

Abstract
Herein, we report a highly sensitive electrocatalytic sensor-cell construct that can electrochemically communicate with the internal environment of immune cells (e.g., macrophages) via the selective monitoring of a particular reactive oxygen species (ROS), hydrogen peroxide. The sensor, which is based on vertically aligned single-walled carbon nanotubes functionalized with an osmium electrocatalyst, enabled the unprecedented detection of a local intracellular “pulse” of ROS on a short second time scale in response to bacterial endotoxin (lipopolysaccharide-LPS) stimulation. Our studies have shown that this initial pulse of ROS is dependent on NADPH oxidase (NOX) and toll like receptor 4 (TLR4). The results suggest that bacteria can induce a rapid intracellular pulse of ROS in macrophages that initiates the classical innate immune response of these cells to infection.
Keywords: intracellular sensor, electrocatalytic sensor, carbon nanotubes, reactive oxygen species, hydrogen peroxide, macrophage cells
Introduction
Reactive oxygen species (ROS) production or dysregulation are implicated in many human pathologies, including inflammation, atherosclerosis, cancer, diabetes, and neurodegeneration.1 ROS is a collective term referring to oxygen-derived species, including superoxide anion radical (O2•–) and hydrogen peroxide (H2O2). In addition to their classical cytotoxic and antimicrobial effects, ROS take part in a diverse array of biological signaling events and are thus critical in control of cell function. Macrophages play a central role in host defense by producing ROS via the NADPH oxidase enzyme complex (NOX).2−4 NOX is stimulated to produce ROS, when lipopolysaccharide (LPS) binds to the Toll Like Receptor 4 (TLR4).5,6 NOX is a multicomponent enzyme that is dormant in unstimulated cells but can be activated by various stimuli. In the activated form, the NADPH oxidase complex mediates the transfer of electrons from cytosolic NADPH to O2 to produce superoxide anion (O2–),7 which is rapidly converted to hydrogen peroxide (H2O2) and subsequently other ROS including the hydroxyl radical (OH•). The ROS response might either be involved in ’signaling’ or “antibacterial” functions depending on the amount generated and duration of the ROS burst.2 This behavior may also depend on location and stimuli. A “short sharp” ROS response early after interaction of an immune cell with bacteria, would indicate a signaling response and may be important for priming the cells for subsequent antibacterial responses.
Despite the important role ROS play in determining cellular fate, technology for monitoring selective, spatial, and temporal production of ROS within cells is currently lacking. Development of such technology will enable a more thorough understanding of the role that ROS play in physiological and disease states and allow translation of this information for clinical benefit.8
Although a variety of sensors for cellular ROS assessment have been developed, most rely heavily on the use of ROS-sensitive fluorescent probes. These probes suffer from a number of limitations: (i) they lack specificity for a particular ROS; (ii) they cannot be targeted to specific intracellular compartments; and (iii) they can produce ROS upon light exposure and can therefore lead to false positives.9 Another technique, electron paramagnetic resonance (EPR), also known as electron spin resonance (ESR) spectroscopy, is a definitive method for detecting and characterizing free radicals including ROS. However, because of the short half-life of ROS coupled with their low concentration in biological systems, EPR detection of these species at ambient temperatures requires chemical “spin traps” to provide long-lived stable radical species.10 The possible disturbance to cellular systems from such traps together with a lack of sensitivity at the single cell level, restricts the use of EPR spectroscopy for intracellular ROS detection.
Electrochemical technology offers a promising platform to enable the development of new tools to tackle the challenge of measuring cellular chemistry on the nanoscale. For example, it has recently been shown that conducting nanostructures can be used to access the inside of cells. However, most only show proof of principle that electrochemical intracellular sensing is possible11−14 and no detailed insight into new biological phenomena has been gained. Furthermore, no reports exist on using electrocatalytic modified nanostructures to sense intracellular events, although others have alluded to their use,15 to allow for investigations into intracellular signaling. Therefore, by combining nanostructured electrodes, that have dimensions sufficient to span the plasma membrane, with electrocatalytic functionalization a sensing platform emerges as a front runner in providing a new approach to characterize intracellular events.
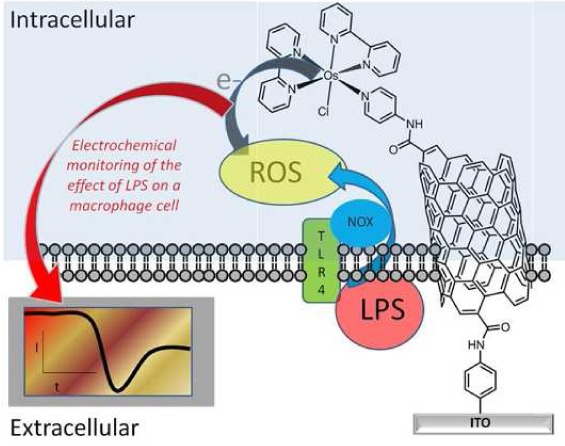
Here we demonstrate, for the first time, an intracellular electrocatalytic sensor that can selectively detect one particular ROS, hydrogen peroxide (H2O2), intracellularly in situ on an unprecedented time scale. The strategy of the ROS detection via the sensor-cell construct can be seen in Scheme 1. The intracellular electrocatalytic sensor has allowed the first directed approach to selectively monitor ROS and delineate the hierarchy of the redox mediated events controlling early ROS production on a short time scale. This provides new biological insights into early bacterial stimulation of a macrophage immune response and provides a new tool for investigating such local and spatial cell signaling events.
Scheme 1. Diagrammatic Representation of a Sensor-Cell Construct and the Strategy for Investigating Early Reactive Oxygen Species (ROS) Generation from Macrophages in Response to Lipopolysaccharide (LPS) Stimulation.

NOX: NADPH oxidase.
Results and Discussion
The strategy for fabricating surfaces to enable the sensing of H2O2 involved three main steps. First, an indium tin oxide (ITO) conducting substrate was modified with an aryl amine tether layer using well characterized diazonium chemistry.16 The aryl amine was then used as an anchor for tethering carboxylated SWCNTs by carbodiimide chemistry.17,18 Finally, the ITO electrode nanostructured with SWCNTs was further modified with an osmium bipyridine (Osbpy) by using carbodiimide chemistry forming an ITO-SWCNT-Osbpy sensor (Scheme 1). Electrodes modified with osmium complexes have previously been used as bioelectrocatalysts and were shown to be biocompatible.19,17,20,21 However, there are no examples of using such sensors for monitoring cellular chemistry as described in these investigations. Cyclic voltammetry was initially performed to confirm the presence of osmium using the ITO-SWCNT-Osbpy in 50 mM PBS alone and a typical cyclic voltammogram obtained can be seen in Figure 1A red curve. Note that the electrochemical area for all the experiments reported herein was controlled via use of an O-ring with a diameter of 4 mm. A redox couple was observed that can be attributed to the redox behavior of OsII/OsIII.17,19 A reduction peak was obtained at 207 mV (±1SD 34.0 n = 3) (Epc) and an oxidation peak at 307 mV (±1SD 42.7 n = 3) (Epa) obtained using CVs in Figure 1S. This is the potential at which the current is at its highest for the reduction and oxidation. Normally with a surface confined electrochemical process it would be expected that the peak separation be zero. However, Epa (anodic peak potential) is larger than Epc (cathodic peak potential), and this behavior could be due to the electron transfer rate being relatively slow.
Figure 1.

(A) Typical cyclic voltammograms obtained in PBS (red) and 100 mM H2O2 (blue) at an ITO-SWCNTs-Osbpy electrode at a scan rate of 5 mV s–1. (B) Typical fixed potential amperograms obtained for addition of 1 mM aliquots of H2O2 to PBS using an ITO-SWCNT-Osbpy electrode poised 400 mV (I) to establish the presence of oxidative processes and at 150 mV (II) to investigate the reductive processes. (C, D) Typical cyclic voltammograms obtained for (C) PBS and in (D) 100 mM H2O2 solutions at ITO-SWCNT-Osbpy electrode at 5, 10, 20, 50, 100, and 200 mV s–1.
A cyclic voltammogram was subsequently performed with a 100 mM solution of H2O2 in 50 mM PBS to ascertain the ability of the osmium modified sensor to interact with H2O2. A typical cyclic voltammogram obtained using the ITO-SWCNT-Osbpy sensor can be seen in Figure 1A red curve. In the presence of H2O2 there was a change in the behavior with an increase in oxidation and reduction peak currents Figure 1A blue curve. Additionally, there was an approximate 30 mV shift to less negative potentials for the reduction peak in the presence of the H2O2 and the oxidation catalytic peak is shifted by 60 mV more positive. We envisaged this resulted from catalytic reaction(s) of H2O2 with the Osbpy. Control cyclic voltammograms were performed with ITO-SWCNT modified electrodes in the presence and absence of H2O2 and no redox couple was observed (Figure 2S) in PBS alone. This suggests that the Osbpy is causing the disproportionation of the hydrogen peroxide under the experimental conditions (pH and relative concentrations of Osbpy and H2O2). The scan rate of 5 mV s–1 was chosen (Figure 1A) to compare the electrochemical behavior in the presence and absence of H2O2 because this is the scan rate at which the difference in current obtained in the absence of the H2O2 compared to its presence, when catalysis was occurring at the osmium modified surface, is the greatest. This is due to the rate limiting step being the interaction of the osmium with the H2O2, and explains why at faster scan rates the difference in peak current associated with catalysis is negligible when compared to the osmium electrochemistry without the presence of H2O2. To note, prior to running any scans on the fabricated electrodes, the stability was established by sonicating for 5 min in ethanol then running 200 consecutive cyclic voltammograms until a relative steady state was observed and this can be seen in Figure 1S.
To look at this catalytic behavior more closely, we obtained chronoamperograms (Figure 1B). These clearly show that the addition of H2O2 increases the oxidation current at an applied potential of 400 mV and increases the reduction current at an applied potential of 150 mV. At 400 mV the redox reaction must be oxidation of OsII and at 150 mV, reduction of OsIII (no response to H2O2 was observed in the absence of the Osbpy complex). These findings indicate that H2O2 can reduce OsIII to OsII (current at 400 mV due to reoxidation of OsII to OsIII and that H2O2 can oxidize OsII to OsIII (current at 150 mV due to rereduction of OsIII to OsII).The corresponding reactions resulting in this behavior are as follows, which is similar to iron induced H2O2 disproportionation.22
| 1 |
| 2 |
The applied potentials used were chosen from the cyclic voltammograms in Figure 1A red curve to be above the potential in which peak currents were obtained to ensure sufficient thermodynamic driving force for the redox event to occur as governed by the Nernst equation.
A concentration of 1 mM was used as the rate of catalytic process begins to reach a maximum as demonstrated by the beginning of the plateau in the concentration profile in Figure 2B. These results confirm that the redox behavior observed in Figure 1 in the presence of H2O2 is derived from the modification of SWCNT with Osbpy. Other workers have modified electrodes with osmium polymers and investigated the ability of osmium derivatives, that are different from the one used in these investigations, to interact with H2O2.23,24 They reported that H2O2 could interact with their modified electrodes but via an electrocatalytic reductive mechanism. In particular, they provided evidence that the mechanism was not via the disproportionation of H2O2.23,24 To the best of our knowledge, the result reported here is the first example of an osmium complex that catalyzes the disproportionation of H2O2. However, the reason for this difference from that reported in the literature toward the mechanisms of interaction of osmium complexes and H2O2 is not clear and requires further investigation. Moreover, using chronoamperograms from Figure 1B, we estimate that the reduction current begins to rise 0.1 s after the addition of H2O2, showing the sensing is fast with a sub-second detection limit. It is worth noting that although the oxidation signal obtained is twice the magnitude of the reduction signal, as can be seen from Figure 1B, for the cellular studies, it was decided to use the reduction signal for monitoring H2O2. The justification for this is because this occurs at lower potentials and, consequently, it was envisaged this would avoid the possibility of other molecules interfering with the signal and giving rise to false positives.
Figure 2.
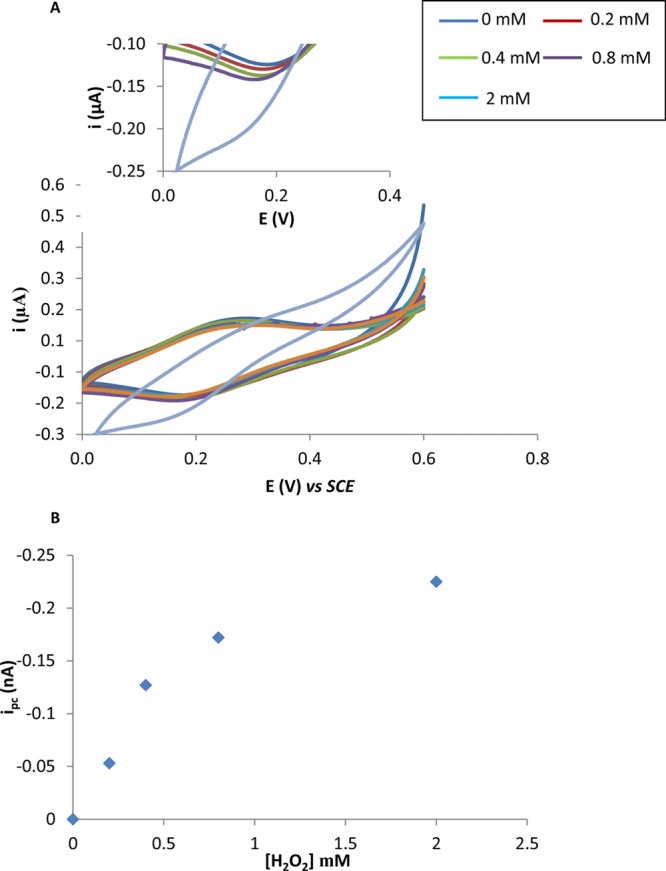
(A) Cyclic voltammograms performed with PBS solution containing 0.2, 0.4, 0.8, and 2 mM of H2O2 at ITO-SWNT-Osbpy electrodes at a scan rate of 5 mV s–1. Insert is enlarged version of cathodic currents obtained. (B) Peak current versus concentration of H2O2.
A scan rate study was performed to look at the electrochemical characteristics of the osmium redox couple in more detail and typical cyclic voltammograms obtained over the range 5–200 mV s–1 can be seen in Figure 1C and in the presence of H2O2 in Figure 1D. Mean peak currents were measured by extrapolating the baseline and measuring the peak height from this point and plotted against the scan rate (Figure 3SA, B). This showed that the redox couple of the osmium process was a surface controlled process as current was proportional to scan rate when CVs were obtained in PBS alone. On addition of H2O2, there is a deviation from this linearity with scan rate. A current function plot and a plot of scan rate versus peak current in the presence of H2O2 provided further mechanistic insight in to the rate limiting step. A positive slope (Figure 3SC) was observed and therefore indicates that the rate limiting step controlling the rate of H2O2 disproportionation is the interaction and electron transfer of the H2O2 with the Osbpy.23 The apparent rate constant (kapp) was calculated using the data obtained from a series of cyclic voltammograms (Figure 1) with different scan rates (e.g., 0.05–0.2 V s–1). The data were analyzed on the basis of Laviron’s method, using the following simplified equation.
| 1 |
where m is the peak separation obtained from the CVs, n is the number of electrons transferred, υ is the scan rate, F is the Faraday constant, and the other symbols have their usual meaning. Thus, the rate constant can be obtained from the slope of the plot of m vs 1/υ. We estimate the calculated rate transfer coefficient of 6.7 s–1 and in the presence of the hydrogen peroxide a rate constant of 0.97 s–1.
Prior to performing cell experiments, it was important to assess the ability of the sensors to detect varying concentrations of H2O2. We performed a cyclic voltammetric study on solutions of H2O2 ranging from 0 to 2 mM and typical voltammograms obtained at 5 mV s–1 can be seen in Figure 2A. A plot of the concentration of H2O2 versus peak current yields a logarithmic relationship with a maximum catalytic velocity reached by a concentration of 2 mM H2O2. It is worth mentioning that the electrocatalytic reductive current seen in Figure 2A is similar to that obtained for 100 mM in Figure 1A. This is explained by the fact the reaction between the H2O2 and Osbpy has reached a maximal velocity and therefore any further increase in H2O2 after 2 mM results in no further enhancement of the electrocatalytic signal. The data in Figure 2 and Figure 4SB show that the magnitude of reduction current is proportional to H2O2 concentration and thus we proposed to utilize the sensors ability to monitor H2O2 production in cells in response to stimulation by the bacterial molecule lipopolysaccharide (LPS). However, it was important to show that the SWCNT-Osbpy nanostructures were entering the cell. In order to investigate this, two experiments were performed. First, the biological stain, methylene blue, was introduced into the cells using the protocol recently developed by our group and used as a fingerprint to identify if the nanostructures have accessed the cytoplasm.18 Briefly, cells were exposed for 30 min to 50 μM methylene blue prior to either being centrifuged or drop coated (no external force applied) on to ITO-SWCNTs-Osbpy electrodes. Fresh PBS solution was added to the cell solution prior to drop coating and centrifugation, ensuring no methylene blue was left in solution. Thus, any electrochemical signal observed could be attributed to methylene blue being up taken by the cells. A redox couple typical of methylene blue was observed for centrifuged samples that can be viewed in Figure 6S. The cells that were drop-coated resulted in no redox couple over the potential range studies. Thus, we infer that the SWCNTs-Osbpy cannot access the intracellular environment without application of an external force to allow the SWCNT-Osbpy nanostructures to cross the plasma membrane. This result is consistent with our previous findings on using chemically assembled SWCNTs.18 Second, EPR spectroscopy was used to validate our findings and will be discussed in further detail in the succeeding section. Additionally, scanning electron microscopy images of CNTs modified surfaces (Figure 8S) were taken which show bundles of CNTs in the nanoscale dimension with sufficient height to penetrate the plasma membrane, which is typically 5–10 nm.
It was also important to assess the viability of cells on insertion of nanostructures. This study was done with trypan blue dye. Cells were wired to the ITO-SWCNT-Osbpy electrodes using centrifugal force and an aliquot of trypan blue was placed on the chips. Those cells whose membrane was intact remained unstained whereas those that were damaged or dead took up the dye and were stained blue. Cells on these chips had a viability of 77% (±5.4% error is ±1SE of mean, n = 9) and controls, in which centrifugation of cells was performed on ITO modified with the arylamine tether layer only, gave a mean cell viability of (74% ± 7.6% error is ±1SE of mean n = 9; p value of >0.81 not significant). This confirms that the nanostructures are not inducing cell death.
Figure 3 shows typical amperograms obtained for RAW 264.7 macrophages wired at ITO-SWCNT-Osbpy (Figure 3A–C) and ITO modified with SWCNTs only (Figure 3D). LPS is known to induce ROS production, including H2O2 in macrophages.25 On addition of 1 μg/mL LPS to cells wired to ITO-SWCNT-Osbpy sensors, a total mean current drop of 682.5 pA ± 64.1 pA (n = 5) was obtained within 5 s of exposure to LPS (Figure 3A). The average of H2O2 produced per cell was calculated as described in detail in the Supporting Information, and we estimate that a single cell produces 26.59 zmoles of H2O2 in response to LPS stimulation. This value is significantly smaller than the ROS response observed for calcium ionophore stimulated macrophage,26 which has been reported to produce 10 fmoles per single cell.26 The difference between our observation and previous work is to be expected as calcium ionophore would stimulate the classical induction of the respiratory production of ROS. The classical induction is expected to be significantly higher than that used for cell signaling purposes in which the ROS produced for signaling are localized and at low concentrations. To the best of our knowledge, this is the earliest reported detection of ROS production in response to LPS stimulation of immune cells.
Figure 3.

Typical current versus time amperograms obtained at ITO-SWCNT-Osbpy sensors interfaced with RAW 264.7 cells at an applied potential of 150 mV in 50 mM PBS containing (A–C) 0.1 M KCl and (D) ITO-SWCNTs only. All 4 wired electrodes had LPS aliquots placed in the electrochemical cells at approximately 2 s. Traces A and D were recorded with wired cells which had not been exposed to any ROS inhibitor. Trace B was obtained with cells exposed to the ROS inhibitor 10 mM N-acetyl-l-cysteine (NAC) prior to the electrochemical assay. Trace C was exposed to 10 μM NADPH oxidase inhibitor diphenyleneiodonium (DPI). Traces EI and EII show typical curves in the absence (green) and presence (blue trace) of LPS obtained for cells that were drop coated (no centrifugation).
A possible explanation for our ability to observe, this previously unseen ROS pulse, can be found in the unique characteristics that occur when performing electrochemistry at the nanoscale.27 For instance, nanoelectrodes can measure ultrafast electron-transfer kinetics. Faster rates of mass transport occur due to a change from planar diffusion, observed at macro electrodes, to radial diffusion at nanoelectrodes. This is particularly pertinent for ROS which degrade quickly and thus diffusion is not a limitation of the sensing. These factors coupled with reduced ohmic drop leads to higher signal-to-noise ratio and explains why we are able to see the unprecedented generation of ROS on the observed second time-scale. Previous reports have indicated times of within 2 min28 and for reactive nitrogen species (RNS) within 5 h29 and both appear to reach a maximum after 24 h on stimulation with LPS. To confirm that our technology could detect these longer bursts of ROS, cells were drop coated on to the electrode surfaces. Amperograms were run for 4000 s on untreated controls (Figure 3 (EI)) and cells exposed to LPS (Figure 3 (EII)). Importantly, as predicted, we see a much larger decrease in mean current of approximately 1990 nA (±715) pA in those cells exposed to LPS. It is worth highlighting that this latter signal, associated with classical macrophage immune defense, is significantly larger and longer-lived than the initial short “pulse” of ROS detected on initial exposure to LPS. This suggests that their role and origin are different.
Investigations were performed to assess the effect of other common ROS, including O2•–, and reactive nitrogen species (RNS) including peroxynitrite (OONO–), NO (nitric oxide) and nitrite anion (NO2–) on the analytical output, as described in more detail in the Supporting Information. These ROS and RNS were generated in situ while performing fixed potential amperometry as previously demonstrated by Amatore and co-workers.14 Aliquots of stock solutions of peroxynitrite or KO2 (which decomposes to give O2•–) or DEANONOate (which decomposes to give NO and NO2–)30 were added to an electrochemical cell, at which an applied potential of 150 mV was applied. It would be envisaged that if any of these ROS and RNS were contributing to the increases in reduction current detected when cells were exposed to LPS, then an increase in cathodic current should occur on exposure of these to the sensor. However, this was not the case and no observed interference from peroxynitrite was obtained (Figure 7S). Interestingly, in the presence of in situ generated O2•–, NO and NO2– an increase in anodic current was observed. Clearly, if these were responsible for the signal observed in LPS treated cells then it would be envisaged that an increase in oxidative current would occur, which is contrary to the experimental observation of an increase in reduction current. Therefore, these studies provide strong evidence that the signal arising inside the cell is a result of the production of H2O2 in response to LPS stimulation.
The change in current in response to LPS stimulation occurs as Osbpy is catalyzing the degradation of H2O2. When the ITO-SWCNT-Osbpy sensors were placed in PBS in the absence of cells and an applied potential of 150 mV, the background signal was cathodic. Spiking in 25 μL standard addition of a 100 mM stock solution of H2O2 to 5 mL of electrolyte, giving a standard additions that equated to 500 μM, an increase in this cathodic (reductive) current was observed as expected (Figure 4SA). When performing cellular studies the current was anodic rather than cathodic as seen for acellular controls. To ascertain what causes this change in behavior a study was performed in which the ITO-SWCNT-Osbpy sensors were exposed to culture medium alone and then amperometry was performed which results in an anodic current (Figure 5S). Thus, this change occurs due to exposure to the culture medium. We propose that at this potential other oxidative electrochemical processes must be occurring at the electrodes and the resulting current can be defined as the net oxidative current. We suggest that on addition of LPS, the ROS formed oxidizes the Osbpy, which is then reduced instantly at the potential applied. Consequently, this reduces the net oxidative process, resulting in a drop in the current as observed on exposure to LPS.
To ascertain if the drop in signal was due to ROS, we utilized a well-known antioxidant compound, N-acetyl cysteine (NAC), which has previously been shown to inhibit ROS production in response to LPS.31,32 A typical amperogram obtained for cells exposed to 10 mM NAC for 1.5 h prior to spiking in 1 μg/mL LPS can be seen in Figure 3B, and after exposure to LPS, no decrease in signal was observed. This provides evidence that the decrease in signal observed in Figure 3A occurs as a result of ROS production in response to LPS stimulation. Additionally, Figure 3D confirms that the Osbpy selectively monitors the ROS at the potential applied as no decrease was observed with cells wired to ITO-SWCNTs sensors when compared to ITO-SWCNT-Osbpy electrodes (Figure 3A).
Our detection of a short pulse of ROS within seconds of the macrophage exposure to LPS suggests a novel signaling mechanism that might control cell responses. Thus, we wanted to determine the origin of this rapid ROS response and if it depended on the LPS receptor TLR4. It was assumed that a membrane associated NADPH oxidase (NOX) was responsible for the ROS production although this enzyme usually requires assembly of a complex of subunits to become active.33 To determine if NOX was involved in the short LPS-induced ROS burst, we used the well-known NOX inhibitor diphenyleneiodonium (DPI).34 We exposed the cells to 10 μM DPI for 90 min prior to running the electrochemical assay. On addition of 1 μg/mL of LPS to wired cells exposed to DPI no significant decrease in current was observed (Figure 3C), suggesting that the initial ROS pulse we detect is generated by a NADPH oxidase.
Evidence that the signal generated in the electrochemical studies was due to intracellular generation of ROS and that the ROS did not enter the extracellular environment was confirmed by EPR spectroscopy. In order to detect any extracellular ROS, the spin trap [5-(diisopropoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DIPPMPO) was used. EPR spectroscopy was performed by adding 100 mM DIPPMPO to cells that were untreated (Figure 4A) or exposed to 1 μg/mL LPS (Figure 4B). EPR spectra were collected for up to 30 min. No characteristic spectrum due to ROS adducts of DIPPMPO was observed over the time scale of our LPS experiments, suggesting that no ROS was produced extracellularly over this time. Positive controls were run using OH• and OOH• generated in situ from TiO2 in PBS and H2O2 exposed to UV light, respectively35,36 (Figure 4C, D). These results imply that the initial ROS we detect in the RAW cells stimulated with LPS are intracellular and do not escape to the extracellular environment.
Figure 4.
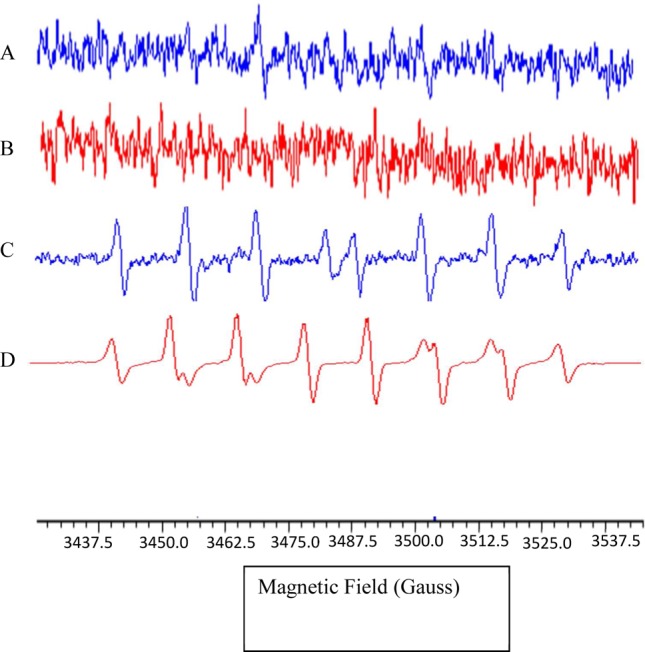
EPR spectroscopy to detect extracellular ROS using the DIPPMPO spin trap. Typical EPR spectra from (A) 2 × 106 RAW 264.7 cells in 100 mM DIPPMPO solution, (B) RAW 264.7 cells treated with 1 μg/mL LPS. Positive controls for EPR spectra due to OH and OOH adducts of DIPPMPO were obtained from (C) DIPPMPO–OH generated from TiO2 suspension in PBS, after exposure to UV light and (D) DIPPMPO-OOH generated from TiO2 suspension in PBS with added H2O2, after exposure to UV light. EPR spectra in A and B were run at 100x the gain used in spectrum D.
Previous reports have suggested that ROS are important for activating TLR4 signaling,37 while others have suggested that TLR4 may stimulate ROS production.38 However, no reports have shown ROS production in response to LPS stimulation on such a short time scale as we report here. It is well understood that ROS generation is implicated in TLR cell signaling responses.38,39 However, it was shown that MyD88, the downstream adaptor of TLR4, controls NADPH oxidase function in primary macrophages.34 Thus, it is not clear where NADPH oxidase is located in the hierarchical order of TLR4 signaling pathways. Consequently, investigations were undertaken to ascertain if the ROS pulse observed in response to LPS on this unprecedented time scale was dependent on ligation of TLR4. In addition, it was important to determine that this short ROS response was not unique to RAW 264 macrophages. To address both points, we formed a sensor-cell construct using a newly described nontransformed mouse macrophage line (MPI cells) that has characteristics of primary macrophages.40 To elucidate the role of TLR4 in generating the fast ROS response, we made use of wild type MPI cells and those that had been derived from TLR4-deficient mice (TLR4–/−).40 For normal (wild type) MPI cells stimulated with LPS, a decrease in current similar to that observed with RAW 264.7 cells was seen (Figure 5A). However, when MPI cells lacking TLR4 (TLR4–/−) (Figure 5B) were stimulated with LPS, no current decrease was observed. This was shown to be significant by plotting the data with a moving average (Figure 6). These findings demonstrate that TLR4 is indeed required for the ROS pulse and that MPI cells also produce a rapid short burst of ROS in response to LPS stimulation.
Figure 5.

Typical current versus time amperograms obtained at ITO-SWCNT-Osbpy sensors interfaced with (A) wild type macrophage cells (MPI) (B) that are TLR 4 deficient (TLR 4 – /−) and that were exposed to 1 μg/mL LPS. (C) Wild type cells and (D) TLR4 deficient cells that were exposed to 5 μg/mL phorbol 12-myristate 13-acetate (PMA). All amperograms were performed at an applied potential of 150 mV in 50 mM PBS containing 0.1 M KCl.
Figure 6.
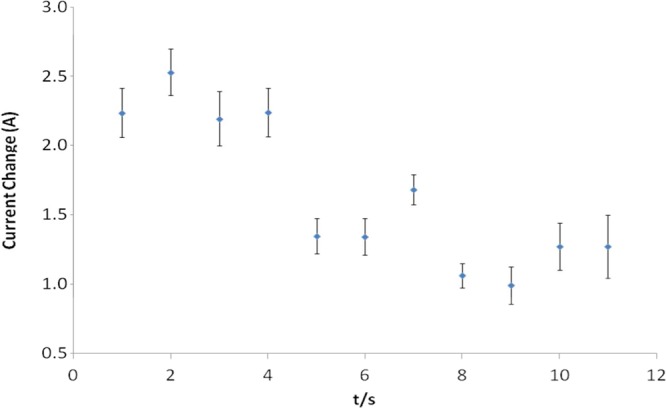
Moving average of the current change observed for a typical response obtained (Figure 5A) for wild type MPI cells exposed to LPS.
It was also important to show that the sensor cell constructs could detect ROS produced by other cell stimuli. When we used the soluble stimulant phorbol 12-myristate 13-acetate (PMA), a large response was detected in both wild type (Figure 5C) and TLR4–/– cells (Figure 5D) because of a rapid production of ROS characteristic of PMA stimulation that is not dependent on TLR4.41
Thus, we can conclude that the rapid short “pulse” of ROS generation in response to LPS stimulation in macrophages is mediated by TLR4. With this information in mind and in conjunction with our findings that NADPH oxidase is responsible for this early ROS burst, the hierarchy of components involved in this early ROS burst is elucidated. Our data show that LPS must bind to TLR4 and immediately trigger ROS generation from an associated NOX. This is concluded because of the time frame of the pulse. TLR4 has previously been shown to associate with NOX4 isoform38 and this would be a candidate for the short time scale ROS burst detected here. However, our results suggest an immediate generation of ROS from the NOX (within 5 s), whereas ROS generated in response to microbial stimulation in phagocytes typically takes 10–30 min due to assembly of an active NADPH oxidase complex.42,43 We speculate that this immediate ROS “pulse” may initiate downstream signaling and cell activation.
Conclusion
In conclusion, we demonstrate that by developing new nanoelectrochemical technology it is possible to electrochemically communicate with the internal environment of immune cells. More importantly, this provides a generic platform to allow detailed mechanistic studies on how early ROS signals are produced and are involved in controlling cell behavior. To this aim, we have established that macrophages produce an early pulse of ROS on stimulation with LPS. The time scale and kinetics observed for their production indicates that this ROS pulse is likely used as an initiator signal activating the downstream signaling pathways associated with inflammation. Additionally, our studies indicate that there is a pool of assembled NADPH oxidase that can be rapidly activated by LPS to facilitate the production of superoxide and subsequently the downstream production of mediators such as H2O2 associated with an innate immune response. We show that this NADPH oxidase production of ROS is, however, dependent on the TLR4 activation by LPS. The electrocatalytic intracellular sensor electrode provides an alluring platform which can be tailored to sense short-lived localized cell signaling events, shedding new light on the role ROS play in controlling cell function.
Methods
Electrode Preparation
Initially, ITO deposited on Corning low alkaline earth boro-alumino silicate glass with ITO coated on one surface (resistivity = 20–25 Ω) purchased from Delta Technologies Limited was rinsed with HPLC grade ethanol and then ultrahigh purity H2O, exposed to UV light for an hour, and then rinsed in acetone and isopropyl alcohol. The strategy for preparation of functionalized SWCNTs covalently attached to ITO was based on using an electrografted tether layer, which then acted as an anchor for the SWCNTs. ITO samples were modified in situ with p-phenylenediamine using the method we recently reported.18 A 10 mM p-phenylenediamine solution was rapidly added to 1 M NaNO2 equivalent solution and allowed to react for 3 min. The reaction solution was then poured into the electrochemical cell, and the resultant diazonium cation was electrografted to the ITO scanning from 0.4 to −0.6 V followed by fixed potential deposition at −0.6 V for 2 min and finally a further scan from 0.4 to −0.6 V to confirm passivation (and hence grafting) of the electrode.
Uncut SWCNTs (NanoLab, Inc.) were acid-treated by adding 25 mg to 27 mL of a 3:1 mixture of concentrated H2SO4 and HNO3 and sonicating for 12 h. Following sonication, the contents were poured into 1000 mL of distilled water and left to settle overnight. The SWCNTs were then filtered through a 0.22 μm hydrophilic PVDF filter (Millipore) under suction, with washing until the rinsewater was close to pH 7. The disks containing wet SWCNTs were placed in an oven at 65 °C and dried overnight. The SWCNT formed a mat which was then scraped off the filter paper. Suspensions of cut SWCNTs were prepared by sonication of dried SWCNTs mats in dimethyl sulfoxide (DMSO). SWCNTs were coupled to arylamine modified ITO samples by submerging modified ITO samples in a 0.2 mg mL–1 DMSO suspension of cut SWCNTs containing 0.5 mg mL–1 dicyclohexyl carbodiimide (DCC). The reactants were sonicated for 30 min and then heated to 65 °C for 24 h in a closed cell. After preparation, SWCNT modified electrodes were sonicated in acetone for 2 min and isopropyl alcohol for 10 s and finally rinsed in Milli-Q water. The electrodes were dried with argon gas between each washing step.
Details of the synthesis of osmium(II)bis-2,2-bypyridine(p-aminomethylpyridine)chlorido hexafluorophosphate was previously reported by Rawson et al.17 Osbpy was coupled to the carboxylic acid-terminated tips of immobilized SWCNTs by first submerging the SWCNT-modified ITO samples in a 40 mM aqueous solution of 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDC) and 10 mM N-hydroxysuccinamide (NHS) solution for 1 h and then transferring the samples to a 2.5 mM solution of the osmium complex dissolved in pH 6.3 phosphate buffer and ethanol (7:3). The electrodes were allowed to react for 24 h before being removed and washed with Milli-Q water and dried with nitrogen.
Electrochemical Characterization at ITO-SWCNT-Os Electrodes
All electrochemical studies were carried out with a Gamry 600 potentiostat and data acquisition software (Gamry electrochemistry software version 5.61a) and a three-electrode cell consisting of a saturated calomel reference electrode, Pt counter electrode, and then the working electrode of either bare ITO modified with SWCNT or ITO modified with SWCNTs functionalized with Osbpy. The electrochemical area was controlled via use of an O-ring with a diameter of 4 mm.
Cyclic voltammograms were performed using ITO-SWCNT-Osbpy electrodes in 50 mM PBS in the presence and absence of varying concentrations of H2O2. Scan rate studies were performed over a range from 5 mV- 200 mV s–1. All cyclic voltammograms were performed from a starting potential of 0 V and a switching potential of 0.6 V and an end potential of 0 V and carried out in triplicate.
Fixed potential amperometry was performed at ITO-SWCNTs-Osbpy electrodes using 50 mM PBS solutions to investigate the redox behavior of osmium/H2O2 interaction. Initial oxidative processes were studied by applying a potential of 400 mV for 150 s prior to spiking in an aliquot of 100 mM stock H2O2, giving a final assay concentration of 1 mM. Subsequently, reductive processes were investigated by applying a potential of 150 mV for 150 s and subsequently spiking in to give a final assay concentration of 1 mM H2O2.
Method for Assaying Electrochemical Intracellular Response to LPS in RAW 264.7
2.5 × 106 RAW 264.7 cells were seeded in three 75 cm2 flasks containing 18 mL DMEM (10% FBS, 1% of penicillin/streptomycin, 2.4% glutamate, and 2.4% HEPES). These cells were grown for 2 days at 37 °C in a 5% CO2 atmosphere and reached an approximate 80% confluence. The old DMEM was aspirated off and 3 mL of fresh DMEM was placed in each flask and cells were detached using a cell scraper. The cell suspensions were pooled giving a total volume of 9 mL. ITO electrodes modified with single-walled carbon nanotube (SWCNTs) and SWCNTs with bound osmium bipyridine were placed in 50 mL centrifugation tubes and 2 mL of cell suspension at 1 × 106 cell/mL was placed in each tube. These were then centrifuged at 3000 rpm forcing the electrodes modified with SWCNTs and osmium bipyridine (ITO-SWCNT-Osbpy) through the plasma membrane. Prior to centrifugation, a batch of 2 mL of cell suspension at a concentration of 1 × 106 cell/mL were exposed (treated cells) to 10 mM N-acetyl-l-cysteine (NAC) a ROS inhibitor or diphenyleneiodonium (NADPH oxidase inhibitor) for 1.5 h and then the electrochemistry was performed as usual. Each was carried out in triplicate. Untreated cells were also wired to the ITO-SWCNT-Osbpy electrode.
These tubes were then placed in the incubator until the samples were required for electrochemical analysis. The wired cells were then ready to be investigated for their ability to detect intracellular changes in response to LPS (Escherichia coli 0111:B4) purchased from Sigma-Aldrich. The wired cells were rinsed by placing in a Petri dish containing 50 mM PBS at pH 7.4 and agitated to rinse. After the rinse, the nanostructured indium tin oxide (ITO) acting as the working electrode was placed in an electrochemical cell, under potentiostat control, alongside a platinum counter electrode and saturated calomel reference electrode and fixed potential amperometry was subsequently performed using 5 mL of PBS as the electrolyte. Fixed potential amperometry was used to study the change in current with time on the addition of LPS. A fixed potential of 400 mV was initially applied, ensuring no reduction was occurring, for 10 s and then switched to 150 mV and current was sampled over approximately 1100 s period. When a steady state current was reached, an aliquot of 10 μL of a stock solution of 500 μg mL–1 lipopolysaccharide from Escherichia coli 0111:B4 (LPS) giving a final concentration of 1 μg mL–1 was added to the electrochemical cell solution. This was then repeated for each chip (n = 4). Long-term exposure was assessed by drop coating cells on to electrodes and sampling current using fixed potential amperometry for 4000 s in the presence and absence of 1 μg/mL LPS.
Assay with MPI Cells
Wild type (MPI-2) TLR4 deficient MPI macrophage40 were grown in RPMI medium containing 10% FBS, 1% of penicillin/streptomycin, 2.4% glutamate and 30 ng/mL murine GM-CSF. Cells were grown for 2–3 days at 37 °C in a 5% CO2 atmosphere to reach 80–90% confluency. Adherent cells were detached with 1.0 mM EDTA in PBS, combined with nonadherent cells and stimulated with ultrapure LPS preparations44 as described above for RAW cells.
Electron Paramagnetic Resonance Spectroscopy
For trapping extracellular ROS, we used the spin trap [5-(diisopropoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide; 2-diisopropylphosphono- 2-methyl-3, 4-dihydro-2H-pyrrole-1-oxide] (DIPPMPO) (Enzo Life Science). Cells were harvested to give a final concentration of 2 × 106 cells per assay. Stock solutions of DIPPMPO were prepared in PBS at concentrations of 1 M. These solutions were degassed with oxygen free nitrogen to remove oxygen from the solution. RAW 264.7 cells were harvested to give an assay of 2 × 106 cells which were incubated with the DIPPMPO spin trap (final concentration of 100 mM) for 15 min prior to running EPR. For LPS studies, a final concentration of 1 μg/mL was added and cells were appraised by EPR. EPR spectra were run on a Bruker EMX-Micro spectrometer (Bruker, UK), operating at 9 GHz utilizing gas-permeable tubing for the cell samples and run at 37 °C. Spectra were analyzed with WinEPR software to obtain identification of the radical adducts trapped.
Acknowledgments
We thank the Leverhulme Trust (F/00 094/BD and ECF/2013-603), BBSRC (BB/L017059/1), Royal Society (RG110087), Wellcome Trust (WT091285MA), EPSRC (EP/K027263/1), ERC (Consolidator Grant 614787), and NC3Rs (NC/L00058X/1) for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.5b06493.
Electrochemical data on surface stability, control surfaces, osmium redox couple, the intracellular presence of the SWCNT-Osbpy structures, and selectivity data for the sensor (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Reddy J. K.; Lalwani N. D.; Reddy M. K.; Qureshi S. A. Excessive Accumalation of Auto-Fluorescent Lipofuscin in the Liver During Heptocarcinogenesis by Methyl Cloffernapate and other Hypolipidemic Peroxisome Proliferators. Cancer. Res. 1982, 42, 259–266. [PubMed] [Google Scholar]
- Fialkow L.; Wang Y.; Downey G. P. Reactive Oxygen and Nitrogen Species as Signaling Molecules Regulating Neutrophil Function. Free Radical Biol. Med. 2007, 42, 153–164 10.1016/j.freeradbiomed.2006.09.030. [DOI] [PubMed] [Google Scholar]
- Dröge W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Bogdan C.; Röllinghoff M.; Diefenbach A. Reactive Oxygen and Reactive Nitrogen Intermediates in Innate and Specific Immunity. Curr. Opin. Immunol. 2000, 12, 64–76 10.1016/S0952-7915(99)00052-7. [DOI] [PubMed] [Google Scholar]
- Lu Y.-C.; Yeh W.-C.; Ohashi P. S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Beutler B. Tlr4: Central Component of the Sole Mammalian LPS Sensor. Curr. Opin. Immunol. 2000, 12, 20–26 10.1016/S0952-7915(99)00046-1. [DOI] [PubMed] [Google Scholar]
- Russell D. G.; VanderVen B. C.; Glennie S.; Mwandumba H.; Heyderman R. S. The Macrophage Marches on its Phagosome: Dynamic Assays of Phagosome Function. Nat. Rev. Immunol. 2009, 9, 594–600 10.1038/nri2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson B. C.; Chang C. J. Chemistry and Biology of Reactive Oxygen Species in Signaling or Stress Responses. Nat. Chem. Biol. 2011, 7, 504–511 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanaraman B.; Darley-Usmar V.; Davies K. J. A.; Dennery P. A.; Forman H. J.; Grisham M. B.; Mann G. E.; Moore K.; Roberts L. J. II; Ischiropoulos H. Measuring Reactive Oxygen and Nitrogen Species with Fluorescent Probes: Challenges and Limitations. Free Radical Biol. Med. 2012, 52, 1–6 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. K.: James P. E. In Biomedical Applications of EPR Spectroscopy; Gilbert B. C.; Murphy D.; Davies M., Eds.; Royal Society of Chemistry: Cambridge, U.K., 2008; Vol. 21, Chapter 1, pp 15–32. [Google Scholar]
- Duan X.; Gao R.; Xie P.; Cohen-Karni T.; Qing Q.; Choe H. S.; Tian B.; Jiang X.; Lieber C. M. Intracellular Recordings of Action Potentials by an Extracellular Nanoscale Field-Effect Transistor. Nat. Nanotechnol. 2011, 7, 174–179 10.1038/nnano.2011.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R.; Park J.-H.; Choi Y.; Heo C.-J.; Yang S.-M.; Lee L. P.; Yang P. Nanowire-Based Single-Cell Endoscopy. Nat. Nanotechnol. 2012, 7, 191–196 10.1038/nnano.2011.226. [DOI] [PubMed] [Google Scholar]
- Gao R.; Strehle S.; Tian B.; Cohen-Karni T.; Xie P.; Duan X.; Qing Q.; Lieber C. M. Outside Looking In: Nanotube Transistor Intracellular Sensors. Nano Lett. 2012, 12, 3329–3333 10.1021/nl301623p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Noël J.-M.; Velmurugan J.; Nogala W.; Mirkin M. V.; Lu C.; Guille Collignon M.; Lemaître F.; Amatore C. Nanoelectrodes for Determination of Reactive Oxygen and Nitrogen Species Inside Murine Macrophages. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11534–11539 10.1073/pnas.1201552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausmeyer J.; Actis P.; López Córdoba A.; Korchev Y.; Schuhmann W. Nanosensors for the Detection of Hydrogen Peroxide. Electrochem. Commun. 2014, 40, 28–30 10.1016/j.elecom.2013.12.015. [DOI] [Google Scholar]
- Brooksby P. A.; Downard A. J. Electrochemical and Atomic Force Microscopy Study of Carbon Surface Modification via Diazonium Reduction in Aqueous and Acetonitrile Solutions. Langmuir 2004, 20, 5038–5045 10.1021/la049616i. [DOI] [PubMed] [Google Scholar]
- Rawson F. J.; Garrett D. J.; Leech D.; Downard A. J.; Baronian K. H. R. Electron Transfer from Proteus Vulgaris to a Covalently Assembled, Single Walled Carbon Nanotube Electrode Functionalised with Osmium Bipyridine Complex: Application to a Whole Cell Biosensor. Biosens. Bioelectron. 2011, 26, 2383–2389 10.1016/j.bios.2010.10.016. [DOI] [PubMed] [Google Scholar]
- Rawson F. J.; Yeung C. L.; Jackson S. K.; Mendes P. M. Tailoring 3D Single-Walled Carbon Nanotubes Anchored to Indium Tin Oxide for Natural Cellular Uptake and Intracellular Sensing. Nano Lett. 2013, 13, 1–8 10.1021/nl203780d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson F. J.; Gross A. J.; Garrett D. J.; Downard A. J.; Baronian K. H. R. Mediated Electrochemical Detection of Electron Transfer from the Outer Surface of the Cell Wall of Saccharomyces Cerevisiae. Electrochem. Commun. 2012, 15, 85–87 10.1016/j.elecom.2011.11.030. [DOI] [Google Scholar]
- Vostiar I.; Ferapontova E. E.; Gorton L. Electrical “Wiring” of Viable Gluconobacter Oxydans Cells with a Flexible Osmium-Redox Polyelectrolyte. Electrochem. Commun. 2004, 6, 621–626 10.1016/j.elecom.2004.04.017. [DOI] [Google Scholar]
- Hasan K.; Patil S. A.; Leech D.; Hägerhäll C.; Gorton L. Electrochemical Communication Between Microbial Cells and Electrodes via Osmium Redox Systems. Biochem. Soc. Trans. 2012, 40, 1330–1335 10.1042/BST20120120. [DOI] [PubMed] [Google Scholar]
- Shigehara K.; Anson F. C. Electrocatalytic Activity of Three Iron Porphyrins in the Reduction of Dioxygen and Hydrogen Peroxide at Graphite Cathodes. J. Phys. Chem. 1982, 86, 2776–2783 10.1021/j100211a043. [DOI] [Google Scholar]
- Prévoteau A.; Mano N. Oxygen Reduction on Redox Mediators May Affect Glucose Biosensors Based on “Wired” Enzymes. Electrochim. Acta 2012, 68, 128–133 10.1016/j.electacta.2012.02.053. [DOI] [Google Scholar]
- Salimi A.; Kavosi B.; Hallaj R.; Babaei A. Fabrication of a Highly Sensitive Glucose Biosensor Based on Immobilization of Osmium Complex and Glucose Oxidase onto Carbon Nanotubes Modified Electrode. Electroanalysis 2009, 21, 909–917 10.1002/elan.200804486. [DOI] [Google Scholar]
- Park S. Y.; Ji G. E.; Ko Y. T.; Jung H. K.; Ustunol Z.; Pestka J. J. Potentiation of Hydrogen Peroxide, Nitric Oxide, and Cytokine Production in RAW 264.7 Macrophage Cells Exposed to Human and Commercial Isolates of Bifidobacterium. Int. J. Food Microbiol. 1999, 46, 231–241 10.1016/S0168-1605(98)00197-4. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Arbault S.; Chen Y.; Crozatier C.; Tapsoba I. Electrochemical Detection in a Microfluidic Device of Oxidative Stress Generated by Macrophage Cells. Lab Chip 2007, 7, 233–238 10.1039/B611569A. [DOI] [PubMed] [Google Scholar]
- Oja S. M.; Wood M.; Zhang B. Nanoscale Electrochemistry. Anal. Chem. 2013, 85, 473–486 10.1021/ac3031702. [DOI] [PubMed] [Google Scholar]
- Hsu H.-Y.; Wen M.-H. Lipopolysaccharide-Mediated Reactive Oxygen Species and Signal Transduction in the Regulation of Interleukin-1 Gene Expression. J. Biol. Chem. 2002, 277, 22131–22139. [DOI] [PubMed] [Google Scholar]
- Pekarova M.; Kralova J.; Kubala L.; Ciz M.; Lojek A.; Gregor C.; Hrbac J. Continuous Electrochemical Monitoring of Nitric Oxide Production in Murine Macrophage Cell Line RAW 264.7. Anal. Bioanal. Chem. 2009, 394, 1497–1504 10.1007/s00216-009-2813-x. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Arbault S.; Bouret Y.; Cauli B.; Guille M.; Rancillac A.; Rossier J. Nitric Oxide Release During Evoked Neuronal Activity in Cerebellum Slices: Detection with Platinized Carbon-Fiber Microelectrodes. ChemPhysChem 2006, 7, 181–187 10.1002/cphc.200500202. [DOI] [PubMed] [Google Scholar]
- Pahan K.; Sheikh F. G.; Namboodiri A. M. S.; Singh I. N-Acetyl Cysteine Inhibits Induction of No Production By Endotoxin or Cytokine Stimulated Rat Peritoneal Macrophages, C6 Glial Cells and Astrocytes. Free Radic. Free Radical Biol. Med. 1998, 24, 39–48 10.1016/S0891-5849(97)00137-8. [DOI] [PubMed] [Google Scholar]
- Kimura K.; Ito S.; Nagino M.; Isobe K.-i. Inhibition of Reactive Oxygen Species Down-Regulates Protein Synthesis in RAW 264.7. Biochem. Biophys. Res. Commun. 2008, 372, 272–275 10.1016/j.bbrc.2008.05.036. [DOI] [PubMed] [Google Scholar]
- Lambeth J. D. NOX Enzymes and the Biology of Reactive Oxygen. Nat. Rev. Immunol. 2004, 4, 181–189 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Wong S. W.; Kwon M.-J.; Choi A. M. K.; Kim H.-P.; Nakahira K.; Hwang D. H. Fatty Acids Modulate Toll-like Receptor 4 Activation through Regulation of Receptor Dimerization and Recruitment into Lipid Rafts in a Reactive Oxygen Species-dependent Manner. J. Biol. Chem. 2009, 284, 27384–27392 10.1074/jbc.M109.044065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd N. J. F.; Jha A. N. Titanium Dioxide Induced Cell Damage: A Proposed Role of the Carboxyl Radical. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2009, 660, 79–82 10.1016/j.mrfmmm.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Reeves J. F.; Davies S. J.; Dodd N. J. F.; Jha A. N. Hydroxyl Radicals (OH) are Associated with Titanium Dioxide (TiO2) Nanoparticle-Induced Cytotoxicity and Oxidative DNA Damage in Fish Cells. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2008, 640, 113–122 10.1016/j.mrfmmm.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A.; Saegusa K.; Noguchi T.; Sadamitsu C.; Nishitoh H.; Nagai S.; Koyasu S.; Matsumoto K.; Takeda K.; Ichijo H. Ros-Dependent Activation of the Traf6-Ask1-P38 Pathway is Selectively Required for TTL4-Mediated Innate Immunity. Nat. Immunol. 2005, 6, 587–592 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- Park H. S.; Jung H. Y.; Park E. Y.; Kim J.; Lee W. J.; Bae Y. S. Cutting Edge: Direct Interaction of TLR4 with NAD(P)H Oxidase 4 Isozyme Is Essential for Lipopolysaccharide-Induced Production of Reactive Oxygen Species and Activation of NF-κB. J. Immunol. 2004, 173, 3589–3593 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- Bedard K.; Krause K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Fejer G.; Wegner M. D.; Györy I.; Cohen I.; Engelhard P.; Voronov E.; Manke T.; Ruzsics Z.; Dölken L.; Prazeres da Costa O.; Branzk N.; Huber M.; Prasse A.; Schneider R.; Apte R. N.; Galanos C.; Freudenberg M. A. Nontransformed, GM-CSF–Dependent Macrophage Lines Are a Unique Model to Study Tissue Macrophage Functions. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E2191–E2198 10.1073/pnas.1302877110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan E. G.; Savage J. C.; Hise A. G.; Landreth G. E. CD14 and Toll-Like Receptors 2 and 4 Are Required for Fibrillar Aβ-Stimulated Microglial Activation. J. Neurosci. 2009, 29, 11982–11992 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrichon L.; Picciocchi A.; Debeurme F.; Defendi F.; Beaumel S.; Jesaitis S.; Dagher M. C.; Stasia M. J. Characterization of Superoxide Overproduction by the D-LoopNox4-Nox2 Cytochrome b558 in Phagocytes—Differential Sensitivity to Calcium and Phosphorylation Events. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 78–90 10.1016/j.bbamem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bekay R.; Alvarez M.; Carballo M.; Martın-Nieto J.; Monteseirı J.; Pintado E.; Bedoya F.; Sobrino F. Activation of Phagocytic Cell NADPH Oxidase by Norfloxacin: a Potential Mechanism to Explain its Bactericidal Action. J. Leukocyte Biol. 2002, 71, 255–261. [PubMed] [Google Scholar]
- Galanos C.; LÜDeritz O. Electrodialysis of Lipopolysaccharides and Their Conversion to Uniform Salt Forms. Eur. J. Biochem. 1975, 54, 603–610 10.1111/j.1432-1033.1975.tb04172.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.