

Abstract
The regulator of G protein signaling 10 (RGS10) protein is a GTPase activating protein that accelerates the hydrolysis of GTP and therefore canonically inactivates G proteins, ultimately terminating signaling. Rheb is a small GTPase protein that shuttles between its GDP- and GTP-bound forms to activate mTOR. Since RGS10 suppression augments ovarian cancer cell viability, we sought to elucidate the molecular mechanism. Following RGS10 suppression in serum-free conditions, phosphorylation of mTOR, the eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), p70S6K and S6 Ribosomal Protein appear. Furthermore, suppressing RGS10 increases activated Rheb, suggesting RGS10 antagonizes mTOR signaling via the small G-protein. The effects of RGS10 suppression are enhanced after stimulating cells with the growth factor, lysophosphatidic acid, and reduced with mTOR inhibitors, temsirolimus and INK-128. Suppression of RGS10 leads to an increase in cell proliferation, even in the presence of etoposide. In summary, the RGS10 suppression increases Rheb-GTP and mTOR signaling in ovarian cancer cells. Our results suggest that RGS10 could serve in a novel, and previously unknown, role by accelerating the hydrolysis of GTP from Rheb in ovarian cancer cells.
Keywords: Regulator of G protein Signaling 10 protein (RGS10), lysophosphatidic acid, mTOR, 4E-BP1 and Rheb
1. INTRODUCTION
When external agonists bind to GPCRs, signal transduction occurs through heterotrimeric G-proteins. This critical event must be effectively regulated, which is the canonical role of Regulators of G protein signaling (RGS) proteins – to attenuate signaling. Upon agonist binding to a GPCR, its structural conformation changes, which elicits intracellular signaling cascades. This occurs through G protein alpha subunits, which exchange bound GDP for GTP and dissociate from G protein beta and gamma subunits to transmit signals. RGS proteins are GTPase activating proteins (GAPs) that accelerate the hydrolysis of GTP from G alpha protein subunits, acting as ‘off’ switches for GPCR-mediated signaling cascades. Therefore, RGS proteins control the master switch that regulates signal duration. When they inactivate G proteins they are preventing endless intracellular signaling. The canonical function of the RGS10 protein is to increase the hydrolysis of GTP from the activated forms of Gαi3, Gαo and Gαz, but not Gαs (1), and terminate signaling.
Herein, we observed a previously unknown function of RGS10 as a regulator of an important intracellular signaling event, which does not directly involve G-alpha proteins. Indeed, other studies have documented non-canonical roles of RGS proteins. For example, these alternative roles include acting as guanine nucleotide exchange factors (RGS-GEFs) and serving as a signaling liaison between tyrosine kinases and G-proteins of both the G-alpha and Ras superfamilies (2). Other studies suggest RGS proteins have GAP-independent functions in undefined cellular processes (3). Thus, the role of RGS proteins is likely more multifaceted than currently appreciated.
Rheb, the Ras homolog enriched in brain protein, is a member of the small GTPase superfamily of monomeric proteins. The function of Rheb is to bind to and activate mTOR (4) and thus, it is essential for coupling all upstream signals to this pathway (5,6). In this regard, Rheb also facilitates the phosphorylation of eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), ultimately regulating cell growth (7). Rheb is regulated in part by the tuberous sclerosis complex 2 (TSC2) protein (8), which inhibits Rheb using its GAP activity (9). TSC2 is a known tumor suppressor that forms a complex with TSC1 and negatively regulates mTORC1. Experimental data linking mutations to critical residues in the proteins suggests that Rheb is only able to activate mTOR when in its GTP-bound state (10). Thus, any protein with GAP activity that facilitates the rapid hydrolysis of GTP from Rheb is an important regulator of the mTOR signaling pathway.
The mTOR protein, which is an abbreviation for the mammalian (or mechanistic) target of rapamycin, is a serine/threonine-protein kinase in the phosphatidylinositol 3-kinase (PI3-K)/Akt signaling pathway. As such, mTOR is a leading signal integrator and thus a master regulator of cell physiology. It also forms the catalytic subunit of two intracellular complexes, mTORC1 (mTOR-Raptor-GβL) and mTORC2 (mTOR-Rictor-GβL), which regulate the processes of growth, survival and protein translation. The mTORC1 complex also phosphorylates 4E-BP1, which allows it to govern multiple cellular processes, such as growth. In addition, mTOR can also activate Akt, which results in an increase in cell survival and proliferation.
In more recent years, highly sophisticated inhibitors are being used to target proteins that are involved in key translational complexes. Rapamycin and its analogues temsirolimus and everolimus are inhibitors of mTOR that rapidly prevent the activity of mTORC1 and also cause a progressive shutdown of mTORC2. Inhibition of mTOR signaling has shown promise as a cancer therapeutic in several different cancer types. A dual mTORC1/2 inhibitor, INK128, inhibits cancer cell migration by decreasing the expression of four critical pro-invasion genes: Y-box binding protein, vimentin, metastasis associated 1 and CD44 (11–13).
We recently showed that the suppression of RGS10 expression is involved in mediating chemoresistance in ovarian cancer cells (14). In this earlier study we demonstrate that chemotherapy-induced cell toxicity is significantly altered by RGS10 reduction, which allows cells to survive at much higher drug concentrations compared to cells with unmodified RGS10. Furthermore, the suppression of RGS10 expression occurs through epigenetic modulation via histone de-acetylation in tumorigenesis and DNA methylation in chemoresistance (15). Taken together, our previous studies imply that cancer cells have the ability to epigenetically modify RGS10 protein expression under toxic insults for enhanced viability and ultimately cell survival.
Most notably, the molecular mechanism explaining RGS10’s influence over cell signaling pathways that enhance viability and survival was previously unknown. Herein we provide evidence of RGS10 binding to Rheb and thereby affecting the mTOR signaling pathway. Upon suppression of RGS10 there is a significant increase in activated Rheb bound to GTP, which results in enhanced mTORC1 activity and phosphorylation of 4E-BP1, mTOR, p70S6K and the ribosomal protein S6. The functional outcome of RGS10 suppression in the short term (<48 hours) is an enhancement of cell proliferation and growth quantified by significantly larger cells when stimulated with lysophosphatidic acid. Since the canonical function of RGS10 is to regulate G alpha subunits proceeding from GPCR signaling, this is the first study to demonstrate a new facet and mechanistic role of RGS10 and propose it regulates mTORC1 by serving as a GAP, or an ‘off switch’ for Rheb.
2. MATERIALS AND METHODS
2.1 Materials
Ascites-isolated SKOV-3 ovarian cancer cells were purchased from American Type Culture Collection (Manassas, VA) and cultured at 37° C in the presence of 5% CO2 in DMEM medium supplemented with 10% fetal bovine serum (FBS) and antibiotics (Mediatech Inc., Manassas, VA). HeyA8 cells were kindly gifted from Dr. Isaiah J. Fidler (The University of Texas M.D. Anderson Cancer Center, Houston, TX) and cultured in RPMI medium supplemented with 10% FBS (Mediatech). Cells isolated from the ovary, OVCAR-3 were purchased from the American Type Culture Collection and cultured in RPMI supplemented with 10% FBS. INK-128 and temsirolimus were purchased from Cayman Chemical (Ann Arbor, MI). Lysophosphatidic acid (LPA, 18:1, 1-oleoyl-2-hydroxy-sn-glycero-3-phosphate) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and reconstituted in charcoal-stripped, 0.1% fatty acid free BSA immediately prior to use.
2.2 Reducing RGS10 expression
SKOV-3 and HeyA8 ovarian cancer cells were plated in 6-well dishes at 120,000 cells/well and 100,000 cells/well, respectively. The plated cells were incubated for approximately 18 h at 37°C in 5% CO2 and then transfected with siGENOME RISC-free control (siRISC) or Dharmacon SmartPools siRNA targeting RGS10 (ThermoFisher Scientific), following manufacturer’s recommended protocol. The siRISC is chemically modified to impair processing and uptake by RISC, which isolates cellular effects related to transfection, but unrelated to siRGS10. In other experiments requiring transfection in a 96-well plate, 100 nM concentration of siRNA and 0.25 μL of Dharmafect 1 transfection reagent (ThermoFisher Scientific) were used per well. Transfection medium was replaced with DMEM medium with 10% FBS after 8 h. Transfected cells were incubated for another 30 h and all assays were performed approximately 48 h post-transfection. In other experiments, stable cell lines were created with shGFP vector and shRNA for RGS10 in HeyA8 Parental cells using SureSilencing shRNA Plasmid for Human RGS10 (SABiosciences, Qiagen, Valencia, CA). HeyA8 cells were transfected with shRNA constructs using Fugene (Promega, Madison, WI) at a 3:1 plasmid to transfection reagent ratio.
2.3 Immunoblotting
Transfected cells were incubated for an additional 24 h and then serum-starved overnight. After 48 h post-transfection, specified conditions were treated with lysophosphatidic acid (10 μM), temsirolimus (10 μM) or INK128 (10 μM) as indicated. The cells were lysed in buffer containing protease/phosphatase inhibitor cocktail (Cell Signaling Technology, Danvers, MA) and processed for SDS-PAGE. After transferring the denatured proteins to nitrocellulose membranes, the blots were probed with primary antibodies for either total mTOR, phospho-mTOR (Ser2448) XP Rabbit mAb #5536, total 4E-BP-1, phospho-4E-BP1 (Thr37/46) #2855, GAPDH, β-Actin (8H10D10) Mouse mAb #3700, phospho-Akt (Ser473) XP Rabbit mAb #4060, phospho-p70 S6 kinase (Thr389) Rabbit mAb #9234, phospho-p70 S6 kinase (Ser371) antibody #9208, phospho-S6 ribosomal protein (Ser235/236) Rabbit mAb #4858, and phospho-eIF2α (Ser51) XP Rabbit mAb #3398 (all from Cell Signaling Technology) or RGS10 (Santa Cruz Biotechnology, Dallas, TX). After blotting for p4E-BP1, the membrane was washed extensively with TBS-T and re-probed with total 4E-BP1. All other stripping occurred with 30 min incubation in buffer (2% SDS, 62.5 mM Tris-HCl (pH 6.8) and 100 mM 2-mercaptoethanol). For confirmation purposes, non-phosphorylated as well as phosphorylated p70 S6 kinase control cell extracts from MCF-7 cells (+/− insulin treatment) #9203 were used as positive and negative controls that were probed with phospho-p70 S6 kinase (Thr389) and phospho-(Ser371) primary antibodies. Blots were incubated for 4 h with either total- or phospho-mTOR or total- or phospho-AKT diluted 1:1000 in 5% w/v BSA in 1X TBS-T at room temperature. The membrane was washed with TBS-T and probed with secondary anti-rabbit HRP conjugated antibody (Amersham ECL detection kit, GE Health Life Sciences, Piscataway, NJ) diluted 1:2000 in 2% milk TBS-T and incubated for 2 h at room temperature. Other membranes were incubated for 2.5 h with phospho-4E-BP1 primary diluted 1:1000 in 5% w/v BSA at room temperature. Following primary incubation, the blot was washed with TBS-T and probed with secondary anti-rabbit HRP conjugated antibody diluted 1:5000 in 2% w/v BSA and incubated for 2.5 h at room temperature. For detection of RGS10, membranes were incubated for 1.5 h with goat anti-RGS10 primary antibody diluted in 5% w/v BSA in 1X TBS-T at room temperature. Secondary donkey anti-goat HRP conjugated antibody was diluted in 1% w/v BSA in 1X TBS-T and the membrane was incubated for 1.5 h at room temperature. For detection of total and phospho-p70 S6, blots were incubated for 1.5 h at room temperature in the primary antibody diluted 1:1000 in 5% w/v BSA in 1X TBS-T. Other blots were washed with TBS-T and probed with anti-mouse HRP conjugated secondary antibodies (Amersham) diluted 1:2000 in 2.5% milk TBS-T and incubated for approximately 2 h or anti-rabbit HRP conjugated secondary antibodies diluted 1:2000 in 2% milk w/v in 1X TBS-T and incubated for 1.5 h at room temperature. The proteins were detected using a Fluorchem HD2 chemiluminescent imaging system (Protein Simple, Santa Clara, CA) with SuperSignal™ West Dura Extended Duration Substrate (ThermoFisher Scientific). Protein bands were quantified using Image J (National Institutes of Health, Bethesda, MD). Representative blots are shown and these reflect data that was repeated at least four times.
2.4 Evaluation of RGS10 expression
RNA was collected from cells using TRIzol® Reagent (Life Technologies, Gran Allen, NY) according to manufacturer’s protocol. Afterward, RNA was quantified using a NanoDrop2000 Spectrophotometer (ThermoFisher Scientific) and then reverse-transcribed into cDNA using iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Following cDNA synthesis, the cDNA template was then used in a PCR reaction with primers for RGS10 and ImmoMix (Bioline USA Inc., Taunton, MA). The following primers were used for RGS10 forward: 5′- AAC CGC ACC CTC TGA TGT TC -3′ and reverse: 5′- GGC TGT AGC TGT CGT ACT TCA -3′; β2-microglobulin forward: 5′- GTG GCC TTA GCT GTG CTC G -3′ and reverse: 5′- ACC TGA ATG CTG GAT AGC CTC -3′ based on algorithm-generated sequences from Primer Bank (16). To quantify PCR results and visualize peaks, the PCR products were then loaded into an Agilent DNA 1000 chip in gel-dye matrix and analyzed using the Agilent 2100 Bioanalyzer System and software (Agilent Technologies, Santa Clara, CA), which automatically generated peaks.
2.5 Rheb activation assay
SKOV-3 cells were plated in 10 cm dishes at a density of approximately 400,000 cells per dish and incubated overnight at 37°C. The cells were transfected with siRGS10, siRISC, pcDNA, and RGS10 plasmid, where indicated. After 48 h, the culture media was removed and cells were rinsed with ice-cold PBS. Then, 1 mL of ice-cold lysis buffer containing protease and phosphatase inhibitors was added to each dish. Plates were placed on ice for 10–20 min with agitation every 5 min. Lysates were cleared by centrifugation for 10 min at 12,000 × g at 4°C. The protein supernatant was collected and stored at −80°C until quantified by BCA assay. Next, 0.5–1 mL of cell lysate was aliquotted to microcentrifuge tubes and steps were performed according to the manufacturer’s protocol for the Rheb Activation Assay (NewEast Biosciences, Malvern, PA). Briefly, the assay utilizes anti-active Rheb mouse monoclonal antibodies (specifically recognizes Rheb-GTP from vertebrates) during the cell lysate incubation with gentle agitation. Thus, bound and active Rheb is pulled down by protein A/G agarose and precipitated active Rheb is detected using immunoblot analysis with anti-Rheb rabbit polyclonal antibody. For the GTPγS/GDP protein loading, 20 μl of 0.5 M EDTA (20 mM final concentration) was added to each microcentrifuge tube prior to the addition of 5 μl of 100 X GTPγS (positive control) or GDP (negative control) to the appropriate individual tubes, which were then incubated for 30 min at 30°C with agitation. Loading was stopped by putting tubes on ice and adding 32.5 μl of 1 M MgCl2. In a similar experiment, 1 μg of purified human full-length Rheb protein (Abcam, Cambridge, MA) was treated with GDP or GTPγS prior to immunoprecipitation with anti-active Rheb and immunoblotting with total Rheb. Following these procedures, electrophoresis was performed using approximately 20 μL/well of the pull-down supernatant loaded onto a polyacrylamide gel (17%) and protein bands were resolved by immunoblotting on a nitrocellulose membrane. Proteins were detected by ECL using SuperSignal West Pico Chemiluminescent Substrate. Results were repeated and observed three times.
2.6 Measurement of free phosphate generation
In order to assess the RGS10 GAP function on Rheb non-radioactively, we used a malachite green reagent assay to detect free phosphate generation as previously demonstrated (17). Ten μL of assay buffer (50 mM HEPES at pH 7.5, 100 mM NaCl, 5 mM EDTA, 10 mM MgCl2) and 10 μL of purified RGS10 full-length protein (Abcam) were added into a clear 96-well plate. The final concentration of RGS10 was 1 μM. After a 30 min incubation, 10 μL of purified full-length Rheb protein (Abcam) was added. The final concentration of Rheb was 1.625 μM. After a 5 min incubation, 10 μl of GTPγS or GDP were added, where indicated. The final concentration of GTPγS or GDP were 600 μM. After 120 min incubation, the Malachite Green Phosphate assay was performed following the protocol provided by the manufacturer (Cayman). After a 20 min incubation, the absorbance of each well and the phosphate standards, which are provided by the kit, were determined by SpectraMax M2 plate reader (Molecular Devices, Sunnyvale, CA) at 620 nm.
2.7 Immunofluorescence
SKOV-3 cells were seeded on 12 mm coverslips in a 24-well plate at a density of 50,000 cells per well. After 24 h, cells were transfected with siRGS10 or siRISC as indicated. After 24 h, cells were serum starved overnight, then left untreated or treated with 10 μM temsirolimus for approximately 16 h, and then pulse treated with lysophosphatidic acid for 30 min at a concentration of 10 μM per well. After pulse treatment, cells were fixed with 3.7% formaldehyde in PBS for 15 min. The cells were then washed at least once with PBS and permeabilized with 0.1% Triton X-100 in PBS for 15 min. After permeabilization, cells were washed twice with PBS prior to exposure to the Whole Cell Stain Green solution (ThermoFisher Scientific) for 30 min in the dark at room temperature. Subsequently, cells were washed twice with PBS and the coverslips were mounted on the slides. Images were taken using the Zeiss LSM 710 Confocal Microscope (Zeiss, Thornwood, NY). Each condition had duplicates, and ≥5 fields were analyzed per coverslip using ZEN 2011 software. Experiments were repeated multiple times and the data reflects pooled results.
2.8 Quantification of cell perimeter and number
SKOV-3 cells were seeded in a 96-well plate at a density of 3,000 cells per well. After 24 h, cells were transfected with siRGS10 or siRISC as indicated. After 30 h, cells were either left untreated or treated with temsirolimus for approximately 16 h and then pulse treated with lysophosphatidic acid for 30 min at a concentration of 10 μM per well. After pulse treatment with LPA, cells were fixed with 50 μL of 3.7% formaldehyde in PBS for 15 min. The cells were then washed once with 50 μL of PBS and permeabilized with 50 μL of 0.1% Triton X-100 in PBS for 15 min. After permeabilization, cells were washed twice with 50 μL PBS prior to exposure to 50 μL of the Whole Cell Stain Solution (ThermoFisher Scientific) for 1 h in the dark at room temperature. Subsequently, cells were washed twice with 50 μL of PBS and then treated with 50 μL of DAPI in PBS (1:2000) for 20 min. After 20 min, DAPI was aspirated, 50 μL of PBS was added to each well, and the plate was sealed for high-throughput scanning using the Cellomics ArrayScan VTI High Content Analysis Reader (ThermoFisher Scientific). Each condition had at least three or more replicates, and ≥5 fields were analyzed per well using high content screening analysis software. Experiments were repeated multiple times and the data reflects pooled results. The automated software determines the average cell colony perimeter and the number of valid cells per well. The results were graphed with GraphPad Prism (LaJolla, CA) as the average. The cell area data was averaged and normalized to display only the change in area compared to control conditions. The assessment of cell count was measured as previously described (18).
2.9 Assessment of active cell proliferation
Approximately 4,000 HeyA8 and 5,000 SKOV-3 cells were plated into 96-well plates in the presence of 10% fetal bovine serum. The plated cells were incubated for 18 h at 37°C in 5% CO2 and then transfected with siRNA for RISC or RGS10. Transfected cells were incubated for an additional 24 h prior to treatment with etoposide, nacodazole, aphidicolin, paclitaxel or BrdU, where indicated. For the BrdU treatment, cells were pulse treated for 1 h with a BrdU analog, whereas all other conditions were treated with drugs for approximately 4 h. Cells were then stained according to the protocol provided by the manufacturer (Millipore, Billerica, MA). Plates were scanned using Cellomics ArrayScan (ThermoFisher Scientific) and the number of cells was automatically captured. Each condition had 6 replications and 8 fields per well were analyzed using high content scanning software. Representative images are shown reflecting data captured from one of multiple experiments that produced identical results. The data was retrieved from the manufacturer’s software and results were plotted with GraphPad Prism.
2.10 Evaluation of nascent protein synthesis
Approximately 3000 HeyA8 or 2000 SKOV-3 cells were plated into 96-well plates in complete media overnight prior to siRNA transfection for 24 h. The Click-iT® Plus OPP Alexa Fluor® 647 Protein Synthesis Assay Kit (Life Technologies, Grand Allen, NY) was used as a nonradioactive method for the detection of protein synthesis utilizing fluorescence microscopy and high-content imaging. Cells were either untreated or treated with LPA (10μM) for 60 min prior to fixation and subsequent following of the protocol provided by the manufacturer. Approximately 200 (SKOV-3) or >500 (HeyA8) total cells were automatically quantified for the Alexa Fluor® 647 fluorescence intensity, which corresponds to protein synthesis, using high-content imaging. The data is presented as the average protein synthesis in bar graphs and representative images of HeyA8 cells are depicted in the figure.
In other experiments, approximately 3000 SKOV-3 cells were plated into 96-well plates with methionine/serum-free medium for 18 h prior to siRNA transfection and another 24 h incubation. The Click-iT® AHA Alexa Fluor® 488 Protein Synthesis HCS Assay Kit (Life Technologies) was used as a method for the detection of nascent protein synthesis utilizing fluorescence microscopy and high-content imaging. Essentially, an amino acid analog of methionine containing an azido moiety, which is similar to 35S-methionine but not radioactive, is fed to cultured cells in 96-well plates. The following day, approximately 48 h post-transfection, cells were treated with LPA (10μM) for 30 min or left untreated. Nascent proteins were detected with the green-fluorescent Alexa Fluor® 488 alkyne and imaged using the Cellomics ArrayScan (ThermoFisher Scientific). Data is presented as supplementary images.
2.11 Statistics
The statistical differences were analyzed using an analysis of variance (ANOVA) test, followed by Bonferroni’s multiple comparison tests between groups using GraphPad Prism. When comparing only two groups, the Student’s t-test was used. Where it is indicated in the figures, *p < 0.05 **p < 0.01 and ***p < 0.001 indicate the levels of significance.
3. RESULTS
The goal of our study was to identify the molecular mechanism linking RGS10 suppression to cell viability and resistance to chemotherapy-induced cell toxicity. To commence the investigation, we measured the phosphorylation of proteins downstream of major signaling pathways after RGS10 modification under serum-free, low nutrient conditions. The purpose was to determine what is specifically altered as a consequence of RGS10 suppression in microenvironments with low nutrients. Knock down of RGS10 using siRNA resulted in an increase in 4E-BP1 phosphorylation in multiple ovarian cancer cell lines (Fig. 1A). This reproducible increase of ~2.5-fold in HeyA8 cells and ~3.5-fold in SKOV-3 cells among 4E-BP1 phosphorylation is significant in the absence of nutrients and/or agonist stimulation (Fig. 1B). Since OVCAR-3 cells have relatively low levels of endogenous RGS10 protein (Fig. 1C) in comparison with HeyA8 and SKOV-3 cells (Fig. 1D), we did not use these in subsequent experiments.
Figure 1. RGS10 suppression induces the phosphorylation of 4E-BP1 in ovarian cancer cells under low nutrient conditions.
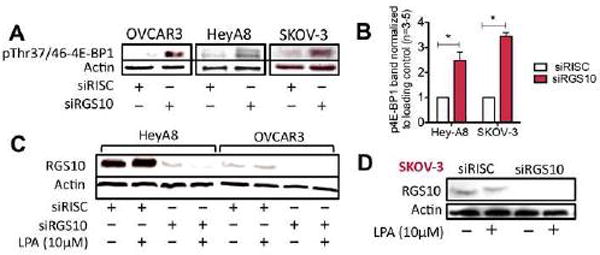
(A) OVCAR-3, HeyA8 and SKOV-3 cells were transfected with the siGENOME RISC-Free control siRNA (siRISC) or siRGS10 and kept in serum-free medium prior to immunoblotting detection for phospho-4E-BP1 at Thr37/46 and actin. (B) Repeated experiments (n=3–5) of phospho-4E-BP1 in HeyA8 and SKOV-3 cells were quantified using ImageJ. A significant increase in the phosphorylation among siRGS10 cells (*p<0.05 vs siRISC) is observed. (C) HeyA8, OVCAR-3 or (D) SKOV-3 cells were processed for detection of RGS10 protein expression and actin.
Since differences in 4E-BP1 phosphorylation were observed in unstimulated conditions, we evaluated 4E-BP1 phosphorylation with external stimulation of the mTOR pathway. It is well established that growth factors activate mTOR through the PI3K/Akt and ERK/MAPK signaling pathways (19); thus, we selected the growth factor lysophosphatidic acid to activate mTOR and phosphorylate 4E-BP1 (20) for all subsequent experiments (SFig. 1). HeyA8 cells were starved of serum overnight and then stimulated with lysophosphatidic acid for 5, 30 or 60 min, prior to the detection of protein phosphorylation (20). Immunoblotting experiments show RGS10 knockdown without any agonist stimulation enhances the phosphorylation of 4E-BP1(Thr37/46), p70S6K(Thr389) and S6 ribosome(Ser235/236) (Fig. 2A). Furthermore, the phosphorylation of mTOR(Ser2448) was also moderately increased with RGS10 suppression and without agonist (Fig. 2B).
Figure 2. Lysophosphatidic acid activates the mTOR signaling pathway, enhancing the effects of RGS10 suppression-induced phosphorylation changes.

(A and B) Hey-A8 cells transfected with siRISC or siRGS10 were treated with 10 μM of lysophosphatidic acid (LPA) for 0, 5, 30 or 60 min prior to immunoblotting of total and phospho-4E-BP1(Thr37/46), RGS10, phospho-eIF2a(Ser51), phospho-S6 ribosomal (S6 Ribo) protein(Ser235/236), total and phospho-p70S6K(Thr389), total and phospho-mTOR(Ser2448), total and phospho-Akt(Ser473), or GAPDH. (C) Hey-A8 cells were transfected with siRISC or siRGS10 and treated with LPA (10 μM, 30 min) or pre-treated with INK-128 (10 μM, 18 h) prior to immunoblotting analysis for phospho-4E-BP1(Thr37/46), phospho-mTOR(Ser2448) or actin.
In contrast, comparisons of Akt(Ser473) and eIF2a(Ser51) phosphorylation between the siRISC (control) and siRGS10 (experimental) conditions show that lysophosphatidic acid stimulation enhanced protein phosphorylation under conditions of RGS10 suppression. Although these experiments were performed in HeyA8 cells, the phosphorylation of mTOR and 4E-BP1 after knockdown of RGS10 was confirmed in other ovarian cancer cell lines (SFig. 2). Taken together, these results corroborate that the mTOR pathway is activated after RGS10 suppression.
Although it is well known that the mTORC1 complex phosphorylates 4E-BP1, we evaluated p4E-BP1 after chemical inhibition of mTOR to verify that the signal was exclusively originating from mTOR to activate 4E-BP1. As previously observed, RGS10 suppression or lysophosphatidic acid stimulation (10 μM) for 30 min elicited phosphorylation of 4E-BP1 and mTOR. In the presence of temsirolimus (10 μM), a modest inhibitor of mTOR kinase activity that binds to the FKBP12-rapamycin binding domain (FRB) (21), both pmTOR and p4E-BP1 are reduced (SFig. 1B and C), but not absent. However, treating the cells with INK128, a dual mTORC1/2 ATP-competitive inhibitor that also blocks p4E-BP1 (12,13), completely abolished p4E-BP1 and reduced pmTOR with RGS10 suppression (Fig. 2C). This eliminates the possibility that the phosphorylation of 4E-BP1 comes from an unidentified source and confirms the role of mTOR.
In order to identify the molecular mechanism explaining how RGS10 suppression influences the phosphorylation of 4E-BP1 and other proteins in the mTOR signaling pathway, we assessed direct regulators of mTOR signaling and observed Rheb activity changes. We repeatedly detected an abundance of activated Rheb bound to GTP when RGS10 is suppressed (Fig. 3A, 1.4 vs 1.0). In comparison to RISC-free control siRNA conditions (siRISC), after suppression of RGS10 and incubation with GTPγS, we observed an increase in the bound GTPγS upon immunoprecipitation of active Rheb (Fig. 3B, ~2.3). Similarly, purified Rheb protein incubated with GTPγS or GDP prior to immunoprecipitation of active, GTP-Rheb and assessment of the total protein, showed an increase (Fig. 3C). This substantiates the specificity of the Rheb antibodies and system used, which was also reported by other groups (22,23). To confirm our results, we performed the reciprocal set of experiments using an RGS10-specific antibody. After RGS10 suppression, the association with Rheb is dramatically reduced in comparison to siRISC control and/or increased with RGS10 expression (Fig. 3D).
Figure 3. Evaluation of the GAP activity of RGS10.
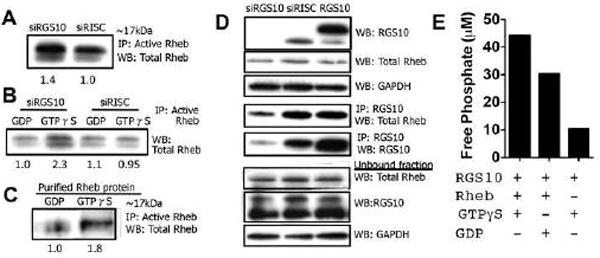
(A) SKOV-3 cells were transfected with either siRISC or siRGS10 prior to immunoprecipitation with an active Rheb monoclonal antibody and immunoblotting with total Rheb rabbit polyclonal antibody. (B) In a separate experiment, cells were treated as indicated above, but GTPγS and GDP were added to the cell extracts in vitro and incubated for 30 min prior to the pull-down of active Rheb and blot for total Rheb (for details see Methods). (C) In the reciprocal experiment, purified, full-length Rheb protein was incubated with GTPγS or GDP prior to immunoprecipitation with active Rheb and immunoblot for total Rheb. (D) Cells were treated as previously described and also transiently transfected with an RGS10 expression vector prior to immunoprecipitation. Following transfection, cell extracts were probed for RGS10, total Rheb and Rheb. Other extracts were immunoprecipitated with anti-RGS10 goat polyclonal antibody and blotted with anti-total Rheb rabbit polyclonal antibody. Unbound fraction is also shown. (E) A non-radioactive method using a malachite green reagent kit was utilized to measure inorganic free phosphate in a cell-free, aqueous solution and assess the GAP activity of RGS10. Purified proteins RGS10 and Rheb were incubated in the presence of GTPγS or GDP, where indicated, for 120 min. When RGS10 induces the hydrolysis of GTP to GDP, free phosphate is released into the solution, which is detected by the formation of a green molybdophosphoric acid complex and measured in a 96-well plate using absorbance that is directly related to the free phosphate release (17).
In addition we used a cell-free system and a non-radioactive, malachite green-based assay to measure the GAP activity of RGS10 (17). Herein we incubated purified RGS10, purified Rheb and GTPγS together, in comparison to GDP incubation or without Rheb in solution. We observed an increase in the amount of free phosphate released with all three (Fig. 3E), which indicates the hydrolysis of GTP to GDP when RGS10, Rheb and GTPγS are together. These data suggest that RGS10 influences the de-activation of Rheb, which also elucidates how RGS10 affects the mTOR signaling pathway.
This pathway is known to regulate cell proliferation. Indeed, when cells were transfected with siRISC or siRGS10, the latter significantly increased the average number of cells observed (Fig. 4A, ***p<0.001). To verify this was active cell proliferation, we constructed a stable HeyA8 cell line expressing either shRGS10 or shGFP in order to visually observed active proliferation using BrdU incorporation (Fig. 4B). Quantification demonstrated more active proliferation among shRGS10-expressing HeyA8 cells (Fig. 4C, ***p<0.001 vs shGFP), which are also unaffected by the presence of etoposide and aphidicolin, but not nocodazole or paclitaxel. Similar results were achieved in SKOV-3 cells (data not shown). This suggests that RGS10 suppression endows cells with a proliferative advantage in the presence of some, but not all, types of chemotherapy. As a consequence, the loss of RGS10 could translate into the acquisition of chemoresistance, which corroborates our previous studies (14).
Figure 4. RGS10 suppression enhances ovarian cancer cell proliferation and protein synthesis.
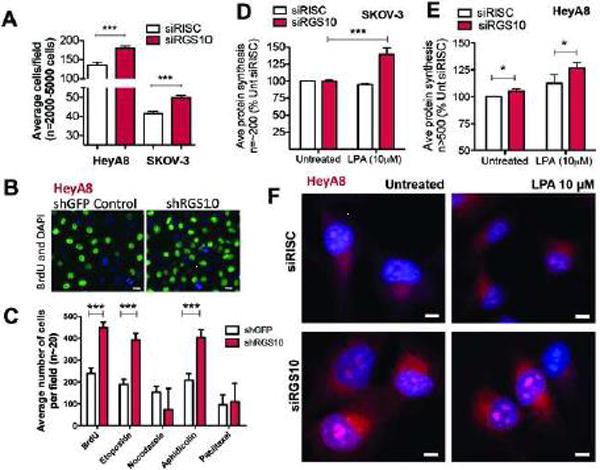
(A) HeyA8 and SKOV-3 cells were transfected with either siRISC or siRGS10 and stained with DAPI. Cells were automatically counted using a high-throughput fluorescence imager and results are quantified as the average number of cells per field. The number of cells (n=2,000 – 5,000) observed among fields (n=15–100) across multiple experiments (n=3–6) were pooled together and displayed as a bar graph using GraphPad Prism. (B) Representative shGFP and shRGS10 HeyA8 cells were pulse-treated for one hour with a BrdU analog. Cells were fixed and then stained with DAPI and an anti-BrdU antibody prior to high-throughput imaging. The representative image shown here visualizes the fluorescence from HeyA8 cells and the difference between active proliferation among control cells and suppression of RGS10. (C) In other experiments, shGFP or shRGS10 stably-expressing HeyA8 cells were treated with different drugs or a BrdU analog prior to fixation, immunofluorescence staining and automatic cell counting. The average number of cells per field is shown and represents ~20 fields and thousands of total cells. ***p<0.001. Approximately (D) 2000 SKOV-3 cells or (E) 3000 HeyA8 were plated in complete media prior to siRNA transfection. The Click-iT® Plus OPP Alexa Fluor® 647 Protein Synthesis Assay Kit was used to measure protein synthesis. Cells were treated with LPA (10μM) for 60 min or left utreated. Approximately 200 (SKOV-3) or >500 (HeyA8) total cells were automatically quantified for fluorescence intensity, which corresponds to protein synthesis, and the data is presented as bar graphs normalized to siRISC untreated conditions (100%). *p<0.05, ***p<0.001. (F) Representative images from the previous experiment to measure protein synthesis are shown in HeyA8 cells.
Since mTOR also regulates factors such as protein synthesis, we next measured nascent protein synthesis after transfection with siRISC or siRGS10, with and without treatment of lysophosphatidic acid. As anticipated, there is an increase in nascent protein synthesis after RGS10 suppression with and/or without lysophosphatidic acid stimulation of mTOR (Fig. 4D, E and F). Because cells usually couple growth and proliferation, the results of RGS10 suppression, corroborate each other.
We next sought to determine whether cells manifested altered morphology in response to RGS10 suppression and lysophosphatidic acid stimulation, especially since mammalian cell size is usually restricted by 4E-BP1 and mTOR. Confocal imaging of cells with or without RGS10 suppression in the presence or absence of lysophosphatidic acid showed visible differences in cell size morphology (Fig. 5A). The bottom right panel shows SKOV-3 cells with RGS10 suppression after 30 min of lysophosphatidic acid treatment, in comparison with siRISC control cells.
Figure 5. RGS10 suppression alters morphology in lysophosphatidic acid-stimulated ovarian cancer cells.
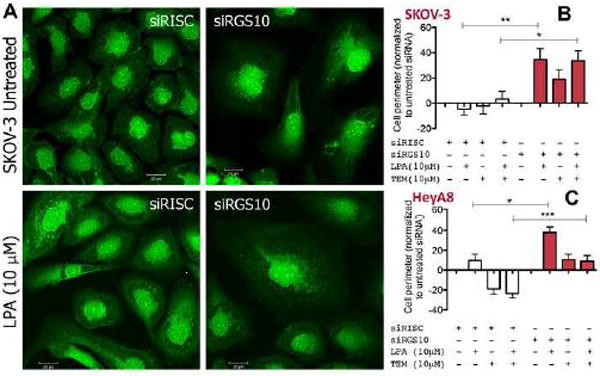
(A) SKOV-3 cells plated on glass coverslips were transfected with either siRISC or siRGS10 and treated with lysophosphatidic acid (10μM) for 30 min prior to preparation for confocal microscopy using the Whole Cell Stain Solution (ThermoFisher Scientific). (B) SKOV-3 or (C) HeyA8 cells were transfected with either siRISC or siRGS10 prior to the automated computer assessment of cell perimeter. Cells were treated with lysophosphatidic acid (LPA, 10 μM, 30 min) or temsirolimus (10 μM, ~16 h) where indicated. Cells were prepared for immunofluorescence using the Whole Cell Stain Solution and automatically scanned with high-throughput software to calculate cell perimeter. Data shows the average from 62–146 different fields per condition, which measures thousands of cells, from a series of replicate experiments and is normalized to each untreated condition (i.e. siRISC-white bars or siRGS10-shaded bars). ***p<0.001, **p<0.01 and *p<0.05 where indicated.
Using high-throughput imaging, computer-automated quantification and immunofluorescence, we measured alterations in cell perimeter that occur with RGS10 suppression in SKOV-3 (Fig. 5B) and HeyA8 cells (Fig. 5C). In these studies, we quantified a change in the perimeter of RGS10-suppressed cells (red bars), which was significantly increased after stimulation with lysophosphatidic acid. In contrast, among siRISC conditions (white bars), treatment with lysophosphatidic acid (30 min, 10μM) did little to change the cell perimeter.
We also observed that HeyA8 cells were comparatively more responsive to inhibition with temsirolimus, than SKOV-3 cells. This is a reflection of the PI3KCA activating mutation carried by SKOV-3 cells that causes constitutively active PI3K signaling due to the continuous phosphorylation of Akt (24). The PI3KCA mutation in SKOV-3 cells diminishes the ability to significantly inhibit this pathway using a modest mTOR inhibitor, in contrast to the more responsive HeyA8 cells, which do not have a PI3KCA mutation and demonstrate a reduction in cell perimeter with temsirolimus treatment. Taken together, this data provides an additional functional outcome governed by the mTOR/4E-BP1 pathway that is recapitulated by stimulation of RGS10-suppressed cells.
4. DISCUSSION
Herein we show that suppression of RGS10 leads to an increase in activated Rheb bound to GTP, which results in the phosphorylation of mTOR, 4E-BP1, p70S6K and ribosomal protein S6. To our knowledge, this is the first report in the literature documenting the association of RGS10 with Rheb. The canonical functions of RGS proteins most often occur at the plasma membrane to inactivate G alpha proteins stimulated by agonist-induced GPCRs. In contrast, we report an effect of RGS10 that would not occur along the plasma membrane, nor is it dependent upon agonist stimulation of a GPCR.
In this study, we repeatedly demonstrate that RGS10 suppression increases protein phosphorylation within the mTOR signaling pathway. As a functional consequence of this, ovarian cancer cells increase proliferation, nascent protein synthesis and cell perimeter when RGS10 is suppressed and/or lysophosphatidic acid stimulates the cells. The results also further corroborate the ability of RGS10 suppression to override inhibitory effects of specific types of chemotherapy, such as etoposide, which extends the observations we previously reported (14).
We observed that RGS10 suppression in low nutrient conditions (incubation in serum-free medium), triggered the phosphorylation of 4E-BP1 among SKOV-3, HeyA8 and OVCAR-3 ovarian cancer cells and thus, produced signaling through the mTOR pathway without traditional agonist stimulation. Extending these findings suggests the autonomous ability of ovarian cancer cells to augment growth and viability, even in the absence of ‘sufficient’ nutrients within the tumor microenvironment, analogous to hypoxic conditions. Thus, loss of RGS10 expression could endow cells with a significant survival and viability advantage.
No reports in the literature to date have proposed that RGS10 regulates mTOR signaling through binding to Rheb. Nor has any report characterized the functional outcomes in cancer cells resultant from RGS10 suppression. Thus, our study fills a major gap in our understanding of how mTOR signaling is regulated and the unique role of RGS10. This knowledge is important because the mTOR signaling pathway remains crucial and often functionally dysregulated among cancer cells, especially since it is linked to PI3K signaling. Signaling through PI3K/mTOR is critical because it regulates the cell’s ability to grow, survive and proliferate; these functional outcomes are all at the crux of delineating the difference between a normal and a cancerous cell. Since we have previously investigated the relationship between RGS10 suppression and Akt activation (14), we refrained from further studies with PI3K herein.
Our prior studies are the only other reports found in the literature that describes the role of RGS10 in ovarian cancer cells (14,15). In this regard, we previously found that the suppression of RGS10 is acquired through epigenetic changes (15) as chemoresistance develops. Furthermore, this change allows ovarian cancer cells to survive in the presence of significantly higher chemotherapy doses than usual (14). Herein we have uncovered the specific molecular mechanism that explains these prior observations.
Because the PI3K/mTOR signaling pathway is one of the most frequently dysregulated among cancer, it represents an enormous target of inhibition. Currently there are several FDA-approved mTOR signaling inhibitors, everolimus and temsirolimus, which are used clinically to manage patients with renal cell carcinoma and HER2-negative breast cancer (everolimus). These agents owe their existence to the soil bacteria living on Easter Island where their parent compound, rapamycin or sirolimus, was discovered (25). Other similar agents are under development, although there have been issues with safety and a lack of overall survival advantage, which are major concerns preventing further development.
The obvious connection between our current study and our previous work is the idea of using mTOR inhibition as a mechanism to alleviate chemoresistance in ovarian cancer. Indeed, other studies have already shown that mTOR inhibition represents a strategy to overcome chemoresistance in a variety of different types of cancers (26–30), especially with combination therapy, which is the mainstay of treating nearly every cancer type. This also insinuates that our results could be adaptable to other subtypes of cancer, beyond our focus on ovarian cancer cells. Among ovarian tumors, serous epithelial ovarian adenocarcinoma frequently displays enhanced mTOR phosphorylation; additionally, this occurs particularly among cisplatin-resistant ovarian cancer cells, in comparison to their matched parental cisplatin-sensitive control cells, which also translates to a greater sensitivity to mTOR inhibition (31). The study cited here, although not specifically focused on RGS10, is nevertheless directly aligned with our findings and interpretation of results.
In order to directly test this theory in vivo, a tumor model with controlled suppression of RGS10 is necessary. Our repeated attempts to maintain such a model long-term were thwarted by the inability to continuously passage cells with suppressed RGS10. Although we show HeyA8 cells herein with stable shRGS10, we observed that cells in culture quickly regain RGS10 expression. Using shRNA constructs, time after time this happened, even though we have successfully manipulated the expression of other RGS proteins ((32) and data not shown). However, since we hypothesize that the net effect of this model would be the continual activation of Rheb, similar work has already been produced (33) and our results would most likely be a reiteration of these. In this study, Jiang and Vogt describe a constitutively active Rheb that phosphorylates 4E-BP1, produces larger cells with more protein and generally induces oncogenic transformation.
It is fascinating that no agonist is needed by ovarian cancer cells to achieve phosphorylated mTOR and 4E-BP1 with suppression of one small GAP protein – RGS10. The implication of our findings with tumor biology is that the loss of RGS10 would have drastically negative consequences on a dysplastic cell and more so on any cell without a properly functioning TSC1/TSC2 complex. Biological systems with critical functions often have redundant control mechanisms in place for proper regulation. For example, the cell cycle has abundant proteins to serve as checkpoint controls. Thus, it is not surprising to learn that regulation of mTOR activation through Rheb could have more than one proteins involved: TSC1/TSC2 complex and RGS10. Molecularly, if two proteins exist to regulate Rheb and thus control mTOR activation, then there would be a total loss in the regulatory capability without functional expression of TSC1/TSC2 and RGS10. Could rescuing these help compensate for aberrant signaling? Future studies will address such questions and predictions. The answers are currently unknown.
The main conclusion of our study is that RGS10 suppression causes an increase in activated, GTP-bound Rheb and results in mTOR pathway activation among ovarian cancer cells. We proposed that RGS10 is a GAP that accelerates the hydrolysis of GTP from Rheb, acting as a master “off” switch that regulates signal duration of mTOR and 4E-BP1 phosphorylation. Although the role of RGS10 was previously unappreciated, based on our data herein, we propose a new role for RGS10 in signaling (Fig. 6). This is significant for our understanding of the molecular complexities that evolve chemoresistance because serous carcinoma exhibits enhanced mTOR phosphorylation, especially among cisplatin-resistant cells. Therefore, this discovery is important to developing ideas for successfully targeting chemoresistance since refractory disease is the clinical problem preventing cure of this malignancy.
Figure 6. Model integrating RGS10 into the mTOR signaling pathway.
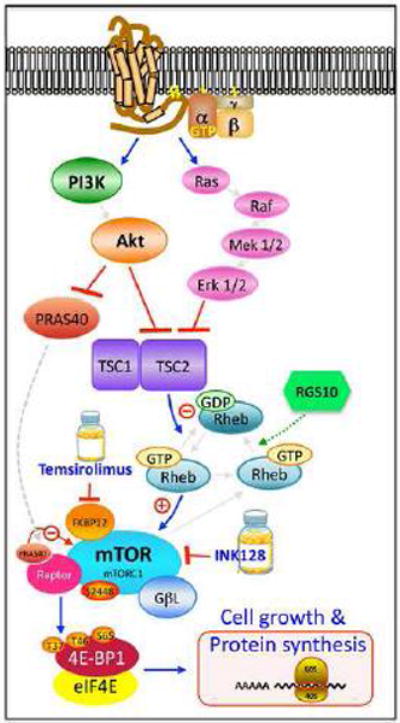
A G protein-coupled receptor initiates a signaling cascade upon activation at the cell surface. The signal is transmitted through the G proteins to initiate ERK/MAPK and PI3K/Akt/mTOR, in the case of growth factor signaling. Activation affects the inhibition of the TSC1/TSC2 complex, which regulates Rheb activation. Rheb-GTP relieves mTORC1 inhibition. Phosphorylated mTOR phosphorylates 4E-BP1, which regulates cell growth and nascent protein synthesis. We propose that RGS10 binds and accelerates the hydrolysis of GTP from Rheb, which is especially apparent under low nutrient conditions and minimized upon treatment with the lipid growth factor, lysophosphatidic acid. The data shown herein supports this since RGS10 suppression increases the amount of Rheb-GTP in agonist-free cell systems as well as increasing the phosphorylation of 4E-BP1 and mTOR. RGS10 is depicted in green with a dashed line.
Supplementary Material
Highlights.
RGS10 suppression triggers phosphorylation of mTOR, 4E-BP1, p70S6K and rProtein-S6
Rheb-GTP is increased in ovarian cancer cells after RGS10 suppression
Suppression of RGS10 increases cell proliferation in the presence of chemotherapy
Effects of RGS10 suppression occur in serum-free conditions with low nutrients
Lysophosphatidic acid stimulation of ovarian cancer cells minimizes RGS10’s effects
Acknowledgments
This work was supported by research grants from the National Institutes of Health (1R15CA151006-01 American Recovery and Reinvestment Act and 1R15CA176653-01A1), a Research Scholar Grant 120634-RSG-11-269-01-CDD from the American Cancer Society and the Georgia Research Alliance. Core facilities that helped generate data are funded by the National Institutes of Health National Cancer Institute CA16672.
Abbreviations
- 4E-BP1
(eukaryotic translation initiation factor 4E binding protein 1)
- GAP
(GTPase activating protein)
- GTP
(Guanosine-5′-triphosphate)
- LPA
(lysophosphatidic acid)
- mTOR
(mammalian or mechanistic target of rapamycin)
- RGS10
(regulator of G protein signaling 10)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest
Contributor Information
Molly K. Altman, Email: altmanmk@uga.edu.
Ali A. Alshamrani, Email: ali86@uga.edu.
Wei Jia, Email: wjia10@uga.edu.
Ha Nguyen, Email: nguyen99@uga.edu.
Jada M. Fambrough, Email: jfambrough@gru.edu.
Sterling K. Tran, Email: stran@uga.edu.
Mihir Patel, Email: mbpatel@uga.edu.
Pooya Hoseinzadeh, Email: pooya94@uga.edu.
Aaron M. Beedle, Email: beedlea@uga.edu.
References
- 1.Hunt TW, Fields TA, Casey PJ, Peralta EG. RGS10 is a selective activator of G alpha i GTPase activity. Nature. 1996;383:175–177. doi: 10.1038/383175a0. [DOI] [PubMed] [Google Scholar]
- 2.Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005;1:51–66. doi: 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sethakorn N, Yau DM, Dulin NO. Non-canonical functions of RGS proteins. Cell Signal. 2010;22:1274–1281. doi: 10.1016/j.cellsig.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Current biology: CB. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 5.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22:169–176. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goorden SM, Hoogeveen-Westerveld M, Cheng C, van Woerden GM, Mozaffari M, Post L, Duckers HJ, Nellist M, Elgersma Y. Rheb is essential for murine development. Molecular and cellular biology. 2011;31:1672–1678. doi: 10.1128/MCB.00985-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes & development. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nature cell biology. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 9.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Current biology: CB. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 10.Tee AR, Blenis J, Proud CG. Analysis of mTOR signaling by the small G-proteins, Rheb and RhebL1. FEBS Lett. 2005;579:4763–4768. doi: 10.1016/j.febslet.2005.07.054. [DOI] [PubMed] [Google Scholar]
- 11.Weinberg R. The Biology of Cancer, Second Edition. Garland Science; 2014. [Google Scholar]
- 12.Guo Y, Kwiatkowski DJ. Equivalent benefit of rapamycin and a potent mTOR ATP-competitive inhibitor, MLN0128 (INK128), in a mouse model of tuberous sclerosis. Molecular cancer research: MCR. 2013;11:467–473. doi: 10.1158/1541-7786.MCR-12-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hooks SB, Callihan P, Altman MK, Hurst JH, Ali MW, Murph MM. Regulators of G-Protein signaling RGS10 and RGS17 regulate chemoresistance in ovarian cancer cells. Molecular cancer. 2010;9:289. doi: 10.1186/1476-4598-9-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ali MW, Cacan E, Liu Y, Pierce JY, Creasman WT, Murph MM, Govindarajan R, Eblen ST, Greer SF, Hooks SB. Transcriptional suppression, DNA methylation, and histone deacetylation of the regulator of G-protein signaling 10 (RGS10) gene in ovarian cancer cells. PloS one. 2013;8:e60185. doi: 10.1371/journal.pone.0060185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic acids research. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monroy CA, Mackie DI, Roman DL. A high throughput screen for RGS proteins using steady state monitoring of free phosphate formation. PloS one. 2013;8:e62247. doi: 10.1371/journal.pone.0062247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia W, Eneh JO, Ratnaparkhe S, Altman MK, Murph MM. MicroRNA-30c-2* expressed in ovarian cancer cells suppresses growth factor-induced cellular proliferation and downregulates the oncogene BCL9. Molecular cancer research: MCR. 2011;9:1732–1745. doi: 10.1158/1541-7786.MCR-11-0245. [DOI] [PubMed] [Google Scholar]
- 19.Alberts B. Molecular Biology of the Cell. Sixth. Garland Science; 2015. [Google Scholar]
- 20.Kam Y, Exton JH. Role of phospholipase D1 in the regulation of mTOR activity by lysophosphatidic acid. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2004;18:311–319. doi: 10.1096/fj.03-0731com. [DOI] [PubMed] [Google Scholar]
- 21.Shor B, Zhang WG, Toral-Barza L, Lucas J, Abraham RT, Gibbons JJ, Yu K. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer research. 2008;68:2934–2943. doi: 10.1158/0008-5472.CAN-07-6487. [DOI] [PubMed] [Google Scholar]
- 22.Wang CC, Held RG, Hall BJ. SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PloS one. 2013;8:e83941. doi: 10.1371/journal.pone.0083941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Rivera E, Jayaraman P, Parikh F, Davies MA, Ekmekcioglu S, Izadmehr S, Milton DR, Chipuk JE, Grimm EA, Estrada Y, Aguirre-Ghiso J, Sikora AG. Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of TSC2. Cancer research. 2014;74:1067–1078. doi: 10.1158/0008-5472.CAN-13-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beaufort CM, Helmijr JC, Piskorz AM, Hoogstraat M, Ruigrok-Ritstier K, Besselink N, Murtaza M, van IWF, Heine AA, Smid M, Koudijs MJ, Brenton JD, Berns EM, Helleman J. Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PloS one. 2014;9:e103988. doi: 10.1371/journal.pone.0103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. The Journal of antibiotics. 1975;28:721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 26.Grunwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB, Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer research. 2002;62:6141–6145. [PubMed] [Google Scholar]
- 27.Lu CH, Wyszomierski SL, Tseng LM, Sun MH, Lan KH, Neal CL, Mills GB, Hortobagyi GN, Esteva FJ, Yu D. Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:5883–5888. doi: 10.1158/1078-0432.CCR-06-2837. [DOI] [PubMed] [Google Scholar]
- 28.Beeram M, Tan QT, Tekmal RR, Russell D, Middleton A, DeGraffenried LA. Akt-induced endocrine therapy resistance is reversed by inhibition of mTOR signaling. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2007;18:1323–1328. doi: 10.1093/annonc/mdm170. [DOI] [PubMed] [Google Scholar]
- 29.Tsurutani J, West KA, Sayyah J, Gills JJ, Dennis PA. Inhibition of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway but not the MEK/ERK pathway attenuates laminin-mediated small cell lung cancer cellular survival and resistance to imatinib mesylate or chemotherapy. Cancer research. 2005;65:8423–8432. doi: 10.1158/0008-5472.CAN-05-0058. [DOI] [PubMed] [Google Scholar]
- 30.Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10519–10524. doi: 10.1073/pnas.0800939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mabuchi S, Kawase C, Altomare DA, Morishige K, Sawada K, Hayashi M, Tsujimoto M, Yamoto M, Klein-Szanto AJ, Schilder RJ, Ohmichi M, Testa JR, Kimura T. mTOR is a promising therapeutic target both in cisplatin-sensitive and cisplatin-resistant clear cell carcinoma of the ovary. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:5404–5413. doi: 10.1158/1078-0432.CCR-09-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altman MK, Nguyen DT, Patel SB, Fambrough JM, Beedle AM, Hardman WJ, Murph MM. Regulator of G-Protein Signaling 5 Reduces HeyA8 Ovarian Cancer Cell Proliferation and Extends Survival in a Murine Tumor Model. Biochemistry research international. 2012;2012:518437. doi: 10.1155/2012/518437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang H, Vogt PK. Constitutively active Rheb induces oncogenic transformation. Oncogene. 2008;27:5729–5740. doi: 10.1038/onc.2008.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.