

Abstract
The neurodegenerative synucleinopathies, which include Parkinson disease, multiple system atrophy, and Lewy body disease, are characterized by the presence of abundant neuronal inclusions called Lewy bodies and Lewy neurites. These disorders remain incurable and a greater understanding of the pathologic processes is needed for effective treatment strategies to be developed. Recent data suggest that pathogenic misfolding of the presynaptic protein, α-synuclein (α-syn), and subsequent aggregation and accumulation is fundamental to the disease process. It is hypothesized that the misfolded isoform is able to induce misfolding of normal endogenous α-syn, much like what occurs in the prion diseases. Recent work highlighting the seeding effect of pathogenic α-syn has largely focused on the detergent-insoluble species of the protein. In this study we performed intracerebral inoculations of the sarkosyl-insoluble or sarkosyl-soluble fractions of human Lewy body disease brain homogenate and show that both fractions induce CNS pathology in mice at 4 months post-injection. Disease-associated deposits accumulated both near and distal to the site of the injection suggesting a cell-to-cell spread via recruitment of α-syn. These results provide further insight into the prion-like mechanisms of α-syn and suggest that disease-associated α-syn is not homogenous within a single patient but might exist in both soluble and insoluble isoforms.
Keywords: α-Synuclein, Lewy body disease, Parkinson disease, Prion, Prion-like, Soluble prion
INTRODUCTION
Prions are infectious and self-replicating protein entities that are intrinsically involved in the pathogenesis of the transmissible spongiform encephalopathies, or prion diseases. These disorders affect both humans and animals and are considered a unique subset of neurodegenerative disorders because they not only arise via familial or idiopathic routes but may additionally arise via infection through exposure to exogenous prions both experimentally and naturally (1). It is becoming increasingly clear, however, that proteins implicated in other neurodegenerative disorders, including α-synuclein (α-syn) and tau of Parkinson disease (PD) and Alzheimer disease (AD), respectively, may self-propagate in a prion-like manner. As such, the umbrella of prion disorders is expanding and the term ‘prion’ is no longer restricted to describing the unusual behavior of the rogue isoform of cellular prion protein.
The first data to suggest a prion-like mechanism for synucleinopathies emerged simultaneously from 2 of 3 groups studying embryonic nigral tissue that had been grafted into the brains of patients with PD some 15 years earlier (2-4). The presence of tau immunoreactivity as well as the presence of ubiquitin and phosphorylated α-synuclein (p-syn)-positive Lewy bodies in the grafts strongly suggested that pathology in the host had spread to healthy donor tissue. In vitro studies have since implicated a cell-to-cell mode of transmission; α-syn can be transmitted from affected neurons to neighboring healthy neurons via endocytosis and exosomal uptake and release, and it is implicated in the accumulation process through a failure of protein quality control systems (5-9). Furthermore, in vivo studies have demonstrated the ability of α-syn pathology to be experimentally transmissible. Intracerebral inoculation of synthetic preformed α-syn fibrils was shown to initiate a progressive neurodegenerative α-synucleinopathy in both transgenic and non-transgenic mice (10, 11). In addition, multiple system atrophy “prions” were shown to induce a lethal disease phenotype in mice hemizygous for the mutant A53T α-syn transgene following a 100-day incubation period (12). By contrast, only minor deposition of Lewy body-like inclusions was observed following inoculation with sarkosyl-insoluble brain homogenate from a patient with dementia with Lewy bodies in non-transgenic mice following an extended 450-day incubation period (13). Several studies have investigated the prion-like properties of low-spin brain homogenates, recombinant fibrils or insoluble fractions of synucleinopathy brain homogenates but little efforts have focused on the soluble components. It is clear from the prion field that preparations of soluble samples from prion-infected brains are infectious (14). Moreover, soluble α-syn has been described in brain homogenates from patients with multiple system atrophy, dementia with Lewy bodies, and PD and may be involved in the pathogenesis of such synucleinopathies (15).
Here, we demonstrate that intracerebral inoculation of both sarkosyl-soluble and sarkosyl-insoluble fractions of brain homogenate preparations from a patient with transitional Lewy body disease (LBD) induces rapid (~120 days) prion-like seeding of α-syn in the brains of mice expressing the human α-syn transgene.
MATERIALS AND METHODS
Mice
Six 8-week-old male or female BDF1 transgenic mice that over-express human wild type α-syn under the regulatory control of the platelet-derived growth factor (PDGF-β) promoter (Line D) and 6 non-transgenic littermates (Charles River, Wilmington, MA) were used for experimental injections (16). Line D mice expressing human α-syn have an age-dependent progressive accumulation of α-syn-immunoreactive inclusions. Five transgenic or non-transgenic littermates (non-transgenic littermates do not express human α-syn and do not have a progressive accumulation of α-syn inclusions) were used for control injections. Housing and procedures were performed according to the National Institute of Health Guide for the Care and Use of Experimental Animals, and were approved and conducted in accordance with Mayo Clinic Institutional Animal Care and Use.
Behavioral Tests
A behavioral battery was performed consisting of open field test, elevated plus maze test, light/dark chamber exploration test, and wire hang test. Mice were acclimated to the room of testing 2 hours prior to testing and all tests were performed during the first half of the light cycle. Behavioral equipment was cleaned with 30% ethanol between each animal.
Elevated Plus Maze Test
This anxiety/exploration test was performed in a maze elevated 50 cm from the floor and consisting of 4 arms (50 × 10 cm), with 2 of the arms enclosed with roofless gray walls (35 × 15 cm, L × H). Mice were placed in the center of the maze facing an open arm, and tracked for 5 minutes with an overhead camera and AnyMaze software.
Open Field Test
Mice were placed in the center of an open-field arena (40 × 40 × 30 cm, W × L × H) and allowed to roam freely for 15 minutes. Side-mounted photobeams raised 7.6 cm above the floor measured rearing, and an overhead camera tracked movement with AnyMaze software (Stoelting Co., Wood Dale, IL). Mice were analyzed for multiple measures, including average speed, time mobile, total distance traveled, and distance traveled in the center zone (20 × 20 cm).
Light/Dark Chamber Exploration Test
This test of anxiety/exploration was conducted in a light/dark chamber (40 × 40 × 30) divided into 2 compartments with a small opening joining the light and dark areas. The dark compartment was covered and mice were placed in them at the far end of the light chamber facing away from the dark chamber. Their activity was tracked for 10 minutes with the overhead camera and AnyMaze software.
Wire Hang Test
This is a test of motor function, strength and deficit. Each mouse was hung from a wire 3 mm thick and 60 cm in length by its forepaws, at a height of 20 cm above a protective pillow. The time for which the wire was held was recorded (s), with a maximum time allowed of 120 seconds.
Preparation of Sarkosyl-Soluble and Sarkosyl-Insoluble LBD Inocula
Samples were taken from the putamen and amygdala of a patient with pathologically confirmed transitional LBD (Fig. 1i B). On the morning of stereotaxic surgery, the samples were pooled and a 10% (weight/volume, w/v) brain homogenate was prepared in sterile phosphate-buffered saline (PBS) containing EDTA-free complete protease inhibitor (Roche Diagnostics, Basel, Switzerland), with 1% Triton X-100, by mechanical homogenization for 5 minutes on ice. The homogenate was centrifuged for 10 minutes at 3000 × g at 4°C, and the supernatant was collected. A 2.5% sarkosyl solution was prepared and used to dilute the supernatant to 2% w/v. The homogenate was next centrifuged at high speed (50,000 × g) for 1 hour at 4°C, and the supernatant was collected and used for the sarkosyl-soluble injections. The pellet was resuspended in 30 μl of sterile PBS by vigorous mixing and 7.5 μg of total protein was used for the sarkosyl-insoluble and sarkosyl-soluble injections.
Figure 1.

(i) (A-D) The amygdalae of the Lewy body disease (LBD) patient (B) (highlighted in green on low-power image, D) and of the control subject (A) (highlighted in green on low-power image, C) were immunostained with anti-α synuclein antibody LB509. An abundance of Lewy bodies and Lewy neurites were present in the amygdala of the LBD patient but not control. (ii) (A, B) Electron microscopy image showing the soluble (A) and insoluble (B) fraction. The soluble fraction typically contained tubular-like fibrils of ~70 nm in length and 12 to 15 nm width (red arrow). The insoluble fraction contained large fibrils of around 250 nm in length and 6-8 nm width (red arrow) and also frequently contained annular (black arrow) and tubular structures. (iii) (A-C) Western blotting using the phosphorylated-α synuclein antibody (p-syn) of sarkosyl-soluble (A) and sarkosyl-insoluble (B, and C; enhanced contrast) fractions of LBD brain homogenate reveal distinct low (17 kDa) and high (70 kDa) molecular weight bands, respectively. Scale bars: i, 50 μm; ii, 50 nm.
Intra-Cerebral Inoculations
Groups of 6 mice were stereotaxically injected at 8 weeks old unilaterally into the caudate putamen with 7.5 μg of total protein of brain homogenate pooled from the amygdala and putamen of a control subject or a patient with transitional LBD. Inoculations were performed using the following co-ordinates from Bregma: anteroposterior (AP) +0.2 mm, lateral (L) −2.0 mm, dorsoventral (DV) −2.6 mm. The inoculum was injected at a rate of 0.2 μl/minute and the needle was left at the site of injection for 10 minutes. Animals were killed under general anesthesia (ketamine/xylazine, 120/20 mg/kg) by terminal perfusion via the right ventricle using 60 ml of saline followed by 60 ml of neutral buffered formalin (10%; ~4% formaldehyde), at 120 days post-injection.
Histology and Immunohistochemistry
Immunohistochemistry was performed in 2 Mayo Clinic Florida laboratories using either the free-floating technique (McLean laboratory) or the paraffin-embedded technique (Dickson laboratory). Paraffinized tissue was deparaffinized with 3 5-minute changes of xylene, and rehydrated. Free-floating sections were washed 3 times for 5 minutes in PBS. Tissue sections were either processed with or without the use of a mouse on mouse (M.O.M.) blocking reagent (Vector Laboratories, Burlingame, CA). Primary antibodies were added to the tissue and incubated overnight at 4°C, or for 1 hour at room temperature when using the M.O.M. reagent. Antibodies were used at the following concentrations: anti-phosphorylated α-syn (p-syn; Wako Chemicals USA, Richmond, VA), which is specific for human α-syn with a phosphorylated Ser129, (1:500), the disease-associated anti-human α-syn 5G4, which is of the IgG1κ isotype, and recognizes the linear epitope sequence (E)GVVHGV(A), corresponding to amino acid sequence 46-53 of human α-syn (17) (generous gift from Gabor Kovacs, Medical University of Vienna, Austria, and purchased from EMD Millipore, Princeton, NJ) (1:500), the 4B12 anti-human α-syn (epitope lies within 103-108 of α-syn [Covance, San Diego, CA]) (1:1000), and LB509, which reacts with an epitope within the region encoded by amino acids 115-122 of human α-syn (ThermoFisher Scientific, Grand Island, NY) (1:500). Slides were treated with biotinylated anti-mouse or anti-rabbit secondary antibody (Thermo Scientific Pierce, Rockford, IL) (1:200) and incubated for 30 minutes at room temperature. Samples were next rinsed with PBS and incubated with Vectastain ABC Reagent (Vector Laboratories) for 30 minutes. Tissue sections were covered with diaminobenzidine at room temperature, and after gentle washing with water, the samples were next immersed in Mayer's Hematoxylin. Samples were dehydrated with ethanol and xylene and mounted with Super Mount (Innogenex, San Ramon, CA) and cover slipped. Reactive astrocytes were measured by a similar procedure, but using anti-glial fibrillary acidic protein ([GFAP], Abcam, Cambridge, MA) (1:300) antibody. NeuN antibody (Abcam) (1:300) was also used. Samples were analyzed using a Leica DMI6000 B microscope (Wetzlar, Germany).
Proteinase K Resistance
Following deparaffinization, mouse brain slices were incubated for 20 minutes at room temperature with 10 μg/μl Proteinase K (Sigma Aldrich, St Louis, MO). Immunohistochemistry was performed with the LB509 antibody, as described.
Human Subjects
One LBD patient and 1 control without neurodegenerative disorders were investigated (Table 1). The cases were selected for short postmortem intervals (PMI), because a long PMI has been shown to lead to decreased α-syn expression in PD patients relative to controls (18). Further, minimizing AD-type pathology was necessary to reduce the possibility of cross-seeding of tau and α-syn, or of amyloid-β and α-syn. Based on these criteria, a 76-year-old male with limbic (transitional) LBD was chosen (19). The control was matched as closely as possible for age, gender, AD-type pathology, and PMI (Table 1). Patients or their proxies had signed informed consent for autopsy according to the Institutional Review Board guidelines.
Table 1.
Human Patient Neuropathological Characterization
| Control Brain | Lewy Body Disease Brain | |
|---|---|---|
| Age, years | 75 | 76 |
| Gender | Male | Male |
| Race | Caucasian | Caucasian |
| PMI, hours | < 6 | < 6 |
| Pathological diagnosis | Normal | Transitional Lewy body disease |
| Braak NFT stage | III | III |
| Thal amyloid phase | 1 | 1 |
PMI, Postmortem interval; NFT, neurofibrillary tangles
Characterization of Human Brain Homogenate
Electron Microscopy
Control and LBD tissue samples prepared for injection were examined by electron microscopy. Samples were diluted to 0.01–0.10 mg/ml in 1X Tris-buffered saline, and 10 μl of each sample was adsorbed onto a 400-mesh carbon/Formvar-coated copper grid (EM Sciences Inc., Hatfield, PA) for 60 seconds and then stained with 2% uranyl acetate for 60 seconds. Electron micrographs were captured using a Phillips EM208S electron microscope and Gatan Orius 831 digital camera.
Western Blotting
Samples to be electrophoresed were diluted in 7.5 μl of sample buffer and 3 μl of reducing agent. The total volume was made up to 30 μl with water; the samples were then heated at 95°C for 5 minutes and run on a 4% to 12% Tris-Glycine gel (Sigma Aldrich) at 200V for 1 hour. Following electrophoresis, gels were transferred onto a PVDF membrane (Sigma Aldrich) at 33V for 1.5 hours. The membranes were incubated for 1 hour at room temperature in a blocking solution, and then rinsed in PBS-tween (0.05%) (Sigma Aldrich). The primary antibody (p-syn; Wako) was added at a dilution of 1:1000 and incubated for 1 hour at room temperature. Following 4 washes of 10 minutes each, the membranes were then incubated in anti-mouse IgG horseradish peroxidase-conjugated antibody diluted at 1:3000. The membranes were washed as above and developed using the Hybond-chemiluminescence (ECL) system (GE Healthcare, Umatilla, FL), according to the manufacturer's instructions.
RESULTS
Human Neuropathology
A patient with transitional LBD and a control subject were selected for inclusion in the study based on their suitability to the study selection criteria (Table 1). At autopsy, the control subject had mild non-specific neurodegenerative changes characterized by sparse neurofibrillary tangles in the entorhinal cortex, hippocampus and amygdala. The calculated whole brain weight of the control subject was 1320 grams. No Lewy body pathology was reported.
The calculated whole brain weight of the LBD patient at autopsy was 1340 grams. Many dystrophic neurites were detected in the CA2/3 region with anti-α-syn immunohistochemistry (LB509). A few Lewy bodies and Lewy neurites were also detected in the endplate, CA1 and subiculum. The basal nucleus of Meynert had severe neuronal depopulation, but no neurofibrillary tangles were detected by thioflavin S fluorescent microscopy. Lewy bodies and Lewy neurites were numerous in the basal forebrain. A few senile plaques and sparse neurofibrillary tangles were present in the amygdala. There was mild spongiform change in the amygdala and many Lewy bodies and Lewy neurites were present (Fig. 1i B, D). The basal ganglia had α-syn-immunoreactive spheroids in the globus pallidus, as well as numerous dot-like and curvilinear neurites and glial cytoplasmic inclusions in the putamen. There were no senile plaques or neurofibrillary tangles in the putamen or globus pallidus. The substantia nigra had moderate to severe neuronal loss with extraneuronal neuromelanin, gliosis and Lewy bodies in residual neurons. The neuronal loss was most marked in the ventrolateral cell group, but also affected the medial cell group. A pathological diagnosis of limbic (transitional) LBD was made.
Characterization of LBD Brain for Intracerebral Injection in Mice
The LBD brain used for intracerebral injections in mice was characterized by Western blotting, immunohistochemistry and electron microscopy. LB509 immunostaining revealed the presence of many Lewy bodies and Lewy neurites (30-35 per 20x field) in the amygdala of the LBD brain (Fig. 1i B, D); no Lewy bodies or Lewy neurites were evident in the amygdala of the control brain (Fig. 1i A, C).
Electron microscopy of the amygdala revealed shorter 70-nm-long tubular fibrils in the soluble fraction and longer 250-nm fibrils in the insoluble fraction (Fig. 1ii A, B). The insoluble LBD fraction also contained annular structures resembling protofibrils, which were previously described as particularly abundant in recombinant mutant α-syn preparations (20). Western blotting was performed using the anti-p-syn antibody (Fig. 1iii). Gel electrophoresis revealed the presence of 2 distinct high- and low-molecular weight bands. The sarkosyl-insoluble fraction (Fig. 1iii B, C) produced a relatively weak band at around 69 kDa; whereas the sarkosyl-soluble fraction (Fig. 1iii A) produced a strong band at around 17 kDa. These low- and high-molecular weight bands suggest the presence of both soluble monomeric p-syn and insoluble oligomeric p-syn within the brain homogenate. Because previous studies have demonstrated cross-reactivity of other anti-phosphorylated α-syn antibodies with neurofilament including pSer129/81A (22), we immunostained healthy non-transgenic mice with pSer129/81A and p-syn (Supplementary Fig. 1). Cross-reactivity with neurofilament was observed with pSer129/81a, as previously reported, but no cross-reactivity with the p-syn antibody was observed.
Prion-like Seeding of LBD Pathology in Mice
To determine if the soluble or insoluble fractions of LBD brain homogenate from a patient with LBD can seed and propagate human α-syn pathology in mice, transgenic mice expressing wild-type α-syn under the control of the PDGF promoter (Line D mice [16]) were intracerebrally inoculated at the level of the dorsal neostriatum with 3 μl (7.5 μg of total protein) of sarkosyl-insoluble or sarkosyl-soluble fraction of LBD brain homogenate, or the control brain homogenate. Mice were killed at 4 months post-injection by terminal perfusion and brains were analyzed by immunohistochemistry for the presence of disease-associated α-syn reactivity using the disease-specific antibody 5G4 (Fig. 2A-I), or a p-syn antibody (Fig. 2J-L). The 5G4 antibody binds aggregated α-syn preparations and α-syn from patients with synucleinopathies but not from control patients (23). Specificity of the antibodies was investigated in-house using immunohistochemistry and immunoblotting (Supplementary Fig. 2). Key results are summarized in Table 2.
Figure 2.

Sarkosyl-insoluble Lewy body disease (LBD) brain homogenate induces seeding in transgenic mice. (A-L) Disease-specific deposits were detected by 5G4 antibody (A-I) or phosphorylated α-synuclein (p-syn) antibody (J-L). 5G4 immunohistochemistry revealed heavy neuropil-like staining in the CA2 and CA3 regions of the hippocampus (B, and higher magnification C), the pons (E, and higher magnification F) and medulla (H, and higher magnification I). Littermate transgenic control mice injected with the insoluble-fraction of an age-matched control human brain were unreactive to the 5G4 antibody (A, D, G). Staining with the p-syn antibody revealed the presence of immunoreactive deposits in the hippocampus of transgenic mice injected with the insoluble LBD brain homogenate (K). Insets indicate region of hippocampus in the box (black lines) of the low-power view and immunostained deposits in the indicated box (broken red lines) at high power. Mice injected with the insoluble fraction of the control brain remained free of p-syn immunoreactivity (J). Sparse p-syn-immunoreactive inclusions were observed in the motor cortex of transgenic mice injected with the insoluble LBD brain homogenate (L). Scale bars = 20 μm.
Table 2.
Seeding of Lewy Body Disease Pathology in Mice
| Transgenic Mice | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inocula | LBD | Cnt | LBD | Cnt | LBD | Cnt | LBD | Cnt | LBD | Cnt | LBD | Cnt | LBD | Cnt |
| Fraction | Insoluble | Soluble | Insoluble | Soluble | Soluble | Insoluble | Soluble | |||||||
| Antibody | 5G4 | 5G4 | 5G4 | 5G4 | psyn | psyn | psyn | psyn | 4B12 | 4B12 | LB509 | LB509 | LB509 | LB509 |
| CA2 | +++ | - - - | - - - | - - - | +++ | - - - | - - - | - - - | ++ | ++ | +++ | + | + | + |
| CA3 | +++ | - - - | - - - | - - - | +++ | - - - | - - - | - - - | ++ | ++ | +++ | + | + | + |
| Dentate Gyrus | +++ | - - - | - - - | - - - | +++ | - - - | - - - | - - - | + | + | +++ | - - - | + | - - - |
| Pons | +++ | - - - | - - - | - - - | - - - | - - - | - - - | - - - | + | + | - - - | - - - | - - - | - - - |
| Medulla | +++ | - - - | - - - | - - - | - - - | - - - | - - - | - - - | + | + | - - - | - - - | - - - | - - - |
| Motor Cortex | - - - | - - - | - - - | - - - | + | - - - | - - - | - - - | +++ | + | + | + | + | + |
| Striatum | ++ | - - - | - - - | - - - | ++ | - - - | - - - | - - - | ++ | + | + | + | + | + |
| PK-res | Yes | No | No | No | ||||||||||
Line D human α-synuclein transgenic mice were inoculated at the level of the dorsal neostriatum with 3 μl (7 μg total protein) of sarkosyl-soluble or sarkosyl-insoluble Lewy body disease (LBD) brain homogenate, or with an age-matched control (Cnt) brain homogenate prepared as sarkosyl-soluble or sarkosyl-insoluble. Mice were killed at 4 months post-injection by terminal perfusion and brains were analyzed by immunohistochemistry for the presence of phosphorylated α-synuclein (psyn) or disease-associated α-synuclein using the 5G4 and LB509 antibodies. Transgenic mice inoculated with soluble LBD brain homogenate and unreactive to 5G4 or psyn antibodies were additionally stained using the 4B12 antibody. Mice injected with insoluble LBD were found to have 5G4, LB509 and psyn-reactive deposits in various brain regions, and were proteinase K (PK)-resistant. Mice injected with soluble LBD were unreactive to 5G4 and psyn antibodies, but were found to have few LB509-reactive deposits in the dentate gyrus that were PK-sensitive. Transgenic mice injected with soluble LBD were found to have more abundant 4B12 deposits than mice injected with control brain homogenate, suggesting that soluble LBD seeds human transgenic α-synuclein in this host. The degree of staining of antibody is indicated by the plus sign (+, slight staining; ++, moderate staining; +++, abundant staining). An absence of staining is indicated by minus (- - -) signs.
Insoluble LBD Brain Homogenate
5G4 immunohistochemistry revealed regional-specific staining in 6 out of 6 transgenic mice injected with the insoluble LBD fraction. Little staining was evident proximal to the injection site but an abundance of neuropil-like staining was observed in the hippocampal formation. Stain burden varied between individual mice but 5G4-positive structures consistently appeared in the radiatum layer of the molecular layer of the dentate gyrus, and the CA1 and CA2 regions (Fig. 2B, C). 5G4 also revealed a heavy cytoplasmic stain in neurons of the pons (Fig. 2E, F) and medulla (Fig. 2H, I). Transgenic mice injected with control brain homogenate had no 5G4 immunopositive structures in the hippocampal formation (Fig. 2A), and the neurons of the pons and medullar of control-injected transgenic mice also remained clear of 5G4 positive staining (Fig. 2D, G, respectively).
Transgenic mice injected with the sarkosyl-insoluble fraction of the LBD brain homogenate were also immunoreactive to p-syn antibody in the hippocampal formation (Fig. 2K); sparse p-syn-immunoreactive intracytoplasmic inclusions (1 per 20x field) were observed in the motor cortex (Fig. 2L). Control-injected transgenic mice had no p-syn immunoreactivity (Fig. 2J).
To determine if insoluble α-syn could induce α-syn pathology in non-transgenic mice, non-transgenic littermates were injected with the sarkosyl-insoluble fraction of LBD brain homogenate. These mice were found to have p-syn immunoreactivity at 4 months post-injection in various brain regions, including the caudate putamen and piriform cortex, but were not reactive to other anti-α-syn antibodies (Supplementary Fig. 3A, B, D, Supplementary Table 1).
Eosinophilic cytoplasmic inclusions (0–3 per 20X field) were observed in the brains of 2 out of 6 transgenic mice receiving sarkosyl-insoluble LBD brain homogenate (but not in mice receiving control brain homogenate) following hematoxylin and eosin staining (Fig. 3A). The inclusions were frequently present in the thalamus and hypothalamus. In non-transgenic mice, 2 out of 6 injected with sarkosyl-insoluble LBD brain homogenate also had spheroidal cytoplasmic deposits in the thalamus (Supplementary Fig. 4). Five out of 5 transgenic mice injected with control brain homogenate were negative for eosinophilic cytoplasmic inclusions.
Figure 3.

Eosinophilic cytoplasmic inclusions in transgenic mice injected with the sarkosyl-insoluble fraction of a Lewy body disease (LBD) brain homogenate preparation. (A, B) Transgenic mice inoculated with insoluble LBD had eosinophilic cytoplasmic inclusions in the hypothalamic nucleus. These cytoplasmic inclusions were spheroidal (B). (C, D) This morphology was distinct from the elliptical morphology of extracellular eosinophilic structures not associated with disease, which appeared in all mice including mice injected with control brain homogenate, and non-transgenic mice. Scale bars = 20 μm.
Soluble LBD Brain Homogenate
In the sarkosyl-soluble cohort, a seeding effect was not detected in transgenic mice with 5G4 or p-syn antibodies. However, upon further investigation, seeding could be observed in 6 out of 6 transgenic mice using the anti-human α-syn antibody 4B12 (Fig. 4). Further, a very low burden of immunoreactivity (0–3 inclusions per 20X field) was observed with the anti-α-syn antibody LB509 (Fig. 5C).
Figure 4.

Sarkosyl-soluble Lewy body disease brain homogenate induces seeding in transgenic mice. Transgenic mice inoculated with sarkosyl-soluble LBD brain or sarkosyl-soluble human control brain were analyzed by anti-human α-synuclein (α-syn) (4B12) immunohistochemistry. In mice receiving LBD brain (B) the secondary motor cortex had a visibly higher burden of 4B12-positive neurons than mice receiving control brain (A). Near the site of injection at the dorsal neostriatum neurons in mice receiving LBD brain homogenate (D) appeared more heavily stained than neurons at the same site in mice receiving control brain (C). A strong neuropil-staining was also observed in this area in mice receiving the LBD brain (D). In mice receiving the LBD brain a pale core could be observed frequently (F, medial parietal association cortex (MPtA); G, hippocampus) and cell bodies were frequently dystrophic in the cortex (F, MPtA; H, primary somatosensory cortex) with heavy dendritic and axonal staining (I, dorsal neostriatum); in contrast, neurons of mice receiving the control brain typically appeared healthy (E, neostriatum). Scale bars = 20 μM.
Figure 5.

Sarkosyl-insoluble Lewy Body disease brain homogenate induces the deposition of PK-resistant α-synuclein in transgenic mice. The dentate gyri of transgenic mice intracerebrally inoculated with the sarkosyl-insoluble or sarkosyl soluble fraction of a LBD brain were analyzed for the presence of proteinase-K (PK) resistant α-synuclein (α-syn) using LB509 immunohistochemistry. Mice receiving the sarkosyl-insoluble fraction (A) had abundant LB509 immunoreactivity which was partially resistant to PK-degradation (B). Mice receiving the sarkosyl-soluble fraction (C) had few LB509-immunoreactive α-syn deposits (arrows). Following PK-treatment, the LB509 immunoreactivity was diminished (D). Transgenic mice receiving the sarkosyl-insoluble fraction from the control brain homogenate (E) had no LB509-immunoreactive deposits at 4 months post-injection. Scale bar = 200 μM.
Brodmann areas including the secondary motor cortex (Fig. 4B) of transgenic mice injected with the sarkosyl-soluble fraction of LBD brain homogenate had a visibly heavier burden of 4B12-positive neurons compared to mice injected with the sarkosyl-soluble fraction of a control patient (Fig. 4A). The caudate putamen near the site of the injection was heavily stained in mice receiving the LBD sarkosyl-soluble fraction (Fig. 4D), but staining was less pronounced in the caudate putamen of transgenic mice receiving control brain homogenate (Fig. 4C; and higher magnification of a proximal area, 4E). Neuronal cell bodies appeared swollen in the caudate putamen of sarkosyl-soluble LBD-injected mice (Fig. 4D, I), and staining was additionally present in the dendrites and axons (Fig. 4I). Few to-no cells had dendritic or axonal staining in mice receiving control inocula (Fig. 4C, E). Perinuclear and cytoplasmic staining with a pale core (Fig. 4F, G) and dystrophic neurons (Fig. 4H) appeared more frequently in brains of mice receiving the sarkosyl-soluble fraction of the LBD homogenate than brains of control mice. Transgenic mice receiving the sarkosyl-soluble fraction did not have eosinophilic cytoplasmic inclusions.
Nontransgenic mice injected with the soluble LBD fraction did not display a seeding effect upon 4B12 immunohistochemistry (data not shown), but did display p-syn immunoreactivity at 4 months post-injection in the caudate putamen near the site of injection (Supplementary Fig. 3C, Supplementary Table 1).
Proteinase-K Resistance
To determine if α-syn seeded in transgenic mice is proteinase-K (PK) resistant, transgenic mouse brains were immunostained with LB509 and incubated with PK at 10 μg/μl for 15 minutes. The dentate gyri were chosen as the area of interest because a heavy burden of LB509 immunoreactivity was observed in this area in 4 out of 6 transgenic mice that had received the sarkosyl-insoluble fraction (Fig. 5A). Following PK-treatment, a large population of PK-resistant α-syn was apparent, demonstrating that α-syn seeded by the sarkosyl-insoluble fraction is partially PK-resistant (Fig. 5B). Six out of 6 transgenic mice injected with the sarkosyl-soluble fraction had a low burden of LB509 immunoreactivity (Fig. 5C). Following PK treatment, mice receiving the sarkosyl-soluble fraction had none to few LB509-immunoreactive deposits remaining (Fig. 5D). Five out of 5 transgenic mice receiving control brain homogenate had no LB509-positive deposits in the dentate gyri at 4 months post-injection (Fig. 5E).
Other Effects of Intracerebral Injection of LBD Brain Homogenate
To determine if mice injected with the sarkosyl-soluble or sarkosyl-insoluble fractions exhibited inflammatory changes following intracerebral injection, the brains of transgenic mice were stained with anti-ionized calcium-binding adapter molecule 1 (Iba1) or -GFAP, markers of microgliosis and astrogliosis, respectively, at 4 months post-injection. Brain slices were also stained with anti-NeuN antibody, a marker of neuronal integrity, and with an apoptosis kit. Four out of 6 transgenic mice injected with sarkosyl-insoluble LBD homogenate appeared to display a marked and widespread increase in GFAP immunoreactivity (Fig. 6A) compared to transgenic mice receiving human control brain homogenate (Fig. 6B). The substantia nigra of mice injected with the insoluble fraction showed marked GFAP immunoreactivity in 2 mice; other areas of intense immunoreactivity include the amygdala-hippocampal area, the hippocampal formation, the cerebellum, medulla and pons (Fig. 6A). Sarkosyl-soluble LBD did not induce an increase in GFAP immunoreactivity in transgenic mice and there was no discernable difference in Iba1 immunoreactivity between control mice and LBD injected mice (data not shown). Non-transgenic mice injected with the sarkosyl-insoluble LBD fraction displayed an apparent increase in GFAP immunoreactivity in the thalamus, and non-transgenic mice injected with the soluble LBD fraction appeared to display an apparent loss of NeuN reactivity in the hippocampus (Supplementary Fig. 5).
Figure 6.
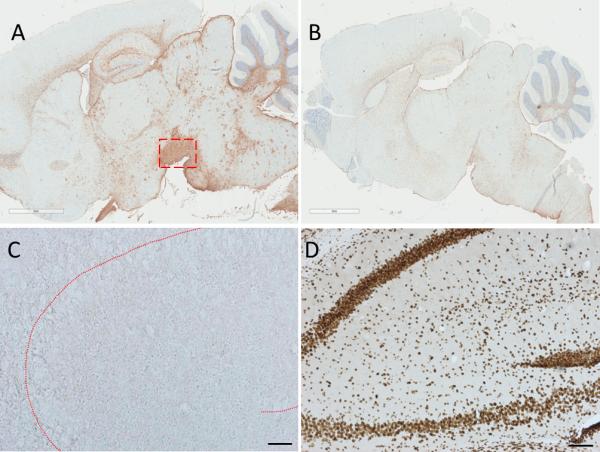
Glial fibrillary acidic protein (GFAP) and apoptosis. Transgenic mice injected with sarkosyl-insoluble Lewy body disease (LBD) brain homogenate displayed a widespread increase in GFAP immunoreactivity (A) with intense staining at the substantia nigra (boxed area), compared with transgenic mice injected with sarkosyl-insoluble control brain which appeared to have lower GFAP burdens (B). Transgenic mice injected with sarkosyl-insoluble LBD brain homogenate did not show evidence of apoptosis using an apoptosis kit (C, hippocampus). The proprietary positive control demonstrated staining in treated hippocampal slices (D). Scale bars (black) = 300 μM; (white) = 2 mm.
None of the transgenic or non-transgenic mice injected with either sarkosyl-soluble or -insoluble LBD brain homogenate (Fig. 6C) displayed apoptosis, which was demonstrated in the hippocampal slice treated with proprietary positive control ex vivo (Fig. 6D).
Behavioral Characterization of Injected Mice
All mice were subjected to a battery of behavioral tests including open field, elevated plus-maze, and wire-hang prior to being euthanized at 4 months post-injection. No behavioral phenotype was detected in any of the mice (Supplementary Fig. 6, Supplementary Table 2).
DISCUSSION
Our data demonstrate that a single intracerebral inoculation of either sarkosyl-soluble or sarkosyl-insoluble fraction of a human LBD brain homogenate is sufficient to induce central nervous system (CNS) pathology in α-syn transgenic and non-transgenic mice. To our knowledge these data demonstrate for the first time that both the soluble and insoluble preparations of brain homogenate of a synucleinopathy patient are able to cross the human-mouse species barrier and induce CNS pathology in mice. In transgenic mice we found that the sarkosyl-insoluble LBD fraction was robust at inducing phosphorylated and disease-specific (p-syn-, 5G4- and LB509-reactive) α-syn deposits that were partially PK-resistant. Only a mild seeding effect of PK-sensitive and LB509-reactive α-syn was observed in some (but not all) transgenic mice receiving the sarkosyl-soluble fraction. Therefore, we additionally investigated any potential seeding with the anti-human α synuclein antibody 4B12 and found that the sarkosyl-soluble fraction seeded 4B12-reactive α-syn, which was apparent by a visibly heavier burden of 4B12-positive neurons compared to transgenic mice that were injected with control brain homogenate. This is in line with previous work by Luk and colleagues, who demonstrated an apparent increase in α-syn-positive neurons in transgenic mice (expressing the human α-syn A53T mutation) that had received inoculations of pre-formed fibrils compared to control transgenic mice (10). In non-transgenic mice, p-syn pathology was induced following the intracerebral injection of either the sarkosyl-soluble or sarkosyl-insoluble LBD fractions but the deposits were not reactive to 5G4, LB509 or 4B12 anti-α-syn antibodies. We speculate that endogenous mouse α-syn is recruited during prion-like seeding in the non-transgenic mice such that the pathology is not recognized by human-specific anti-α-syn antibodies. Conversely, in transgenic mice, seeding is apparent by human specific anti-α-syn antibodies because transgenic human α-syn is seeded.
In line with the data presented here, mice injected with nigral Lewy body-enriched fractions (insoluble only) displayed diffuse neuronal inclusions of α-syn at 120 days post-injection (24). Interestingly, that study reported the presence of nigrostriatal dopaminergic neurodegeneration in mice that was associated with the α-syn pathology whereas we found no evidence of loss of tyrosine hydroxylase-positive fibers and no apoptosis (data not shown). We observed apparent NeuN loss in the hippocampus of 2 non-transgenic mice injected with the soluble LBD fraction (supplementary data); however, this was not statistically significant upon quantification (data not shown), most likely because the experiment was underpowered for stereology. No cell loss was observed in transgenic mice or with the insoluble homogenate fraction. In addition to the Recasens study (24), recombinant fibrillar and recombinant soluble α-syn induced CNS pathology in mice expressing human wild type α-syn by 8 months post-injection following a single intracerebral inoculation (25). Moreover, recombinant sarkosyl-insoluble α-syn as well as sarkosyl-insoluble α-syn from the brains of patients with dementia with Lewy bodies induced p-syn pathology in wild type mice following intracerebral inoculation; the corresponding sarkosyl-soluble fractions did not (13). It is not clear why our sarkosyl-soluble fractions, which were injected within a similar dose range (5–10 μg) were able to induce CNS pathology when the soluble fractions used in the study by Masuda-Suzukake did not; we propose this may be due to the presence of an EDTA-free protease inhibitor in our preparations, in combination with our strict selection criteria, which brains with short post-mortem intervals were chosen for our study. Postmortem protease digestion of α-syn may reduce its seeding effect, particularly of soluble α-syn, which is more vulnerable to protease digestion than insoluble. Indeed, α-syn expression in the frontal cortex of PD patients was shown to decrease as the postmortem interval increased, but remained constant in controls (26). EDTA is a chelating agent that was included in the homogenate preparations in the Masuda-Suzakake study, and which may contribute to reduced prion-like transmissibility of sarkosyl-soluble α-syn because it sequesters metal ions and interferes with the activity of metalloenzymes, both of which are required by many proteins to be functionally active (26, 27). Indeed, metalloenzymes appear to play a pivotal role in the progression of PD pathology by modulating the aggregation state of α-syn (28-30). Furthermore, the soluble fractions used by the Masuda-Suzakake team were recombinant; indeed, the study does not present data regarding the seeding propensity of soluble fractions from the brains of human patients, which, if tested, may have seeded α-syn. Our protocol also took into consideration the possibility that regional-specific strains of α-syn might exist, similar to specific conformations of α-syn oligomers that are present in distinct regions of A53T α-syn transgenic mice (31). In that case, strains purported to be specific to the amygdala might not seed efficiently in the putamen; therefore, we pooled areas from the amygdala and putamen of the LBD patient in order to facilitate optimal seeding in the mouse putamen. However, it should be noted that these distinct conformational differences may not occur in all α-syn strains but might rather reflect the so-called ‘chameleon property’ of α-syn whereby it is able to adopt different conformational structures depending on its immediate environment (32). Interestingly, however, Bousset et al generated 2 different high-molecular weight assemblies (i.e. fibrils and ribbons) from the same precursor α-syn, and found that the fibrils were capable of inducing biochemical modifications within cells that led to apoptotic events to a much higher extent than the ribbons, suggesting that distinct strains had been generated (33). In depth in vivo studies are needed to evaluate strain properties including possible variations in incubation periods to strengthen the case for true α-syn strains.
We found that p-syn pathology in transgenic or non-transgenic mice injected with the sarkosyl-insoluble fraction appears bilaterally, which is in accordance with the study of Masuda-Suzakake et al. In all mice receiving either the soluble or insoluble LBD fractions, the hippocampus and striatum had the highest burden of α-syn (p-syn, 5G4 or LB509) immunoreactivity, suggesting that α-syn seeds injected into the striatum most efficiently transversed the striatal-hippocampal pathway, or alternatively, that endogenous α-syn in the hippocampal neurons is more vulnerable to seeding by exogenous soluble and insoluble LBD fractions. We observed that transgenic Line D mice inoculated with control brain homogenate (or not injected) first express human α-syn most abundantly in the hippocampus and striatum, which may reflect the propagation pathway in mice injected with LBD brain homogenates. In agreement with our data, others have demonstrated moderate to severe grading of α-syn pathology predominantly in the hippocampus and striatum following intracerebral inoculation of α-syn fibrils in the substantia nigra of non-transgenic mice (13), with little spread to the contralateral side. Other studies have revealed a more widespread phenotype; indeed, intracerebral inoculation of α-syn filaments lateral to the lateral ventricle in the cerebrum of transgenic mice expressing human wild type α-syn induced bilateral rostral-caudal spreading of α-syn pathology (34). Inclusions were observed in the cortex, hippocampus, midbrain, brainstem and spinal cord. In addition, Luk et al demonstrated abundant α-syn pathology in various brain regions (particularly the cortex) at 90 days following intra-striatal and intra-neocortex injection in young asymptomatic M83 mice, using brain lysate from M83 transgenic mice or α-syn pre-formed fibrils (10). The pathology was found to spread along telencephalic axonal pathways whereas our data show spread along the striatal-hippocampal pathway. Interestingly, the hippocampus was relatively spared in the Luk study, which is in contrast to our study. It is clear, therefore, that different mouse models, different α-syn preparations, and different injection sites induce distinct pathological phenotypes.
Seeding to non-neuronal cells has been investigated as well as propagation to neuronal cells. Several studies have described uptake of α-syn by non-neuronal cells including oligodendrocytes, with dynamin playing a key role in this uptake process (35-37). Indeed glial cells do not express α-syn mRNA and it is likely that α-syn is taken up by glial cells during cell-to-cell spread of α-syn in multiple system atrophy, which is characterized by the presence of glial cytoplasmic inclusions that are not present in other synucleinopathies. Interestingly, Reyes et al demonstrated that α-syn monomers and oligomers are more readily taken up by oligodendroglial cells than α-syn fibrils in vitro (35). Substantial amounts of both detergent-soluble and detergent-insoluble α-syn were detected in the brains of PD patients and patients with dementia with Lewy bodies; however, multiple system atrophy patients were found to have high levels of soluble α-syn but not insoluble α-syn (15), suggesting that neuronal and non-neuronal cells may process α-syn differently and thus may harbor different forms (insoluble vs. soluble) of α-syn. Future studies will evaluate the uptake of soluble and insoluble α-syn by non-neuronal cells in vitro and in vivo.
Seeding by the sarkosyl-soluble LBD fraction was detected in transgenic mice by 4B12 immunohistochemistry. Mild seeding was also observed with LB509 immunohistochemistry, but not with anti-p-syn. It is possible that the sarkosyl-soluble fraction might display a delayed incubation period in transgenic mice compared to the insoluble fraction, which induced robust p-syn deposition. We hypothesize that transgenic mice receiving the soluble fraction might have p-syn deposition at a later time point, (e.g. >4 months post-injection) but we did not investigate time-points beyond 4 months. Intriguingly, the sarkosyl-soluble fraction induced p-syn deposition in non-transgenic mice at 4 months post-injection, similar to the sarkosyl-insoluble fraction. Thus, comparing transgenic with non-transgenic mice, we observed that the α-syn substrate (endogenous mouse α-syn vs. human transgenic α-syn) plays a more pivotal role in the observed pathologic phenotype than the seed. We also show that like-conformations were not apparent in transgenic and non-transgenic mice receiving the same LBD fractions, as LB509 reactivity was not consistent between transgenic and non-transgenic mice. Indeed, distinct species were propagated in the insoluble cohort defined as phosphorylated and LB509-reactive in transgenic mice, vs. phosphorylated- and LB509-unreactive in non-transgenic mice. Future studies in which α-syn is depleted from each LBD fraction using anti-α-syn antibodies will confirm that α-syn is the active seed.
Our study is the first to show a transmissible component of the sarkosyl-soluble fraction of a brain homogenate preparation from a synucleinopathy patient. Precedent for a transmissible component in sarkosyl soluble fractions is found in detergent-soluble transmissible spongiform encephalopathies prions. Preparations of soluble prions from mice infected with the ovine prion disease, scrapie, are infectious upon intracerebral inoculation in hamsters with a delay in incubation period of just 15.9 to 25.6 days compared to hamsters injected with the insoluble fraction (14). Furthermore, prion infectivity in rodents and sheep is associated with soluble components in the blood plasma, and soluble prions are detected in the blood of variant Creutzfeldt-Jakob patients (38-40). In addition, protease-sensitive prion diseases have been identified within the last decade, and pathogenic mutations of the prion protein lead to the formation of soluble, protease-sensitive prion species with increased lethality (41-44). In agreement with the prion field, our data suggest that both insoluble and soluble α-syn proteins from the brain of a LBD patient are transmissible under experimental conditions. Osterberg et al demonstrated progressive aggregation of α-syn in transgenic mice expressing green fluorescent protein (GFP)-tagged wild-type human α-syn (GFP-α-syn) following intra-cerebral inoculation of mouse sequence pre-formed fibrils (45). At 2.5 to 4 months post-injection, immature somatic and neuritic inclusions were observed on the ipsilateral side, but by 4 to 13 months post-injection mature somatic inclusions were present on both ipsilateral and contralateral sides. Interestingly at the later stage, Osterberg et al noted a decrease in soluble cytoplasmic and nuclear GFP-α-syn staining, which may reflect the recruitment of soluble α-syn by the pre-formed fibrils and subsequent aggregation. thus emphasizing a key role for soluble α-syn in the pathogenic process.
Partial resistance to PK-digestion is characteristic of most prions and this feature is routinely exploited for the diagnoses of transmissible spongiform encephalopathies. In the synucleinopathies, growing evidence suggests that partial PK-resistance is also a feature of some disease-associated α-syn. PK-treatment was shown to enhance LBD α-syn immunoreactivity with a C-terminal antibody, but an N-terminal antibody showed diminished reactivity (46). Therefore, we used the C-terminal, Lewy body-specific anti-α synuclein antibody LB509 to detect α-syn in transgenic mice that had received sarkosyl-soluble or -insoluble LBD preparations. In our study, transgenic mice receiving control brain homogenate were unreactive to LB509, and non-transgenic mice receiving LBD homogenates were also unreactive to LB509 but the sarkosyl-insoluble LBD fraction seeded abundant PK-resistant α-syn in transgenic mice. In contrast, the sarkosyl-soluble fraction seeded few LB509 reactive α-syn deposits, making PK-sensitivity difficult to discern in this cohort. We conclude that the insoluble LBD fraction is more robust at inducing PK-resistant and LB509-reactive CNS pathology in transgenic mice than the soluble LBD fraction and that transgenic mice are more susceptible to prion-like seeding of PK-resistant LBD fractions than non-transgenic mice. These data may demonstrate the presence of a species-barrier whereby seeding of healthy α-syn by pathological exogenous α-syn is most efficient in hosts of the same species (47).
The data presented herein support the hypothesis that heterogenous species of soluble and insoluble prion-like α-syn from LBD brain can experimentally induce seeding in transgenic mice expressing human α-syn, and are transmissible to non-transgenic mice. These data are a novel insight into the enigmatic prion-like mechanisms of α-syn pathogenesis in human LBD.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Gabor Kovacs for the generous gift of 5G4 antibody and Dr. Eliezer Masliah for the Line D mice, Ms. Linda Rousseau, Virginia Phillips, and Monica Castenedes for excellent immunohistological support.
This work was supported by NIH NS073740 to Pamela J. McLean.
Footnotes
Disclosures: The authors have nothing to disclose.
REFERENCES
- 1.Weissmann C, Enari M, Klohn PC, et al. Transmission of prions. J Infect Dis. 2002;186(Suppl 2):S157–65. doi: 10.1086/344575. [DOI] [PubMed] [Google Scholar]
- 2.Kordower JH, Chu Y, Hauser RA, et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–6. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 3.Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–3. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 4.Mendez I, Vinuela A, Astradsson A, et al. Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nat Med. 2008;14:507–9. doi: 10.1038/nm1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danzer KM, Kranich LR, Ruf WP, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SJ, Desplats P, Lee HJ, et al. Cell-to-cell transmission of alpha-synuclein aggregates. Methods Mol Biol. 2012;849:347–59. doi: 10.1007/978-1-61779-551-0_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez-Erviti L, Seow Y, Schapira AH, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42:360–7. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danzer KM, Ruf WP, Putcha P, et al. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. Faseb J. 2011;25:326–36. doi: 10.1096/fj.10-164624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luk KC, Kehm VM, Zhang B, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–86. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luk KC, Kehm V, Carroll J, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watts JC, Giles K, Oehler A, et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci USA. 2013;110:19555–60. doi: 10.1073/pnas.1318268110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136:1128–38. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berardi VA, Cardone F, Valanzano A, et al. Preparation of soluble infectious samples from scrapie-infected brain: a new tool to study the clearance of transmissible spongiform encephalopathy agents during plasma fractionation. Transfusion. 2006;46:652–8. doi: 10.1111/j.1537-2995.2006.00763.x. [DOI] [PubMed] [Google Scholar]
- 15.Campbell BC, McLean CA, Culvenor JG, et al. The solubility of α-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson's disease. J Neurochem. 2001;76:87–96. doi: 10.1046/j.1471-4159.2001.00021.x. [DOI] [PubMed] [Google Scholar]
- 16.Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–9. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 17.Kovacs GG, Wagner U, Dumont B, et al. An antibody with high reactivity for disease-associated α-synuclein reveals extensive brain pathology. Acta Neuropathol. 2012;124:37–50. doi: 10.1007/s00401-012-0964-x. [DOI] [PubMed] [Google Scholar]
- 18.Dumitriu A, Moser C, Hadzi TC, et al. Postmortem interval influences α-synuclein expression in Parkinson disease brain. Parkinsons Dis. 2012;2012:614212. doi: 10.1155/2012/614212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosaka K, Yoshimura M, Ikeda K, et al. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree--a new disease? Clin Neuropathol. 1984;3:185–92. [PubMed] [Google Scholar]
- 20.Lashuel HA, Petre BM, Wall J, et al. α-Synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 21.Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–10. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sacino AN, Brooks M, Thomas MA, et al. Amyloidogenic α-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014;127:645–65. doi: 10.1007/s00401-014-1268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner U, Dumont B, Pikkarainen M, et al. An antibody with high reactivity for disease-associated α-synuclein reveals extensive brain pathology. Acta Neuropathol. 2012;124:37–50. doi: 10.1007/s00401-012-0964-x. [DOI] [PubMed] [Google Scholar]
- 24.Recasens A, Dehay B, Bove J, et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–62. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 25.Sacino AN, Brooks M, McGarvey NH, et al. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol Commun. 2013;1:38. doi: 10.1186/2051-5960-1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carver PL. Metal ions and infectious diseases. an overview from the clinic. Met Ions Life Sci. 2013;13:1–28. doi: 10.1007/978-94-007-7500-8_1. [DOI] [PubMed] [Google Scholar]
- 27.Waldron KJ, Robinson NJ. How do bacterial cells ensure that metalloproteins get the correct metal? Nat Rev Microbiol. 2009;7:25–35. doi: 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]
- 28.Lee EJ, Woo MS, Moon PG, et al. α-Synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J Immunol. 2010;185:615–23. doi: 10.4049/jimmunol.0903480. [DOI] [PubMed] [Google Scholar]
- 29.Choi DH, Kim YJ, Kim YG, et al. Role of matrix metalloproteinase 3-mediated α-synuclein cleavage in dopaminergic cell death. J Biol Chem. 2011;286:14168–77. doi: 10.1074/jbc.M111.222430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sung JY, Park SM, Lee CH, et al. Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J Biol Chem. 2005;280:25216–24. doi: 10.1074/jbc.M503341200. [DOI] [PubMed] [Google Scholar]
- 31.Tsika E, Moysidou M, Guo J, et al. Distinct region-specific α-synuclein oligomers in A53T transgenic mice: implications for neurodegeneration. J Neurosci. 2010;30:3409–18. doi: 10.1523/JNEUROSCI.4977-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uversky VN. A protein-chameleon: conformational plasticity of α-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn. 2003;21:211–34. doi: 10.1080/07391102.2003.10506918. [DOI] [PubMed] [Google Scholar]
- 33.Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two α-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sacino AN, Brooks M, McGarvey NH, et al. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol Commun. 2013;1:38. doi: 10.1186/2051-5960-1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reyes JF, Rey NL, Bousset L, et al. α-Synuclein transfers from neurons to oligodendrocytes. Glia. 2014;62:387–98. doi: 10.1002/glia.22611. [DOI] [PubMed] [Google Scholar]
- 36.Kisos H, Pukass K, Ben-Hur T, et al. Increased neuronal α-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling α-synucleinopathies. PLoS One. 2012;7:e46817. doi: 10.1371/journal.pone.0046817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konno M, Hasegawa T, Baba T, et al. Suppression of dynamin GTPase decreases α-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol Neurodegener. 2012;7:38. doi: 10.1186/1750-1326-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown P, Rohwer RG, Dunstan BC, et al. The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion. 1998;38:810–6. doi: 10.1046/j.1537-2995.1998.38998408999.x. [DOI] [PubMed] [Google Scholar]
- 39.Hunter N, Foster J, Chong A, et al. Transmission of prion diseases by blood transfusion. J Gen Virol. 2002;83:2897–905. doi: 10.1099/0022-1317-83-11-2897. [DOI] [PubMed] [Google Scholar]
- 40.Edgeworth JA, Farmer M, Sicilia A, et al. Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet. 2011;377:487–93. doi: 10.1016/S0140-6736(10)62308-2. [DOI] [PubMed] [Google Scholar]
- 41.Gambetti P, Dong Z, Yuan J, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jansen C, Head MW, van Gool WA, et al. The first case of protease-sensitive prionopathy (PSPr) in The Netherlands: a patient with an unusual GSS-like clinical phenotype. J Neurol Neurosurg Psych. 2010;81:1052–5. doi: 10.1136/jnnp.2009.175646. [DOI] [PubMed] [Google Scholar]
- 43.Head MW, Knight R, Zeidler M, et al. A case of protease sensitive prionopathy in a patient in the UK. Neuropathol Appl Neurobiol. 2009;35:628–32. doi: 10.1111/j.1365-2990.2009.01040.x. [DOI] [PubMed] [Google Scholar]
- 44.Coleman BM, Harrison CF, Guo B, et al. Pathogenic mutations within the hydrophobic domain of the prion protein lead to the formation of protease-sensitive prion species with increased lethality. J Virol. 2014;88:2690–703. doi: 10.1128/JVI.02720-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osterberg VR, Spinelli KJ, Weston LJ, et al. Progressive aggregation of α-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015;10:1252–60. doi: 10.1016/j.celrep.2015.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda A, Hashimoto M, Mallory M, et al. Abnormal distribution of the non-Abeta component of Alzheimer's disease amyloid precursor/α-synuclein in Lewy body disease as revealed by proteinase K and formic acid pretreatment. Lab Invest. 1998;78:1169–77. [PubMed] [Google Scholar]
- 47.Hagiwara K, Hara H, Hanada K. Species-barrier phenomenon in prion transmissibility from a viewpoint of protein science. J Biochem. 2013;153:139–45. doi: 10.1093/jb/mvs148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.