michalakis S., Muller M., Biel M., Carell T. (2010) Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew Chem. Int. Ed. Engl., 49, 5375–5377. [DOI] [PubMed] [Google Scholar]
- 20.Song C.X., Szulwach K.E., Fu Y., Dai Q., Yi C., Li X., Li Y., Chen C.H., Zhang W., Jian X. et al. (2011) Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotech., 29, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khare T., Pai S., Koncevicius K., Pal M., Kriukiene E., Liutkeviciute Z., Irimia M., Jia P., Ptak C., Xia M. et al. (2012) 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Biol., 19, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Mahdawi S., Virmouni S.A., Pook M.A. (2014) The emerging role of 5-hydroxymethylcytosine in neurodegenerative diseases. Front. Neurosci., 8, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mellen M., Ayata P., Dewell S., Kriaucionis S., Heintz N. (2012) MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell, 151, 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhubi A., Chen Y., Dong E., Cook E.H., Guidotti A., Grayson D.R. (2014) Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry, 4, e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villar-Menendez I., Blanch M., Tyebji S., Pereira-Veiga T., Albasanz J.L., Martin M., Ferrer I., Perez-Navarro E., Barrachina M. (2013) Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington's disease. Neuromolecular Med., 15, 295–309. [DOI] [PubMed] [Google Scholar]
- 26.Wang F., Yang Y., Lin X., Wang J.Q., Wu Y.S., Xie W., Wang D., Zhu S., Liao Y.Q., Sun Q. et al. (2013) Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington's disease. Hum. Mol. Genet., 22, 3641–3653. [DOI] [PubMed]
- 27.Chouliaras L., Mastroeni D., Delvaux E., Grover A., Kenis G., Hof P.R., Steinbusch H.W., Coleman P.D., Rutten B.P., van den Hove D.L. (2013) Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol. Aging., 34, 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strauss K.A., Puffenberger E.G., Huentelman M.J., Gottlieb S., Dobrin S.E., Parod J.M., Stephan D.A., Morton D.H. (2006) Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med., 354, 1370–1377. [DOI] [PubMed] [Google Scholar]
- 29.Alarcon M., Abrahams B.S., Stone J.L., Duvall J.A., Perederiy J.V., Bomar J.M., Sebat J., Wigler M., Martin C.L., Ledbetter D.H. et al. (2008) Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet., 82, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arking D.E., Cutler D.J., Brune C.W., Teslovich T.M., West K., Ikeda M., Rea A., Guy M., Lin S., Cook E.H. et al. (2008) A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet., 82, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bakkaloglu B., O'Roak B.J., Louvi A., Gupta A.R., Abelson J.F., Morgan T.M., Chawarska K., Klin A., Ercan-Sencicek A.G., Stillman A.A. et al. (2008) Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet., 82, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vernes S.C., Newbury D.F., Abrahams B.S., Winchester L., Nicod J., Groszer M., Alarcon M., Oliver P.L., Davies K.E., Geschwind D.H. et al. (2008) A functional genetic link between distinct developmental language disorders. N. Engl. J. Med., 359, 2337–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott-Van Zeeland A.A., Abrahams B.S., Alvarez-Retuerto A.I., Sonnenblick L.I., Rudie J.D., Ghahremani D., Mumford J.A., Poldrack R.A., Dapretto M., Geschwind D.H. et al. (2010) Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci. Transl. Med., 2, 56ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poliak S., Gollan L., Martinez R., Custer A., Einheber S., Salzer J.L., Trimmer J.S., Shrager P., Peles E. (1999) Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron, 24, 1037–1047. [DOI] [PubMed] [Google Scholar]
- 35.Poliak S., Salomon D., Elhanany H., Sabanay H., Kiernan B., Pevny L., Stewart C.L., Xu X., Chiu S.Y., Shrager P. et al. (2003) Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol., 162, 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penagarikano O., Abrahams B.S., Herman E.I., Winden K.D., Gdalyahu A., Dong H., Sonnenblick L.I., Gruver R., Almajano J., Bragin A. et al. (2011) Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell, 147, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coufal N.G., Garcia-Perez J.L., Peng G.E., Yeo G.W., Mu Y., Lovci M.T., Morell M., O'Shea K.S., Moran J.V., Gage F.H. (2009) L1 retrotransposition in human neural progenitor cells. Nature, 460, 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Secco D., Wang C., Shou H., Schultz M.D., Chiarenza S., Nussaume L., Ecker J.R., Whelan J., Lister R. (2015) Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife., 4, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Persico A.M., D'Agruma L., Maiorano N., Totaro A., Militerni R., Bravaccio C., Wassink T.H., Schneider C., Melmed R., Trillo S. et al. (2001) Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry, 6, 150–159. [DOI] [PubMed] [Google Scholar]
- 40.Kim H.G., Kishikawa S., Higgins A.W., Seong I.S., Donovan D.J., Shen Y., Lally E., Weiss L.A., Najm J., Kutsche K. et al. (2008) Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet., 82, 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bienvenu T., Chelly J. (2006) Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet., 7, 415–426. [DOI] [PubMed] [Google Scholar]
- 42.Gharani N., Benayed R., Mancuso V., Brzustowicz L.M., Millonig J.H. (2004) Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol. Psychiatry, 9, 474–484. [DOI] [PubMed] [Google Scholar]
- 43.Won H., Mah W., Kim E. (2013) Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front. Mol. Neurosci., 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kerin T., Ramanathan A., Rivas K., Grepo N., Coetzee G.A., Campbell D.B. (2012) A noncoding RNA antisense to moesin at 5p14.1 in autism. Sci. Transl. Med., 4, 128ra140. [DOI] [PubMed] [Google Scholar]
- 45.Qureshi I.A., Mehler M.F. (2012) Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci., 13, 528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohadi M., Mirabzadeh A., Esmaeilzadeh-Gharehdaghi E., Rezazadeh M., Hosseinkhanni S., Oladnabi M., Firouzabadi S.G., Darvish H. (2012) Novel evidence of the involvement of calreticulin in major psychiatric disorders. Prog. Neuro-psychopharmacol. Biol. Psychiatry, 37, 276–281. [DOI] [PubMed] [Google Scholar]
- 47.Scobie K.N., Hall B.J., Wilke S.A., Klemenhagen K.C., Fujii-Kuriyama Y., Ghosh A., Hen R., Sahay A. (2009) Kruppel-like factor 9 is necessary for late-phase neuronal maturation in the developing dentate gyrus and during adult hippocampal neurogenesis. J. Neurosci., 29, 9875–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duncan J., Johnson S., Ou X.M. (2012) Monoamine oxidases in major depressive disorder and alcoholism. Drug Discov. Ther., 6, 112–122. [PubMed] [Google Scholar]
- 49.Mahishi L., Usdin K. (2006) NF-Y, AP2, Nrf1 and Sp1 regulate the fragile X-related gene 2 (FXR2). Biochem. J., 400, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiocchetti A.G., Kopp M., Waltes R., Haslinger D., Duketis E., Jarczok T.A., Poustka F., Voran A., Graab U., Meyer J. et al. (2015) Variants of the CNTNAP2 5′ promoter as risk factors for autism spectrum disorders: a genetic and functional approach. Mol. Psychiatry, 20, 839–849. [DOI] [PubMed] [Google Scholar]
- 51.Woo T.U., Walsh J.P., Benes F.M. (2004) Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch. Gen. Psychiatry, 61, 649–657. [DOI] [PubMed] [Google Scholar]
- 52.Jamain S., Quach H., Betancur C., Rastam M., Colineaux C., Gillberg I.C., Soderstrom H., Giros B., Leboyer M., Gillberg C. et al. (2003) Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet., 34, 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., Senman L. et al. (2007) Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet., 39, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durand C.M., Betancur C., Boeckers T.M., Bockmann J., Chaste P., Fauchereau F., Nygren G., Rastam M., Gillberg I.C., Anckarsater H. et al. (2007) Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet., 39, 25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berkel S., Marshall C.R., Weiss B., Howe J., Roeth R., Moog U., Endris V., Roberts W., Szatmari P., Pinto D. et al. (2010) Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet., 42, 489–491. [DOI] [PubMed] [Google Scholar]
- 56.Sato D., Lionel A.C., Leblond C.S., Prasad A., Pinto D., Walker S., O'Connor I., Russell C., Drmic I.E., Hamdan F.F. et al. (2012) SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Gen., 90, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jamain S., Betancur C., Quach H., Philippe A., Fellous M., Giros B., Gillberg C., Leboyer M., Bourgeron T. (2002) Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry, 7, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O'Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K. et al. (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science, 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmead B., Trapnell C., Pop M., Salzberg S.L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kharchenko P.V., Tolstorukov M.Y., Park P.J. (2008) Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotech., 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuan P.F., Chung D., Pan G., Thomson J.A., Stewart R., Keles S. (2009) A statistical framework for the analysis of ChIP-Seq data. J. Am. Stat. Assoc., 106, 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Welch R., Keles S. (2015), R package, in press.
- 64.Robinson M.D., McCarthy D.J., Smyth G.K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carlson M. (2015) Bioconductor, R. (ed.), in press.
- 66.Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E. (2001) Segmental duplications: organization and impact within the current human genome project assembly. Genome Res., 11, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen L., Shao N.Y., Liu X., Maze I., Feng J., Nestler E.J. (2013) diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS One., 8, e65598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu G., Wang L.G., Han Y., He Q.Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. Omics., 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Geifman N., Monsonego A., Rubin E. (2010) The Neural/Immune Gene Ontology: clipping the Gene Ontology for neurological and immunological systems. BMC Bioinformatics, 11, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H. et al. (2014) JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucl. Acids Res., 42, D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grant C.E., Bailey T.L., Noble W.S. (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics, 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bailey T.L., Johnson J., Grant C.E., Noble W.S. (2015) The MEME Suite. Nucl. Acids Res., 43, W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S. (2007) Quantifying similarity between motifs. Genome Biol., 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.