

Abstract
Type 1 Gaucher disease is an inherited lysosomal enzyme deficiency with variable age of symptom onset. Common presenting signs include thrombocytopenia, anemia, hepatosplenomegaly, bone abnormalities, and, additionally in children, growth failure. Fifty-seven patients aged 3–62 years at the baseline of two phase III trials for velaglucerase alfa treatment were enrolled in the single extension study. In the extension, they received every-other-week velaglucerase alfa intravenous infusions for 1.2–4.8 years at 60 U/kg, although 10 patients experienced dose reduction. No patient experienced a drug-related serious adverse event or withdrew due to an adverse event. One patient died following a convulsion that was reported as unrelated to the study drug. Only one patient tested positive for anti-velaglucerase alfa antibodies. Combining the experience of the initial phase III trials and the extension study, significant improvements were observed in the first 24 months from baseline in hematology variables, organ volumes, plasma biomarkers, and, in adults, the lumbar spine bone mineral density Z-score. Improvements were maintained over longer-term treatment. Velaglucerase alfa had a good long-term safety and tolerability profile, and patients continued to respond clinically, which is consistent with the results of the extension study to the phase I/II trial of velaglucerase alfa. EudraCT number 2008-001965-27; http://www.clinicaltrials.gov identifier NCT00635427. Am. J. Hematol. 90:584–591, 2015. © 2015 Wiley Periodicals, Inc.
Introduction
Gaucher disease (GD) is a rare genetic disease caused by mutations in GBA, which encodes a hydrolytic, lysosomal enzyme (glucosylceramidase). When this enzyme is deficient, the major natural glucocerebroside substrates of the enzyme accumulate in cells, notably macrophages, which are most responsible for the visceral pathophysiology in type 1 GD. Type 1 GD can present at variable ages with thrombocytopenia, anemia, hepatosplenomegaly, bone marrow infiltration, and various focal and generalized bone abnormalities, and additionally in children, with growth failure [1,2].
Current GD-specific treatment, enzyme replacement therapy (ERT) and substrate reduction therapy, is beneficial but not curative, and so patients usually have regular continual dosing. Valuable research into disease outcomes, with and without treatment, has been conducted through GD registries [3–5]. There is, however, also a need for clinical trial evidence for long-term drug efficacy and safety, and this should include data in pediatric patients.
Velaglucerase alfa is available as an ERT for type 1 GD. Two randomized, parallel-group phase III trials of velaglucerase alfa were conducted between 2007 and 2009 in treatment-naïve patients, including children: study identifiers TKT032 (a parallel assessment of two doses) and HGT-GCB-039 (a noninferiority study comparing velaglucerase alfa with imiglucerase). No patient discontinued from these studies because of an adverse event (AE), and improvements in hemoglobin concentration, platelet count, spleen volume, liver volume, biomarkers [6,7], and bone mineral density (BMD) were observed. Patients who completed the initial trials were enrolled in an open-label, phase III extension study for up to 5 years of follow-up, to evaluate the long-term safety and efficacy of velaglucerase alfa.
Patients and Methods
The extension study was an open-label study conducted between March 2008 and December 2012 into which patients who completed the trial TKT032 (53 weeks) or HGT-GCB-039 (41 weeks) were enrolled. As previously described, the diagnosis of GD was confirmed by an enzyme assay (deficient leukocyte glucocerebrosidase activity) before the TKT032 and HGT-GCB-039 trials, and patients’ genotypes were analyzed [6,7].
Patients were ineligible to enroll into the extension study if they had a significant comorbidity (e.g., malignancies, hepatic cirrhosis, autoimmune liver disease, etc.); were pregnant, lactating, or unable to comply with the protocol; or had received treatment with an investigational drug other than velaglucerase alfa in the 30 days before study entry.
Eleven clinical sites participated in one or both of the initial trials; nine sites in eight countries enrolled patients into the extension study as well. Each site had the protocol, protocol amendments, and patient consent forms approved by an independent ethics committee or institutional review board. The study was conducted in compliance with the International Conference on Harmonisation (ICH) Guideline for Good Clinical Practice and the US Code of Federal Regulations, Title 21, Part 56. Each patient or their legally authorized representative(s) gave written informed consent.
All patients received their first three infusions of the extension study at a clinical site. If patients did not experience an infusion-related AE or drug-related serious AE, subsequent infusions could be administered at home by trained medical personnel per the discretion and direction of the investigator. An infusion-related AE was defined as an AE that was considered related to the study drug and began within 12 hrs of the infusion.
In the trials TKT032 and HGT-GCB-039, patients were randomly assigned to one of the two treatment arms; to maintain blinding, all patients who were enrolled in the extension study after completing either of these trials were assigned 60 U/kg/infusion of velaglucerase alfa regardless of the treatment or dose received in the initial trial. According to the extension study protocol, velaglucerase alfa was to be administered in a 60-min intravenous infusion every other week (EOW).
Once patients completed a cumulative 24-month period of ERT from their first infusion in the initial trial, investigators could increase or decrease their dose by 15 U/kg once every 12 months based on the achievement of therapeutic goals [8], but each patient's dose had to stay within the range of 15–60 U/kg.
Patients could continue to receive velaglucerase alfa in the extension study until commercial velaglucerase alfa became available to them individually, in their respective countries, or the study was terminated (patients for whom the drug was not marketed in their country at the time that they left the study nonetheless continued to receive treatment through a charitable access program).
Although all patients were naïve to ERT at the start of the core studies, they received different ERTs for the first 9 months. Therefore, two main analysis groups were predefined for the safety and efficacy evaluations based on the drug received in the initial trials: velaglucerase alfa or imiglucerase. No comparative analyses were performed.
Safety assessments
Patients were monitored continuously for AEs. Vital signs were measured, and concomitant medication use was recorded at every infusion visit (EOW). Approximately every 3 months, physical examinations were performed, safety-related laboratory tests were conducted, and blood samples were screened for the presence of anti-velaglucerase alfa antibodies using an electrochemiluminescent bridge immunoassay. The antibody testing methods are described in detail elsewhere [9,10], but in brief, if the antibody screening test was positive, radioimmunoprecipitation and electrochemiluminescent assays were used to confirm the presence of immunoglobulin G (IgG) and IgE antibodies, respectively. Samples that met confirmatory antibody-positive cut-point criteria were tested for enzyme neutralizing activity using a neutralizing antibody assay.
A urine pregnancy test was carried out before every infusion in female patients of child-bearing potential. If the test was positive, the infusion was to be held, and the result confirmed with a serum human chorionic gonadotropin test. If the serum result was also positive, the investigator was to contact the Shire medical monitor to determine the appropriate course of action.
Efficacy assessments
Blood samples were taken approximately every 3 months to measure hemoglobin concentration, platelet count, chemokine (C–C motif) ligand 18 (CCL18) level, and chitotriosidase activity, and blood samples were taken yearly for type I collagen telopeptides N- and C-terminal cross-links (NTx and CTx; markers of bone turnover). CCL18 and chitotriosidase were measured according to the methods described by Boot et al. [11] and Aguilera et al. [12], respectively. CTx and NTx were measured using an enzyme-linked immunosorbent assay.
Patients underwent quantitative abdominal magnetic resonance imaging at yearly intervals to assess their spleen and liver volumes. Organ volumes were measured by a single independent reviewer who was blinded to patient identity and the timing of the imaging in relation to the study. The measurements were normalized to the patient's body weight (% BW). A spleen volume corresponding to 0.2% BW or a liver volume corresponding to 2.5% BW was equal to 1 multiple of normal.
The BMD of the lumbar spine and the femoral neck was assessed in patients over 18 years of age using dual-energy X-ray absorptiometry (DXA). Different DXA scanners were used between patients and to address differences in calibrations between various machines from the same manufacturer, the BMIL Quality Assurance/Quality Control phantom (BioMedical Imaging Laboratory, Dayton, OH) [13] was measured on all the scanners, and cross-calibration equations were developed.
Most scanners were made by Hologic, Inc. (Bedford, MA) and the others were by GE Lunar (Madison, WI). A between-manufacturer calibration using published cross-calibration equations derived from patient data [14,15] was carried out to account for systematic differences between these manufacturers. GE Lunar BMD values were translated into the Hologic scale.
Drifts in calibration (gradual trends up or down) can occur in individual scanners over time, as can break points (jumps) because of machine repairs, for example. To address this, longitudinal quality assurance data were collected from the scanners, and corrective calculations were made if needed; an assumption of no drift was made for any sites without data for their machines.
One reviewer at a central laboratory (Wright State University, Dayton, OH) analyzed the phantom and patient data; the reviewer was blinded to the treatment each patient had received. The analysis was overseen and checked by another reviewer.
A database of reference values by Hologic was used to convert BMD measurements to Z- and T-scores. Sex- and race-specific reference values were used to calculate Z-scores, whereas T-scores for all adults were based on Caucasian female reference data [16]. Z-scores represent the number of standard deviations (SDs) difference from age-matched means, and T-scores represent the number of SDs difference from young normal means. The T-scores were used to categorize BMD measurements as normal (T-score ≥ –1), osteopenic (>–2.5 to <–1), or osteoporotic (≤–2.5) [17].
Safety and efficacy analyses
The primary objective of the extension study was to evaluate the long-term safety of velaglucerase alfa treatment. Safety results were evaluated in all patients who received at least one dose of velaglucerase alfa in the extension study. AEs were summarized in tables by type or category.
Efficacy data were analyzed in the efficacy analysis population, which was defined as all patients who were enrolled in the extension study. During the extension study, two subjects were withdrawn following confirmation that they did not have type 1 GD but were in fact carriers who had been misdiagnosed due to false-positive laboratory test results. These two misdiagnoses have previously been described [7]. It was then specified that the efficacy analysis population included only the enrolled patients with type 1 GD. So, the carriers were included in the safety analyses, but excluded from the efficacy analysis population.
The extension study population was split into two according to the study drug received in the initial trial: one group who received imiglucerase in HGT-GCB-039 and one group who received velaglucerase alfa in either TKT032 or HGT-GCB-039 (Fig. 1).
Figure 1.
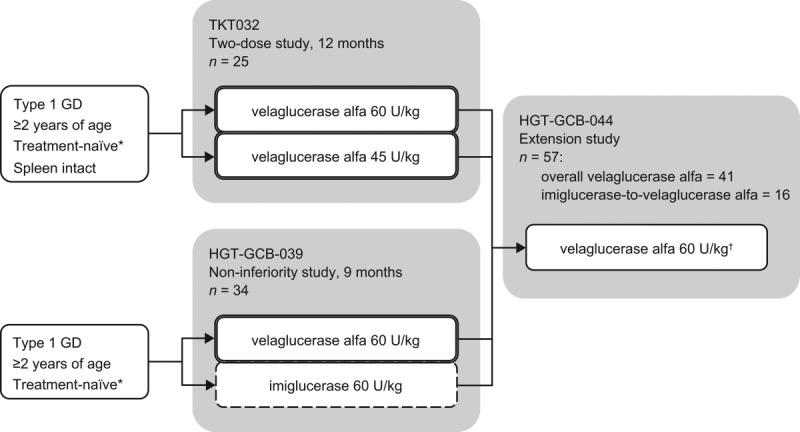
Outline of the two initial trials and the extension study. Two of 34 patients treated in HGT-GCB-039 did not enroll in the extension study. The overall velaglucerase alfa group = patients from the three treatment arms outlined by a double line. The imiglucerase-to-velaglucerase alfa group = patients from the treatment arm outlined by a dashed line. *Never received Gaucher disease-specific treatment or at least none received in the 30 months (TKT032) or 12 months (HGT-GCB-039) before study entry. †Investigators could adjust doses within the range of 15–60 U/kg.
Efficacy variables
The secondary objectives of the extension study were to evaluate the effects of velaglucerase alfa treatment on hemoglobin concentration, platelet count, and spleen and liver volumes (the primary efficacy variables). Plasma chitotriosidase and CCL18 (GD biomarkers), serum markers of bone turnover, BMD, and assessments of growth and development in patients under 18 years of age were all evaluated as exploratory objectives (data specific to children are reported elsewhere).
Summary statistics at protocol-defined time points were calculated. Mean changes and mean percentage changes from baseline were also calculated, together with 95% confidence intervals to assist interpretation. Baseline was defined as before the first dose in the initial trials.
Mean changes at 24 months were calculated, and, to evaluate changes over the entire study period, a longitudinal data analysis was also performed using linear mixed models.
The linear mixed models took all available data into account and were adjusted for age at baseline, the baseline value of the clinical variable, splenectomy status (for platelet count only), and sex (for hemoglobin concentration only).
Intermittent missing data were imputed for the change-from-baseline analysis at 24 months: last observation carried forward for missing post-baseline data, and, while extremely rare, missing baseline data were replaced with the next available measurement.
The BMD analyses were conducted in all adults, and then, since bisphosphonates affect bone turnover [18], they were repeated in the subset who were not treated concomitantly with bisphosphonates. Summary statistics were calculated for the CTx and NTx levels at baseline and post-baseline time points, and Pearson's correlation was used to investigate a potential linear relationship between BMD Z-scores and CTx or NTx values.
Results
Analysis groups
All 25 patients who participated in TKT032 were enrolled in the extension study. Thirty-two of 34 patients who received at least one ERT infusion in HGT-GCB-039 were enrolled in the extension study (Fig. 1): 16 from each treatment arm of HGT-GCB-039.
All 57 patients received velaglucerase alfa in the extension study (Table I). The 16 patients who received imiglucerase in HGT-GCB-039 were the imiglucerase-to-velaglucerase alfa group. All other patients received velaglucerase alfa in the initial trial, and they were pooled and called the overall velaglucerase alfa group (Fig. 1); this group of 41 patients included the two GD carriers who were excluded from the efficacy analysis population.
TABLE I.
Efficacy Analysis Population Before First Dose in Initial Trials
| Overall velaglucerase alfa (n = 39) | Imiglucerase-to-velaglucerase alfa (n = 16) | |
|---|---|---|
| Median age, years (range) | 29 (6, 62) | 26 (3, 58) |
| Children, n (%) | 8 (21) | 5 (31) |
| Male, n (%) | 21 (54) | 7 (44) |
| GBA genotype, n (%) | ||
| N370S/N370S | 13 (33) | 2 (13) |
| N370S/84GG | 1 (3) | 0 |
| N370S/L444P | 2 (5) | 1 (6) |
| L444P/L444P | 2 (5) | 2 (13) |
| N370S/Other | 13 (33) | 6 (38) |
| L444P/Other | 3 (8) | 0 |
| F213I/F213I | 0 | 2 (13) |
| Other/Other | 5 (13) | 3 (19) |
| Splenectomy status, n (%) | ||
| Intact | 30 (77) | 6 (38) |
| Splenectomized | 9 (23) | 10 (63) |
| Chitotriosidase gene 24-base pair allele, n (%) | ||
| Wild-type gene | 20 (51) | 10 (63) |
| Heterozygous | 18 (46) | 5 (31) |
| Homozygous (enzyme deficient) | 1 (3) | 1 (6) |
| Efficacy variables, median (range) | ||
| Hemoglobin, g/dL | 10.90 (7.1, 14.4)a | 10.65 (8.1, 13.1) |
| Platelet count ×109/L | 82.5 (13, 310) | 190.5 (63, 430) |
| Spleen volume, MN | 13.60 (4.8, 65.1)b | 21.45 (3.1, 44.4) |
| Liver volume, MN | 1.50 (0.8, 3.2)a | 1.60 (0.7, 2.8) |
| Plasma chitotriosidase, nmol/mL/hrc | 43,769 (12,678, 99,393)d | 36,319 (11,330, 112,777) |
| Plasma CCL18, ng/mL | 1,789.0 (731, 4,065) | 1,799.5 (806, 5,902) |
| BMD Z-score, patients ≥18 years | ||
| Lumbar spine | −1.73 (−4.20, 0.78) | −1.54 (−2.68, 3.22) |
| Femoral neck | −0.59 (−2.77, 2.37) | 1.71 (−1.83, 4.49) |
| BMD T-score category, patients ≥18 years, n | ||
| Lumbar spine: OPO, OPN, NOR | 11, 15, 5 | 3, 3, 5 |
| Femoral neck: OPO, OPN, NOR | 1, 10, 20 | 0, 1, 10 |
The genotyping methods used in the initial trials (for GBA and the chitotriosidase gene) have been described in Refs.[6–7].
n = 38.
n = 29.
Chitotriosidase-deficient patients not included; laboratory measurement multiplied by two in patients heterozygous for the 24-base pair chitotriosidase gene mutation.
In addition to one chitotriosidase-deficient patient, one patient with low baseline activity (<5,000 nmol/mL/hr) was excluded from this group.
MN: multiple of normal; BMD: bone mineral density; OPO: osteoporosis; OPN: osteopenia; NOR: normal.
Nineteen of 57 patients completed the extension study. Thirty-eight patients discontinued from the study, mostly (34 patients) due to the termination of the trial by the sponsor. Three patients withdrew consent for personal reasons and one patient died.
Velaglucerase alfa and concomitant drugs
In the efficacy analysis population, the overall velaglucerase alfa group (39 patients) received between 1.5 and 4.8 years of ERT with velaglucerase alfa in the extension study (median time, 3.7 years). The imiglucerase-to-velaglucerase alfa group received 1.2–4.1 years of velaglucerase alfa treatment (median time, 3.6 years).
Eight of 39 patients in the overall velaglucerase alfa group and two patients in the imiglucerase-to-velaglucerase alfa group had up to three stepwise reductions in dose from 60 U/kg. In both groups, the median dose received in the extension study was 60.0 U/kg per EOW infusion (range, 38.0–60.5 U/kg).
Only one patient (overall velaglucerase alfa group) took pre-infusion medication (diphenhydramine and dexamethasone) to prevent an infusion reaction. Bisphosphonates were used during the initial trial, the extension study, or both by four adults in the overall velaglucerase alfa group and by three adults in the imiglucerase-to-velaglucerase alfa group.
The two GD carriers who were excluded from the efficacy analysis population were included in the safety population, and they received velaglucerase alfa for a total of 3.4 years and 3.1 years before they were withdrawn from the extension study.
Safety (extension study)
Almost all patients experienced an AE during the extension study (Table II). The vast majority of events were Grade 1 or 2 in intensity, i.e., mild or moderate; 12 of 431 AEs reported in the overall velaglucerase alfa group and nine of 189 AEs in the imiglucerase-to-velaglucerase alfa group were graded severe.
TABLE II.
Extension Study AE Summary in Safety Population
| Overall velaglucerase alfa (n = 41) |
Imiglucerase-to-velaglucerase alfa (n = 16)) |
|||
|---|---|---|---|---|
| Patient experience | Number of patients (%) | Number of events | Number of patients (%) | Number of events |
| Any AE | 38 (93) | 431 | 15 (94) | 189 |
| ≥1 drug-related AE | 9 (22) | 35 | 7 (44) | 21 |
| ≥1 infusion-related AE | 5 (12) | 25 | 1 (6) | 1 |
| ≥1 serious AE | 6 (15) | 11 | 4 (25) | 8 |
| Life-threatening AE | 0 | 0 | ||
| Death | 0 | 1 (6) | ||
| ≥1 serious and drug-related AE | 0 | 0 | ||
| Discontinued from study due to AE | 0 | 0 | ||
Sixteen of 57 patients experienced AEs that were deemed possibly or probably related to velaglucerase alfa treatment (Table II). Of the 56 drug-related AEs reported in total, the only events that were experienced by more than one patient were hypertension (two patients in the overall velaglucerase alfa group) and headache (one patient in each analysis group). The drug-related AEs of hypertension (two events) were infusion-related.
Six of 57 patients experienced infusion-related AEs (Table II). The percentage of patients experiencing infusion-related AEs in every 3-month interval in the extension study was low or zero (Supporting Information Fig. S1). In the imiglucerase-to-velaglucerase alfa group, there was no change in infusion-related AEs around the time of switching to velaglucerase alfa treatment (Supporting Information Fig. S1). Sixteen of 57 patients had at least one home infusion during the extension study. No infusion-related AEs happened during their home infusions.
Nineteen serious AEs were reported (Supporting Information Table SI), including a spontaneous first-trimester abortion in a patient with a history of miscarriages and suspected anti-phospholipid syndrome and a patient death following a convulsion. No serious AEs were considered to be related to velaglucerase alfa treatment.
The death reported was of a 6-year-old male patient (GBA genotype F213I/F213I) from India who was 3 years old at the time of enrollment into study HGT-GCB-039. Twelve days after the last velaglucerase alfa infusion, the patient was reported to have had involuntary movements and breathlessness; he then stopped breathing and died while at home. The seizure and death were considered by the investigator to be unrelated to velaglucerase alfa and related to GD progression. The patient was diagnosed with type 1 GD by his treating physician, but experienced seizures and gait disturbance during the study that may have indicated type 3 GD. He participated in the extension study for 24 months.
Two patients became pregnant during the extension study. One patient chose to continue velaglucerase alfa treatment and signed a pregnancy informed consent form. The patient did not report any AEs during the pregnancy, but infusions were discontinued less than 3 months after the pregnancy was confirmed because of the termination of the extension study (the patient started to receive infusions again after velaglucerase alfa become commercially available in Russia); she delivered at 39 weeks, and the neonate was described as normal. One patient discontinued infusions when the pregnancy was confirmed. The patient had a spontaneous first-trimester abortion (not related to study drug) and resumed velaglucerase alfa infusions approximately 2 months after the abortion. Two male patients fathered children during the study; their female partners both delivered after full-term pregnancies, and the neonates were described as healthy and normal.
There were no clinically significant trends in vital sign measurements or physical examination findings that suggested an increased risk of harm with velaglucerase alfa (data not shown).
Anti-velaglucerase alfa antibodies
One patient tested positive for IgG anti-velaglucerase alfa antibodies in the extension study, which was transient. The first positive test was at week 53 (end of study visit for the initial trial and first dose in the extension study) and tests at weeks 65, 77, and 89 were also positive; tests from week 101 until the patient discontinued were negative. The sera had neutralizing activity, but there were no changes in the patient's hemoglobin concentration or platelet count deemed to suggest altered drug efficacy, and no drug-related AEs were reported. No other patients tested positive for anti-velaglucerase alfa antibodies, including three patients who were positive for anti-imiglucerase antibodies and switched to velaglucerase alfa after 9 months of imiglucerase treatment [6].
Efficacy
0–24 months
The mean increase in hemoglobin concentration was 2.75 g/dL (26%) in the overall velaglucerase alfa group, and there was a 120% mean increase in the platelet count compared with baseline; a 64% mean decrease in spleen volume and a 27% mean decrease in liver volume were also observed. The mean levels of the biomarkers chitotriosidase and CCL18 decreased by around 80%, and the BMD Z-scores assessed at the lumbar spine and femoral neck increased by means of 0.62 SD and 0.12 SD respectively, but the 95% confidence interval around the mean change in the femoral neck Z-score included zero (no statistically significant change). The results in the imiglucerase-to-velaglucerase alfa group were similar (Supporting Information Table SII).
At baseline, the median serum CTx and NTx levels in adults were 0.473 (range, 0.20–1.64) ng/mL and 12.5 (range, 6.5–41.2) nmol bone collagen equivalents/L, respectively, in the overall velaglucerase alfa group and 0.424 (range, 0.20–0.83) ng/mL and 10.5 (range, 6.5–17.8) nmol bone collagen equivalents/L, respectively, in the imiglucerase-to-velaglucerase alfa group. The mean CTx level increased in the overall velaglucerase alfa group between baseline and 24 months, and then it decreased slightly in subsequent months despite continued treatment (statistical significance was not tested). The mean CTx level in the imiglucerase-to-velaglucerase alfa group also increased at first, during 9 months of imiglucerase ERT, and then it returned to the baseline level with subsequent velaglucerase alfa treatment in the extension study. The trends in NTx levels were similar. No significant correlation was found between levels of CTx and NTx and BMD Z-scores (all P-values >0.05).
Longitudinal analysis
The results of this analysis also indicated that there were significant clinical improvements in the first 24 months. Over longer-term treatment, the improvements were either maintained or continued at an attenuated rate (Fig. 2).
Figure 2.

Mean changes and mean percentage changes in efficacy variables over time estimated from linear mixed models. Changes compared with baseline (BL) values are plotted (baseline was before the first dose in the initial trials). Error bars show 95% confidence intervals. Dotted line at 9 months indicates the time that the imiglucerase-to-velaglucerase alfa group switched to ERT with velaglucerase alfa. BMD: bone mineral density.
Discussion
Velaglucerase alfa had a good long-term safety profile overall that was consistent with the phase I/II extension study of velaglucerase alfa conducted in a group of 10 adult patients [19]. This study also confirmed that infusions can be safely administered at home by qualified and trained medical personnel.
Only one patient required pre-infusion medication, and only six (11%) patients experienced an infusion-related AE during the extension study. The percentage of patients experiencing infusion-related events seems to be highest in the first year of treatment and decreases over time, which is similar to other ERTs [20,21].
In the group who switched to velaglucerase alfa when they enrolled in the extension study, there was no increase in infusion-related AEs around the time of switching. In fact, only one infusion-related AE occurred in the 16 patients who switched to velaglucerase alfa over 1.2–4.1 years. Switching from imiglucerase to velaglucerase alfa has been shown to be safe in previous reports, both in patients who transitioned without a break or dose reduction [10,22] and in those who experienced a break or dose adjustment beforehand [23–25].
There was one death following a serious AE of convulsion that the investigator considered related to GD progression and unrelated to treatment. The patient was a 3-year-old male at time of enrollment into the primary study, HGT-GCB-039, and he was 6 years of age at time of death. The patient met the eligibility criteria at study entry, as his treating physician diagnosed him as having type 1 GD. During the course of study participation, the patient developed neurologic symptoms that are suggestive of type 3 GD, and his GBA genotype, F213I/F213I, is also consistent with type 3 GD [26]. Velaglucerase alfa is under investigation for type 3 GD (http://ClinicalTrials.gov identifier NCT01685216), but published clinical data in patients with type 3 GD are so far limited to a single case report [27].
Pregnancy was an exclusion criterion, and patients were required to use a medically acceptable form of contraception during the extension study, but two patients in our analysis population became pregnant. One of these women continued receiving velaglucerase alfa infusions during pregnancy. The physician determined that it was in the patient's best interest to continue infusions. The Shire medical monitor determined that it would be acceptable for the patient to continue infusions and remain in the study; the patient elected to do so and signed a pregnancy informed consent form. The results of these two women did not cause the sponsor to revise the risk-benefit profile of velaglucerase alfa, which, under the US Food and Drug Administration pregnancy categories, is a Category B drug (“animal reproduction studies have failed to demonstrate a risk to the fetus and there are no adequate and well-controlled studies in pregnant women”) [28,29]. We know that other women with GD have used velaglucerase alfa in pregnancy; in a retrospective record review of 21 women and 25 pregnancies, no safety signals were identified [30].
One of the 57 patients tested positive for anti-drug antibodies, which suggests that developing antibodies against velaglucerase alfa is uncommon. An apparently low immunogenic potential has also been found in other velaglucerase alfa trials [10,19]. The percentages of patients who have developed anti-drug antibodies with other approved ERTs are 15% [31] or, in taliglucerase alfa trials in type 1 GD patients, 53% and 13%. However, various factors, including assay sensitivity and specificity, timing of sampling, underlying disease, and concomitant medication, can affect antibody test results. So, the incidences of antibody production associated with each drug may not be comparable [32].
The patient who tested positive for anti-velaglucerase alfa antibodies was transiently positive; there was no apparent effect on the clinical efficacy of velaglucerase alfa, even though the patient's sera were reported as having neutralizing activity, and no drug-related AEs were reported. The clinical relevance of developing anti-ERT (including velaglucerase alfa) antibodies is unknown.
The primary efficacy variable findings indicated that patients continued to respond to ERT in the extension study. Levels of plasma chitotriosidase and CCL18 both decreased over time, reflecting an ongoing fall in the total burden of Gaucher cells in the body [11,33]. These biomarkers are being investigated for their potential to predict long-term clinical complications, such as osteonecrosis [34,35].
Lumbar spine BMD Z-scores in adults improved by 24 months, which was encouraging given the prevalence of osteopenia in the GD population and retrospective registry-based research suggesting a greater risk of spinal and nonspinal fractures with lumbar spine Z-scores ≤–1 compared with Z-scores >–1 [36,37]. BMD was also assessed as an exploratory variable in the phase I/II study of velaglucerase alfa, and there was consistent improvement over time [38]. In this study, mean Z-score changes in the femoral neck were much smaller than in the lumbar spine (and without nominal significance), but also, most patients (>64%) at baseline were no more than 1 SD below peak bone density at the femoral neck, which is considered normal.
Although the changes in BMD suggest bone remodeling, there was no significant correlation between BMD Z-scores and the CTx and NTx values. This may have been because of the wide range of ages in the analysis population, from young adults with developing bone mass to postmenopausal women and elderly men. The attrition in patient numbers after 24 months and the use of bisphosphonates by some patients may also have affected the results.
To the best of our knowledge, this is the first time in a study of GD that a cross-calibration of DXA scanners has been done. Cross-calibration reduces differences between the BMD measurements of different scanners [39]. In this study, multiple scanners made by two different manufacturers were used. The cross-calibration enabled the calculation of average absolute changes in BMD as well as the conversion of the BMD data to Z-scores based on a single database of reference values. BMD values can be converted to Z- and T-scores using manufacturer-specific reference datasets, but the scores obtained from different manufacturers’ systems are not always comparable [40–42].
Ten patients had their dose reduced at least once. Zimran et al. [19] reported continued clinical improvements in the phase I/II study of velaglucerase alfa despite dose reduction in almost all patients. Less than one-fifth of patients had their dose reduced in this study. None of them subsequently had their dose increased, but the duration of follow-up after dose reduction was different between patients and, although dose adjustments were permitted per the study protocol, this study was not designed to evaluate dosing variation. According to the standards of care, the recommended starting dose of velaglucerase alfa for ERT-naïve patients is 60 U/kg (and patients switching from imiglucerase can be switched to the same U/kg dose) [29].
Study limitations
All patients received at least 24 months of ERT, but relatively few patients reached 60 months. Because there were fewer subjects at later time points, the longer-term data set was incomplete.
The cross-calibration of DXA scanners was post hoc; nonetheless, with available longitudinal phantom data, adequate cross-calibration was possible for the duration of the study. Cross-calibration was considered necessary to pool study data that had been collected from multiple centers and increases the likelihood of detecting a possible treatment effect. Where sites were missing quality assurance data for their machines, an assumption of no drift (up or down) was made, and this may have affected the accuracy of the DXA results.
Several efficacy assessments were exploratory; although they supported the direction of the results in hemoglobin concentration, platelet count, spleen volume, and liver volume and also provide information for future research, they have less evidentiary value.
Conclusion
Velaglucerase alfa had a good safety and tolerability profile in this long-term study, and patients continued to demonstrate a satisfactory clinical response. This is consistent with the data reported from the extension study of the phase I/II trial.
Acknowledgments
The clinical studies were sponsored by Shire. The authors acknowledge the other investigators for their considerable contributions to the clinical study, including Marie-Françoise Ben Dridi, MD, and Hadhami Ben Turkia, MD, of La Rabta Hospital (Tunis, Tunisia) and Neerja Gupta of the All India Institute of Medical Sciences (New Delhi, India). They acknowledge the compliance and dedication of the patients and their families during the course of these trials and the professionalism and assistance of all the nurses who were involved in the trials. They thank Yune Kunes, PhD, of Shire, for her bioanalytical expertise in the preparation of this article and Clare Guni, BMBS, of Excel Scientific Solutions, who provided medical writing services funded by Shire.
Author Contributions
DAH, DEG, EAL, AM, MK, DE, IK, PG, AB, and AZ were investigators in the clinical trials. TNH oversaw the analysis of the DXA data, designed the calibration procedures, checked all calibration equations, including their proper application to the patient data, and ran consistency checks on the final DXA data. NW fact-checked all data in the article. EC was the medical monitor for the extension study. All authors reviewed drafts and approved the final version of the article.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- 1.Cox TM. Gaucher disease: Clinical profile and therapeutic developments. Biologics. 2010;4:299–313. doi: 10.2147/BTT.S7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mistry PK, Zimran A. Type 1 Gaucher disease—clinical features. In: Futerman AH, Zimran A, editors. Gaucher Disease. Boca Raton, FL: Taylor & Francis Group; 2007. p. 155–173. [Google Scholar]
- 3.Wenstrup RJ, Kacena KA, Kaplan P, et al. Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res. 2007;22:119–126. doi: 10.1359/jbmr.061004. [DOI] [PubMed] [Google Scholar]
- 4.Weinreb NJ, Goldblatt J, Villalobos J, et al. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J Inherit Metab Dis. 2013;36:543–553. doi: 10.1007/s10545-012-9528-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersson H, Kaplan P, Kacena K, et al. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008;122:1182–1190. doi: 10.1542/peds.2007-2144. [DOI] [PubMed] [Google Scholar]
- 6.Ben Turkia H, Gonzalez DE, Barton NW, et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. Am J Hematol. 2013;88:179–184. doi: 10.1002/ajh.23382. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez DE, Ben Turkia H, Lukina EA, et al. Enzyme replacement therapy with velaglucerase alfa in Gaucher disease: Results from a randomized, double-blind, multinational, phase 3 study. Am J Hematol. 2013;88:166–171. doi: 10.1002/ajh.23381. [DOI] [PubMed] [Google Scholar]
- 8.Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41:4–14. doi: 10.1053/j.seminhematol.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Séllos-Moura M, Barzegar S, Pan L, et al. Development of a panel of highly sensitive, equivalent assays for detection of antibody responses to velaglucerase alfa or imiglucerase enzyme replacement therapy in patients with Gaucher disease. J Immunol Methods. 2011;373:45–53. doi: 10.1016/j.jim.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 10.Zimran A, Pastores GM, Tylki-Szymanska A, et al. Safety and efficacy of velaglucerase alfa in Gaucher disease type 1 patients previously treated with imiglucerase. Am J Hematol. 2013;88:172–178. doi: 10.1002/ajh.23383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boot RG, Verhoek M, de Fost M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: A novel surrogate marker for assessing therapeutic intervention. Blood. 2004;103:33–39. doi: 10.1182/blood-2003-05-1612. [DOI] [PubMed] [Google Scholar]
- 12.Aguilera B, Ghauharali-van der Vlugt K, Helmond MT, et al. Transglycosidase activity of chitotriosidase: Improved enzymatic assay for the human macrophage chitinase. J Biol Chem. 2003;278:40911–40916. doi: 10.1074/jbc.M301804200. [DOI] [PubMed] [Google Scholar]
- 13.Hangartner TN. A study of the long-term precision of dual-energy X-ray absorptiometry bone densitometers and implications for the validity of the least-significant-change calculation. Osteoporos Int. 2007;18:513–523. doi: 10.1007/s00198-006-0280-1. [DOI] [PubMed] [Google Scholar]
- 14.Genant HK, Grampp S, Gluer CC, et al. Universal standardization for dual X-ray absorptiometry: Patient and phantom cross-calibration results. J Bone Miner Res. 1994;9:1503–1514. doi: 10.1002/jbmr.5650091002. [DOI] [PubMed] [Google Scholar]
- 15.Hanson J. Standardization of femur BMD. J Bone Miner Res. 1997;12:1316–1317. doi: 10.1359/jbmr.1997.12.8.1316. [DOI] [PubMed] [Google Scholar]
- 16.Schousboe JT, Shepherd JA, Bilezikian JP, et al. Executive Summary of the 2013 International Society for Clinical Densitometry Position Development Conference on Bone Densitometry. J Clin Densitom. 2013;16:455–466. doi: 10.1016/j.jocd.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 17.World Health Organization. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO study group. World Health Organ Tech Rep Ser. 1994;843:1–129. [PubMed] [Google Scholar]
- 18.Eekman DA, Bultink IE, Heijboer AC, et al. Bone turnover is adequately suppressed in osteoporotic patients treated with bisphosphonates in daily practice. BMC Musculoskelet Disord. 2011;12:167. doi: 10.1186/1471-2474-12-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010;115:4651–4656. doi: 10.1182/blood-2010-02-268649. [DOI] [PubMed] [Google Scholar]
- 20.Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13:95–101. doi: 10.1097/GIM.0b013e3181fea459. [DOI] [PubMed] [Google Scholar]
- 21.Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75:65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serratrice C, Bengherbia M, Alessandrini M, et al. Effects of switching from imiglucerase to velaglucerase alfa without dose reduction nor wash out in type 1 Gaucher disease. Blood Cells Mol Dis. 2014;53:94–96. doi: 10.1016/j.bcmd.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Pastores GM, Rosenbloom B, Weinreb N, et al. A multicenter open-label treatment protocol (HGT-GCB-058) of velaglucerase alfa enzyme replacement therapy in patients with Gaucher disease type 1: Safety and tolerability. Genet Med. 2014;16:359–366. doi: 10.1038/gim.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Dussen L, Cox TM, Hendriks EJ, et al. Effects of switching from a reduced dose imiglucerase to velaglucerase in type 1 Gaucher disease: Clinical and biochemical outcomes. Haematologica. 2012;97:1850–1854. doi: 10.3324/haematol.2011.059071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elstein D, Altarescu G, Maayan H, et al. Booster-effect with velaglucerase alfa in patients with Gaucher disease switched from long-term imiglucerase therapy: Early Access Program results from Jerusalem. Blood Cells Mol Dis. 2012;48:45–50. doi: 10.1016/j.bcmd.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Park JK, Orvisky E, Tayebi N, et al. Myoclonic epilepsy in Gaucher disease: Genotype-phenotype insights from a rare patient subgroup. Pediatr Res. 2003;53:387–395. doi: 10.1203/01.PDR.0000049515.79882.94. [DOI] [PubMed] [Google Scholar]
- 27.Vairo F, Netto C, Dorneles A, et al. Enzyme replacement therapy in a patient with Gaucher disease type III: A paradigmatic case showing severe adverse reactions started a long time after the beginning of treatment. JIMD Rep. 2013;11:1–6. doi: 10.1007/8904_2013_214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.U.S. Food and Drug Administration. 2006. CFR - Code of Federal Regulations Title 21 - Food and Drugs. Part 201 - Labeling § 201.57. Accessed July 1, 2014. Available at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=201.57.
- 29.Shire Human Genetic Therapies, Inc. 2013. VPRIV® (velaglucerase alfa for injection): Highlights of prescribing information. Accessed July 1, 2014. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/022575s012lbl.pdf.
- 30.Elstein D, Hughes D, Goker-Alpan O, et al. Outcome of pregnancies in women receiving velaglucerase alfa for Gaucher disease. J Obstet Gynaecol Res. 2014;40:968–975. doi: 10.1111/jog.12254. [DOI] [PubMed] [Google Scholar]
- 31.Genzyme Corporation. 2005. CEREZYME® (imiglucerase for injection). Accessed December 1, 2014. Available at: http://www.cerezyme.com/~/media/CerezymeUS/Files/pdf/cerezyme_pi.pdf.
- 32.Pfizer Inc. 2014. ELEYSO™ (taliglucerase alfa) for injection: Highlights of prescribing information. Accessed December 1, 2014. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022458s003s006lbl.pdf.
- 33.Hollak CE, van Weely S, van Oers MH, et al. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest. 1994;93:1288–1292. doi: 10.1172/JCI117084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Dussen L, Hendriks EJ, Groener JE, et al. Value of plasma chitotriosidase to assess non-neuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J Inherit Metab Dis. 2014;37:991–1001. doi: 10.1007/s10545-014-9711-x. [DOI] [PubMed] [Google Scholar]
- 35.Pavlova EV, Deegan PB, Cox TM. Biomarkers for osteonecrosis in Gaucher disease. Expert Opin Med Diagn. 2012;6:1–13. doi: 10.1517/17530059.2012.626402. [DOI] [PubMed] [Google Scholar]
- 36.Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160:2835–2843. doi: 10.1001/archinte.160.18.2835. [DOI] [PubMed] [Google Scholar]
- 37.Khan A, Hangartner T, Weinreb NJ, et al. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: A study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res. 2012;27:1839–1848. doi: 10.1002/jbmr.1680. [DOI] [PubMed] [Google Scholar]
- 38.Elstein D, Foldes AJ, Zahrieh D, et al. Significant and continuous improvement in bone mineral density among type 1 Gaucher disease patients treated with velaglucerase alfa: 69-month experience, including dose reduction. Blood Cells Mol Dis. 2011;47:56–61. doi: 10.1016/j.bcmd.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Choi YJ, Lee BJ, Lim HC, et al. Cross-calibration of iDXA and Prodigy on spine and femur scans in Korean adults. J Clin Densitom. 2009;12:450–455. doi: 10.1016/j.jocd.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 40.Faulkner KG, Roberts LA, McClung MR. Discrepancies in normative data between Lunar and Hologic DXA systems. Osteoporos Int. 1996;6:432–436. doi: 10.1007/BF01629574. [DOI] [PubMed] [Google Scholar]
- 41.Pocock NA, Sambrook PN, Nguyen T, et al. Assessment of spinal and femoral bone density by dual X-ray absorptiometry: Comparison of Lunar and Hologic instruments. J Bone Miner Res. 1992;7:1081–1084. doi: 10.1002/jbmr.5650070911. [DOI] [PubMed] [Google Scholar]
- 42.Laskey MA, Crisp AJ, Cole TJ, et al. Comparison of the effect of different reference data on Lunar DPX and Hologic QDR-1000 dual-energy X-ray absorptiometers. Br J Radiol. 1992;65:1124–1129. doi: 10.1259/0007-1285-65-780-1124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information