

Abstract
Background and Aims
Although patients infected by genotype-1b hepatitis C virus (HCV) with Q70 and/or M91 core gene mutations have an almost five-fold increased risk of developing hepatocellular carcinoma (HCC) and increased insulin resistance, the absence of a suitable experimental system has precluded direct experimentation on the effects of these mutations on cellular gene expression.
Methods
Huh-7 cells were treated long-term with human serum to induce differentiation and to produce a model system for testing high-risk and control HCV. For clinical validation, profiles of infected cells were compared to each other and to those of liver biopsies of patients with early-stage HCV-related cirrhosis followed prospectively for up to 23 years (n=216).
Results
Long-term culture in human serum produced growth-arrested, hepatocyte-like cells whose gene profile overlapped significantly with that of primary human hepatocytes. High-risk (Q70/M91) and control (R70/L91) viruses had dramatically different effects on gene expression of these cells. The high-risk virus enhanced expression of pathways associated with cancer and type II diabetes, while the control virus enhanced pathways associated with oxidative phosphorylation. Of special clinical relevance, the transcriptome of cells replicating the high-risk virus correlated significantly with an HCC high-risk profile in patients (Bonferroni-corrected P=0.03), whereas no such association was observed for non-HCC-related clinical outcomes.
Conclusions
The cell-based system allowed direct head-to-head comparison of HCV variants and provided experimental support for previous clinical data indicating an oncogenic effect of core gene mutations. This simple experimental system distinguished HCV variants and will enable future mechanistic analysis and exploration of interventional approaches.
Keywords: HCC, HCV, core-mutations, human serum
Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of site-specific cancer-related death worldwide [1] and the most rapidly increasing cause of cancer-related death in the United States (US) [2]. Chronic hepatitis C virus (HCV) infection is a leading risk factor for HCC in the US, Japan, and other parts of the world [3]. HCV will remain a major cause of HCC for the foreseeable future despite the introduction of highly effective direct-acting antiviral drugs that increase the percentage of patients who achieve a sustained virological response (SVR) [4]. Many barriers to HCV treatment exist, including the high percentage of patients who are unaware of their infection and the high cost of treatment. Moreover, for poorly understood reasons, successful HCV treatment does not eliminate the HCC risk [5]. Patients with advanced fibrosis/cirrhosis who achieve an SVR are advised to continue life-long screening for HCC [6]. Currently, many patients with HCV-related HCC are diagnosed at a late stage. Only 12% of patients with HCV-related HCC are diagnosed through surveillance in the US [7]. Methods for stratifying HCC risk would allow HCV treatment and HCC surveillance to be focused on the individuals most likely to benefit.
Data about factors modulating the risk of other types of cancer might provide clues about risk stratification for HCC. Approximately 12% of human cancers are associated with oncogenic viruses [8]. Experimental studies suggest that the HCV core gene has oncogenic potential. Expression of the core gene in transgenic mice leads to insulin resistance, hepatic steatosis, and liver cancer [9, 10]. In cell culture systems, the core gene causes cellular transformation and disruption of several growth control pathways [11].
In the case of human papillomavirus, the cancer risk varies by viral strain [12], demonstrating the importance of sequence- and strain-specific differences in viral oncogenesis. In the case of HCV, genotype-1b is reported to increase the HCC risk [13]. Among patients infected by genotype-1b HCV, those with virus harboring mutations in codons 70 and/or 91 in the core gene have the highest risk [14]. Patients infected with viruses carrying a mutation in codon 70 (Q70) and/or codon 91 (M91) had a 4.65-fold increased occurrence of HCC (P=0.017) [14]. The close association between codon 70 mutations and HCC risk has been observed in several independent patient cohorts [15-22], primarily in Asian (Japanese) populations. The impact in non-Asian population is unclear. Significantly, a recent study showed that the elevated HCC risk among patients with Q70 persisted even after they achieved an SVR [25]. The Q70/M91 mutations are also associated with insulin resistance [23] and failure of interferon-based therapies [24].
Despite clinical data showing an association between Q70/M91 mutations and HCC, the absence of supporting laboratory data has raised concern that it is an artifact of uncontrolled confounding in the clinical data. To address this concern, we developed a cell culture system for testing the impact of high-risk HCV on the expression of cancer-related molecular pathways. The model utilizes Huh-7 cells cultured long-term in media containing human serum (HS), as was done previously for Huh-7.5 cells [26]. Under these conditions, Huh-7 cells acquire the capacity for contact inhibition and they express many gene pathways that are typical of primary human hepatocytes (PHH). These growth-arrested, hepatocyte-like Huh-7 cells support replication of full-length HCV. Matched viruses with and without the high-risk mutations were indistinguishable in their virological properties; however, cells replicating the high-risk variant had enhancement of gene pathways associated with oncogenesis and insulin resistance compared to cells replicating the control HCV, which had enhancement of pathways associated with normal metabolism, such as oxidative phosphorylation. In addition, cells replicating the high-risk virus had a global transcriptome that correlated significantly with a high-HCC risk profile among 216 HCV-infected patients with early-stage cirrhosis who were followed prospectively for up to 23 years to determine clinical outcome. The transcriptome of cells replicating the control virus did not correlate with HCC or any other clinical outcome. Thus, we introduce a simple experimental system that distinguished high-risk and control HCV. When combined with each other, the clinical and experimental data emphasize the importance of HCV mutations as prognostic indicators of liver cancer risk. The simple experimental system will enable detailed biochemical investigation of cellular pathways disrupted by high-risk HCV strains and provide a platform for the discovery of drugs that reverse these disruptions.
Materials and Methods
Huh-7 cultures
Huh-7 cells cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (FBS) are referred to as FBS/Huh-7 cells. To induce differentiation, Huh-7 cells were maintained in media containing 2% AB human serum (HS) (Corning-Mediatech), without sub-culturing and with regular media changes as previously described [26] and are referred to as HS/Huh-7 cells. Additionally, a range of dimethyl sulfoxide (DMSO) concentrations (1% - 2.5%) (Sigma-Aldrich) was also tested [27]. Primary human hepatocytes (PHH) were cultured as described in Supplementary methods.
Quantification of hepatocyte differentiation markers and innate antiviral gene expression
RNeasy Plus Mini kits (Qiagen) were used to purify RNA. Reverse transcription (RT) was performed using SuperScript III First-Strand Synthesis (Invitrogen) and random hexamers. RT products were then used for quantitative-PCR (q-PCR) using the LightCycler 480 SYBR Green I Master kit. Expression of hepatocyte-specific genes, albumin and A1AT, and innate immune antiviral genes was determined and calculated using the ΔΔCt method. RPS11 was used for normalization. Primer sets were previously reported [27, 28]. Statistical analysis of RNA quantitation was performed using Student's t-Test. A two-tailed P value < 0.05 was regarded as statistically significant.
Viral constructs
APP144, a Con1/JFH-1 chimeric construct (ApathLLC), has five enhancing mutations (L831F, I857T, I1312V, K2073R and I2266L) and core, E1, E2, P7 and part of NS2 from Con1 (genotype 1b; accession number: AJ238799) and other regions from JFH-1 (genotype 2a; accession number: AB047639). APP144 has Q70 (glutamine) and L91 (leucine). Mutagenesis was performed as previously described [29], creating constructs with R70/L91 and Q70/M91, which were verified by sequencing.
Infectivity assays
Infectious titers were determined by a tissue culture infectious dose 50 assay (TCID50) previously described [30]. Standard methods were used for preparation of viral stocks, intracellular HCV RNA quantitation, Western blotting and flow cytometry to assess percentage of infected cells; detailed in Supplementary methods.
Gene-expression profiling and bioinformatics analysis
Genome-wide gene expression profiling was performed using the Human-HT-12 Expression Beadchip (Illumina); 200 ng/sample. Hybridized chips were scanned using HiScan Array Scanner, raw data were extracted using Genome Studio software ver.3 (Illumina), and normalized by cubic spline algorithm implemented in Illumina Normalizer module of GenePattern genomic analysis toolkit (www.broadinstitute.org/genepattern). The dataset is available at NCBI Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo, accession number GSE64605). The transcriptome profile of PHH was previously described [31] (www.ncbi.nlm.nih.gov/geo, accession number GSE29907, samples GSM740485 and GSM740486).
The global transcriptome profile of HCV-infected cells was compared to that of liver biopsy specimens from 216 patients with early stage HCV-associated cirrhosis and no prior history of HCC or complications of cirrhosis. These patients were followed longitudinally for a median of 10 years to identify patients who developed HCC, died from any cause, progressed in Child-Pugh class, and/or experienced hepatic decompensation. The cohort and the transcriptome profile of the liver specimens have been described previously (NCBI Gene Expression Omnibus accession number: GSE15654) [32]. For each gene in the dataset, the association with each clinical outcome was calculated using the Cox score, as previously described [33]. Vectors of Cox scores were merged to create a matrix, and the association between HCV core gene variants and clinical outcomes were assessed by using a Subclass Mapping algorithm, a bi-directional gene signature enrichment-based subclass association determination method, as previously described [34]. A mutation-outcome association with a Bonferroni-corrected p-value <0.05 was regarded as statistically significant.
Differential modulation of molecular pathways in the Kyoto Encyclopedia of Genes and Genomes database (KEGG) (http://www.genome.jp/kegg/) was determined by using Gene Set Enrichment analysis (GSEA) [35]. MicroRNA and reactome databases were also investigated (http://www.broadinstitute.org/gsea/msigdb/collections.jsp). Gene sets with a false discovery rate (FDR) <0.05 were regarded as statistically significant.
Results
Differentiation of Huh-7 cells by long-term culture in human serum
We used human serum (HS) to induce differentiation of Huh-7 cells, as described previously for Huh-7.5 cells [26]. Cells were plated in media containing 10% FBS and cultured until they reached 60% to 70% confluence. Cells were then cultured in media containing 2% HS for three weeks or more to produce monolayers of growth-arrested HS/Huh-7 cells (Fig. 1A). The HS/Huh-7 cells exhibited contact inhibition and grew in a cobble-stone pattern, unlike FBS/Huh-7 cells (Fig. 1B).
Fig. 1. Differentiation of Huh-7 cells by long-term culture in human serum.
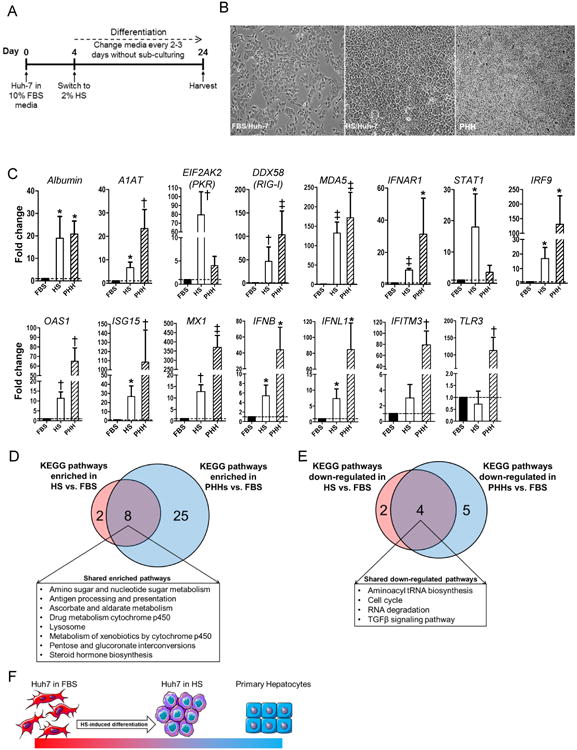
(A) Experimental conditions for Huh-7 cells cultured in media containing 2% HS (HS/Huh-7). (B) Phase contrast microscopy of FBS/Huh-7 (left), HS/Huh-7 (middle) and primary human hepatocytes (PHHs) (right). (C) Expression of hepatocyte-specific genes and antiviral genes in HS/Huh-7 cells and PHHs relative to that in FBS/Huh-7 cells. Shared metabolic pathways significantly (D) enhanced or (E) down-regulated in HS/Huh-7 cells and PHHs compared to FBS/Huh-7 cells. (F) Model of the impact of long-term culture of Huh-7 cells in HS. * P<0.05, † P< 0.01 and ‡ P< 0.001.
The HS/Huh-7 cells had increased levels of hepatocyte-specific mRNAs compared to FBS/Huh-7 cells, including an approximately 20-fold increase in albumin mRNA (P = 0.03) and an approximately 6-fold increase in alpha-1 antitrypsin (A1AT) mRNA (P = 0.01) (Fig. 1C). The level of albumin mRNA in HS/Huh-7 cells was comparable to that of cultured PHH (shown in Fig. 1C); the levels were 19- and 20.8-fold higher than in FBS/Huh-7 cells, respectively. Compared to FBS/Huh-7 cells, HS/Huh-7 cells expressed up to 120-fold higher levels of antiviral genes, such as DDX58 (encoding RIG-I) (P = 0.001), MDA5 (P = 0.001), IFNAR1 (P = 0.0001), STAT1 (P = 0.04), IRF9 (P = 0.02), EIF2AK2 (encoding PKR) (P = 0.006), OAS1 (P = 0.005), ISG15 (P = 0.02), MX1 mRNA (P = 0.002), IFNB (P = 0.02) and IFNL1 (P = 0.01) (Fig. 1C). FBS and HS cells had similar levels of TLR3 mRNA, as expected due to a known lesion in this gene [36]. They also had similar levels of IFITM3 mRNA. Cultured PHH expressed higher levels of several antiviral genes, but lower levels of PKR and STAT1. The robust antiviral response accords with data implicating IFN signaling in HCV control [37].
To investigate global similarities between HS/Huh-7 cells and primary human hepatocytes (PHH), we analyzed the KEGG pathways differentially regulated in both HS/Huh-7 cells and PHH compared to FBS/Huh-7 cells. Eight of the ten pathways enhanced in HS/Huh-7 cells versus FBS/Huh-7 cells were also enhanced in PHH versus FBS/Huh-7 cells (Fig. 1D and table 1). Many of the shared pathways carry out hepatocyte functions, such as steroid biosynthesis and drug metabolism by cytochrome P450. Conversely, four of the six pathways down-regulated in HS/Huh-7 cells versus FBS/Huh-7 cells were also down-regulated in PHH versus FBS/Huh-7 cells (Fig. 1E and table 2). These down-modulated pathways included cell cycling, aminoacyl tRNA biosynthesis, TGF-β signaling, and RNA degradation. The cobble-stone morphology of the HS/Huh-7 cells combined with their capacity for contact inhibition and their gene expression profile indicate that long-term culture in HS produced cells with many features typical of PHH (Fig. 1F). Two different lots of HS induced similar cell growth properties and similar enhancement of the expression of hepatocyte-specific genes in HS/Huh-7 cells, establishing the consistency of the approach (Supplementary Fig. 1).
Table 1.
Gene Set Enrichment Analysis (GSEA) of significantly enriched gene sets of the Kyoto Encyclopedia of Genes and Genomes database (KEGG) in PHHs and HS/Huh-7 cells when compared to FBS/Huh-7 cells.
| Enriched in PHH compared to FBS/Huh-7 | Enriched in HS/Huh-7 compared to FBS/Huh-7 | ||
|---|---|---|---|
|
|
|
||
| Gene set | FDR* | Gene set | FDR* |
| AMINO SUGAR AND NUCLEOTIDE SUGAR METABOLISM | 0.019 | AMINO SUGAR AND NUCLEOTIDE SUGAR METABOLISM | 0.01 |
| ANTIGEN PROCESSING AND PRESENTATION | 0.012 | ANTIGEN PROCESSING AND PRESENTATION | 0.003 |
| ASCORBATE AND ALDARATE METABOLISM | <0.001 | ASCORBATE AND ALDARATE METABOLISM | 0.03 |
| DRUG METABOLISM CYTOCHROME P450 | <0.001 | DRUG METABOLISM CYTOCHROME P450 | 0.002 |
| LYSOSOME | 0.017 | LYSOSOME | 0.003 |
| METABOLISM OF XENOBIOTICS BY CYTOCHROME P450 | <0.001 | METABOLISM OF XENOBIOTICS BY CYTOCHROME P450 | 0.006 |
| PENTOSE AND GLUCURONATE INTERCONVERSIONS | <0.001 | PENTOSE AND GLUCURONATE INTERCONVERSIONS | 0.008 |
| STEROID HORMONE BIOSYNTHESIS | <0.001 | STEROID HORMONE BIOSYNTHESIS | 0.01 |
|
| |||
| ALLOGRAFT REJECTION | 0.034 | STEROID BIOSYNTHESIS | 0.002 |
| APOPTOSIS | 0.019 | TERPENOID BACKBONE BIOSYNTHESIS | 0.007 |
| ARACHIDONIC ACID METABOLISM | <0.001 | ||
| AUTOIMMUNE THYROID DISEASE | 0.02 | ||
| BETA ALANINE METABOLISM | 0.042 | ||
| BUTANOATE METABOLISM | 0.047 | ||
| COMPLEMENT AND COAGULATION CASCADES | <0.001 | ||
| CYTOSOLIC DNA SENSING PATHWAY | 0.018 | ||
| DRUG METABOLISM OTHER ENZYMES | <0.001 | ||
| FRUCTOSE AND MANNOSE METABOLISM | 0.029 | ||
| GLYCEROLIPID METABOLISM | 0.018 | ||
| GLYCOLYSIS GLUCONEOGENESIS | 0.016 | ||
| GLYCOSAMINOGLYCAN DEGRADATION | 0.008 | ||
| GRAFT VERSUS HOST DISEASE | 0.013 | ||
| INTESTINAL IMMUNE NETWORK FOR IGA PRODUCTION | 0.018 | ||
| LINOLEIC ACID METABOLISM | <0.001 | ||
| PENTOSE PHOSPHATE PATHWAY | 0.019 | ||
| PEROXISOME | 0.017 | ||
| PORPHYRIN AND CHLOROPHYLL METABOLISM | 0.006 | ||
| PPAR SIGNALING PATHWAY | 0.034 | ||
| PRIMARY BILE ACID BIOSYNTHESIS | 0.018 | ||
| PRION DISEASES | 0.012 | ||
| RETINOL METABOLISM | <0.001 | ||
| TOLL LIKE RECEPTOR SIGNALING PATHWAY | 0.007 | ||
| TRYPTOPHAN METABOLISM | 0.017 | ||
False discovery rate (FDR) <0.05 were regarded as statistically significant. Shared gene sets are in boldface type. Click gene set for details.
Table 2.
GSEA of significantly down-regulated gene sets of the KEGG in PHHs and HS/Huh-7 cells when compared to FBS/Huh-7 cells.
| Enriched in FBS/Huh-7 compared to PHH | Enriched in FBS/Huh-7 compared to HS/Huh-7 | ||
|---|---|---|---|
|
|
|
||
| Gene set | FDR* | Gene set | FDR* |
| AMINOACYL TRNA BIOSYNTHESIS | 0.026 | AMINOACYL TRNA BIOSYNTHESIS | <0.001 |
| CELL CYCLE | 0.001 | CELL CYCLE | 0.01 |
| RNA DEGRADATION | 0.028 | RNA DEGRADATION | 0.013 |
| TGF BETA SIGNALING PATHWAY | 0.005 | TGF BETA SIGNALING PATHWAY | <0.001 |
|
| |||
| OOCYTE MEIOSIS | 0.017 | NOD LIKE RECEPTOR SIGNALING PATHWAY | 0.043 |
| DNA REPLICATION | 0.006 | WNT SIGNALING PATHWAY | 0.029 |
| RIBOSOME | 0.005 | ||
| SPLICEOSOME | 0.032 | ||
| MISMATCH REPAIR | 0.034 | ||
False discovery rate (FDR) <0.05 were regarded as statistically significant. Common gene sets are in boldface type. Click gene set for details.
DMSO was tested as an alternative method for inducing differentiation of Huh-7 cells [27]. Similar to the process used to create HS/Huh-7 cells, cultures were initiated in media containing 10% FBS. When the cells reached 60% -70% confluence, they were switched to media containing 1% - 2.5% DMSO (Fig. 2A). The only concentration that yielded growth-arrested cells with hepatocyte-like morphology was 2% (Fig. 2B). Similar to HS/Huh-7 cells, the 2% DMSO/Huh-7 cells had significantly increased mRNA levels of hepatocyte-specific markers and antiviral genes (Fig. 2C) compared to FBS/Huh-7 cells. The induction of albumin was only 3.2-fold, which was significantly lower than for HS/Huh-7 cells (P=0.048) (Figs. 2C, 1C). The global transcriptome of the DMSO/Huh-7 cells was not investigated.
Fig. 2. Differentiation of Huh-7 cells by long-term culture in DMSO.
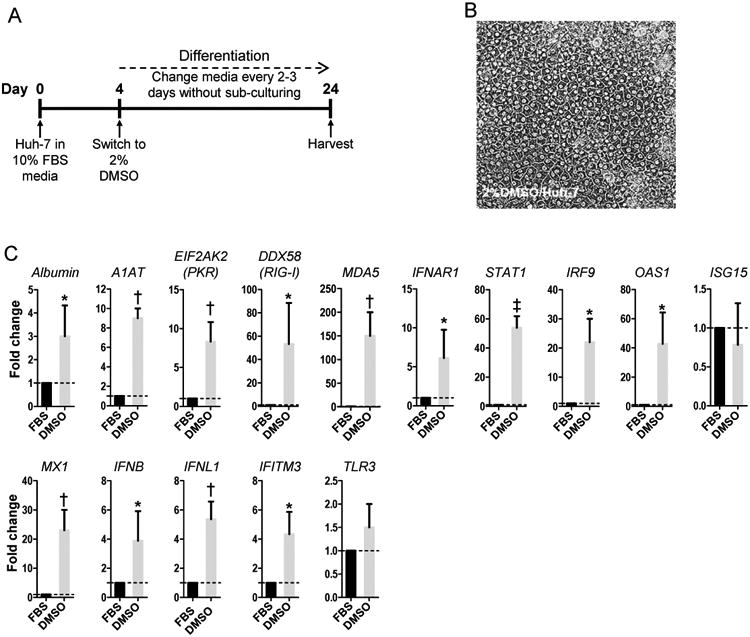
(A) Experimental conditions for Huh-7 cells cultured in media containing 2% DMSO (DMSO/Huh-7). (B) Phase contrast microscopy of differentiated DMSO/Huh-7 cells. (C) Expression of hepatocyte-specific genes and antiviral genes in DMSO/Huh-7 cells compared to that in FBS/Huh-7 cells. * P<0.05, † P< 0.01 and ‡ P< 0.001.
HCV replication and infectious particle production
Huh-7 cells grown under a variety of conditions were tested for their susceptibility to APP144, a chimeric virus with a genotype 1b core gene (Fig. 3A-F). Cells were either infected after they had been cultured in HS or DMSO (Fig. 3C) or they were infected prior to culture in HS or DMSO, with subsequent differentiation (Fig. 3D). The percentage of infected cells was determined by flow cytometry using an antibody (9E10) directed against the HCV NS5A protein. FBS/Huh-7 cells served as controls (Fig. 3B). The percentage of positive cells ranged from 93.6% to 1.6% (Fig. 3E). Only about 7% of the cells differentiated in DMSO expressed NS5A, versus 45% of the cells differentiated in HS (Fig. 3E). Although the HS/Huh-7 cells contained less HCV RNA and produced fewer infectious particles than FBS/Huh-7 cells (Fig. 3F), their level of production was high enough to support investigation of high-risk and control HCV variants.
Fig. 3. Susceptibility of differentiated Huh-7 cells to HCV APP144 infection.
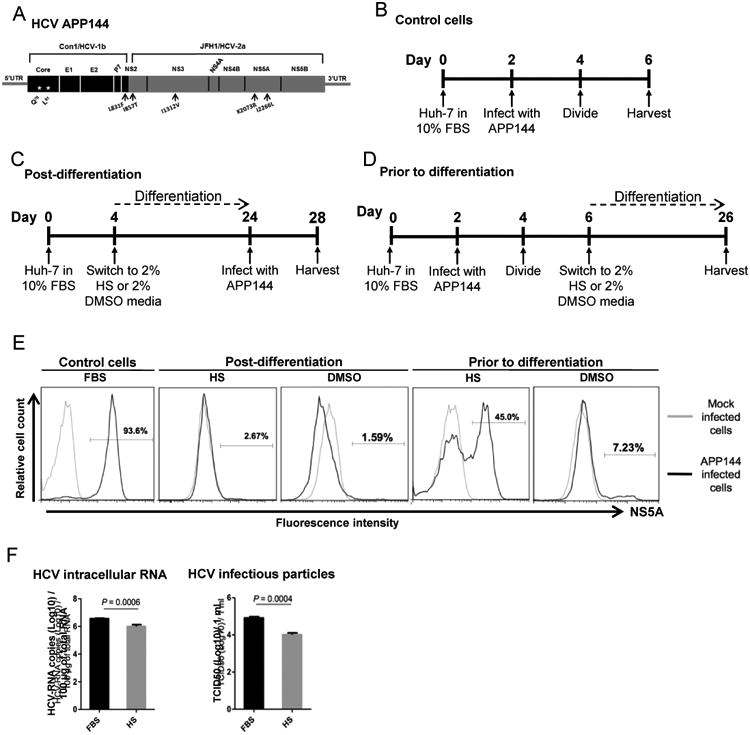
(A) Diagram of the APP144 virus. Black arrows indicate adaptive mutations. (B) Infection of undifferentiated control FBS/Huh-7 cells. (C) Infection of Huh-7 cells cultured in media containing either 2% human serum (HS/Huh-7) or 2% DMSO (DMSO/Huh-7) post-differentiation. (D) Infection of HS/Huh-7 or DMSO/Huh-7 cells prior to differentiation with subsequent exposure to differentiating conditions. (E) Flow cytometry analysis of the proportion of NS5A positive cells among control FBS/Huh-7 cells, HS/Huh-7 or DMSO/Huh-7 cells that were infected either post-differentiation or prior to differentiation with subsequent exposure to differentiating conditions. (F) Levels of intracellular HCV RNA replication and the amount of infectious particles released into the media within a 24 hour window were determined 4 days post infection in FBS/Huh-7 cells and 24 days post infection in HS/Huh-7 cells.
High-risk and control viruses have similar fitness in HS/Huh-7 cells
A pair of matched viruses was derived from APP144. One harbored high-risk core gene mutations, Q70/M91; the other had R70/L91 (Fig. 4A). FBS/Huh-7 cells were infected, cultured for four days to allow the infection to spread, and then they were switched to HS-containing media and cultured for an additional three weeks to produce differentiated HCV-infected cells (Fig. 4B). In assays of virological fitness, the Q70/M91 and R70/L91 viruses were indistinguishable. They persisted in a similar percentage of cells (44.7% versus 44.2%) (Fig. 4C), produced similar levels of intracellular HCV RNA (6.1 log10 versus 6.0 log10 copies/100 ng) (Fig. 4D), released similar quantities of infectious particles (4.0 log10 versus 4.1 log10 TCID50/ml) (Fig. 4E), expressed similar levels of intracellular HCV p21 core protein (Fig. 4F) and, as shown by immunofluorescence/confocal microscopy, had similar subcellular localization of the core protein (data not shown).
Fig. 4. Viral fitness of the high-risk and control viruses in HS/Huh-7 cells.
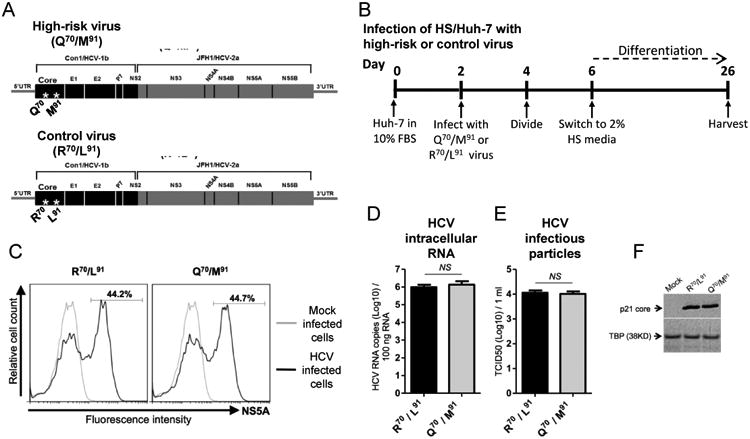
(A) Diagrams of the high-risk (Q70/M91) and the control (R70/L91) viruses. (B) Infection of HS/Huh-7 cells. (C) Flow cytometry analysis of the proportion of NS5A positive cells among HS/Huh-7 cells infected either with R70/L91 or Q70/M91 viruses. (D) Levels of intracellular HCV RNA replication and (E) amount of released infectious particles of R70/L91 and Q70/M91 viruses were determined in HS/Huh-7 cells 24 days after infection. (F) Expression levels of p21 core protein of R70/L91 and Q70/M91 viruses were determined in HS/Huh-7 cells 24 days after infection. NS; not significant.
Differential impact of core gene mutations on the global gene profile of HS/Huh-7 cells
The two viruses had very different effects on cellular gene expression, despite their similar virological fitness. Of great interest, the global transcriptomic profiles of cultures replicating the high-risk virus (Q70/M91) were significantly correlated with those of liver biopsies of patients with early-stage HCV-related cirrhosis (n=216) who had an increased incidence of HCC when followed prospectively for up to 23 years [32] (Bonferroni-corrected P=0.03) (Fig. 5A). The transcriptome of the cells replicating the control virus (R70/L91) did not correlate with increased HCC incidence (Bonferroni-corrected P= 0.60) or any of the other clinical outcomes that were monitored (overall death, progression of Child-Pugh class from A to B or C, and development of symptoms of hepatic decompensation).
Fig. 5. Differential gene signature in HS/Huh-7 cells replicating either the high-risk virus or the control virus.
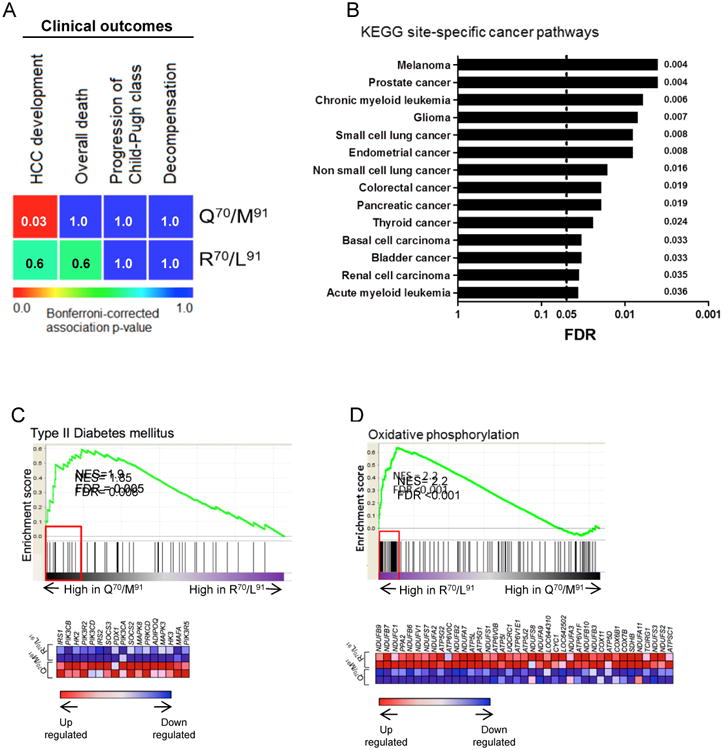
(A) Heatmap of association between global transcriptome of HS/Huh-7 cells that were infected either with the high-risk (Q70/M91) or the control (R70/L91) viruses with long-term clinical outcomes of HCV-related early-stage cirrhosis patients. Bonferroni-corrected p-values are indicated inside each box. (B) GSEA of site-specific cancer pathways in the KEGG set in HS/Huh-7 cells infected with the high-risk virus (Q70/M91) compared to cells infected with the control virus (R70/L91). All 14 gene sets were significantly enhanced in HS/Huh-7 cells replicating high-risk virus while none in cells replicating control virus. GSEA for differential expression of gene sets related to (C) type II diabetes mellitus and (D) Oxidative phosphorylation between HS/Huh-7 cells infected either with the high-risk or control viruses. Genes contributed to pathway enrichment (leading edge genes) are enclosed in red rectangle with the heat map of these genes displayed beneath the enrichment plot. NES; normalized enrichment score. False discovery rate (FDR) was regarded as statistically significant. For detailed description of enrichment blot, see Supplementary information.
To obtain additional information about the impact of the high-risk virus on cellular gene expression, the global transcriptome of HS/Huh-7 cells replicating this variant (Q70/M91) was compared to that of HS/Huh-7 cells replicating the control virus (R70/L91). In GSEA, all 14 site-specific cancer-related KEGG pathways were enhanced in the cells replicating the high-risk virus, while none were enhanced in cells replicating the control virus (Fig. 5B and Supplementary table 1).
Additionally, GSEA revealed that HS/Huh-7 cells replicating the high-risk virus (Q70/M91) had greater induction of pathways related to altered metabolism, such as type II diabetes and insulin signaling, than cultures replicating the control virus (R70/L91) (Fig. 5C and Supplementary table 1). Cells replicating the high-risk virus had altered expression of pathways regulated by miR26, miR181, miR202, and miR519 (FDR <0.001; data not shown). By contrast, cells infected by the control virus showed greater expression of the oxidative phosphorylation pathway, which is a feature of normal aerobic metabolism (Fig. 5D and Supplementary table 1). These cells also tended to have greater expression of pathways associated with physiological functions of primary hepatocytes, such as metabolism of xenobiotics by cytochrome P450 (FDR = 0.06), DNA repair (KEGG homologous recombination) (FDR=0.08) and reactome-P53 independent G1-S DNA damage checkpoint (FDR<0.001); data not shown.
Differential impact of core gene mutations on activation of ISGs
Mutations in codons 70 and 91 (Q70/M91) of the genotype 1b core gene predict poor response to IFN-based therapy [24]. To seek evidence of a differential effect of high-risk and control viruses on IFN responses in HS/Huh-7 cells, mRNA levels of five ISGs (MX1, OAS1, IFITM3, STAT1 and ISG15) were determined in the presence or absence of exogenous IFN treatment. In the absence of IFN treatment, HS/Huh-7 cells infected with either variant had higher ISGs expression than uninfected cells (Fig. 6), demonstrating the capacity of the differentiated cells to respond to viral infection by increasing IFN signaling. Cells infected with the high-risk virus had significantly lower expression of MX1 and OAS1 than cells infected with the control virus (Fig. 6A and B). IFN treatment increased ISG expression in cells infected with either variant compared to infected cells without IFN treatment. The high-risk virus blunted the IFN-mediated ISG induction, with the exception of ISG15 (Fig. 6E), consistent with the association between higher ISG15 expression in liver tissue of patients who do not respond to IFN-based treatments [38].
Fig. 6. Differential activation of ISGs in HS/Huh-7 cells infected with either the high-risk or the control virus.

Expression of (A) MX1, (B) OAS1, (C) IFITM3, (D) STAT1 and (E) ISG15 in HS/Huh-7 cells infected with either the high-risk virus (Q70/M91) or the control virus (R70/L91) in the absence or presence of IFN treatment relative to that in uninfected HS/Huh-7 cells. For IFN treatment, infected-differentiated HS/Huh-7 cells were treated with 100 IU/ml of IFN-α for 24 hours before extracting total RNA. The P values of different comparisons are displayed beneath each graph.
Discussion
Our results shed new light on the clinical data that first established an association between point mutations in the HCV core gene and adverse outcomes, such as failure of IFN-based therapy [24], insulin resistance and type II diabetes [23], and HCC [14]. Our study found a remarkable difference between the impact of the high-risk (Q70/M91) and control (R70/L91) viruses on cellular gene expression, although the two viruses are identical in all but two amino acids out of 3,200. These two viruses and the novel cell culture system we developed to study them provide unique tools for dissecting the mechanistic links between altered metabolism and abnormal cellular growth.
Of particular clinical relevance, we demonstrated that the global transcriptome profile of cells replicating the high-risk virus correlated significantly with the profile of liver biopsy specimens from patients with early-stage HCV-related cirrhosis who had an elevated incidence of HCC during longitudinal follow up. The HS/Huh-7 cells infected with the high-risk virus had an induction of many KEGG pathways associated with cancer and altered metabolism. In particular, cultures infected by the high-risk virus had induction of the type II diabetes gene set; whereas cultures infected by the control virus had greater expression of the oxidative phosphorylation pathway. These results might indicate that the high-risk virus promotes the use of glycolytic pathways, recapitulating an emerging hallmark of cancer [39]. Notably, insulin resistance is one of the major risk factors associated with HCC development in HCV patients [40].
The HS/Huh-7 cell system provides a novel and advantageous model for scoring the pathological potential of HCV mutations present in clinical isolates and for investigating the cellular targets of oncogenic HCV strains. In non-1b subtypes, codons 70 and 91 are generally conserved and thus do not vary according to clinical outcomes [41]; however, it is possible that other codons modulate the oncogenic potential of non-1b subtypes. Well-designed clinical and molecular virology studies are needed to explore this possibility. Genotype 2 HCV is much more sensitive to interferon-based therapy than genotype 1 [42] and genotype 3 HCV is associated with steatosis [43]. The HS/Huh-7 cells provide a new experimental system for investigating the molecular basis of these genotypic differences. Simplicity is an important feature of the experimental system.
Increasing evidence indicates that many cancer cells have the plasticity required for reprogramming [44], focusing attention on the tumor microenvironment as a source of regulatory factors. We aimed to attenuate the neoplastic phenotype of Huh-7 cells by culturing them in adult human serum (HS) rather than fetal bovine serum (FBS). Using this approach, the phenotype of Huh-7 cells changed, as demonstrated by an alteration in morphology from spindle-shaped to cobble-stone, by a switch from unrestrained growth to contact inhibition, and by the induction of hepatocyte-specific genes (such as albumin and A1AT) and genes mediating antiviral and antiproliferative responses. The HS/Huh-7 cells had a global gene profile including enhanced expression of pathways typical of PHH and greatly attenuated expression of pathways associated with cell cycling. A unique feature of this experimental system is its use of a single cell type. Notably, this mono-cell system, when replicating high-risk HCV, acquired a gene signature that was significantly associated with that of liver biopsies of patients at high risk for developing of HCC, even though the biopsies contain multiple cell types. This suggests that among different types of liver cells, hepatocytes are likely to be a key source of the oncogenic signature.
We selected HS/Huh-7 cells for our studies after considering several alternatives. Humanized mice, primary human hepatocytes and hepatocyte-like cells induced from pluripotent stem cells were potential options [45]; however, donor-to-donor variability, high-cost and technical complexities make these challenging to use. We investigated DMSO as a differentiating agent. Previously, Bruno et al. [27] demonstrated that 1% DMSO produced differentiated Huh-7 cells that were highly permissive to HCV, however, we were unable to reproduce this result, most likely because of differences between sublines of Huh-7 cells [46]. Thus, we investigated adult human serum as a differentiating agent. Steenbergen et al. used human serum, rather than DMSO, to differentiate Huh-7.5 cells [26]. We followed their important lead, but elected to use the parental, Huh-7, cell line. Huh-7.5 cells are highly permissive to HCV, but they have a mutation in the gene encoding RIG-I [47] and we wanted to avoid this lesion.
Since a suitable experimental system is now in hand, future detailed biochemical experiments can be carried out to determine the molecular basis of the differential oncogenic and diabetogenic effects on cellular gene expression of the high-risk virus compared to the control. In the meantime, several features of the core gene merit discussion. The high-risk mutations could act at either the protein- or RNA-level and they might, or might not, require viral replication. At the protein-level, it is noteworthy that codon 70 lies within the RNA binding domain of p21 [48]. The substitution of arginine (R), which has a positive charge, with glutamine (Q), which is neutral, might reduce the affinity for RNA; however, the mutation did not affect production of infectious particles, suggesting that it had minimal effect on interactions with genomic RNA. It is possible that the newly discovered HCV minicores play a role since minicores 70 and 91 are predicted to have N-termini at amino acid 70 and 91, respectively [29]. Recent data indicate that minicores circulate in blood [49], suggesting a possible role in infectivity and/or in modulation of host responses to infection. RNA-level effects warrant investigation, perhaps with a focus on differential binding to cellular microRNAs.
In summary, we present the first direct evidence for the sequence-specific effects of HCV variants that were first reported in clinical and epidemiological studies. Our findings were made possible by the development of a novel experimental system that combined HS-differentiated cells and full-length infectious HCV harboring disease-associated mutations. The major advantages of this system are its simplicity, low cost, and ability to recapitulate important clinical findings. This system will enable further biochemical investigation of the biology underlying the cellular pathways disrupted by high-risk HCV strains. This experimental model would be a useful platform for high throughput screening of novel anti-HCC therapies.
Supplementary Material
Acknowledgments
The authors thank Dr. Charles M. Rice for providing us Huh-7 and Huh-7.5 cells. Also, we thank Dr. Francis V. Chisari for providing us Huh-7.5.1 cells. We are grateful to Abbott Laboratories for providing the anti-core mAb. We thank Dr. Enrico Galmozzi for critical reading of the manuscript.
Financial Support: This work was supported in part by NIH grants R01 DK090317, R01 DA031095, R21 CA152514 (to A.D.B.), R01 DK099558 (to Y.H.) and 1K08DK101754 (to R.E.S.).
List of abbreviations
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- FBS
fetal bovine serum
- HS
human serum
- A1AT
alpha-1 antitrypsin
- RIG-I
retinoic acid-inducible gene 1
- DMSO
dimethyl sulfoxide
- RPS11
ribosomal protein subunit 11
- TLR3
toll-like receptor 3
- MDA5
melanoma differentiation-associated protein 5
- IFNAR1
interferon alpha receptor 1
- STAT1
signal transducer and activator of transcription 1
- IRF9
interferon regulatory factor 9
- PKR
protein kinase RNA-activated
- OAS1
2′,5′-oligoadenylate synthetase
- ISG15
interferon stimulated gene 15
- MX1
myxovirus resistance 1
- IFNB
interferon beta
- IFNL1
interferon lambda 1
- IFITM3
interferon-induced transmembrane protein 3
- TCID50
tissue culture infectious dose 50
- TBP
TATA-binding protein
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- GSEA
Gene Set Enrichment Analysis
- NES
normalized enrichment score
- FDR
false discovery rate
Footnotes
Conflict of interests: The authors declare no conflict of interests.
Authors' contributions: A.E., F.J.E., E.H.D, A.L.K, and A.D.B. designed the research; A.E., F.J.E., E.H.D., performed experiments; A.S., M.I. and M.C. conducted longitudinal studies of patients; R.E.S. prepared human primary hepatocytes; A.E., F.J.E., E.H.D., X.S., Y.H. and A.D.B analyzed the data and A.E., F.J.E., E.H.D., Y.H. and A.D.B. wrote the manuscript.
Author names in bold designate shared co-first authorship
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ahmed El-Shamy, Email: ahmed.elshamy@mssm.deu.
Francis J. Eng, Email: francis.eng@mssm.edu.
Erin H. Doyle, Email: erin.doyle@mssm.edu.
Arielle L. Klepper, Email: arielle.klepper@mssm.edu.
Xiaochen Sun, Email: xiaochen.sun@mssm.edu.
Angelo Sangiovanni, Email: angelo.sangiovanni@policlinico.mi.it.
Massimo Iavarone, Email: massimo.iavarone@gmail.com.
Massimo Colombo, Email: massimo.colombo@unimi.it.
Robert E. Schwartz, Email: res2025@med.cornell.edu.
Yujin Hoshida, Email: yujin.hoshida@mssm.edu.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Hoshida Y, Fuchs BC, Bardeesy N, Baumert TF, Chung RT. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. Journal of hepatology. 2014;61:S79–S90. doi: 10.1016/j.jhep.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serag HB. Hepatitis C infection and the increasing incidence of hepatocellular carcinoma: a population-based study. Gastroenterology. 2004;127:1372–1380. doi: 10.1053/j.gastro.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt WN, Nelson DR, Pawlotsky JM, Sherman KE, Thomas DL, Chung RT. Direct-acting antiviral agents and the path to interferon independence. Clin Gastroenterol Hepatol. 2014;12:728–737. doi: 10.1016/j.cgh.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aleman S, Rahbin N, Weiland O, Davidsdottir L, Hedenstierna M, Rose N, et al. A risk for hepatocellular carcinoma persists long-term after sustained virologic response in patients with hepatitis C-associated liver cirrhosis. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2013;57:230–236. doi: 10.1093/cid/cit234. [DOI] [PubMed] [Google Scholar]
- 6.Davila JA, Morgan RO, Richardson PA, Du XL, McGlynn KA, El-Serag HB. Use of surveillance for hepatocellular carcinoma among patients with cirrhosis in the United States. Hepatology. 2010;52:132–141. doi: 10.1002/hep.23615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davila JA, Henderson L, Kramer JR, Kanwal F, Richardson PA, Duan Z, et al. Utilization of surveillance for hepatocellular carcinoma among hepatitis C virus-infected veterans in the United States. Annals of internal medicine. 2011;154:85–93. doi: 10.7326/0003-4819-154-2-201101180-00006. [DOI] [PubMed] [Google Scholar]
- 8.de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. The Lancet Oncology. 2012;13:607–615. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 9.Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nature medicine. 1998;4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 10.Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, et al. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. The Journal of general virology. 1997;78(Pt 7):1527–1531. doi: 10.1099/0022-1317-78-7-1527. [DOI] [PubMed] [Google Scholar]
- 11.Banerjee A, Ray RB, Ray R. Oncogenic potential of hepatitis C virus proteins. Viruses. 2010;2:2108–2133. doi: 10.3390/v2092108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLaughlin-Drubin ME, Meyers J, Munger K. Cancer associated human papillomaviruses. Current opinion in virology. 2012;2:459–466. doi: 10.1016/j.coviro.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raimondi S, Bruno S, Mondelli MU, Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. Journal of hepatology. 2009;50:1142–1154. doi: 10.1016/j.jhep.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 14.Akuta N, Suzuki F, Kawamura Y, Yatsuji H, Sezaki H, Suzuki Y, et al. Amino acid substitutions in the hepatitis C virus core region are the important predictor of hepatocarcinogenesis. Hepatology. 2007;46:1357–1364. doi: 10.1002/hep.21836. [DOI] [PubMed] [Google Scholar]
- 15.Nakamoto S, Imazeki F, Fukai K, Fujiwara K, Arai M, Kanda T, et al. Association between mutations in the core region of hepatitis C virus genotype 1 and hepatocellular carcinoma development. J Hepatol. 2010;52:72–78. doi: 10.1016/j.jhep.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Seko Y, Akuta N, Suzuki F, Kawamura Y, Sezaki H, Suzuki Y, et al. Amino acid substitutions in the hepatitis C Virus core region and lipid metabolism are associated with hepatocarcinogenesis in nonresponders to interferon plus ribavirin combination therapy. Intervirology. 2012;56:13–21. doi: 10.1159/000339993. [DOI] [PubMed] [Google Scholar]
- 17.El-Shamy A, Shindo M, Shoji I, Deng L, Okuno T, Hotta H. Polymorphisms of the core, NS3, and NS5A proteins of hepatitis C virus genotype 1b associate With development of hepatocellular carcinoma. Hepatology. 2013;58:555–563. doi: 10.1002/hep.26205. [DOI] [PubMed] [Google Scholar]
- 18.Fishman SL, Factor SH, Balestrieri C, Fan X, Dibisceglie AM, Desai SM, et al. Mutations in the hepatitis C virus core gene are associated with advanced liver disease and hepatocellular carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:3205–3213. doi: 10.1158/1078-0432.CCR-08-2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeyasu M, Akuta N, Suzuki F, Seko Y, Kawamura Y, Sezaki H, et al. Long-term interferon monotherapy reduces the risk of HCV-associated hepatocellular carcinoma. J Med Virol. 2012;84:1199–1207. doi: 10.1002/jmv.23288. [DOI] [PubMed] [Google Scholar]
- 20.Toyoda H, Kumada T, Kaneoka Y, Maeda A. Amino acid substitutions in the hepatitis C virus core region are associated with postoperative recurrence and survival of patients with HCV genotype 1b-associated hepatocellular carcinoma. Annals of surgery. 2011;254:326–332. doi: 10.1097/SLA.0b013e3182263b8e. [DOI] [PubMed] [Google Scholar]
- 21.Hu Z, Muroyama R, Kowatari N, Chang J, Omata M, Kato N. Characteristic mutations in hepatitis C virus core gene related to the occurrence of hepatocellular carcinoma. Cancer science. 2009;100:2465–2468. doi: 10.1111/j.1349-7006.2009.01338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miura M, Maekawa S, Takano S, Komatsu N, Tatsumi A, Asakawa Y, et al. Deep-sequencing analysis of the association between the quasispecies nature of the hepatitis C virus core region and disease progression. J Virol. 2013;87:12541–12551. doi: 10.1128/JVI.00826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akuta N, Suzuki F, Hirakawa M, Kawamura Y, Yatsuji H, Sezaki H, et al. Amino acid substitutions in the hepatitis C virus core region of genotype 1b are the important predictor of severe insulin resistance in patients without cirrhosis and diabetes mellitus. J Med Virol. 2009;81:1032–1039. doi: 10.1002/jmv.21473. [DOI] [PubMed] [Google Scholar]
- 24.Akuta N, Suzuki F, Kawamura Y, Yatsuji H, Sezaki H, Suzuki Y, et al. Predictive factors of early and sustained responses to peginterferon plus ribavirin combination therapy in Japanese patients infected with hepatitis C virus genotype 1b: Amino acid substitutions in the core region and low-density lipoprotein cholesterol levels. J Hepatol. 2007;46:403–410. doi: 10.1016/j.jhep.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 25.Akuta N, Suzuki F, Hirakawa M, Kawamura Y, Sezaki H, Suzuki Y, et al. Amino acid substitutions in hepatitis C virus core region predict hepatocarcinogenesis following eradication of HCV RNA by antiviral therapy. J Med Virol. 2011;83:1016–1022. doi: 10.1002/jmv.22094. [DOI] [PubMed] [Google Scholar]
- 26.Steenbergen RH, Joyce MA, Thomas BS, Jones D, Law J, Russell R, et al. Human serum leads to differentiation of human hepatoma cells, restoration of very-low-density lipoprotein secretion, and a 1000-fold increase in HCV Japanese fulminant hepatitis type 1 titers. Hepatology. 2013;58:1907–1917. doi: 10.1002/hep.26566. [DOI] [PubMed] [Google Scholar]
- 27.Sainz B, Jr, Chisari FV. Production of infectious hepatitis C virus by well-differentiated, growth-arrested human hepatoma-derived cells. J Virol. 2006;80:10253–10257. doi: 10.1128/JVI.01059-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, et al. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology. 2011;54:1913–1923. doi: 10.1002/hep.24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eng FJ, Walewski JL, Klepper AL, Fishman SL, Desai SM, McMullan LK, et al. Internal initiation stimulates production of p8 minicore, a member of a newly discovered family of hepatitis C virus core protein isoforms. Journal of virology. 2009;83:3104–3114. doi: 10.1128/JVI.01679-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, et al. Science. Vol. 309. New York, NY: 2005. Complete replication of hepatitis C virus in cell culture; pp. 623–626. [DOI] [PubMed] [Google Scholar]
- 31.Jung CJ, Iyengar S, Blahnik KR, Ajuha TP, Jiang JX, Farnham PJ, et al. Epigenetic modulation of miR-122 facilitates human embryonic stem cell self-renewal and hepatocellular carcinoma proliferation. PloS one. 2011;6:e27740. doi: 10.1371/journal.pone.0027740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoshida Y, Villanueva A, Sangiovanni A, Sole M, Hur C, Andersson KL, et al. Prognostic gene expression signature for patients with hepatitis C-related early-stage cirrhosis. Gastroenterology. 2013;144:1024–1030. doi: 10.1053/j.gastro.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. The New England journal of medicine. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoshida Y, Brunet JP, Tamayo P, Golub TR, Mesirov JP. Subclass mapping: identifying common subtypes in independent disease data sets. PLoS One. 2007;2:e1195. doi: 10.1371/journal.pone.0001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li K, Chen Z, Kato N, Gale M, Jr, Lemon SM. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. The Journal of biological chemistry. 2005;280:16739–16747. doi: 10.1074/jbc.M414139200. [DOI] [PubMed] [Google Scholar]
- 37.Andrus L, Marukian S, Jones CT, Catanese MT, Sheahan TP, Schoggins JW, et al. Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology. 2011;54:1901–1912. doi: 10.1002/hep.24557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, Borozan I, Sun J, Guindi M, Fischer S, Feld J, et al. Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology. 2010;138:1123–1133. e1121–1123. doi: 10.1053/j.gastro.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Veldt BJ, Chen W, Heathcote EJ, Wedemeyer H, Reichen J, Hofmann WP, et al. Increased risk of hepatocellular carcinoma among patients with hepatitis C cirrhosis and diabetes mellitus. Hepatology. 2008;47:1856–1862. doi: 10.1002/hep.22251. [DOI] [PubMed] [Google Scholar]
- 41.El-Shamy A, Hotta H. Impact of hepatitis C virus heterogeneity on interferon sensitivity: an overview. World J Gastroenterol. 2014;20:7555–7569. doi: 10.3748/wjg.v20.i24.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. The New England journal of medicine. 2009;361:580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- 43.Rubbia-Brandt L, Leandro G, Spahr L, Giostra E, Quadri R, Male PJ, et al. Liver steatosis in chronic hepatitis C: a morphological sign suggesting infection with HCV genotype 3. Histopathology. 2001;39:119–124. doi: 10.1046/j.1365-2559.2001.01208.x. [DOI] [PubMed] [Google Scholar]
- 44.Illmensee K, Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proceedings of the National Academy of Sciences of the United States of America. 1976;73:549–553. doi: 10.1073/pnas.73.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu X, Robotham JM, Lee E, Dalton S, Kneteman NM, Gilbert DM, et al. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012;8:e1002617. doi: 10.1371/journal.ppat.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sainz B, Jr, Barretto N, Uprichard SL. Hepatitis C virus infection in phenotypically distinct Huh7 cell lines. PLoS One. 2009;4:e6561. doi: 10.1371/journal.pone.0006561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santolini E, Migliaccio G, La Monica N. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J Virol. 1994;68:3631–3641. doi: 10.1128/jvi.68.6.3631-3641.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El-Shamy A, Eng FJ, Doyle EH, Andreo U, Klepper AL, Muerhoff S, Schiano T, Dieterich D, Harty A, Im G, Perumalswami P, Rice CM, Branch AD. Newly-Discovered HCV Minicores Circulate in Human Blood. Hepatology. 2014;60(Suppl):LB32. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.