

Abstract
The N-methyl-D-aspartate receptor (NMDAR) antagonists: MK-801, phencyclidine and ketamine are open-channel blockers with limited clinical value due to psychotomimetic effects. Similarly, the psychotomimetic effects of the dextrorotatory opioids, dextromethorphan and its metabolite dextrorphan, derive from their NMDAR antagonist actions. Differences in the use dependency of blockade, however, suggest that the binding sites for MK-801 and dextrorphan are distinct. In the absence of exogenous glutamate and glycine, the rate of association of [3H]MK-801 with wild-type NR1-1a/NR2A receptors was considerably slower than that for [3H]dextrorphan. Glutamate individually, and in the presence of the co-agonist glycine, had substantial effects on the specific binding of [3H]MK-801, while the binding of [3H]dextrorphan was not affected. Mutation of residues N616 and A627 in the NR1 subunit had a profound effect on [3H]MK-801 binding affinity, while that of [3H]dextrorphan was unaltered. In contrast, NR1 residues, W611 and N812, were critical for specific binding of [3H]dextrorphan to NR1-1a/NR2A complexes with no corresponding influence on that of [3H]MK-801. Thus, [3H]dextrorphan and [3H]MK-801 have distinct molecular determinants for high-affinity binding. The ability of [3H]dextrorphan to bind to a closed channel, moreover, indicates that its recognition site is shallower in the ion channel domain than that of MK-801 and may be associated with the extracellular vestibule of the NMDAR.
Keywords: NMDA receptor, Dextrorphan, MK-801, Glutamate, Binding, Mutagenesis
1. Introduction
Dextrorphan (DX), the O-demethylated dextrorotatory opioid metabolite of dextromethorphan (DXM), is known to have a high-affinity binding site associated with the NMDAR (Franklin and Murray, 1992). [3H]DX binding studies using rat brain membrane homogenates and slide-mounted brain sections revealed a Kd of between 56 and 70 nM depending on the region of the brain examined (Franklin and Murray, 1992; Roth et al., 1996). Holtzman (1980) demonstrated that DX acts as a full substitute for PCP in rats trained to discriminate between phencyclidine (PCP) and saline. Stereotyped behavioral responses normally associated with PCP in (Spraguee-Dawley) rats are duplicated by the administration of DX (Ishmael et al., 1998). The order of potency necessary to induce stereotyped behavior was: MK-801 > PCP > (±) cyclazocine > DX > (±) ketamine (Ket) > DXM. The rank order of activity for these antagonists correlates well with their respective affinities for NMDAR.
NMDAR are thought to assume either a tetrameric or a pentameric structure resulting from the combination of any two NR1 splice variants interacting with two or three NR2 encoded subunits to form an ionotropic channel consisting of cytosolic and extracellular vestibules as well as a transmembrane pore (Fig. 1A and B) (Hawkins et al., 1999; Dingledine et al., 1999). Each NMDA receptor subunit harbors four hydrophobic domains, three of these are true transmembrane spanning regions (M1, M3 and M4 (not shown); Fig. 1A) and one (M2; Fig. 1A) forms a reentrant loop (Sprengel et al., 2001). NMDAR subunit composition is a determinant of the pharmacologic characteristics of noncompetitive NMDAR antagonists (Avenet et al., 1996; Lynch et al., 1995; Yamakura et al., 1993).
Fig. 1.
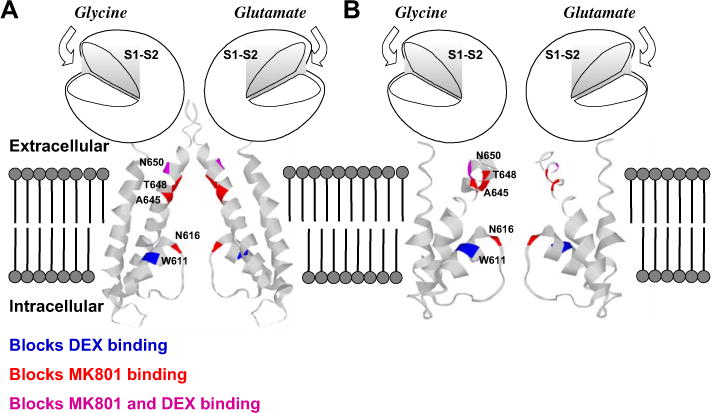
(A) Schematic representation of NR1 and NR2 subunits of the rat NMDA receptor shown with hypothetical binding domains illustrated. For clarity, only two of five assumed subunits are shown from a homology model of NR1/NR2A superimposed on the closed conformation of a bacterial (Streptomyces lividans) K+ channel (KcsA) structure (Doyle et al., 1998); the amino terminal domain is not shown. S1 and S2 are regions containing the binding sites for the co-agonists glutamate (NR2) and glycine (NR1). As there is no homologous M4 region in the KcsA structure, it is not possible to superimpose an equivalent component of NR1. The regions studied in this investigation and their effect on [3H]MK-801 and [3H]DX binding are color-coded for identification; amino acid numbering is indicated on one subunit. (B). Similar schematic diagram except that the homology model of NR1/NR2A was superimposed on the open conformation of the Methanobacterium thermoautotrophicum K+ channel (MthK) (Jiang et al., 2002).
A highly conserved asparagine (N616) in the M2 region of each NMDAR subunit (Fig. 1A and B) is believed to contribute to blockade by both Mg2+ and noncompetitive antagonists (Burnashev et al., 1992; Mori et al., 1992; Yamakura et al., 1993; Sakurada et al., 1993; Wollmuth et al., 1998). NR1-1a (N616Q) and NR2B (N616Q) mutants both attenuated Mg2+ blockade, while the double mutant NR1-1a (N616Q)/NR2B (N616Q) was most effective in abrogating MK-801 antagonism (Mori et al., 1992). Similarly, NR1-1a mutants (N616Q/R) both affect MK-801 antagonist activity with the arginine mutant demonstrating the most dramatic change (Sakurada et al., 1993). Three essential structural requirements for NMDAR blockade by MK-801 and dextromethorphan as described by Kroemer et al. (1998) are: a hydrogen bond between a protonated amine in the ligand and a suitable residue, a hydrophobic center of the ligand that is not so bulky as to interfere with interaction at a hydrophobic region of the receptor and a hydrophobic interaction with the receptor involving an aromatic ring of the ligand (Fig. 3A and B).
Fig. 3.
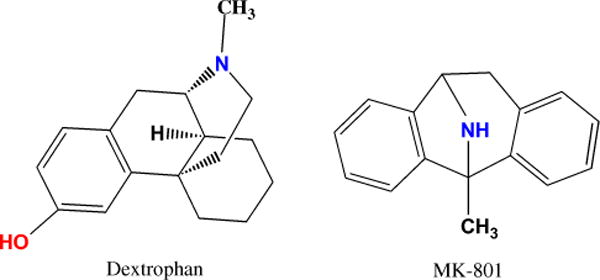
Graphical representation of dextrorphan and MK-801 illustrating critical regions of the proposed pharmacophore as reported by Kroemer et al. (1998). The site of the proposed hydrophobic point is red and the protonated amine blue. The representations were produced using ChemDraw (CambridgeSoft Corporation, 100 CambridgePark Drive, Cambridge, MA 02140, USA).
MK-801, along with Ket and PCP, act as open-channel blockers of the NMDAR (Huettner and Bean, 1988). These compounds have the antecedent requirement that the receptor be in the activated, or open, state in order for ligand binding to its high-affinity site to occur (Fig. 1B) (Huettner and Bean, 1988). Although the binding of DX and MK-801 has been shown to be mutually exclusive at equilibrium, their respective binding sites are likely to contain unique recognition elements. In contrast to MK-801, DX has been shown to display less use dependency in producing noncompetitive inhibition of NMDA receptor-mediated depolarization in a hippocampal slice (Cole et al., 1989). These results are consistent with the possibility of DX acting at a site relatively exterior to that of MK-801 in the channel domain. The regulation of [3H]DX binding by Mg2+ and polyamines also appears to distinguish [3H]DX binding from both [3H]MK-801 and [3H]TCP (Franklin and Murray, 1992).
Evidence for overlapping and/or multiple recognition sites for noncompetitive antagonists of the NMDAR has been provided previously. Yamakura et al. (1993) demonstrated that the contribution of the conserved M2 asparagine residue contributed variably to the interaction with the noncompetitive antagonists, MK-801, PCP, Ket and SKF-10,047. Ket antagonism was least influenced by asparagine → glutamine mutations, while SKF-10,047 was most influenced. More recently, these authors reported that an array of racemic opioids block NMDAR at a site which at least partially overlaps with that of Ket and MK-801 (Yamakura and Shimoji, 1999). Kashiwagi et al. (2002) has reported differential effects of mutations on the blockade of three open-channel blockers of the NMDAR. Evaluation of residues associated with the M2 region of the NMDAR revealed that mutation of the critical asparagine (N616) reduced the block of memantine, MK-801, N1-N4-N8 tribenzyl-spermidine and Mg2+ (Kashiwagi et al., 2002). Conversely, many mutations in the pre-M1, M1, M3 and post-M3 regions had little or no effect on the block by memantine, while reducing that of MK-801 and TB-3-4 (Kashiwagi et al., 2002).
The objective of the current investigation was to compare the binding characteristics of the two noncompetitive NMDAR antagonists, DX and MK-801, to heterologously expressed NMDAR complexes. Examination of association kinetics and agonist dependency of [3H]DX and [3H]MK-801 binding to wild-type NMDAR distinguished these recognition sites. Furthermore, the influence of mutations associated with the M2, M3 and M4 domains of the NR1 subunit on the binding affinity of [3H]DX and [3H]MK-801 support the contention that the sites of ligand–receptor interaction for the two antagonists contain distinct molecular determinants.
2. Materials and methods
2.1. Cell culture
COS-7 cells were maintained in high glucose Dulbecco’s Modified Essential Media (DMEM) (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum, 100 units penicillin, 100 μg streptomycin and 2.2 g/l NaHCO3. Cells were grown in a 5% CO2 atmosphere at 37 °C. Confluent cultures in T-75 flasks (Corning Inc., Corning, NY) were split 1 flask:4 plates into 150 mm Nunclon plates (Nalge Nunc International, Rochester, NY) for transfection. Cells were allowed to reach 70–90% confluency for transfection via cationic lipid reagent.
2.2. Site-directed mutagenesis of NMDA NR1
2.2.1. Constructs
A fragment containing the entire open reading frame of NR2A (kindly provided by Professor P. Seeburg, accession number M91561) was subcloned into the eukaryotic expression vector pTL2 (generously provided by Dr. T. Lufkin) and used for transient transfection experiments. The construction of pTL1/NR1-1a (from the pN60 generously provided by Dr. S. Nakanishi, accession number X63255) has been described previously (Ishmael et al., 1996). Amino acid residues of the NR1 subunit are numbered from the initiator methionine as in the seminal paper reporting the sequence of NR1 (Moriyoshi et al., 1991). Consequently, residue NR1 (N616) in this paper would correspond to residue NR1 (598) in reports using the alternative numbering system adopted by other investigators (Burnashev et al., 1992; Yamazaki et al., 1992). The differences in the two systems are a result of the removal of an 18 residue signal peptide to form the mature peptide. Site-directed mutagenesis was carried out using the QuikChange system (Stratagene, La Jolla, CA). Briefly, oligonucleotides containing the desired base changes and a silent mutation were synthesized and allowed to anneal with the vector containing the appropriate NMDA receptor DNA sequence. The silent mutation introduced a new diagnostic restriction site to facilitate mutant screening. The mutation was amplified via 18 extension cycles and the product was further processed as per the kit instructions (Stratagene, La Jolla, CA). The presence of the desired mutation was determined by restriction enzyme: NR1-1a N616Q (PstI), N616R (BglII), W611A (NaeI), N650A (Eco47III), T789A (Eco47III) and N812A (NaeI).
2.2.2. Sequence analysis
The entire coding region of all mutant constructs, as well as the parental clone, was sequenced (MWG Biotech, High Point, NC) with an ABI capillary DNA sequencer (Applied Biosystems, Foster City, CA). The sequences of all mutations were compared by alignment to the parental construct to further ensure fidelity of the amplification (VectorNTI, InfoMax, Frederick, MD).
2.2.3. Transient transfection of COS-7 cells
On the day of transfection, cells were washed with 10 ml serum and antibiotic-free DMEM. Each plate received 15 ml of serum and antibiotic-free media. All transfection and expression media contained 200 μM Ket (Sigma, St. Louis, MO) as a protectant from glutamate activation of transfected NMDAR complexes. Plates were transfected with a mixture of DNA and lipid (VennNova, Pompano Beach, FL) in serum and antibiotic-free DMEM with each 150 mm plate receiving 37 μg of DNA and 148 μg of the cationic lipid reagent. Optimum DNA to lipid ratio in the system used was determined by β-galactosidase expression using a pSV-β-Gal vector (Promega, Madison, WI) (data not shown). The transfection ratio of the NMDA receptor subunit plasmids that resulted in the highest level of complex expression was empirically determined to be 1:1 (data not shown) by measuring specific binding of 1 nM [3H]MK-801. Additionally, 15% of the DNA added was a vector encoding for the large T antigen to enhance activation of the SV40 promoter associated with the pTL vector. The ratios of all DNA transfected into host cells was then 42.5% for NR1-1a (wild-type or mutant), 42.5% NR2A and 15% large T antigen. Lipid and DNA were diluted individually to equal volumes (2 ml/150 mm plate) of room temperature serum and antibiotic-free DMEM supplemented with 200 μM Ket. The two mixtures were then combined and allowed to incubate for 15–45 min at room temperature. The transfection reaction (4 ml) was added in a drop-wise manner to each plate. The transfection reaction was allowed to proceed for 24 h in a 5% CO2 incubator at 37 °C. At 24 h the transfection media was replaced with complete high glucose DMEM (10% FBS; 10 units penicillin and 10 μg streptomycin per liter) (Atlanta Biologicals, Norcross, GA) and 200 μM Ket. Transfected cells were then harvested at 48 h.
2.3. Buffers
All binding reactions, with the exception of the association and co-agonist protocols (see below), were carried out in 5 mM HEPES (pH 7.4) buffer. Reactions involving MK-801 were carried out in 5 mM HEPES (pH 7.4) buffer containing 100 μM glutamate and 100 μM glycine, while those involving DX were performed in the presence of a 3-μM concentration of each co-agonist. All incubations were at room temperature and, with the exception of the association experiments, allowed to proceed for 4 h.
2.4. Membrane preparation and reaction conditions
Cells were scraped with a rubber policeman into ice-cold buffer (5 mM HEPES, 10 mM EDTA, pH 7.4) (Sigma) and homogenized by 10 strokes with a glass Dounce (Kontes, Vineland, NJ) using a tight pestle. Membranes were then centrifuged at 40,000 × g for 20 min at 4 °C. The supernatant was decanted and the pellets frozen at −80 °C for a minimum of 1 h. Membranes were then resuspended in buffer (5 mM HEPES, 10 mM EDTA, 100 μM glutamate, pH 7.4) and allowed to incubate for 30 min at room temperature in order to permit dissociation of the Ket used as a protectant during the transfection. Membranes were then centrifuged as above and resuspended in ice-cold buffer (5 mM HEPES, 10 mM EDTA, pH 7.4) followed by centrifugation. Resuspension and centrifugation were repeated three times in ice-cold buffer followed by freezing the pellet for a minimum of 1 h at −80 °C in order to facilitate fracture of vesicles formed during homogenization. A final wash cycle was followed by resuspending the pellet in buffer for determination of the protein concentration using the method of Lowry et al. (1951). The extensive washing protocol is required to remove endogenous glutamate and glycine thereby producing a preparation that is nominally free of the two co-agonists. Average tissue yield per plate was approximately 0.5 mg. Membranes for all assays were resuspended in ice-cold buffer to a concentration of 0.25–0.5 μg/ml on the day of the experiment. All binding reactions received 300 μl of the resuspended membrane preparation. Specific binding was determined to be linear within the protein concentration range used in this investigation (data not shown). It was also established that ligand depletion was less than 5% for both [3H]MK-801 and [3H]DX (data not shown).
2.5. Liquid scintillation measurement of filter-retained titration
Membranes were harvested by rapid filtration with a 24-well Brandel cell harvester (Brandel Inc., Gaithersburg, MD) in ice-cold buffer onto GF/B filters (Brandel Inc.) saturated with 0.5% (v/v) solution of polyethylenimine. Filters were washed (7 × 3 ml) with ice-cold buffer in order to remove unbound radioligand. Filters were agitated on an orbital shaker at 175 rpm for 8–10 h in scintillation fluid (Cytoscint; ICN, Costa Mesa, CA) to elute receptor/ligand complexes. Tritium retained on filters was quantified via a Beckman LS6000IC scintillation counter (Beckman Coulter Inc., Fullerton, CA).
2.6. [3H]MK-801 and [3H]DX equilibrium competition assay
The minimum concentrations of the unlabeled species required for complete displacement of labeled ligands from specific, high-affinity sites were determined by standard equilibrium competition assays for wild-type and mutant NR1-1a/NR2A receptor complexes expressed in transfected cells. Reactions containing either 1 nM [3H]MK-801 or 32 nM [3H]DX (New England Nuclear) were titrated with concentrations of the unlabeled ligand ranging from 0.01 nM to 10 μM. Reactions were allowed to proceed at room temperature for 4 h and then terminated by harvesting the membrane/receptor complexes. Further pharmacological characterization of the heterologously expressed wild-type NMDAR was achieved by a series of equilibrium competition experiments in which [3H]MK-801 (0.8 nM) or [3H]DX (26 nM) was displaced by phencyclidine (0.01 nM–10 μM), dextromethorphan (0.1 nM–100 μM), or ketamine (0.1 nM–100 μM). Data from the DX and MK-801 self-displacement equilibrium competition assays were also used for the determination of [3H]MK-801 and [3H]DX Bmax and Kd values.
2.7. Association kinetics
Rates of association of [3H]MK-801 and [3H]DX under nominal glutamate/glycine conditions were determined in kinetic studies. Membranes derived from pTL1 (NR1-1a)/pTL2 (NR2A) transfected COS-7 cells were incubated at room temperature in buffer containing either 1 nM [3H]MK-801 or 30 nM [3H]DX for 48 and 6 h, respectively. Non-specific binding was determined by the addition of 300 nM of unlabeled MK-801 or 3 μM DX to reactions containing the corresponding radio-labeled species of each.
2.8. Influence of glutamate and glycine concentration on [3H]MK-801 and [3H]DX affinity
The effect of glutamate and glycine concentration individually, and as co-agonists, on [3H]MK-801 and [3H]DX affinity to heterologously expressed wild-type NMDAR complexes was examined. Reactions consisted of 1 nM [3H]MK-801 or 30 nM [3H]DX and from 300 nM to 10 mM glutamate or glycine. Final volumes of the reactions were 500 μl. Non-specific binding is defined as in association protocol. Reactions were allowed to proceed at room temperature for 4 h and then harvested as described above. Pooled binding data were normalized to represent fold stimulation and plotted as a function of agonist concentration (see Section 2.9).
[3H]MK-801 and [3H]DX specific binding dependence on glutamate in the presence of a fixed concentration of glycine (100 μM) was determined. Results from the individual agonist effects on binding indicated optimal binding of [3H]MK-801 and [3H]DX to be occurring at 100 μM and 3 μM of glycine, respectively. Consequently, glutamate concentration effects on [3H]MK-801 were determined in the presence of 100 μM glycine. In the case of [3H]DX binding, dependence on glutamate in the presence of 3 μM glycine was ascertained. Data were analyzed as in individual agonist effect on [3H]MK-801 and [3H]DX experiments (see Section 2.10).
2.9. [3H]MK-801 and [3H]DX affinity in the presence of glutamate and glycine co-agonists
The affinity of [3H]MK-801 for both wild-type and mutant NMDAR complexes was examined via saturation analysis. Membranes from transfected cells were resuspended using 10 strokes with a glass Dounce in 5 mM HEPES (pH 7.4) to a final concentration of 0.25–0.5 μg/ml. Each reaction contained 100 μM glutamate, 100 μM glycine, 0.03–10 nM [3H]MK-801 and 80–150 μg membranes at a final volume of 500 μl. Nonspecific binding was defined in the presence of 300 nM MK-801. [3H]DX affinity was determined by saturation analysis in the presence of 1.5–125 nM [3H]DX. Nonspecific binding was determined in the presence of 3 μM DX.
2.10. Data analysis
2.10.1. Homologous competition assay
Calculation of IC50 values from homologous competition (self-displacement) experiments, in which minimum concentrations of cold ligand required to define non-specific binding for each radioligand, was achieved by fitting a logistic expression to the data. The expression
where X is the concentration of radioligand and Y is binding, returns the IC50 value. Ki values were then determined after the method of Cheng and Prusoff (1973). All pharmacological and statistical analyses were performed with the GraphPad Prism software package version 4 (GraphPad Software, Inc. San Diego, CA).
2.10.2. Saturation analysis of the homologous competition assay
Saturation data from the homologous competition experiments were generated by adjusting the specific activity of each radioligand as a function of the competitor concentration. The amount of radioligand bound at each concentration of cold ligand was calculated by adjusting the specific activity of the stock solution (SAs) at each concentration (SAx) of cold ligand [L] by a factor representing the ratio of the labeled compound [L*] to total compound in the reaction (SAx = [L*]/([L*]+[L]SAs). Binding parameters were determined by fitting a non-linear regression expression Y = (BmaxX)/(Kd + X), where X represents the concentration of cold ligand in nM, to the data.
2.11. Determination of fractional occupancy
Fractional occupancy was calculated as: F=[L]/([L] +[Kd]) where L is the amount of ligand added to the reaction. Determination of % occupancy by the cold ligand at different concentrations of radioligand was determined by considering the molar ratios of each species in an incubation and calculating the resultant occupancies of the diluted radioligand.
2.12. Pharmacological evaluation of transiently expressed wild-type NR1a/NR2A receptor
Calculation of IC50 values derived from the pharmacological evaluation (heterologous competition) of the heterologously expressed wild-type receptor was achieved by fitting a logistic expression to the data; where X is the logarithm of concentration and Y is the response. Combined data from multiple experiments were fit using shared binding parameters (Hill slopes, total and nonspecific) and unique IC50 values.
2.13. Evaluation of initial rates of association
Initial rates of association were compared by linear transformation of binding data to a natural logarithm plot ln([Beq]/[Beq − Bt]) = kobs(t) where Beq and Bt are the amounts of radioligand bound at equilibrium and time t, respectively. Fitting this equation to the association data generates a linear plot with slope corresponding to kobs (Leid et al., 1988).
2.14. Evaluation of saturation isotherm assays
Binding parameters were determined by fitting a nonlinear regression expression Y = (BmaxX)/(Kd + X), where X represents the concentration of radioligand in nM, to the data.
2.15. Statistical analysis
Kd estimates derived from the saturation isotherm experiments were analyzed by one-way analysis of variance. A Dunnet’s post hoc analysis was performed to compare the Kd values of mutant receptors to those of the wild-type control.
3. Results
This investigation focused on a comparison of the binding properties of two noncompetitive antagonists of the NMDA receptor. Our working hypothesis was that the recognition elements for MK-801 and DX in the ion channel domain of the NMDA receptor are not identical. To this end, we attempted to distinguish the molecular determinants of MK-801 and DX binding. As misincorporations by the Pfu polymerase during the site-directed mutagenesis of the parental NR1-1a (wild-type) subunit would confound results from subsequent experimentation, we determined the fidelity of the procedure. Sequence analyses of all constructs confirmed that only the intended mutations occurred during the mutagenesis of the wild-type NR1-1a subunit (Fig. 2). Consequently, any differences in high-affinity binding to mutant NMDAR, as compared to that of the wild-type receptor can be attributed to the single amino acid change introduced by the site-directed mutagenesis alteration of the parental nucleotide sequence.
Fig. 2.
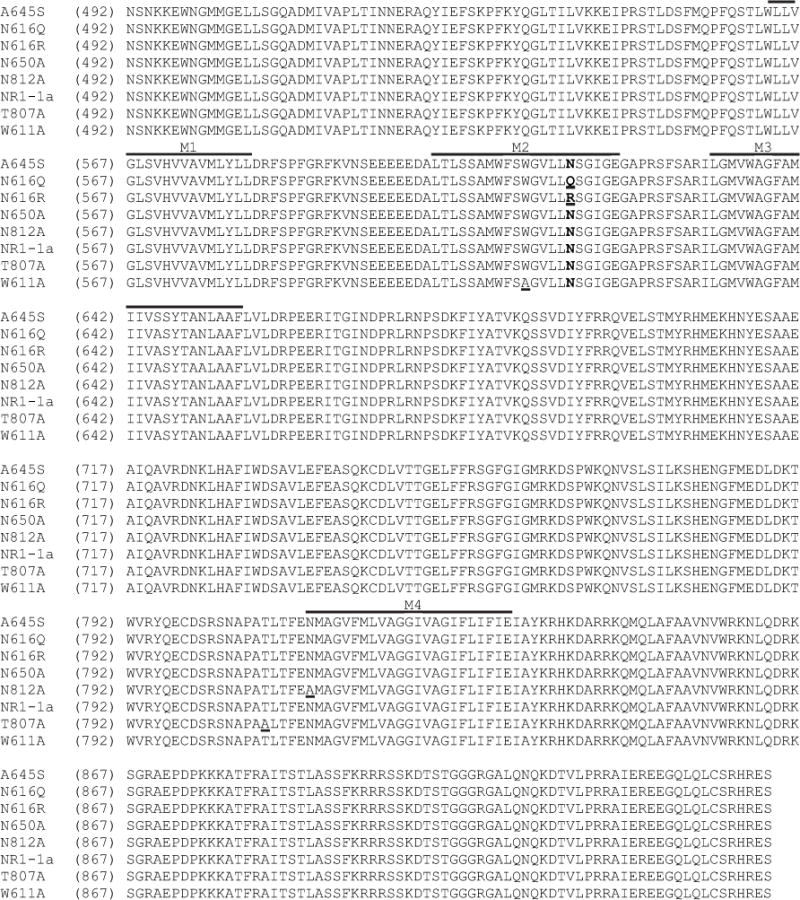
Alignment of the amino acid sequences of wild-type and mutant NR1a subunits of the rat NMDAR used in this investigation. Residues mutated in this study are underlined and segments previously identified as regions M1, M2, M3 and M4 are illustrated by lines above the appropriate sequences (Kuner et al., 1996; Beck et al., 1999). The critical asparagine residues associated with the M2 region of NR1 are in bold type.
In order to test the hypothesis that DX and MK-801 bind to non-identical residues it was critical to precisely define specific binding for each radioligand. As depicted in Fig. 4A specific binding of [3H]MK-801 to membranes derived from COS-7 cells transfected with wild-type NR1-1a and NR2A is eliminated at a concentration of 300 nM MK-801. The minimum concentration of unlabeled DX required to define non-specific binding was also established via competition experiments in the presence of 3 μM glycine and glutamate. Complete inhibition of [3H]DX specific binding is evident at 3 μM of the unlabeled ligand (Fig. 4B). Analysis of three independent self-displacement experiments permitted calculations of IC50 values for both displacers which were 21.6 nM (13.1–35.6 nM, 95% confidence interval [CI]) and 4.36 nM (2.35–8.1 nM 95% CI) for DX and MK-801, respectively. These values are consistent with those determined by heterologous competition assays in which cold MK-801 displaces [3H]DX or the reciprocal experiment (Fig. 5A and B). These data also correlate well with published values for native rat NMDAR binding affinities for these two reference displacers (Franklin and Murray, 1992).
Fig. 4.
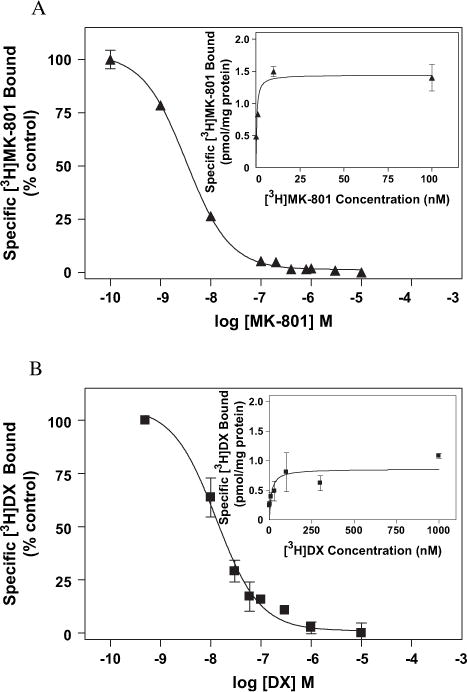
The inhibition of (A) [3H]MK-801 by MK-801 and (B) [3H]DX binding by DX. Increasing concentrations of unlabeled ligand were added to COS-7 membranes expressing wild-type NMDAR. The concentrations of [3H]MK-801 and [3H]DX were 1 nM and 30 nM, respectively. The percentage of ligand bound is normalized with respect to total specific binding. Saturation analysis derived from homologous competition data (see Section 2) for (A, insert) [3H]MK-801 and (B, insert) [3H]DX. Each point represents the pooled values of at least two experiments performed in duplicate. All binding reactions were incubated at room temperature for 4 h in 5 mM HEPES (pH 7.4) containing 100 μM glutamate and 100 μM glycine for MK-801 or 3 μM glutamate and 3 μM glycine for DX. Error bars represent SEM.
Fig. 5.
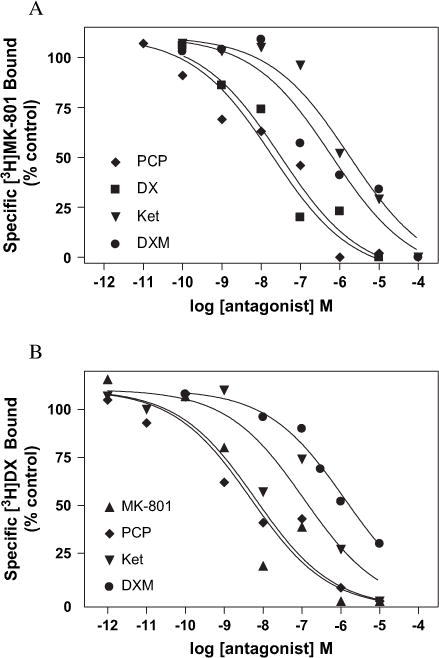
Heterologous competition results demonstrating inhibition of (A) [3H]MK-801 and (B) [3H]DX binding by MK-801, PCP, DX, Ket or DXM. Increasing concentrations of unlabeled ligand were added to COS-7 membranes expressing wild-type NMDAR. The concentrations of [3H]MK-801 and [3H]DX were 1 nM and 30 nM, respectively. The percentage of ligand bound is normalized with respect to total specific binding of each radioligand. Each point represents the pooled values of at least two experiments performed in duplicate. All binding reactions were incubated at room temperature for 4 h in 5 mM HEPES (pH 7.4) containing 100 μM glutamate and 100 μM glycine for [3H]MK-801 or 3 μM glutamate and 3 μM glycine for [3H]DX PCP, DX, Ket or DXM.
Saturation isotherms for both [3H]MK-801 (Fig. 4A, insert) and [3H]DX (Fig. 4B, insert) were generated from the data obtained from the equilibrium self-competition experiments (see Section 2.10). The plots of the saturation analyses represent data pooled from two experiments performed in triplicate. The Kd values for MK-801 (0.44 ± 0.17 nM) and DX (11.2 ± 0.17 nM) calculated from these analyses correlate well with those of previous reports examining native NMDAR in rat forebrain (Franklin and Murray, 1992).
The pharmacological profile of the heterologously expressed wild-type receptors was assessed by a series of equilibrium competition experiments in which [3H]MK-801 and [3H]DX were individually tested against an array of known noncompetitive NMDAR antagonists. The rank order of potency for the NMDAR ligands tested in equilibrium competition analysis was MK-801 > PCP > DX > DXM > Ket for [3H]MK-801 and MK-801 > PCP > DX > Ket > DXM for [3H]DX (Figs. 4A and B, 5A and B). Fitting these equilibrium competition data to a four parameter logistic equation and sharing values for total binding, non-specific binding and Hill slope provided the best fit of the data for all of the NMDAR ligands tested irrespective of the radioligand being displaced (R2 > 0.91) (see Section 2.10). The global analysis revealed a Hill slope of 0.58 ± 0.14 for displacement of [3H]DX and 0.50 ± 0.11 for that of [3H]MK-801 by the array of NMDAR antagonists used in the assays. These results clearly show that the heterologously expressed NMDAR utilized in this investigation has a pharmacological signature that accords well with native receptors as well as heterologous systems developed by others, and moreover was independent of the radioligand used (Franklin and Murray, 1992; Roth et al., 1996; Ferrer-Montiel et al., 1998).
[3H]MK-801 and [3H]DX binding to wild-type NMDAR were further compared by evaluating the kinetics of association of both ligands in well-washed membranes nominally free of endogenous glycine and glutamate. These closed channel conditions were selected both as a prelude to glutamate regulation studies and to test the hypothesis that access to channel binding sites for the two ligands differs. The results of the association of [3H]MK-801 and [3H]DX to wild-type NR1-1a/NR2A complexes expressed in COS-7 membranes are depicted in Fig. 6. These binding data were linearly transformed to a ln(Beq/Beq − Bt) versus time plot. These results indicate that in the presence of nominal agonist and co-agonist concentrations, the association of [3H]DX specific binding is much more rapid than that of [3H]MK-801. Analysis of the linearly transformed data yielded observed association rates of 0.014 h−1 for [3H]MK-801 and 1.45 h−1 for [3H]DX representing greater than a 100-fold difference in on-rates. Thus, in these closed channel conditions, the t1/2 for association of [3H]DX was 0.53 h whereas the t1/2 for association of [3H]MK-801 was 47 h.
Fig. 6.
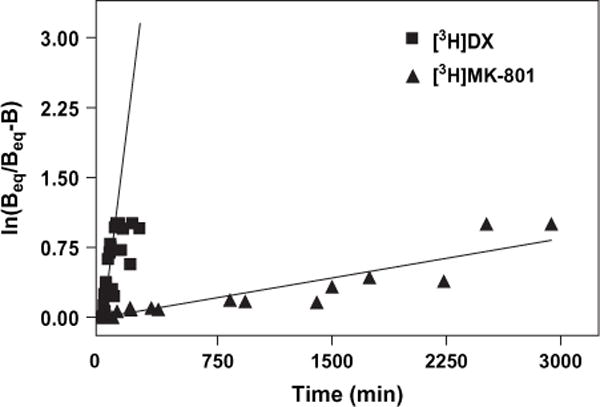
Association kinetics for the specific binding of [3H]MK-801 (filled triangle) and [3H]DX (filled square) to heterologously expressed NMDAR under nominal glycine and glutamate conditions. Association experiments were carried out using 1 nM [3H]MK-801 and 30 nM [3H]DX. Specific binding was determined by the inclusion of either 300 nM MK-801 or 3 mM DX. Each point represents the pooled value of three experiments performed in duplicate. Binding data were linearly transformed to a ln(Beq/Beq−Bt) versus time plot; slopes of the line correspond to kobs. Error bars represent SEM.
Glutamate and related NMDA receptor agonists accelerate the association and dissociation rates of noncompetitive antagonists without changing their equilibrium binding parameters, Kd and Bmax (Kloog et al., 1990). Accordingly, the study of glutamate regulation of noncompetitive antagonist binding must be performed under pre-equilibrium conditions. We therefore compared glutamate and glycine regulation of [3H]DX and [3H]MK-801 binding at the t1/4 of association for each ligand to eliminate differences in the extent of association reaction for the two antagonists as a confounding variable. Incubations with [3H]DX were allowed to proceed for 16 min while those with [3H]MK-801 incubated for 24 h. In addition, both radioligands were used at concentrations that produced identical fractional occupancies of NMDAR. Glycine alone produced a very modest stimulation of both [3H]DX and [3H]MK-801 binding which did not reach the level of statistical significance (Fig. 7A). The influence of glutamate alone also produced a modest increment in [3H]MK-801 binding, but did not affect [3H]DX binding (Fig. 7B). The glutamate concentration response curves were then repeated for each ligand in the presence of a fixed concentration (3 μM) of the co-agonist glycine. In the presence of 3 μM glycine, glutamate (10–100 μM) stimulated [3H]MK-801 binding 2.35-fold while having no significant effect on [3H]DX binding to wild-type NR1-1a/NR2A complexes (Fig. 8).
Fig. 7.
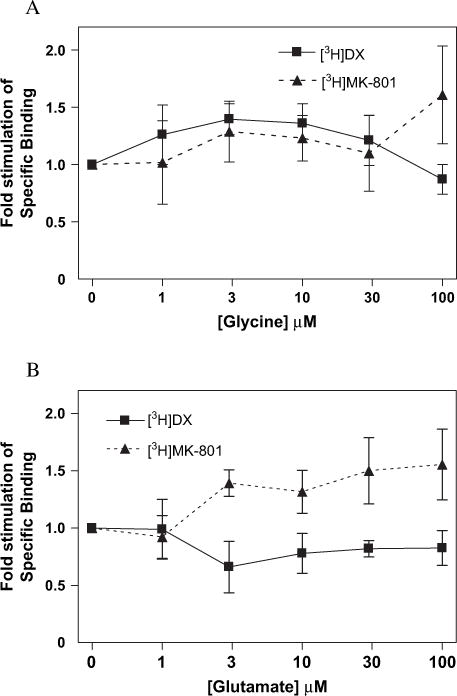
(A) Glycine or (B) glutamate effects on [3H]MK-801 (filled triangle) and [3H]DX (filled square) binding. Values represent the increase in specific binding as compared to control binding in nominal glycine and glutamate conditions. Each point represents pooled values of two ([3H]MK-801) or three ([3H]DX) binding experiments performed in duplicate. Error bars represent SEM.
Fig. 8.
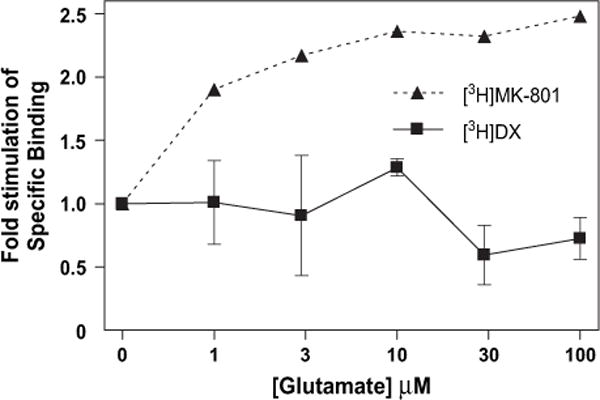
Glutamate concentration response effects on [3H]MK-801 (filled triangle) and [3H]DX (filled square) binding in the presence of the co-agonist glycine. Each data point represents the mean of pooled values of three ([3H]MK-801) or two (3H]DX) binding experiments and represents the increase of specific binding over that which occurs in the presence of glycine alone. Reactions were carried out at room temperature in 5 mM HEPES (pH 7.4) containing either 3 μM ([3H]DX) or 100 μM ([3H]MK-801) glycine. Error bars represent SEM.
Equilibrium saturation analyses with both [3H]MK-801 and [3H]DX were performed using COS-7 cells expressing wild-type NR1-1a/NR2A complexes (Fig. 9A and B). The mean Kd value for [3H]DX binding was 53.9 ± 15.7 nM which is in good agreement with that obtained with native NMDAR complexes in rat fore-brain (Table 1; Franklin and Murray, 1992). The level of expression of [3H]DX sites ranged from 483 to 1025 fmol/mg protein in three independent transfections. Variation in Bmax values of the heterologously expressed wild-type mutant NMDAR complexes is likely the result of differences in the efficiencies of the transient transfection reactions. The equilibrium binding parameters for [3H]MK-801 derived from isotherms run in parallel were Kd = 1.3 ± 0.4 nM and Bmax = 660 ± 243 fmol/mg protein (Table 1). The mean Kd and Bmax values generated from the saturation analysis are similar to those derived from homologous competition analyses. Thus, the Bmax and Kd values reported in this investigation for both MK-801 and DX are therefore the result of analyses performed on data derived from five independent transfections. The concordance of the Bmax values for the two radioligands is consistent with the labeling of an identical population of heterologously expressed receptors. Saturation analysis of non-transfected COS-7 cell membranes revealed no detectable saturable binding associated with either ligand (Fig. 9A and B).
Fig. 9.
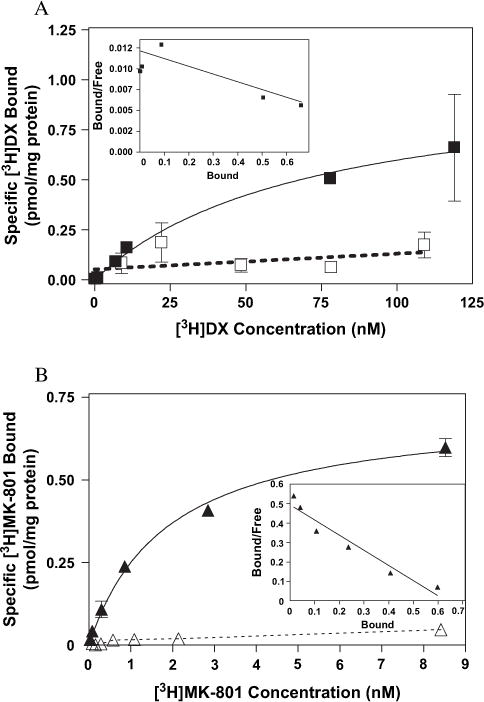
Binding isotherms for (A) [3H]DX and (B) [3H]MK-801 to heterologously expressed wild-type NMDAR (filled square, filled triangle) or non-transfected COS-7 cells (open square, open triangle) as determined by equilibrium saturation analysis. The above figures are representative of three experiments performed in duplicate. Specific binding is defined by the use of unlabeled (A) 3 μM DX or (B) 300 nM MK-801. Reactions were carried out with 30 μM glutamate and glycine for [3H]DX experiments while 100 μM concentrations of the co-agonists were used with [3H]MK-801. Data were fit by non-linear regression as described in Section 2. Error bars represent SEM.
Table 1.
Mean parameter estimates derived from equilibrium saturation isotherms of [3H]DX and [3H]MK-801 binding to heterologously expressed Wild-type and mutant NMDAR
| NR1-1a species | [3H]MK-801
|
[3H]dextrorphan
|
||
|---|---|---|---|---|
| Kd (nM) | Bmax (fmol/mg protein) | Kd (nM) | Bmax (fmol/mg protein) | |
| Wild-type | 1.3 ± 0.4 | 660 ± 243 | 53.9 ± 15.7 | 765 ± 202 |
| N616Q | 22.2 ± 10.3* | 348 ± 81 | 50.3 ± 16.6 | 525 ± 144 |
| N616R | NSB | NSB | 57.1 ± 19.7 | 1034 ± 780 |
| W611A | 3.72 ± 2.12 | 181 ± 15 | NSB | NSB |
| A645S | 8.37 ± 1.84* | 197 ± 23 | 56.5 ± 40.3 | 1370 ± 430 |
| T648A | NSB | NSB | 96.04 ± 102.8 | 2269 ± 1318 |
| N650A | 6.44 ± 2.09* | 837 ± 233 | 215 ± 101* | 3740 ± 248 |
| N812A | 3.72 ± 0.76 | 480 ± 38 | NSB | NSB |
Values are means of two experiments each done with duplicate samples (NSB = no saturable binding, * p ≤ 0.05 when compared to wild-type control).
We then examined the relative contributions of two residues, N616 and W611, both associated with the M2 transmembrane region of NR1, to the equilibrium binding of the two ligands. Two mutations (N → Q and N → R) of the conserved asparagine at position 616 on the NR1-1a subunit were evaluated for influence on [3H]DX and [3H]MK-801 binding (Fig. 10A and B). The mutation of the N616 site to glutamine substantially affected [3H]MK-801 binding affinity while [3H]DX affinity was unaltered (Fig. 10A and B). The N616Q mutation resulted in a 17-fold decrease in the affinity for [3H]MK-801 (Fig. 10B and Table 1). Similarly, substitution of asparagine with arginine (N616R) more dramatically reduced [3H]MK-801 binding inasmuch as no saturable binding could be detected in membranes expressing NR1-1a (N616R)/NR2A complexes (Fig. 10B). In contrast to [3H]MK-801, the equilibrium saturation binding of [3H]DX was not altered by the N616R mutation; the mean Kd value was 57.1 ± 19.7 nM, which was virtually identical to that observed with the wild-type NR1-1a/NR2A receptor (Table 1, Fig. 10A). High-affinity binding of [3H]DX was, however, completely disrupted by mutation (W611A) of the tryptophan residue at position 611, while that of [3H]MK-801 was unaltered (Fig. 11A and B and Table 1).
Fig. 10.
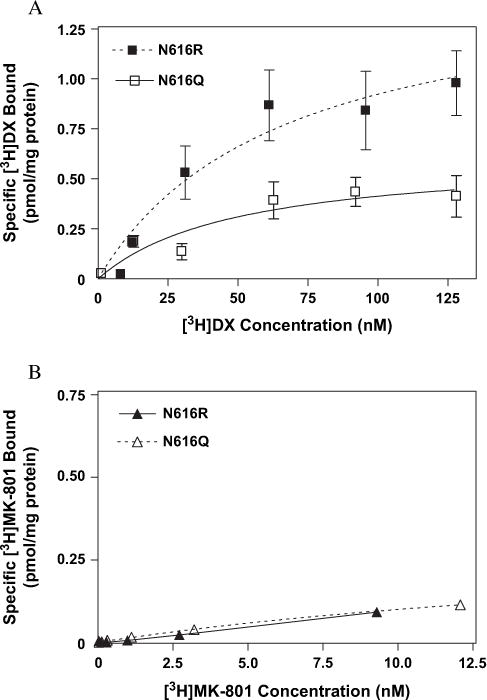
Binding isotherms for (A) [3H]DX and (B) [3H]MK-801 to heterologously expressed mutant NMDAR (NR1-1a[N616Q/R]/NR2A) as determined by equilibrium saturation analysis. The above figures are representative of three experiments performed in duplicate. Error bars represent SEM.
Fig. 11.
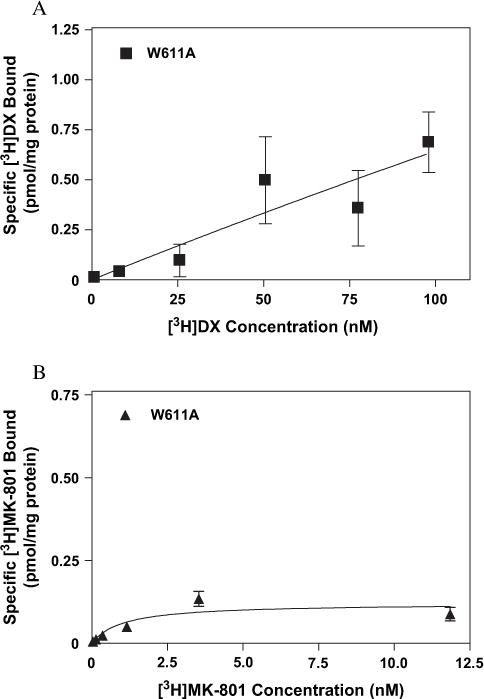
Binding isotherms for (A) [3H]DX and (B) [3H]MK-801 to heterologously expressed mutant NMDAR (NR1-1a[W611A]/NR2A) as determined by equilibrium saturation analysis. The above figures are representative of three experiments performed in duplicate. Error bars represent SEM.
To further explore the nonidentity of sites associated with MK-801 and DX binding we examined various mutations at residues proposed to be part of the extracellular vestibule of the NMDAR. Disruptive mutations at several sites associated with the M3 region of the NR1-1a subunit (A645, T648 and N650) differentially affected the affinities of [3H]MK-801 and [3H]DX. The substitution of serine for alanine at position 645 reduced [3H]MK-801 affinity approximately sixfold with no concomitant effect on that of [3H]DX (Fig. 12A and B and Table 1). Residue T648 has substantial, although differential, effects on high-affinity binding of both [3H]MK-801 and [3H]DX. [3H]MK-801 binding was completely abrogated in NMDAR composed of mutant NR1-1a (T648A)/NR2A complexes, while the affinity of [3H]DX was reduced only twofold (Table 1). The substitution of asparagine 650 by alanine uniformly decreased the affinity of both radioligands by a factor of four to five from values observed with wild-type NMDAR complexes (Table 1). Mutation of residues associated with the M4 region also differentially affected saturable [3H]DX or [3H]MK-801 binding. While conversion of T789 to alanine had no discernable effect on the high-affinity binding of either radioligand (data not shown), the mutation of the non-polar alanine residue to asparagine at site 812 in the M4 transmembrane eliminated [3H]DX, but not [3H]MK-801, binding (Table 1, Fig. 13A and B).
Fig. 12.
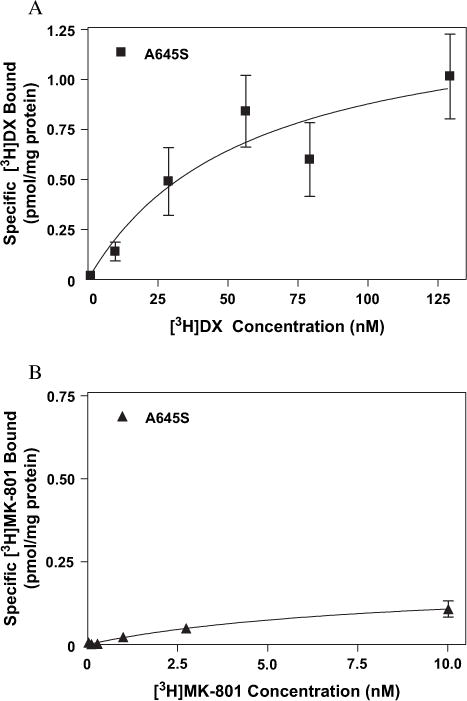
Binding isotherms for (A) [3H]DX and (B) [3H]MK-801 to heterologously expressed mutant NMDAR (NR1-1a[A645S]/NR2A) as determined by equilibrium saturation analysis. The above figures are representative of three experiments performed in duplicate. Error bars represent SEM.
Fig. 13.
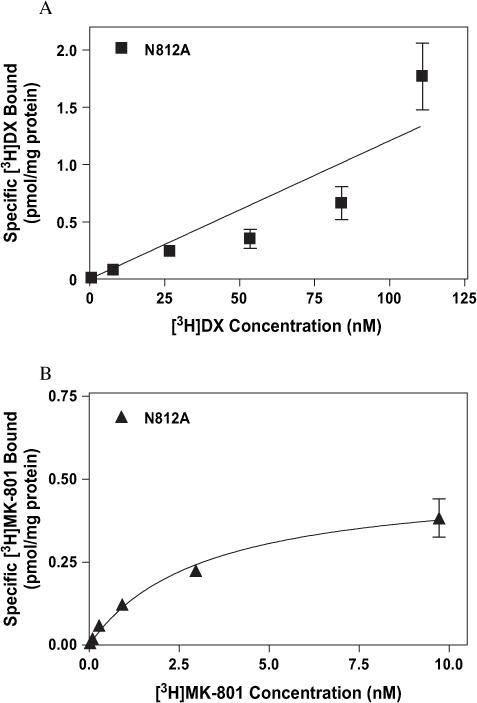
Binding isotherms for (A) [3H]DX and (B) [3H]MK-801 to heterologously expressed mutant NMDAR (NR1-1a[N812A]/NR2A) as determined by equilibrium saturation analysis. The above figures are representative of three experiments performed in duplicate. Error bars represent SEM.
4. Discussion
Critical to this investigation was the ability to precisely define the specific binding of each radioligand. Consequently, the use of unlabeled species of each radioligand was essential to ensure precise delineation of amino acids particular to the high-affinity binding of each radioligand. The low affinity [3H]MK-801 binding site in sham-transfected COS-7 cells reported by Ishmael et al. (1996) using 10 μM MK-801 to define non-specific binding is in contrast to the complete lack of saturable binding we report here for non-transfected cells using the same radioligand with 300 nM cold MK-801 to define non-specific binding. Calculated fractional occupancy of the cold ligands at the highest concentrations of each radioligand used in saturation isotherm assays was greater than 97% for MK-801 and DX. Taken together, it is clear that the use of empirically determined minimum concentrations of unlabeled ligands avoided the generation of spurious displaceable binding of [3H]MK-801 and [3H]DX and as a consequence avoided this confound.
The association of [3H]DX and [3H]MK-801 to heterologously expressed wild-type NR1-1a/NR2A receptor complexes differed dramatically in the presence of nominal concentrations of glutamate and glycine (Fig. 6). We found that [3H]MK-801 associates at a slow rate to heterologously expressed NMDAR complexes in these closed channel conditions. In contrast, the association of [3H]DX occurred at a relatively rapid rate in well-washed C0S-7 membranes nominally free of glutamate and glycine. These profound differences in association rates in the presence of nominal glutamate and glycine concentrations indicate that the accessibility of [3H]DX and [3H]MK-801 to their respective channel recognition sites differs. Similar differences between [3H]DX and [3H]MK-801 association rates have been observed for native receptors in well-washed cerebral cortical membranes where [3H]DX has a t1/2 for association of 0.71 h and [3H]MK-801 a t1/2 of 16 h (data not shown). Rapid association may be a consequence of a readily accessible [3H]DX binding site in the extracellular vestibule of the NMDAR. While others have reported complex association patterns for MK-801 binding, it must be stressed that these earlier experiments examined association and dissociation kinetics with NMDAR in open-channel conditions (Berman and Murray, 1996). Additionally, Berman and Murray (1996) examined [3H]MK-801 binding in membranes derived from cultured cerebellar granule neurons where the NMDAR subunit composition was more heterogeneous than that in the current investigation. The intent of the present investigation was to compare the initial rates of ligand–receptor interaction to a defined NMDAR complex under closed channel conditions. Although the binding sites for [3H]DX and [3H]MK-801 partially overlap, these data are consistent with the lack of coincidence of their respective recognition elements.
The presence of increasing concentrations of glutamate alone increased the specific binding of [3H]MK-801, while having no influence on [3H]DX specific binding. The glutamate-induced stimulation of [3H]MK-801 binding to wild-type NR1-1a/NR2A complexes accords with the requirement for receptor activation and channel opening for blockade by this noncompetitive antagonist to manifest. [3H]DX binding was, however, relatively insensitive to glutamate suggesting less open-channel dependence. The glutamate regulation of [3H]MK-801 and [3H]DX in the presence of a fixed concentration of glycine provided qualitatively similar results, inasmuch as glutamate significantly increased [3H]MK-801, but not [3H]DX, binding. The differential sensitivity of [3H]MK-801 and [3H]DX to glutamate stimulation in the presence of glycine suggests that the recognition elements for these ligands are not coincident. It is therefore reasonable to infer that [3H]DX binds to a site on the heterologously expressed NMDA receptor ion channel which is accessible under closed channel conditions.
Given the essential role of the conserved asparagine residue in the reentrant M2 segment as a structural determinant of noncompetitive antagonist binding to the NMDAR, we assessed the effect of mutation of this site on [3H]DX and [3H]MK-801 binding (Figs. 1A and B, 13A and B). Consistent with previous reports (Mori et al., 1992; Sakurada et al., 1993; Kawajiri and Dingledine, 1993; Ferrer-Montiel et al., 1995; Kashiwagi et al., 2002), we found that both the N616Q and N616R mutants decreased the affinity for MK-801. In sharp contrast, the N616 mutations did not affect the binding affinity of [3H]DX suggesting that this ligand does not derive binding energy from the interaction with N616. The lack of effect of the N616 mutation on [3H]DX binding affinity verifies that the overall structure of the NMDAR channel was not grossly disturbed by these mutations.
To further test our contention that DX binds at a somewhat shallower site than MK-801, we examined the effect of mutations at sites within regions M2, M3 and M4 of the NMDAR (Fig. 1A and B). The disruption of [3H]DX binding by mutation of site W611 to alanine, with no attendant effect on [3H]MK-801 affinity, is similar to the results reported by Ferrer-Montiel et al. (1998). It has been demonstrated that W611 is essential to high-affinity PCP binding, yet has no corresponding effect on that of MK-801 (Ferrer-Montiel et al., 1995). Kashiwagi et al. (2002) demonstrated the insensitivity of MK-801, memantine and Mg2+ blockade to a disruptive mutation of W611. While W611 is not directly involved in MK-801 binding, it may contribute to the overall structure of the pore via an effect on subunit interaction that is essential for PCP binding (Ferrer-Montiel et al., 1995). If the conformation suggested by Kuner et al. (1996) is correct, then W611 would be an unlikely site for the high-affinity interaction of the protonated amine of DX as required by the model proposed by this group (Figs. 1A and B, 3) (1998). The effect of the W611 mutation on the topology of the NR1-1a subunit may influence DX binding in a manner similar to that associated with disruption of PCP binding. This may be a consequence of alteration of a hydrophobic region of the receptor required for high-affinity binding of DX and PCP.
Residues A645, T648 and N650 of NR1 may form part of the extracellular vestibule with T648 and N650 occupying part of a pocket in the extracellular vestibule near N616 (Fig. 1A and B) (Beck et al., 1999; Sobolevsky et al., 2002). The A645S mutation reduced the affinity of [3H]MK-801 by approximately sixfold. This reduction of high-affinity binding of [3H]MK-801 is comparable to the substantial reduction of the MK-801 blockade of inward currents induced by glutamate reported for NR1-1a (A645S)/NR2B receptors (Kashiwagi et al., 2002). Sequence and SCAM analysis has predicted that A645 is part of the α-helical region of M3 with this amino acid associated with the interior of the protein (Sprengel et al., 2001; Beck et al., 1999). A645 may reside at a sufficient depth in the channel to contribute to the pore structure and hence affect the binding of the open-channel antagonist MK-801 through a perturbation of hydrophobic interactions between the ligand and residues close to the pore constriction (Figs. 1A and B, 3). With no corresponding influence on high-affinity binding of [3H]DX, it is evident that A645 does not contribute significant binding energy to this radioligand.
The dramatic difference in the binding parameters of [3H]MK-801 and [3H]DX induced by the T648A mutation is of particular note. The complete loss of high-affinity saturable binding of [3H]MK-801 associated with the T648A mutation is consistent with the predicted pharmacophore, and topology, of the NMDAR (Figs. 1A and B, 3) (Kroemer et al.,1998; Beck et al., 1999; Sprengel et al., 2001). Using SCAM analysis, the accessibility of T648 to MTS reagents in the absence of agonist activation predicts that this residue is likely to be positioned slightly external to the narrow pore constriction of the NMDAR (Fig. 1A and B) (Beck et al., 1999). While it is unlikely that the residue is a site for direct ligand interaction, local conformational changes resulting from the substitution of threonine with the smaller alanine might affect high-affinity binding at the pore constriction (N616) (Beck et al., 1999). Disruptive mutation of N650, a putative residue of the extracellular vestibule, reduced the affinity for both [3H]MK-801 and [3H]DX approximately four- to fivefold. Consistent with our results, a shift of the MK-801 IC50 value from 2 nM to 13 nM for blockade of glutamate-induced currents has been reported for NR1-1a (N650A)/NR2B receptors (Kashiwagi et al., 2002). Since substitution at N650 does not entirely eliminate the binding of either [3H]MK-801 or [3H]DX, it is reasonable to propose that the residue is either a site of interaction for both ligands or confers topology to the pore/vestibule region that influences the affinity of both ligands. The observation that none of the mutations examined in M3 completely disrupt [3H]DX binding indicates that these residues do not represent singular points of contact for [3H]DX. Given the prediction that A645, T648 and N650 of NR1 are reasonably close to the pore constriction, the more robust effect of their mutation on [3H]MK-801 high-affinity binding, relative to that of [3H]DX, may suggest that the hydrophobic interaction of the former is directly influenced by these residues. These residues are part of a region that corresponds to the Lurcher mouse phenotype that is characterized by a constitutively open NMDAR channel (Kohda et al., 2000; Kashiwagi et al., 2002). Recent work has revealed that the proton sensitivity of the NMDAR is reduced by the disruptive mutation of T648 (Low et al., 2003). It has been proposed that T648 is part of a conformational change involving movement of the M3 segment during ligand gating of the NMDAR (Low et al., 2003). Further evidence for a coupling between the pore-forming and Lurcher regions via the gating mechanism is indicated by the disruptive effect of mutations at N616 on proton sensitivity (Kashiwagi et al., 1997). The involvement of T648 in the gating of channel opening is consistent with a mutation-induced conformational change capable of disrupting [3H]MK-801 binding to the N616 residue with no corresponding effect on the binding of [3H]DX.
Residue N812, which is predicted to be near the N-terminal of the transmembrane segment M4, was mutated to alanine and examined for involvement in [3H]MK-801 and [3H]DX binding (Beck et al., 1999). The complete loss of specific [3H]DX binding associated with no concomitant effect on [3H]MK-801 affinity is consistent with overlapping, yet distinct, binding sites for the two ligands. The striking effect on [3H]DX binding argues for direct ligand–receptor interaction occurring in the extracellular vestibule. Interestingly, others have demonstrated that N812 may contribute to a second voltage-independent site for aminoadamantane derivatives (Sobolevsky and Koshelev, 1998). The asparagine residue at site 812, acting as a putative acceptor for the hydrogen associated with the nitrogen “donor atom” of DX, is consistent with the structural requirements for blockade of the NMDAR as previously described (Fig. 3) (Kroemer et al., 1998). Additionally, the accessibility of N812 in the closed state of the NMDAR agrees with our observation that rapid association of [3H]DX is not dependent on receptor activation.
Evidence for overlapping and/or multiple recognition sites for noncompetitive antagonists of the NMDAR have been provided previously. Yamakura et al. (1993) demonstrated that the contribution of the conserved M2 asparagine residue contributed variably to the interaction with the noncompetitive NMDAR antagonists, MK-801, PCP, Ket and SKF-10,047. Ket antagonism was least influenced by asparagine → glutamine mutations, while SKF-10,047 was most influenced. More recently, these authors reported that an array of racemic opioids block NMDAR at a site which at least partially overlaps with that of Ket and MK-801 (Yamakura and Shimoji, 1999). Kashiwagi et al. (2002) has reported differential effects of mutations on the blockade of three open-channel blockers of the NMDAR (Kashiwagi et al., 2002). Evaluation of residues associated with the M2 region of the NMDAR revealed that mutation of the critical asparagine (N616) reduced the block of memantine, MK-801, N1-N4-N8 tribenzyl-spermidine (TB-3-4) and Mg2+ (Kashiwagi et al., 2002). Conversely, many mutations in the pre-M1, M1, M3 and post-M3 regions had little or no effect on the block by memantine while reducing that of MK-801 and TB-3-4 (Kashiwagi et al., 2002). A similar model of two ligands binding to both overlapping and unique residues has also been reported for L-type Ca2+ channels (Hockerman et al., 2000). Mutations to domains IIIS6 and IVS6 revealed a series of residues that differentially influenced the blockade induced by diltiazem, a benzothiazepine, as well as dihydropridines (DHP) and phenylalkylamines (PA) (Hockerman et al., 2000). Thus, distinct yet overlapping determinants critical for different channel ligands have been described for both NMDAR and L-type Ca2+ channels (Yamakura et al., 1993; Kashiwagi et al., 2002; Hockerman et al., 2000).
The possibility of multiple binding sites for noncompetitive antagonists within the NMDAR channel domain has been proposed. The PCP derivative TCP exhibits two qualitatively distinct blocking actions on NMDAR with distinct affinities (Kawajiri and Dingledine, 1993). Only the high-affinity component of blockade was eliminated by mutation of the N616 residue, suggesting that TCP has more than one blocking site on the NMDAR channel. Ketamine has also been demonstrated to block NMDAR by two distinct mechanisms implying multiple binding sites in the ion channel domain (Orser et al., 1997). Additionally, aminoadamantane derivatives are known to have both voltage-dependent and -independent sites of blockade (Sobolevsky and Koshelev, 1998). Thus the existence of multiple binding sites for DX on the NMDAR cannot presently be excluded. A report demonstrating the concentration-dependence of DX blockade of NR1/NR2A channels expressed in Xenopus oocytes found DX to have an IC50value of 30.0 ± 1.0 nM with a Hill slope coefficient of 0.4 ± 0.1 (Ferrer-Montiel et al., 1998). This shallow inhibition curve with a Hill slope less than 1.0 is consistent with multiple DX binding sites within the NMDAR ion channel. Moreover, using the Woodhull analysis to estimate the electrical distance (δ) of the blockade of NR1/NR2A by DX revealed a channel depth of 0.51 ± 0.06 (Ferrer-Montiel et al., 1998). This value is shallower than that typically observed for established channel blockers such as MK-801 and Ket (δ = 0.7–0.8). These data are therefore consistent with at least one DX blocking site occupying a position in the NMDAR channel which is closer to the extracellular surface than that of MK-801. Although Ferrer-Montiel et al. (1998) demonstrated that mutation of the NR1 asparagine in the M2 segment reduced dramatically the DX IC50 value, this was, however, associated with a low Hill slope coefficient (0.6 ± 0.1) which is again consistent with multiple DX binding sites. This is in agreement with the present results derived from global analysis of competition experiments using [3H]DX which were characterized by a Hill slope of 0.5 ± 0.1. It must be stressed that the reduction of the DX blockade reported by Ferrer-Montiel et al. (1998) for an N616Q substitution of the NR1-1a subunit, was derived from analysis of cRNA-injected oocytes at a holding potential of −80 mV. The methods in the current investigation (radioligand binding, equilibrium conditions, depolarized membrane fragments) likely accounts for the differential effect of the N616Q mutation in the two reports.
Considered together these data suggest that the DX and MK-801 binding sites within the NMDAR channel domain comprise distinct amino acids. Additional studies will be required not only to delineate residues in the vestibule region involved in [3H]DX binding, but also to assess the functional significance of these residues in the context of noncompetitive antagonism of the NMDAR.
Acknowledgments
The authors wish to thank Roxanne Armstrong and Kelly Cumuze for excellent technical assistance and Dr. Mark Leid for valuable discussions. Special thanks to Dr. Jeremy Glasner for assistance in the sequencing of constructs used in this investigation.
Supported by National Institute on Drug Abuse grant (DA07218) to T.F.M., an F32 National Research Service Award (DA05728) to J.E.I., and NINDS grant (NS36654) to S.F.T.
References
- Avenet P, Leonardon J, Besnard F, Graham D, Frost J, Depoortere H, Langer SZ, Scatton B. Antagonist properties of the stereoisomers of ifenprodil at NR1A/NR2A and NR1A/NR2B subtypes of the NMDA receptor expressed in Xenopus oocytes. Eur J Pharmacol. 1996;296:209–213. doi: 10.1016/0014-2999(95)00700-8. [DOI] [PubMed] [Google Scholar]
- Beck C, Wollmuth LP, Seeburg PH, Sakmann B, Kuner T. NMDAR channel segments forming the extracellular vestibule inferred from the accessibility of substituted cysteines. Neuron. 1999;22:559–570. doi: 10.1016/s0896-6273(00)80710-2. [DOI] [PubMed] [Google Scholar]
- Berman FW, Murray TF. Characterization of [3H]MK-801 binding to N-methyl-D-aspartate receptors in cultured rat cerebellar granule neurons and involvement in glutamate-mediated toxicity. J Biochem Toxicol. 1996;11(5):217–226. doi: 10.1002/(SICI)1522-7146(1996)11:5<217::AID-JBT2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Burnashev, et al. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science. 1992;257(5075):1415–1419. doi: 10.1126/science.1382314. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cole AE, Eccles CU, Aryanpur JJ, Fisher RS. Selective depression of N-methyl-D-aspartate-mediated responses by dextrorphan in the hippocampal slice in rat. Neuropharmacology. 1989;28:249–254. doi: 10.1016/0028-3908(89)90100-7. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Doyle DA, Morais CJ, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Ferrer-Montiel AV, Sun W, Montal M. Molecular design of the N-methyl-D-aspartate receptor binding site for phencyclidine and dizolcipine. Proc Natl Acad Sci USA. 1995;92:8021–8025. doi: 10.1073/pnas.92.17.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Montiel AV, Merino JM, Planells-Cases R, Sun W, Montal M. Structural determinants of the blocker binding site in glutamate and NMDA receptor channels. Neuropharmacology. 1998;37:139–147. doi: 10.1016/s0028-3908(98)00007-0. [DOI] [PubMed] [Google Scholar]
- Franklin PH, Murray TF. High affinity [3H]dextrorphan binding in rat brain is localized to a noncompetitive antagonist site of the activated N-methyl-D-aspartate receptor-cation channel. Mol Pharmacol. 1992;41:134–146. [PubMed] [Google Scholar]
- Hawkins LM, Chazot PL, Stephenson FA. Biochemical evidence for the co-association of three N-methyl-D-aspartate (NMDA) R2 subunits in recombinant NMDA receptors. J Biol Chem. 1999;274:27211–27218. doi: 10.1074/jbc.274.38.27211. [DOI] [PubMed] [Google Scholar]
- Hockerman GH, Dilmac N, Scheuer T, Catterall WA. Molecular determinants in diltiazem blockade in domains IIIS6 and IVS6 of L-type Ca2+ channels. Mol Pharmacol. 2000;58:1264–1270. doi: 10.1124/mol.58.6.1264. [DOI] [PubMed] [Google Scholar]
- Holtzman SG. Phencyclidine-like discriminative effects of opioids in the rat. J Pharmacol Exp Ther. 1980;214:614–619. [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci USA. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishmael JE, Franklin PH, Murray TF, Leid M. High level expression of the NMDAR1 glutamate receptor subunit in electroporated COS cells. J Neurochem. 1996;67:1500–1510. doi: 10.1046/j.1471-4159.1996.67041500.x. [DOI] [PubMed] [Google Scholar]
- Ishmael JE, Franklin PH, Murray TF. Dextrorotatory opioids induce stereotyped behavior in Spraguee-Dawley and Dark Agouti rats. Psychopharmacology (Berl) 1998;140:206–216. doi: 10.1007/s002130050759. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- Kashiwagi K, Pahk AJ, Masuko T, Igarashi K, Williams K. Block and modulation of N-methyl-D-aspartate receptors by polyamines and protons: role of amino acid residues in the transmembrane and pore forming regions of NR1 and NR2 subunits. Mol Pharmacol. 1997;52:701–713. doi: 10.1124/mol.52.4.701. [DOI] [PubMed] [Google Scholar]
- Kashiwagi K, Masuko T, Nguyen CD, Kuno T, Tanaka I, Igarashi K, Williams K. Channel blockers acting at N-methyl-D-aspartate receptors: differential effects of mutations in the vestibule and ion channel pore. Mol Pharmacol. 2002;61(3):533–545. doi: 10.1124/mol.61.3.533. [DOI] [PubMed] [Google Scholar]
- Kawajiri S, Dingledine R. Multiple structural determinants of voltage-dependent magnesium block in recombinant NMDA receptors. Neuropharmacology. 1993;32:1203–1211. doi: 10.1016/0028-3908(93)90014-t. [DOI] [PubMed] [Google Scholar]
- Kloog Y, Lamdani-Itkin H, Sokolovsky M. The glycine site of the N-methyl-D-aspartate receptor channel: differences between the binding of HA-966 and of 7-chlorokynurenic acid. J Neurochem. 1990;54:1576–1583. doi: 10.1111/j.1471-4159.1990.tb01207.x. [DOI] [PubMed] [Google Scholar]
- Kroemer RT, Koutsilieri E, Hecht P, Liedl KR, Riederer P, Kornhuber J. Quantitative analysis of the structural requirements for blockade of the N-methyl-D-aspartate receptor at the phencyclidine binding site. J Med Chem. 1998;41:393–400. doi: 10.1021/jm9704412. [DOI] [PubMed] [Google Scholar]
- Kohda K, Wang Y, Yuzaki I. Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat Neurosci. 2000;3(4):315–322. doi: 10.1038/73877. [DOI] [PubMed] [Google Scholar]
- Kuner T, Wollmuth LP, Karlin A, Seeburg PH, Sakmann B. Structure of the NMDA receptor channel M2 segment inferred from the accessibility of substituted cysteines. Neuron. 1996;17:343–352. doi: 10.1016/s0896-6273(00)80165-8. [DOI] [PubMed] [Google Scholar]
- Leid M, Schimerlik MI, Murray TF. Characterization of agonist radioligand interactions with porcine atrial A1 adenosine receptors. Mol Pharmacol. 1988;34:334–339. [PubMed] [Google Scholar]
- Low CM, Lyuboslavsky P, French A, Le P, Wyatte K, Thiel WH, Marchan EM, Igarashi K, Kashiwagi K, Gernert K, Williams K, Traynelis SF, Zheng F. Molecular determinants of proton-sensitive N-methyl-D-aspartate receptor gating. Mol Pharmacol. 2003;63(6):1212–1222. doi: 10.1124/mol.63.6.1212. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with Folin phenol reagent. J Biol Chem. 1951;193:266–275. [PubMed] [Google Scholar]
- Lynch DR, Lawrence JJ, Lenz S, Anegawa NJ, Dichter M, Pritchett DB. Pharmacological characterization of heterodimeric NMDA receptors composed of NR 1a and 2B subunits: differences with receptors formed from NR 1a and 2A. J Neurochem. 1995;64:1462–1468. doi: 10.1046/j.1471-4159.1995.64041462.x. [DOI] [PubMed] [Google Scholar]
- Mori H, Masaki H, Yamakura T, Mishina M. Identification by mutagenesis of a Mg2+-block site of the NMDA receptor channel. Nature. 1992;358:673–675. doi: 10.1038/358673a0. [DOI] [PubMed] [Google Scholar]
- Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354(6348):31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- Orser BA, Pennefather PS, MacDonald JF. Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology. 1997;86:903–917. doi: 10.1097/00000542-199704000-00021. [DOI] [PubMed] [Google Scholar]
- Roth JE, Murray TF, Franklin PH. Regional distribution and characterization of [3H]dextrorphan binding sites in rat brain determined by quantitative autoradiography. J Pharmacol Exp Ther. 1996;277:1823–1836. [PubMed] [Google Scholar]
- Sakurada K, Masu M, Nakanishi S. Alteration of Ca2+ permeability and sensitivity to Mg2+ and channel blockers by a single amino acid substitution in the N-methyl-D-aspartate receptor. J Biol Chem. 1993;268:410–415. [PubMed] [Google Scholar]
- Sprengel R, Aronoff R, Volkner M, Schmitt B, Mosbach R, Kuner T. Glutamate receptor channel signatures. Trends Pharmacol Sci. 2001;22(1):7–10. doi: 10.1016/s0165-6147(00)01588-1. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Beck C, Wollmuth LP. Molecular rearrangements of the extracellular vesibule in NMDAR channels during gating. Neuron. 2002;33:75–85. doi: 10.1016/s0896-6273(01)00560-8. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Koshelev S. Two blocking sites of aminoadamantane derivatives in open N-methyl-D-aspartate channels. Biochim Biophys J. 1998;74:1305–1319. doi: 10.1016/S0006-3495(98)77844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth, et al. Intracellular Mg2+ interacts with structural determinants of the narrow constriction contributed by the NR1-subunit in the NMDA receptor channel. J Physiol. 1998;506(Pt 1):33–52. doi: 10.1111/j.1469-7793.1998.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Shimoji K. Subunit- and site-specific pharmacology of the NMDA receptor channel. Prog Neurobiol. 1999;59:279–298. doi: 10.1016/s0301-0082(99)00007-6. [DOI] [PubMed] [Google Scholar]
- Yamazaki, et al. Cloning, expression and modulation of a mouse NMDA receptor subunit. FEBS Lett. 1992;300(1):39–45. doi: 10.1016/0014-5793(92)80160-i. [DOI] [PubMed] [Google Scholar]
- Yamakura T, Mori H, Masaki H, Shimoji K, Mishina M. Different sensitivities of NMDA receptor channel subtypes to noncompetitive antagonists. Neuroreport. 1993;4:687–690. doi: 10.1097/00001756-199306000-00021. [DOI] [PubMed] [Google Scholar]