

SUMMARY
Variation in clinical accuracy of molecular diagnostic methods for cutaneous leishmaniasis (CL) is commonly observed depending on the sample source, the method of DNA recovery and the molecular test. Few attempts have been made to compare these variables. Two swab and aspirate samples from lesions of patients with suspected CL (n = 105) were evaluated alongside standard diagnosis by microscopic detection of amastigotes or culture of parasites from lesion material. Three DNA extraction methods were compared: Qiagen on swab and aspirate specimens, Isohelix on swabs and Boil/Spin of lesion aspirates. Recovery of Leishmania DNA was evaluated for each sample type by real-time polymerase chain reaction detection of parasitic 18S rDNA, and the diagnostic accuracy of the molecular method determined. Swab sampling combined with Qiagen DNA extraction was the most efficient recovery method for Leishmania DNA, and was the most sensitive (98%; 95% CI: 91–100%) and specific (84%; 95% CI: 64–95%) approach. Aspirated material was less sensitive at 80% (95% CI: 70–88%) and 61% (95% CI: 50–72%) when coupled to Qiagen or Boil-Spin DNA extraction, respectively. Swab sampling of lesions was painless, simple to perform and coupled with standardized DNA extraction enhances the feasibility of molecular diagnosis of CL.
Keywords: swab sampling, qPCR, cutaneous leishmaniasis, diagnosis, molecular diagnosis
INTRODUCTION
Parasitic protozoans of the genus Leishmania can cause cutaneous leishmaniasis (CL), a disease manifested by dermal ulcers and sores. The diagnosis of CL remains problematic as the clinical spectrum is broad and may mimic that of other diseases including leprosy, fungal infections, skin cancer, tropical ulcers, mycobacterial ulcers and staphylococcal infections (WHO expert committee 2010). Pentavalent antimonial therapy requiring daily injections for up to 20 days remains the mainstay of treatment for CL in Latin America. Differentiation of CL from other diseases and prevention of overuse of these toxic drugs require diagnostic tests to be highly specific. Sensitive diagnostics are also important because some species of Leishmania can cause chronic dermal manifestations and mucosal involvement, often characterized by scarcity of parasites at the lesion site.
Routine diagnosis of CL is based on demonstration of amastigotes by microscopic examination of the scrapings of skin lesions and in vitro culture of parasites from aspirate material (Faber et al. 2003). Both of these methods require well trained and experienced personnel, laboratory support and quality control programmes; although the specificity should be 100% due to visualization of the parasite, the sensitivity can be variable. We have found that the sensitivity of the diagnostic algorithm can be increased by 8–10% by the inclusion of parasite isolation by culturing the lesion aspirates with microscopy (based on routine diagnosis at the outpatient clinics in Centro Internacional de Entrenamiento e Investigaciones Médicas (CIDEIM). However, culturing presents logistical constrains including cost, availability of appropriate culture media, infrastructure, access to sterile facilities, time to result (which can take up to 2 months) among others. Cultured isolates present the opportunity to discriminate the species of Leishmania using isoenzyme electrophoresis. In Colombia, more than 90% of CL cases are caused by a species of the Leishmania (Viannia) subgenus, of which >75% correspond to Leishmania panamensis infections (Saravia et al. 1998).
Molecular methods have become attractive tools for the diagnosis of CL as they can provide sensitive, specific, rapid and reliable detection of parasites. Currently, these require highly experienced personnel and well-equipped laboratories; however, efforts are being made to simplify these tools. Several molecular amplification methods have been designed for the diagnosis of CL (Ramirez et al. 2000; van der Meide et al. 2008; Espinosa et al. 2009; Adams et al. 2010; Miranda et al. 2012; Hu et al. 2012; Jara et al. 2013) but few have been evaluated for diagnostic accuracy and specificity in studies based on consecutive inclusion and assessment of patients with suspected CL rather than known positive and negative cases (Boggild et al. 2010; Jara et al. 2013). Diagnostic accuracy of molecular assays can vary depending on the sample used and method of DNA recoveryas well as the molecular target and protocol employed. Few attempts have been made to compare the usefulness of different lesion sampling procedures for polymerase chain reaction (PCR) -based diagnosis of CL (Matsumoto et al. 1999), supporting the use of non-invasive methodologies such as filter paper imprints of lesions (Mimori et al. 2002; Boggild et al. 2011). However, thus far simultaneous comparison of DNA recovery methodologies and sample source has not been analysed for the definition of highly sensitive molecular diagnostics for CL. Simple, non-invasive sampling methods coupled to standardized DNA extraction protocols would facilitate reliable diagnosis using molecular tools and obviate invasive and painful sampling procedures such as skin biopsies.
This study is aimed to test non-invasive swab sampling of lesions and conventional aspirate sampling coupled with simple and standardized DNA extraction methods as the basis for optimized sample processing for molecular diagnosis of CL. Clinical specimens were obtained from patients with lesions compatible with suspicion of CL and the diagnostic accuracy of the sample types by quantitative PCR (qPCR) was compared with standard diagnostic methods.
MATERIAL AND METHODS
Study design
This study was designed to evaluate the performance of non-invasive lesion-sampling methodologies coupled to standardized DNA extraction and molecular amplification of Leishmania 18S rDNA as a diagnostic tool for CL. Based on an estimated prevalence of 85% in the suspect population and an expected sensitivity of >90% of the qPCR (van der Meide et al. 2008), a sample size of 96 patients with suspected CL was calculated with a 6% margin of error.
The reference gold standard for parasitological diagnosis of CL was defined as microscopic detection of intracellular amastigotes in Giemsa-stained lesion smears and/or culture isolation of Leishmania from lesion aspirates. Patients with parasitological confirmation of Leishmania infection were referred for treatment according to the standard-of-care therapeutic guidelines provided by the Colombian Ministry of Health and Social Protection (First line: Glucantime®, 20 mg kg−1 weight for 20 days). True and presumptive false positive patients were defined, respectively, as patients with positive reference standard diagnosis and positive PCR, and negative reference standard diagnosis with positive PCR.
Ethics and study population
This study was approved and monitored by the institutional review board for ethical conduct of research involving human subjects of the CIDEIM in accordance with national (resolution 008430, República de Colombia, Ministry of Health, 1993) and international (Declaration of Helsinki and amendments, World Medical Association, Seoul, Korea, October 2008) guidelines. All individuals voluntarily participated in the study and informed consent was obtained from each participant. Patients (aged 2–75 years) with clinical manifestations compatible with active CL were invited to participate. Patients with suspected CL were defined as patients who resided in or had visited known CL endemic areas during the 6 months prior to the onset of the lesion and presented skin lesions clinically compatible with CL that had been present for more than 2 weeks.
Sampling techniques for reference standard diagnosis of CL
Two smears of the lesion scrapings for microscopic evaluation and 4 lesion-aspirate samples were obtained from each patient at the CIDEIM outpatient clinics in Cali and Tumaco, Colombia. Culture of lesion aspirates in Senekjie’s diphasic culture media was performed for all patients; parasite growth was evaluated for 1-month post-inoculation.
Experimental procedures
Two swab samples and two lesion aspirates were taken for molecular diagnosis of CL (Fig. 1). Swab samples were taken by gently rubbing over the ulcer ~10 times (Fig. 2) using commercially available DNA collection swabs (Isohelix SK-1S). For non-ulcerated lesions (nodules, papules or plaques), swab samples were taken from the incision from which lesion scrapings were obtained. Lesion aspirates were obtained from the lesion border as per standard procedure (Figueroa et al. 2009). Samples obtained from CIDEIM-Tumaco were refrigerated using cold packs during transport to CIDEIM-Cali. Swab samples were stored at −20 °C; aspirate samples were put into Qiagen lysis buffer AL1 and then stored at −20 °C; all samples were processed for DNA extraction within 5 days after the samples were taken.
Fig. 1.
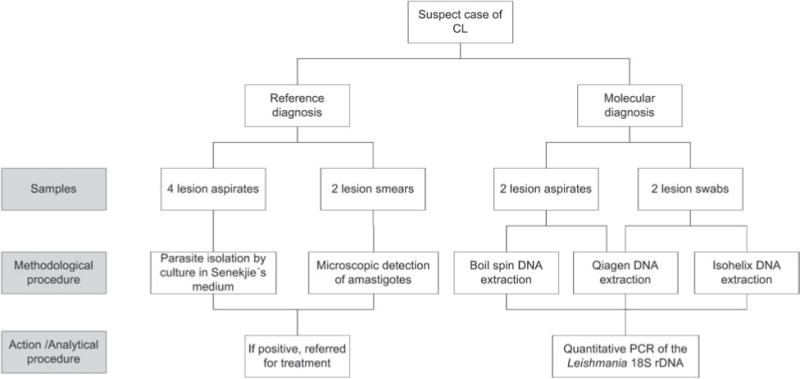
Flow of samples in evaluation. Two lesion smears and 4 aspirates were obtained as part of the reference diagnostic procedure (microscopy and culture). Two lesion swab samples and 2 aspirates were obtained for evaluation by qPCR.
Fig. 2.
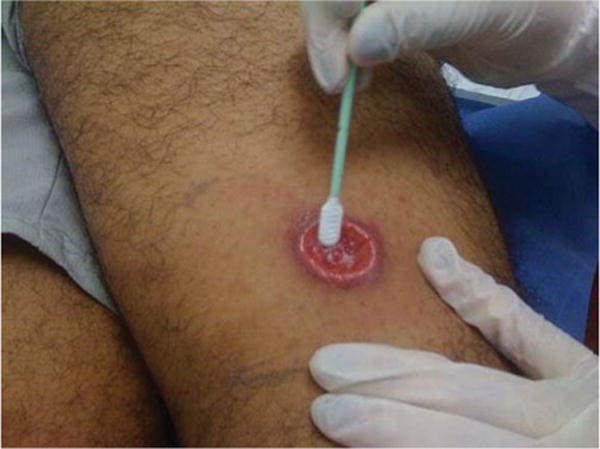
Specimen collection by swab sampling of a cutaneous ulcer of a patient with suspected CL.
DNA extractions
Qiagen DNeasy Blood & Tissue Kit (Qiagen, USA) was used to extract DNA from 1 swab and 1 aspirate sample. Isohelix DNA Isolation Kit (Cell Projects™, Kent, UK) was used to extract DNA from the second swab. A crude ‘Boil-Spin’ method was used for the second aspirate sample. Swab and aspirate replicate samples from each patient were randomly selected for different extraction methodologies. Extractions using Qiagen and Isohelix kits were performed according to manufacturers’ protocols and resultant DNA eluted in 50 μL distilled water. Boil-Spin was processed using a protocol adapted from http://www.finddiagnostics.org/export/sites/default/programs/hat-ond/docs/SOP_RIME_LAMP_kit_template_23MAR12_final.pdf); 40 μL plasma (bovine) plus 60 μL distilled water was added to 100 μL of the aspirate sample and incubated at 90 °C for 10 min. The sample was centrifuged for 3 min at 14000 rpm to eliminate any debris. Two hundred microlitres of the supernatant was removed and used as template DNA. Extraction controls (reagents without DNA sample) were included during every DNA extraction to verify the absence of contamination during the extraction process.
Molecular amplification
qPCR was set up as follows: reactions were conducted in a total volume of 12.5 μL, containing 1.25 μL of the DNA sample, 6.25 μL PCR Mastermix (BioRad), 0.8 μM of each of the two oligonucleotide primers designed to amplify Leishmania 18S rDNA and 0.2 μM of the Leishmania 18S rDNA-specific FAM-labelled TaqMan probe (van der Meide et al. 2008). qPCR was performed in CIDEIM, Cali on a BioRad CFX96 platform as follows: denaturation at 95 °C for 10 min, followed by 35 cycles of denaturation at 95 °C for 15 s and finally at 60 °C for 50 s including FAM detection.
For quantification of parasite load in patient samples, cycle threshold (Ct) values of the samples were extrapolated to a standard curve. Comparisons between experiments were made with a standard curve for 18S rDNA amplification of L. panamensis (MHOM/PA/71/LS94) DNA ranging from 107 to 102 parasites mL−1 in 10-fold dilutions (6 independent replicate experiments with a standard deviation of <0.25% and r2 0.992) and an efficiency of reaction of 101.81%. The baseline threshold was set at 125 in order to compare between different experiments; Ct value was measured for each sample and quantified compared to the standard curve. A negative PCR control and extraction controls were included in each DNA extraction and PCR assay. All samples were analysed double blind to microscopy and culture results.
Species identification
Strains were isolated culturing the needle aspirates of cutaneous lesions and typed by immunoreactivity with monoclonal antibodies. Isoenzyme electrophoresis was performed to identify the species for the strains not accurately typed by reactivity to monoclonal antibodies (Pratt and David, 1981; McMahon-Pratt et al. 1982; Saravia et al. 1998).
Data analysis
Data were entered into EpiData and transferred to STATA for analysis. The diagnostic accuracy of data, including sensitivity and specificity, was calculated for each sample type, and all calculations include 95% confidence intervals (CI). The Kolmogorov—Smirnov test was applied to determine parametric or non-parametric distribution of quantitative data. Kruskal—Wallis 1-way analysis of variance (ANOVA) followed by Dunn’s multiple comparison test was employed for group comparisons. Statistical significance was defined as P<0.05. Data were analysed using Prism 5 software (GraphPad Software, Inc., La Jolla, CA).
Standards for Reporting of Diagnostic Accuracy (STARD) guidelines
This study followed the STARD guidelines (Bossuyt et al. 2003), including blinding of index and reference diagnostic tests.
RESULTS
Patients
A total of 105 suspected patients with lesions compatible with CL were included in this prospective study in two outpatient clinics: CIDEIM-Cali (n = 33) and CIDEIM-Tumaco (N = 72). Characteristics of all enrolled participants and species identity of parasites isolated are summarized in Table 1. In all 90.5% (95 of 105) of patients presented with at least 1 ulcerated lesion. CL was confirmed in 76.2% (n = 80) of the suspected patients by gold standard diagnosis. Fifty-four out of 80 CL patients presented with both positive lesion smear and aspirate cultures; 15 of 80 were only positive for lesion smears and 11 of 80 for culture.
Table 1.
Characteristics of patients with suspected CL.
| Characteristics
|
n (%)
|
|---|---|
| Subjects | n = 105 (%) |
| Diagnosis | |
| Cutaneous leishmaniasisa | 80 (76.2) |
| Lesion smear positive only | 15 (14.3) |
| Culture positive only | 11 (10.5) |
| Lesion smearand Other culture positive | 54 (51.4) |
| Lesion smear and culture negative | 25 (23.8) |
| Age, median (range), years | 23 (3–71) |
| Gender, male (%) | 78 (74.3) |
| Ethnic group, n (%) | |
| Afro-Colombian | 47 (44.84) |
| Mestizo | 51 (47.2) |
| Indigenous | 7 (6.5) |
| White | 1 (0.9) |
| Lesions per subject, median (range) | 1 (1–50) |
| Duration of older lesion, median (range), monthsa | 2 (0.2–240) |
| Lesion characteristics | n = 204 (%) |
| Location of lesion; n (%)a | |
| Upper limbs | 65 (32) |
| Head and neck | 39 (19.2) |
| Trunk | 31 (15.3) |
| Lower limbs | 68 (33.5) |
| Type of lesion; n (%) | |
| Ulcer | 149 (73) |
| Plaque | 28 (13.8) |
| Other | 27 (13.2) |
| Lesion area, median (range), cm2 | 7.28 (0.16–48.7) |
| Leishmania strains isolated | n= 64 (%) |
| Species identification | |
| Leishmania (V) panamensis | 51 (79.7) |
| Leishmania (V) braziliensis | 8 (12.5) |
| Leishmania (V) guyanensis | 3 (4.7) |
| Leishmania amazonensis | 1 (1.6) |
| Leishmania mexicana complex | 1 (1.6) |
One patient was diagnosed with mucocutaneous disease.
Leishmania species
Parasites were isolated and identified in 64% of the participants. At least 4 different Leishmania species pertaining to both the Viannia and Leishmania subgenera caused infections in the participating patient population. The vast majority belonged to species of the Viannia subgenus with L. panamensis predominating overall.
Diagnostic accuracy
The diagnostic accuracy of each sample type coupled with the pre-defined extraction methodology (Fig. 1) was calculated separately against the reference standard diagnostic. Data are summarized in Table 2. The highest diagnostic sensitivity of 98% (95% CI: 90.91–99.61%) and specificity of 84% (95% CI: 64–95.4%) were achieved with qPCR performed on the swab samples coupled with Qiagen DNA extraction. CI of sensitivity overlapped for the swab samples extracted with both Qiagen and Isohelix commercial kits, and CI of specificity overlapped for all extraction methods and sample types (Fig. 3). Of the 10 suspects presenting solely with non-ulcerated lesions, 6 were diagnosed with CL by standard diagnostic procedures; the same 6 patients were also positive for Leishmania by qPCR.
Table 2.
Diagnostic accuracy of swab and aspirate samples compared with the reference diagnostic standard of microscopy and/or culture positive (TP, true positive; FP, false positive; FN, false negative; TN, true negative; CI, confidence interval)
| Sample type | TP | FP | FN | TN | Sensitivity (95% CI) | Specificity (95% CI) |
|---|---|---|---|---|---|---|
| Swab-Qiagen | 78 | 4 | 2 | 21 | 97.5% (CI 91.2–99.6%) | 84% (CI 63.9–95.4%) |
| Swab-Isohelix | 74 | 4 | 6 | 21 | 92.5% (CI 84.4–97.2%) | 84% (CI 63.9–95.4%) |
| Aspirate-Qiagen | 64 | 2 | 16 | 23 | 80% (CI 69.6–88.1%) | 92% (CI 73.9–98.8%) |
| Aspirate-Boil/Spin | 49 | 1 | 31 | 24 | 61.3% (CI 49.7–71.9%) | 96% (CI 79.6–99.3%) |
Fig. 3.
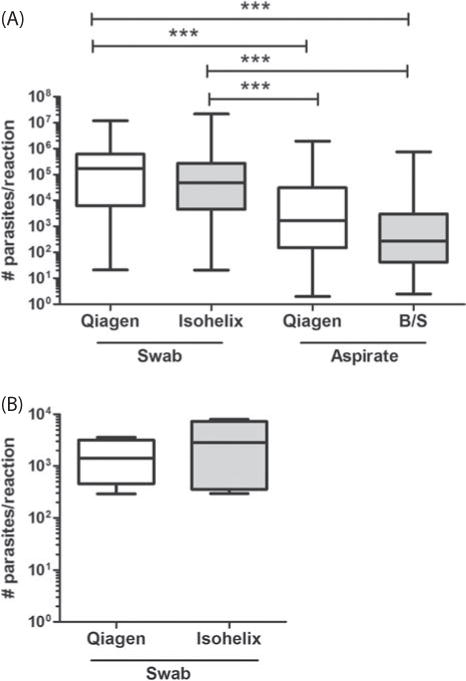
Quantification of parasite loads from lesion swabs and aspirate samples. (A) True positive samples and (B) false positive samples. Parasite number was calculated from a defined Ct threshold of 125 CTU and extrapolated to a parasite DNA standard curve. Data represent number of parasites per reaction (1.25 μL DNA from a total of 50 μL of DNA extraction material) and expressed as median values. Whiskers in box plots show minimum and maximum values. Statistical significance was estimated using the Kruskal–Wallis 1-way ANOVA followed by the Dunn’s test for multiple comparisons. ***: P<0.01. Qiagen and Isohelix: DNA extractions with Qiagen and Isohelix commercial kits, respectively. B/S: Boil-Spin.
Quantification
Parasite loads were estimated by qPCR for all swab and aspirate samples coupled with individual extraction methodologies (Fig. 1). Parasite burden in swab samples extracted with either Qiagen or Isohelix commercial kits was equivalent (Fig. 3A and B). No statistical difference was observed in parasite numbers obtained from aspirate samples extracted with either Qiagen or Boil-Spin methods (Fig. 3A). Importantly, parasite loads quantified from the aspirate material from true positive patients were significantly lower than those obtained from the swab samples (Fig. 3A). Parasite burden in samples from lesions from true and presumptive false positive patients (n = 6) showed that the true positive patients (Fig. 3A) have higher parasite loads compared to putative false positive patients (Fig. 3B), i.e. those only detected by qPCR.
DISCUSSION
Here we describe the sensitive diagnosis by qPCR of CL using non-invasive swab samples from patients with lesions compatible with suspicion of CL coupled with standardized DNA extraction procedures. Swab samples from lesions coupled with Qiagen extraction identified more patients than aspirate samples from lesions coupled with Qiagen extraction. Swab samples were essentially painless to collect compared with lesion scrapings and required less expertise than aspirates or scrapings of lesions. Nevertheless, some challenges for swab sampling should be considered including non-ulcerated lesions such as nodules, plaques or papules, manifestations that present in variable proportions in different endemic settings. For non-ulcerated presentations, either an incision or scraping of the lesion should be performed, ideally followed by swab sampling to access the dermal tissue containing the parasites. Transport of swab samples to reference laboratories for analysis, by ordinary or expedited mail using conventional cold packs allows diagnosis and species identification by molecular methods to be achieved, as required for clinical and public health needs.
Overlapping CI for specificity of DNA extraction methods and sample types suggest that the 18S rDNA is an adequate target for amplification to detect Leishmania DNA from clinical samples. The specificity of qPCR coupled with swab sampling and Qiagen extraction in this consecutive group of patients appeared somewhat low at 84% with wide CI (95% CI: 64–95%). However, 5/6 putatively false positive patients presented with at least 2 positive molecular methods, supporting the interpretation that the index test (qPCR) is more sensitive than the reference test (microscopy and culture). Considering that the samples from the same individuals were independently taken, presumably these samples represent true positive patients undetected by microscopic detection of amastigotes in smears of lesion scrapings and/or parasite isolation from lesion aspirates. Inclusion of clinical characteristics and epidemiological risk factors (Weigle et al. 1993), as well as response to specific therapy in refining the definition of ‘true positives’ would probably diminish the disparity in specificity of conventional parasitological diagnosis and molecular diagnosis of CL.
Quantification of parasite load showed that the parasite burden in true positive patients (diagnosed by the gold standard method and index test) is higher than that of putative false positive patients (only positive by the index test), supporting the likelihood that these patients may not have been detected by microscopy or culture due to the low number of parasites in the active lesion. Parasite loads quantified by qPCR revealed that less parasite DNA was recovered from lesion aspirates compared to swab samples for both the true positive and the false positive samples. This is surprising considering that aspirate samples for parasite isolation are taken from lesion borders where parasites are thought to be more abundant. This finding may reflect the greater efficiency of recovery of tissue material containing parasitic DNA by swab sampling or the greater quantity of parasite DNA in the ulcerated zone of the lesion. Since the CI of the swab sampling coupled to Qiagen or Isohelix DNA extraction overlapped, further exploration of the Isohelix extraction kit would be worthwhile, considering the reduced cost (Isohelix extraction cost two-thirds of the Qiagen extraction), time and technical requirements.
The feasibility of implementing molecular diagnosis of CL beyond well-equipped laboratories requires refinement of sample processing, DNA extraction and the use of simplified molecular diagnostics. Here qPCR was used as a method to assess Leishmania DNA recovery from cutaneous lesions by different sampling methodologies. The recently developed, loop-mediated isothermal amplification (LAMP) is an example of a simplified molecular diagnostic that can be performed within 40 min at 65 °C, and allows a visual read-out. LAMP is currently under development for leishmaniasis and should be evaluated for CL when available (Adams et al. 2010; http://www.finddiagnostics.org/programs/hatond/leishmaniasis/lamp-for-leish.html). Alternative qPCR strategies including multiplex reactions for Leishmania detection at the species level, and differential diagnosis of other infectious agents such as non-tuberculous mycobacteria or Sporothrix, causing similar cutaneous ulcers, could be relevant for clinical decision making. As qPCR detects active infection due to the presence of parasitic DNA, it is worth noting the potential to detect relapse and monitor response to treatment.
Non-invasive swab sampling allows samples to be collected in rural areas and transported to central laboratory facilities where standardized amplification can be efficiently and reliably conducted. Coupling non-invasive sampling to a sensitive and simple molecular diagnostic test provides feasible alternatives for diagnostic challenges such as leishmaniasis in children and complicated manifestations including chronic ulcers and mucosal disease. Furthermore, swab sampling provides storage flexibility as samples can be obtained and stored at ambient temperature or 4 °C, or preserved at −20 °C for longer periods of time. Systematic evaluation of temperature stability needs to be conducted under the conditions of different settings considering that ambient temperature and potentially humidity index could impact the stability of the biological material for DNA amplification.
CONCLUSION
We have demonstrated the feasibility and diagnostic accuracy of a non-invasive alternative to aspirates and biopsies for CL lesion sampling when coupled with standardized DNA extraction and molecular amplification methods. Swab samples are easy to collect, painless for the patient, can be conveniently transported, obviate use of needles, and recovery of DNA from swabs is superior to aspirate samples. We recommend the validation of swab sampling coupled with molecular diagnosis in other epidemiological settings to ratify the ‘fit for purpose’ of this approach to point of care diagnosis of CL.
Acknowledgments
Many thanks to the British Society for Parasitology for hosting the conference on diagnostics. We would like to thank all our patients and their families for taking part in the study. We gratefully acknowledge the support of the personnel of the CIDEIM BioBank and Clinical Unit, Maryori Vidarte, Alejandra Arcos, Jimena Jojoa, Wilson Cortes and Mary Luz Hurtado for their technical assistance in sample procurement, parasite isolation and phenotyping of clinical strains collected in this study.
FINANCIAL SUPPORT: This work was supported by TiPharma, Netherlands (project T4-303).
References
- Adams ER, Schoone GJ, Ageed AF, Safi SE, Schallig HD. Development of a reverse transcriptase loop-mediated isothermal amplification (LAMP) assay for the sensitive detection of Leishmania parasites in clinical samples. American Journal of Tropical Medicine and Hygiene. 2010;82:591–596. doi: 10.4269/ajtmh.2010.09-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggild AK, Valencia BM, Espinosa D, Veland N, Ramos AP, Arevalo J, Llanos-Cuentas A, Low DE. Detection and species identification of Leishmania DNA from filter paper lesion impressions for patients with American cutaneous leishmaniasis. Clinical Infectious Diseases. 2010;50:e1–e6. doi: 10.1086/648730. [DOI] [PubMed] [Google Scholar]
- Boggild AK, Ramos AP, Valencia BM, Veland N, Calderon F, Arevalo J, Low DE, Llanos-Cuentas A. Diagnostic performance of filter paper lesion impression PCR for secondarily infected ulcers and nonulcerative lesions caused by cutaneous leishmaniasis. Journal of Clinical Microbiology. 2011;49:1097–1100. doi: 10.1128/JCM.02457-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, Moher D, Rennie D, de Vet HC, Lijmer JG, Standards for Reporting of Diagnostic Accuracy Group The STARD statement for reporting studies of diagnostic accuracy: explanation and elaboration. The Standards for Reporting of Diagnostic Accuracy Group. Croatian Medical Journal. 2003;44:639–650. [PubMed] [Google Scholar]
- Espinosa D, Boggild AK, Deborggraeve S, Laurent T, Valencia C, Pacheco R, Miranda-Verastegui C, Llanos-Cuentas A, Leclipteux T, Dujardin JC, Buscher P, Arevalo J. Leishmania OligoC-TesT as a simple, rapid, and standardized tool for molecular diagnosis of cutaneous leishmaniasis in Peru. Journal of Clinical Microbiology. 2009;47:2560–2563. doi: 10.1128/JCM.00259-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber WR, Oskam L, van Gool T, Kroon NC, Knegt-Junk KJ, Hofwegen H, van der Wal AC, Kager PA. Value of diagnostic techniques for cutaneous leishmaniasis. Journal of the American Academy of Dermatology. 2003;49:70–74. doi: 10.1067/mjd.2003.492. [DOI] [PubMed] [Google Scholar]
- Figueroa RA, Lozano LE, Romero IC, Cardona MT, Prager M, Pacheco R, Diaz YR, Tellez JA, Saravia NG. Detection of Leishmania in unaffected mucosal tissues of patients with cutaneous leishmaniasis caused by Leishmania (Viannia) species. Journal of Infectious Diseases. 2009;200:638–646. doi: 10.1086/600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu RV, Kent AD, Adams ER, van der Veer C, Sabajo LO, Mans DR, de Vries HJ, Schallig HD, Lai AFRF. First case of cutaneous leishmaniasis caused by Leishmania (Viannia) braziliensis in Suriname. American Journal of Tropical Medicine and Hygiene. 2012;86:825–827. doi: 10.4269/ajtmh.2012.11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jara M, Adaui V, Valencia BM, Martinez D, Alba M, Castrillon C, Cruz M, Cruz I, Van der Auwera G, Llanos-Cuentas A, Dujardin JC, Arevalo J. Real-time PCR assay for detection and quantification of Leishmania (Viannia) organisms in skin and mucosal lesions: exploratory study of parasite load and clinical parameters. Journal of Clinical Microbiology. 2013;51:1826–1833. doi: 10.1128/JCM.00208-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Hashiguchi Y, Gomez EA, Calvopina MH, Nonaka S, Saya H, Mimori T. Comparison of PCR results using scrape/exudate, syringe-sucked fluid and biopsy samples for diagnosis of cutaneous leishmaniasis in Ecuador. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93:606–607. doi: 10.1016/s0035-9203(99)90065-2. [DOI] [PubMed] [Google Scholar]
- McMahon-Pratt D, Bennett E, David JR. Monoclonal antibodies that distinguish subspecies of Leishmania braziliensis. Journal of Immunology. 1982;129:926–927. [PubMed] [Google Scholar]
- Mimori T, Matsumoto T, Calvopina MH, Gomez EA, Saya H, Katakura K, Nonaka S, Shamsuzzaman SM, Hashiguchi Y. Usefulness of sampling with cotton swab for PCR-diagnosis of cutaneous leishmaniasis in the New World. Acta Tropica. 2002;81:197–202. doi: 10.1016/s0001-706x(01)00215-7. [DOI] [PubMed] [Google Scholar]
- Miranda A, Saldana A, Gonzalez K, Paz H, Santamaria G, Samudio F, Calzada JE. Evaluation of PCR for cutaneous leishmaniasis diagnosis and species identification using filter paper samples in Panama, Central America. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2012;106:544–548. doi: 10.1016/j.trstmh.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Pratt DM, David JR. Monoclonal antibodies that distinguish between New World species of Leishmania. Nature. 1981;291:581–583. doi: 10.1038/291581a0. [DOI] [PubMed] [Google Scholar]
- Ramirez JR, Agudelo S, Muskus C, Alzate JF, Berberich C, Barker D, Velez ID. Diagnosis of cutaneous leishmaniasis in Colombia: the sampling site within lesions influences the sensitivity of parasitologic diagnosis. Journal of Clinical Microbiology. 2000;38:3768–3773. doi: 10.1128/jcm.38.10.3768-3773.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravia NG, Segura I, Holguin AF, Santrich C, Valderrama L, Ocampo C. Epidemiologic, genetic, and clinical associations among phenotypically distinct populations of Leishmania (Viannia) in Colombia. American Journal of Tropical Medicine and Hygiene. 1998;59:86–94. doi: 10.4269/ajtmh.1998.59.86. [DOI] [PubMed] [Google Scholar]
- van der Meide W, Guerra J, Schoone G, Farenhorst M, Coelho L, Faber W, Peekel I, Schallig H. Comparison between quantitative nucleic acid sequence-based amplification, real-time reverse transcriptase PCR, and real-time PCR for quantification of Leishmania parasites. Journal of Clinical Microbiology. 2008;46:73–78. doi: 10.1128/JCM.01416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigle KA, Escobar M, Arias AL, Martinez F, Rojas C. A clinical prediction rule for American cutaneous leishmaniasis in Colombia. International Journal of Epidemiology. 1993;22:548–558. doi: 10.1093/ije/22.3.548. [DOI] [PubMed] [Google Scholar]