

Abstract
Osteoarthritis (OA) is a disease characterized by degradation of joints with the development of painful osteophytes in the surrounding tissues. Currently, there are a limited number of treatments for this disease and many of these only provide temporary, palliative relief. In this review, we discuss polymer drug delivery systems that can provide targeted and sustained delivery of imaging and therapeutic agents to OA-affected sites. We focus on technologies such as polymeric micelles and nano- / micro-particles, liposomes, and dendrimers for their potential treatment and/or diagnosis of OA. Several promising studies are highlighted, motivating the continued development of delivery technologies to improve treatments for OA.
1. Introduction
Osteoarthritis (OA) is a debilitating disease that affects joints and their surrounding tissues leading to pain and loss of mobility. While there are many factors involved in the initiation of OA, the full context is not thoroughly understood; it is considered to be a complex disease of the whole joint, rather than a specific cellular or matrix component.[7] Many potential risk factors for OA such as genetic predisposition [9], aging [10], obesity [11], and joint trauma or misalignment [12] [13] have been investigated. While the mechanism of action and pathogenesis of the disease remains incompletely elucidated, it is clear that a combination of both mechanical and biological factors are involved. [14] [7] [15] Figure 1 illustrates the multifaceted, negative impacts that arthritis can have on joints.
Figure 1.
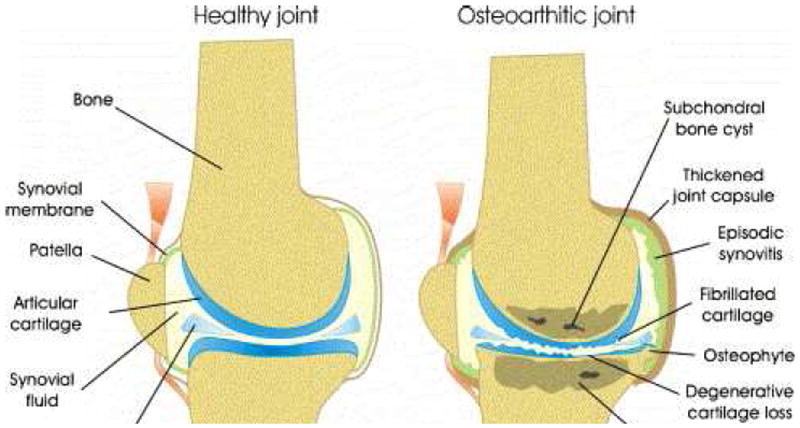
Schematic of a healthy (left) and osteoarthritic (right) knee joint. This schematic shows the detrimental effects caused by the presence of arthritis in a joint. Reproduced with permission from Elsevier. [6]
Because OA is high prevalence and causes significant morbidity, an improved understanding of the pathogenesis of OA and development of improved OA therapies are significant medical needs. As of 2012, over 25% of the United States population over the age of 45 was afflicted with OA [16]. This number is expected to increase to almost 30% by the year 2032. [16] Not only is OA physically incapacitating, it causes a significant financial burden to patients and the healthcare system. Healthcare costs related to OA totaled over $60 billion in 2007, and the aggregate cost of OA is expected to increase to $185.5 billion per year based on data from 2007. [17]
Current treatments of OA are primarily focused on pain alleviation and the improvement of joint function and mobility.[18] These treatments can be classified into three main categories: non-pharmacological, pharmacological, and surgical. Non-pharmacological treatments include reduction of weight on the affected joint or braces that mechanically stabilize the joint. These treatments can be effective, but many patients find them difficult to implement for extended periods of time. Joint replacement surgeries are common in patients with severe symptoms and are generally very effective.[19] However, surgery is often only utilized as a last resort after pharmacological treatments have failed and the patient has experienced debilitating pain for many years.[20]
The primary focus of this review is on pharmacological treatments. Unfortunately, no current treatments address the underlying molecular causes of the disease [14, 20] or are curative in nature. Pharmacological treatments include the administration of analgesics such as non-steroidal anti-inflammatory drugs (NSAID) and in some cases analgesics such as opioids or narcotics.[21] Intra-articular injections and systemic administration of long-acting glucocorticoids can also be effective during flares of inflammation, but these only work temporarily and can have negative consequences for long term use.[22, 23] Other current pharmaceuticals such as hyaluronic acid-based products rely on longitudinal intra-articular injections that each supply only 1-3 months of symptom relief. Injection directly into the joint enables control over dosing the target tissue, but most drugs are quickly cleared from the joint cavity, reducing any potential for long term benefits of treatment.[24] Systemic administration of drugs for OA has been associated with severe side effects with little therapeutic benefit. NSAIDs are known to cause gastrointestinal complications in a significant population of patients and selective cyclooxygenase-2 (COX-2) inhibitors, another class of drug found to be moderately effective at treating symptoms of early-mid stage OA, have been associated with cardiovascular risks. [6] The shortcomings of these conventional therapies motivate not only discovery of new therapeutics but also new delivery systems for targeted and prolonged pharmacological delivery that could lead to better compliance and improved outcomes for patients with OA. Recent studies suggest that inflammatory mediators, proteases, and signaling molecules such as NFκB, ERK1/2, interleukin-1 receptor antagonist, and TLRs may be promising molecular targets for treatment of OA. Without delivery systems, these inflammatory mediators have minimal therapeutic potential because of their lack of persistence at the target site and act to broadly to be effective via oral administration. [14]
The most significant challenge that persists with drug delivery for OA is a lack of vasculature within synovial joints; this is a significant barrier to biodistribution of systemically delivered therapies to the target site. Another challenge is rapid clearance of locally delivered therapeutics due to synovial fluid exchange. The presence of synovial fluid makes the delivery of hydrophobic drugs difficult, as they lack the ability to disperse within the joint. However, hydrophilic therapeutics such as proteins are cleared from the joint via pressure gradients that cause flow of the synovial fluid. Fluid movement within joints is created by ultrafiltration of fluid from the capillaries into the joint cavity and drainage of fluid from the cavity, through the synovial membrane into the lymphatics. [25] Repeated administration of locally-injected therapies on a regular basis is not desirable/feasible; thus this delivery route is not warranted if the benefit is relatively short-lived.[26] This review will survey the utilization of polymeric drug delivery vehicles in the context of OA, including nano- and micro-scale materials. Many nano-scale polymeric materials have been investigated for intravenous delivery and preferential targeting/retention at pathological sites; for example, many of the platforms developed for anticancer agents [27], including polymeric micelles, liposomes, and dendrimers, may also be useful technologies for OA therapy.[28] Micron-scale (microgels, microparticles, etc.) biomaterials made from synthetic and/or natural polymers are also useful for solubilizing and controlling the release of therapeutics for local delivery[29], and their larger size can be beneficial for reducing the diffusivity and rate of clearance from the joint. These technologies enable specific biochemical interactions and/or physicochemical tuning (charge, size, etc) to be utilized to potentially yield longer-acting and more effecive treatments for OA. Prior to reviewing specific OA applications of particulate systems, the general properties and formulation characteristics will be outlined for micelles, liposomes, and solid polymeric particles.
2. Particles for Drug Delivery
2.1. Micelles
Micelles are nanoscale materials comprising amphiphilic polymers that self-assemble in aqueous solvents (Figure 2).[30] Micelle formation is driven by the entropic hydrophobic effect, wherein energetically unfavorable water-cages are excluded from the hydrophobic polymer blocks, followed by self-aggregation of these segments into hydrophobic compartments stabilized by a hydrophilic corona.[31] In order for micelles to form, a critical concentration of the molecule must be present, known as the critical micelle concentration (CMC). For many amphiphilic diblock copolymers, the CMC is low enough to enable micelle stability under dilute conditions (~10-100 ug/mL)[26]. However, a primary challenge in micelle research is reducing susceptibility to premature disassembly in vivo under dilute conditions and/or due to competing interactions with endogenously present serum proteins and cholesterol.[32] Micelle core and shell crosslinking are especially promising for stabilization of these structures.[33, 34]
Figure 2.

Schematic illustrating the multifunctional components that can be utilized in micelle formations [5].
Importantly, the core of the micelles can be utilized to load and solubilize hydrophobic drugs or imaging agents.[35] A primary shortcoming of micelles, on the other hand, is the inability to use hydrophilic drugs unless they are covalently tethered to the micelle. Solvent evaporation methods, emulsion polymerizations, and nano-precipitation have been successfully utilized to encapsulate hydrophobic drugs within the micelle core and enhance colloidal stability of the poorly soluble drugs. Sizes of micelles are dependent on many factors including but not limited to: the polymer chemistry, degree of polymerization, ratio of hydrophobic to hydrophilic polymer block lengths, packing factor, and drug cargo and amount. The size of micelles ranges broadly, but it is generally accepted that sizes from 20-200nm in diameter are ideal for avoiding rapid renal clearance and passively targeting pathological tissues that have leaky vasculature.[36] A smaller diameter facilitates entry into the lymphatic vessels and transport to lymph nodes and improves diffusivity from the vasculature and throughout target tissues.
In addition to core-loading of hydrophobic cargo, micelles can be designed with multiple functionalities. Micelles can also be targeted for preferential uptake by specific cell type through conjugation of peptides, proteins, antibodies, or other targeting ligands onto the hydrophilic corona of the polymer, which is often polyethylene glycol (PEG).[37] [5] PEG is most often employed because it provides micelle shielding or “stealth” from the mononuclear phagocyte (MPS) system, reduces systemic toxicity, and prolongs blood circulation times.[38-41] Even with a PEG corona, micelles, like all nanoparticles, remain susceptible to rapid clearance from the blood by the phagocytic cells of the liver and spleen; this limits the ability to achieve preferential accumulation at pathological sites. Micelles can also be endowed with environmentally-responsive functionalities tuned to trigger drug release or particle uptake in specific pathological environments [27, 37, 42-48] or to provide endosomal escape functions.[49-54] [35]
2.2. Liposomes
Liposomes are aqueous-core vesicles surrounded by a lipid bilayer; figure 3 schematically illustrates the multifunctional components that can comprise liposomes. Unlike micelles, liposomes contain an aqueous core than can carry hydrophilic drug cargo. Similar to micelles, liposomes suffer from removal by the MPS following IV delivery, and this can be at least partially overcome by PEGylation, which can be used to increase stealth and/or provide chemical handles for functionalization with targeting ligands.[56, 57] Liposomes can be formulated using several different techniques resulting in a wide range of sizes from ~ 50 nm up to 5000 nm depending on the buffer, lipid composition, filtration strategy, and number of lipid bilayers that are present. [58] Formulation techniques for drug loaded liposomes can be broken down into passive and active loading techniques.[59] Passive loading, which occurs during liposome formation, can be further divided into mechanical dispersion, solvent dispersion, and detergent removal methods. Mechanical dispersion can be performed via sonication, extrusion, freeze-thaw cycles, lipid film hydration, micro-emulsification, and membrane extrusion. Active loading of liposomes is performed after liposome formation. A common form of active loading utilizes pH gradients to drive water soluble drugs with protonatable amine functionalities into liposomes after formulation. The drug is precipitated out due to raised pH inside of the liposome. [60] This approach enables very high loading efficiency in liposomal products for delivery of drugs such as doxorubicin (Doxil). [61]
Figure 3.
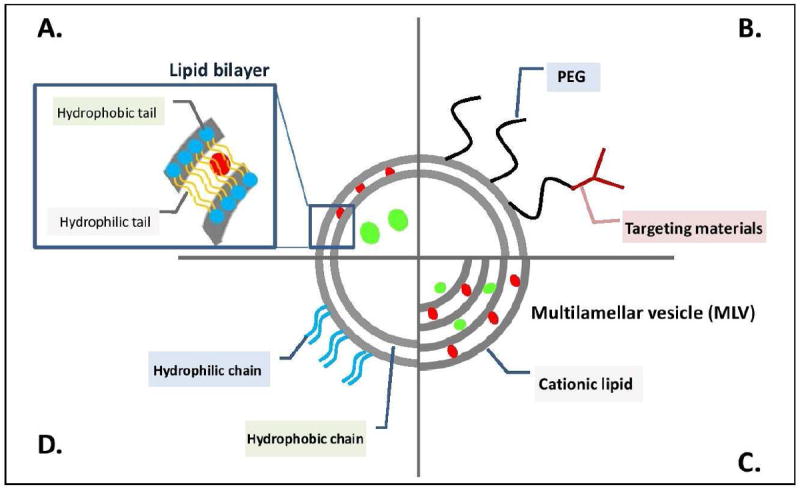
Schematic diagram of various liposomal structures. (a) Illustrates the ability for liposomes to delivery both hydrophilic (green sphere) and hydrophobic (red sphere) drugs either solubilized in the core or embedded within the lipid bilayer. (b)-(d) Represent variations of the traditional liposomes to add targeting ligands or ‘stealth’ using poly(ethylene glycol). Concept of this figure is adapted from Nature Publishing Group. [55]
Liposomes have been the focus of several successful clinical trials, mostly for cancer applications. PEGylated liposomal doxorubicin (Doxil/Caelyx), nonPEGylated liposomal doxorubicin (Myocet), liposomal daunorubicin (DaunoXome), and liposomal cytarabine (Depocyte) are all FDA approved drugs, and there are numerous other anticancer liposomal drugs in advanced clinical development. [62] Liposomes have also commonly been used for imaging applications, for example to encapsulate contrast agents such as In or Gd for MRI.[63] [58] Liposomes for rheumatoid arthritis (RA) have been investigated by Storm et al. concluding that liposomes can functionally improve the therapeutic performance of anti-inflammatory agents for RA through formation of a depot (local administration) or by attaining site specific drug targeting (IV administration). However, at the time of this review, no liposomal therapies for RA have been developed for clinically due to the lack of marketability that is perceived by industry. The benefits of using nanoscale drug delivery systems has not been estimated to outweigh the costs of integrating these new therapies into the clinic for what is estimated to be of marginal benefit for delivery of the same anti-inflammatory compounds currently used. Liposomes would require IV or IA administration while current drugs are routinely delivered orally. In contrast, liposomal delivery of doxorubicin has proven effective for cancer therapies clinically due to the already invasive nature of previous cancer treatments. Although liposomes can supply a prolonged release of the drugs for the treatment of RA, there were not significant changes in the clinical outcome of particle based therapies compared to free drug therapies. [64]
2.3. Dendrimers
Dendrimers are repetitively branched molecules that consist of three components: the initiator core, branched interior, and shell, that latter of which provides surface groups that can be utilized for covalent attachment of cargo or targeting ligands (figure 4). Dendrimers are formed by synthesis of multiple generations that radially branch in 3D from the initiator core. This can be achieved either from a convergent or divergent synthesis approach. [65] The highly branched, generational architecture makes dendrimers flexible platforms for incorporating different types of cargo. They can be readily designed to incorporate small molecule drugs, imaging agents, therapeutic proteins, peptides, or nucleic acids, and targeting agents.[4, 66-68] Moreover, combinations of multiple functionalities can be easily integrated into a single dendrimer design due to the uniqueness of each compartment (initiator, interior, shell) of the molecule.[69] As a results of the sequential nature of dendrimer synthesis, the final product is highly monodispersed compared to many of the other nano- and micro-fabrication techniques used.[70] The surface chemistry of dendrimers is also easily altered and can be optimized to provide ideal properties for a specific application. In a good example of the diversity achievable with dendrimer designs, Tyssen et al. recently performed a high-throughput screen of dendrimers with varying cores, branches, generation numbers, and surface chemistries. Through the screen, they were able to identify a subset of optimally balanced hydrophobic and anionic surface chemistries for the binding and neutralization of HIV-1. Their most promising dendrimer, VivaGel®, is currently in advanced clinical trials. [71] Thus, dendrimers are potentially unique in their ability to be used both as deliver systems and as therapeutics themselves for some select applications (i.e., VivaGel®).
Figure 4.
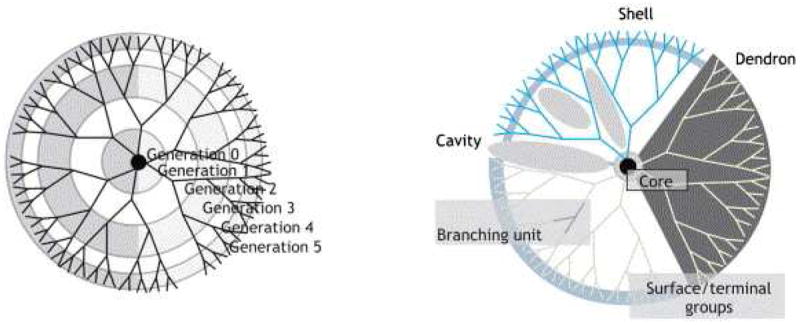
Dendrimer schematic illustrating the well-defined structure that is defined by the number of generations. [4] Figure reproduced with permission from Elsevier.
Polymeric materials are appealing for OA therapies for their ability to supply prolonged drug release. However, a disadvantage of classic dendrimer preparations is the burst release of the encapsulated drug. With dendrimers, upwards of 70% of the encapsulated drugs are often released within a few hours of being reconstituted into saline.[72] This can be overcome by covalently bonding the drug to the dendrimer, or formation of a dendrimeric prodrug. [26] Drug encapsulation within dendrimers has been successfully carried out for anticancer, anti-inflammatory, and anti-microbial drugs by both physical encapsulation and chemical coupling. [73]
The surface chemistry and/or charge of many dendrimers enable incorporation of mechanisms for cellular internalization. Although the mechanism is not entirely elucidated, it is generally thought that cationic nanoparticles bind negatively charged glycosaminoglycans (GAGs) of the cell surface in order to drive internalization.[74] Further, dendrimers can be actively targeted for cell uptake by incorporating a ligand that binds the dendrimer to internalizing cell surface receptors. The ability to penetrate chondrocytes and the extracellular matrix would prove crucial for dendrimers for OA applications.[4, 75, 76]
2.4. Polymeric Nano/Microspheres
Polymeric particles are a more generalized platform that can be formulated in both nano- and micro-scale size ranges and to encapsulate both hydrophilic and hydrophobic drugs, nucleic acids, and proteins. Sizing of these spheres can be tuned by the type of fabrication technique utilized. There are two main types of polymer spheres. The first is a capsule, or polymersome, which consists of a hydrophilic drug reservoir and a polymeric shell; these are analogous to liposomes but are polymer rather than lipid based. The second is solid spheres comprising a homogenous polymeric matrix loaded with dispersed/entrapped drug. Release kinetics of both have been thoroughly studied and are also tunable depending of formulation technique and the chemical composition and molecular weight of the polymer and the drug.
Varying sizes of particles can be obtained depending on the formulation techniques used. Many nano-sized particles are accomplished through spontaneous assembly based on chemistry. [77] Other techniques for size control of these polymeric particles involve solvent and solute concentrations and volumes, emulsifying time, and solvent evaporation methods. A common method of making polymeric particles is the oil-in-water (O/W) emulsion method that utilizes an oil phase with a hydrophobic drug and polymer that is emulsified into a water phase; a surfactant is typically utilized to stabilize the emulsion. The oil phase solvent is then evaporated, trapping the drug inside of polymeric particles. [78] A similar method can also be employed to encapsulate hydrophilic drugs. This method is known as a water-in-oil-in-water (W/O/W) emulsion. In the W/O/W method, a water phase containing the hydrophilic drug or protein is first emulsified into an oil phase containing the polymer. Then, that water and oil emulsification is dropped into a final water phase containing a surfactant and emulsified. The oil solvent is subsequently removed, leaving the hydrophilic therapeutic trapped within the polymeric sphere.[79] Nano-precipitation is another common method for nanoparticle formulation. Nano-precipitation is accomplished through rapid mixing under defined flow parameters. Polymer molecules will nucleate into nanoparticles until colloidal stability is reached during the controlled mixing of solvent and a non-solvent. This transition is made with the use of different solvents dependent upon the polymer solubility. [80, 81] Nano-precipitation can be more elegantly accomplished under fluid control such as micromixers or the use of microfluidic devices. These approaches to nano-precipitation produce less polydispersed particles under more controled batch conditions.[82-86] Nano-sized particles can be used for systemic delivery via intravenous injection while micro-sized particles are typically only utilized for local delivery via injections directly into the target site. For nano-particles to maintain a longer circulation time, they are often surface-modified with functionalities such as PEG to increase hydrophilicity and provide stealth shielding.[87] The vast variety of parameters that can be tuned to control the properties of polymeric micro- and nanospheres makes them a good approach for OA applications.
3. Osteoarthritis Targeting and Drug Delivery
The particle classes summarized above share common characteristics that make them advantageous for OA applications. Polymeric and liposomal particles commonly increase the circulation time and improve the pharmacokinetics of free drugs which can suffer from rapid clearance when delivered systemically. Increased circulation time and systemic persistence effectively increases the probability of targeted or nonspecifically-accumulating drug formulations to accumulate in the inflamed/damaged joints. Generally, polymeric and liposomal particles can be endowed with increased circulation time through surface modification and the addition of PEG.[45, 51, 52, 73, 87-89] For targeting the cartilage matrix, micelles, polymeric nanoparticles, and dendrimers can infiltrate the pores within cartilage due to their nanoscale size; these classes of delivery systems can also be synthesized to have positive surface charge to bind to the inherently negatively charged cartilage matrix. The ability to functionalize the surface of particulate systems with targeting ligands allows for collagen II-binding peptide sequences to be tethered to particles and increase their targeting to cartilage matrix.[90] Polymeric particles also have the ability to target hydroxyapatite that is present in advanced cases of OA when subchondral bone is exposed; this is achieved through the attachment of targeting peptides and bisphosphonates.[26, 91] Polymeric systems can also be optimized for targeting the cartilage surface rather than the underlying matrix by conjugating peptides that bind to epitopes exposed following cartilage degradation such as VDIPEN and NITEGE.[26, 92] Beyond this more generalizable characteristics, the different classes of delivery systems also have unique characteristics that make them especially promising for overcoming different aspects of the delivery barriers present in OA; these more specific applications of each delivery technology are summarized below.
3.1. Micelles
Several applications of micelles have been explored for arthritis (both OA and RA) treatment.[8, 93-96] In these studies, several different hydrophobic, small molecule drugs (indomethacin, dexamethasone, camptothecin, and cyclosporin A) have been formulated into micelles and administered either locally[93, 94] or systemically.[8] Zhang et al. formulated indomethacin into amphiphilic poly(N-isopropylacrylamide)-polyphosphazene micelles containing ethyl 4-aminobenzoate side groups for enhanced loading efficiency of indomethacin.[93] The micellar formulation of indomethacin had enhanced pharmacokinetics (e.g. longer circulation time in blood plasma [97]). Moreover, a single subcutaneous injection of the indomethacin-loaded micelles provided therapeutic efficacy in carrageenan-induced paw edema. In a second model, these micelles significantly reduced swelling of ankle arthritis induced with complete Freund’s adjuvant (CFA) out to 15 days. In a similar approach, Yue Koo et al. developed targeted, sterically-stabilized micelles consisting of a mixture of PEGylated lipids conjugated to the vasoactive intestinal peptide (VIP) targeting moiety.[94] The VIP targets key effector cells, activated T cells, macrophages, and over-proliferating synoviocytes by their overexpression of VIP receptors, most predominately the VPAC2 receptor. The micelles were used to solubilize camptothecin, a drug that serves as a topoisomerase I inhibitor and is thought to be effective at treating arthritis by triggering apoptosis and cell proliferation of key effector cells in the arthritic joint. Camptothecin micelles abrogated collagen induced arthritis (CIA) in mice following a single subcutaneous administration at as little as 0.3 mg/kg dose, which is significantly lower than the usual anti-cancer dose of free camptothecin. Notably, this dose of campothecin in the sterically stabilized micelles completely reversed paw thickening and decreased arthritis scores by half. A recent follow-up study has shown therapeutic efficacy of the sterically stabilized micelles delivering the VIP peptide alone as well.[98] VIP acts therapeutically in the context of RA due to its anti-inflammatory action on T cells and macrophages. VIP causes a shift of the immune reaction toward an anti-inflammatory Th2 type T cell response and the downregulation of pro-inflammatory Th17 subset of the immune response. In the study, VIP-loaded micelles administered intravenously had 13-fold higher uptake in arthritic limbs than free VIP. VIP-loaded micelles (5 nmol/animal) abrogated negative side effects of VIP such as myelosuppression, hematological toxicity, hemorrhagic renal cystitis, and elevations in liver function tests. It also caused functional improvements in terms of decreasing paw thickness and arthritis score by 50% and 75%, respectively, and at the molecular level, this treatment also decreased inflammatory markers in the arthritic joints (TNF-α, IL-1, MMP-2, and MMP-9).
One promising approach to improving performance of micelles is to covalently crosslink them post-assembly in order to improve their stability. In a recent study by Storm et al, core-crosslinked micelles were developed for delivery of the steroidal anti-inflammatory agent dexamethasone (DEX). The authors validated their design in two animal models of arthritis.[8] Figure 6 highlights this micelle design and the results from this study. In this clever approach, thioether ester-containing DEX derivatives were developed in order to allow for core crosslinking within the micelles, which resulted in varying degrees of hydrolytic release based upon oxidation state of the DEX derivative (sulfide, sulfoxide, and sulfone). The sulfone-containing DEX derivative was chosen for evaluation in vivo since it had the fastest release profile; this lead formulation was chosen based on the acute inflammation response characteristic of the animal models tested. Importantly, mice treated with a single intravenous injections of 1, 5, and 10 mg/kg of the DEX-micelles showed significant, dose-dependent reductions in arthritis score and final disease score out to 25 days when compared to the saline control. Free DEX only showed significant reduction in disease score out to day 10 when compared to the saline treated control.
Figure 6.
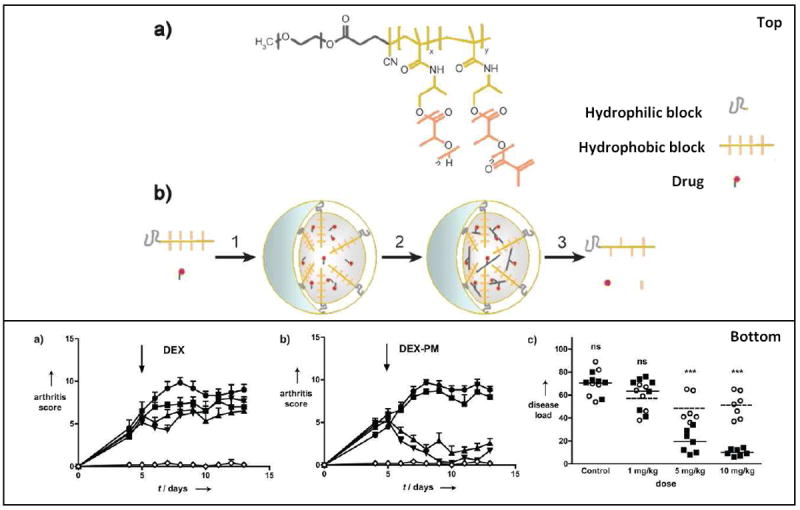
Results and micelle formulation from the study performed by Storm et Al. The top panel is a schematic representation of dexamethasoneloaded core-crosslinked polymeric micelles (DEX-PM). (a) Chemical structure of poly(ethylene glycol)-b-poly(N-(2hydroxypropyl)-methacrylamidelactate) (mPEG-b-pHPMAmLacn) block copolymers. (b) Illustrates the preparation, degradation, and drug release of DEX-PM. The bottom panels (a-b) highlight the results of their study. (a) and (b) show arthritis score after treatment. The mice received an i.v. injection of DEX (a) or DEX-PM (b) dosed at 1 mg/kg (■), 5 mg/kg (▲), or 10 mg/kg (▼). Control mice (●) received PBS or unloaded micelles. The disease load of each individual mouse upon treatment with free DEX (●) or DEX-PM (■). The disease load was defined as the area under the arthritis score curve from treatment (day 5) until the end of the study (day 13). The DEX-PM micelles provided a significant therapeutic effect and reduced both arthritis score and disease load compared to free DEX. Figure reproduced with permission from Wiley. [8]
3.2. Liposomes
Previous reviews have discussed liposomal drug formulations in the context of OA and/or RA, and we refer the reader to these articles for a more in depth review of the early liposomal drug delivery developments in OA.[6, 25, 99-102] A focus of these early reviews is the enhanced pharmacokinetics that liposomes can provide.[25, 99] However, at the time of these reviews, these liposomal drug formulations were first being developed solely as an encapsulation strategy and to increase the retention time of the drugs in the joint after IA injections. Gerwin at el. discusses many different polymer drug formulations for IA delivery specifically for osteoarthritis and concludes that sustained release of OA drugs is essential for seeing therapeutic outcomes. They argue that polymeric systems can provide this sustained release profile both with new OA drugs and current treatments standards. [6] Hoven et al. focuses on liposomal drugs for the treatment of RA, which differs from OA, but can benefit from similar drug delivery technologies and inform the application of techniques toward OA treatment. Unlike some of the early reviews on this topic, Hoven discusses the benefits of targeting, both passive and active targeting, for liposomal delivery of therapeutics. Unfortunately, Hoven draws the conclusion that clinical development of these formulations may be limited due to the high barrier of entry of these types of drug formulations into the current market. [102] This said, several advances in targeting and environmentally-responsive polymers have been made since the publication of these mentioned reviews. These advances in targeting could provide the benefits that are needed to move some of the liposomal technologies from pre-clinical testing to clinical trials. The current review focuses on newer technologies which represent these advancements in targeting and environmental-responsiveness.
Liposomes are particularly well suited for OA drug delivery to the cartilage surface, synovial membrane, and intra articular space. The size of liposomes can be tuned to be optimal for targeting these specific components of a joint. Liposomes can also be formulated from lipids that give them a positive surface charge and make them good candidates for targeting the anionic cartilage surface. [26] Liposomes have also been preclinically tested and shown promise in OA applications. For example, in a rat model of OA induced through intrapatellar ligament injection of monosodium iodoacetate, liposomes containing dexamethasone significantly reduced knee joint inflammation.[103] The multilamellar liposomes used in this study were composed of soybean phosphatidylcholine and dipalmitoyl phosphatidylethanolamine at a molar ratio of 95:5. Local injections (dose: 1mg/kg of drug) of liposomal formulations for diclofenac and dexamethasone were administered to these rats. In the same study, the liposomes were given bioadhesive properties by surface functionalization with either hyaluronan or collagen. In all cases, the liposomal drug formulations decreased inflammation of the rat knee joints down to at least 20% of the initial inflammation volume. The best performing formulation was the bioadhesive hyaluronan-surface functionalized liposomes containing both diclofenac and dexamethasone. These liposomes delivered via local injections reduced injury-induced inflammation volume by 12.9% at 17 days after a single injection of liposomes. Un-targeted liposome formulations were not investigated in this study. [103]
In a very recent study, targeted immunoliposomes (150-250nm in diameter) were developed for early clinical detection of OA. This diagnostic reagent was loaded with a near infrared fluorophore and was surface functionalized with a monoclonal antibody that selectively binds to collagen II (CII) after it is exposed in damaged cartilage.[3] The immunoliposomes bound specifically to damaged cartilage ex vivo and, as detected by fluorescence imaging. Importantly, after intravenous administration, these immunoliposomes also preferentially accumulated in vivo in a spontaneous OA model in guinea pigs at sites of arthritic cartilage. These liposomes, highlighted in Figure 7, show binding to the medial condyles of the older animals where OA initiates. Theranostic liposomes have potential to aid evaluation of therapeutic drug development for OA with small animal models allowing longitudinal studies of individual animals. They have also been used in a mechanical overload in the mouse knee which is a useful model for study of post-traumatic osteoarthritis. [2] The top panel in Figure 7 shows the general use of nanosomes for optical imaging of cartilage damage. Based on strong diagnostic performance in multiple, clinically-relevant OA models, these targeted liposomes show tremendous promise for extension into targeted drug delivery applications.
Figure 7.
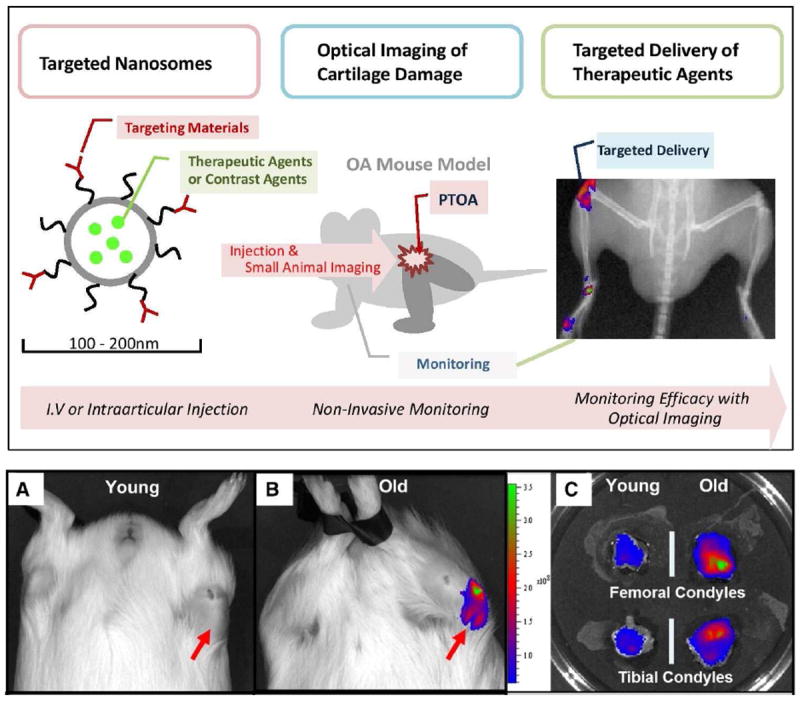
(Top) Schematic diagram of theranostic approach for osteoarthritis-type II collagen antibodies-targeted fluorescent nanosomes. These nanosomes bind on damaged cartilage in an OA animal model, providing a traceable fluorescent signal and delivering therapeutic agents.[2] (Bottom) In a spontaneous OA model in guinea pigs, older animal showed higher accumulation of immunoliposomes. Reproduced with permission from Elsevier. [3]
Liposomes have also been investigated by other groups for applications in RA. Although RA has differnet pathogenesis from osteoarthritis, symptomatic treatment and delivery mechanism of drugs and therapeutics can be similar. [102, 104] Liposomes have shown very promising results for treatment of inflammation caused by OA through the delivery of drugs such as celecoxib [105], methotrexate, and dexamethasone. [102] Targeting liposomes to RA-damaged cartilage has also proven successful through use of targeting ligands such as, prednisolone phosphate [104]; this approach has promising for limiting drug off target side effects. Hofkens at el. investigates liposomal targeting of synovial lining macrophages using prednisolone phosphate. Liposomes were formulated using dipalmitoyl phosphatidylcholine (DPPC), PEG, and cholesterol. The lipid film was hydrated in prednisolone disodium phosphate (PLP), which served as the targeting agent. Free PLP was removed from the liposome solution before treatments were given. Mice were given arthritis using an antigen-induction (AIA) model. The mice were treated 3 days post-induction via IV injection with liposomal PLP or free PLP (both 10mg/kg) or saline. A second mouse model used immune-complex arthritis (ICA) where mice were treated only with the liposomal PLP (10 mg/kg) or saline. Knee joint swelling was measured 1 and 5 days post treatment for the AIA mouse model. Swelling with liposomal PLP decreased by 74% when compared to the saline control and decreased by 64% when compared to the free PLP treatment. By day 5, liposomal PLP almost completely suppressed knee joint swelling. To test the targeting ability of PLP to macrophages in the synovial intima layer, colloidal gold liposomes were injected IV. It was seen via histology that these liposomes were readily taken up by macrophages as they leave the bloodstream and were not taken up by type B synovial fibroblasts. This study then goes into further investigation about macrophage phenotypes in the presences of liposomal PLP. They show that IA and IV injections of liposomal PLP is able to alter suppress M1 synovial macrophage without altering M2 phenotype within the inflamed synovium for both the AIA and ICA mouse models. [104] Thus, this targeting approach may be especially useful for targeting of immunomodulatory compounds.
In addition to targeting macrophages, Vanniisinghe at el. develop a system for synovium-specific targeting that has utility for OA applications. This targeted liposomal drug delivery system was developed to deliver drug cargo to inflamed joints. [106] In this study PEGylated liposomes composed of the lipid DPPC and cholesterol were functionalized with RGD or HAP-1 (SFHQFARATLAS), which targets fibroblast-derived B synoviocytes. The drug cargo that was delivered in this study was a short immunosuppressive peptide (core peptide, CP). CP is a nine amino acid peptide (GLRILLLKV) that is an effective immunosuppressant. Arthritis was induced in rats then the rats were treated with untargeted, RGD, and HAP-1 liposome formulations. Rats were given two IV injections of liposomes two consecutive days (0.5mg drug/0.5 ml/250 g rat/day). The targeted HAP-1 liposomes produced a 10-fold increase in accumulation in the arthritic rat joints compared to its contralateral unaffected joint. The untargeted liposome and the scrambled HAP-1 control only showed a 5-fold increase in fluorescence in the arthritic joint. The RGD targeted liposomes showed a higher fold increase (7-fold higher) than the non-targeted liposomes but this was lower than the HAP-1 targeted liposomes. The delivery of CP via the targeted HAP-1 liposomes significantly reduced paw swelling at day 7 compared to the control paw (>10% reduction in paw swelling) while the delivery in the RGD liposomes and non-targeted liposomes did not significantly decrease paw swelling.[106] This study highlights the significance targeting ligand choice can have on drug delivery via liposomes and highlights the potential for HAP-1.
3.3. Dendrimers
Dendrimers, similar to micelles and liposomes, have the ability to target the major components of healthy and arthritic joints. Sizes of dendrimers span a broad range from tens of nanometers to hundreds of nanometers in size when complexed with one another or with drugs. The smaller diameter dendrimers can target the cartilage matrix and subchondral bone. The end group functionality of dendrimers can be harnessed to tether a targeting ligand or peptide which allows for long retention and greater accumulation of the dendrimers in the joint, leading to prolonged and local drug release. Larger generation dendrimers can also be used for targeting cartilage surface, synovial membranes, and intra articular space, similar to liposomes. [26]
Hayder et al. recently showed the efficacy of using dendrimers for RA treatment after IV injections. They showed that azabisphosphonate (ABP)-capped dendrimers selectively target monocytes and modulate them toward a more anti-inflammatory phenotype. Intravenous injection (10mg/kg) of these dendrimers inhibited the development of inflammatory arthritis in two mouse models indicated by a reduction in inflammatory cytokines and absence of cartilage destruction. The first mouse model was an IL-1ra knockout mouse that spontaneously develops arthritis. Treatment with ABP dendrimers reduced the arthritic score by 80% compared to untreated mice. The second mouse model was a K/BxN serum transfer model, which involves transferring serum from an autoimmune K/BxN mouse to a BALB/c A mouse, resulting in an inflammatory immune response and arthritis. With the treatment of ABP, there was a significant decrease in paw swelling and arthritic score compared to the control. Paw swelling decreased by about 30% and arthritic score decrease by almost 70%. It was also shown that ABP dendrimers do not cause off target effects; this was determined though IV injections of ABP once per week for 8 weeks. Histology was performed on the spleen, kidney, lung, liver, and aorta showing no differences between dendrimer treated mice and non-treated mice. The dendrimer treatments also did not cause changes in body weight of treated mice.[91] In this case, the dendrimer itself has inherent therapeutic/anti-inflammatory and targeting capabilities without requiring loading of any additional drug; this and other studies have shown that screening of dendrimer chemistries can uncover entities with desirable function, yielding simpler, more translational systems. [75, 91, 107]
3.4. Polymer Nano/Microspheres
Polymeric particles are generally thought to be better suited for targeting the cartilage surface, synovial membrane, and intra articular space.[26] The ability to synthesize polymeric particles with larger sizes gives them an advantage over smaller particles, as they are not as easily cleared from the intra-articular space since their diffusion is more limited. A study by Singh et al. demonstrated that larger particles of approximately 900nm were retained locally for a significantly longer time than comparable smaller particles. [108] In this study, poly(2-hydroxyethyl methacrylate)-pyridine (pHEMA-pyridine) was used to create polymeric microspheres at 500nm and 900nm containing fluorescent bovine serum albumin (BSA). In healthy rats, these particles were injected IA, and particle retention over time was measure via fluorescence. The half-life of the 500nm (1.9d) and 900nm particles (2.5d) was significantly higher than the free protein (0.63d). The plateau, measured out to 14 days for the 900nm (30% at 14d) particles was significantly higher than both the retention by the 500nm particles (~5% at 14d) and the free protein (<5% at 14d).
Both natural and synthetic polymers have been used in the creation of spherical polymeric particles. Natural polymers are more disposed towards being immunogenic, which is especially undesirable for OA which is exacerbated by local inflammation. It is also more difficult to achieve reproducibility in production of particles made from natural polymers. [109] Chitosan and gelatin have shown the most promise for drug delivery in OA models and have produced desirable outcomes. [110] For example, chitosan has been used to incorporate Flurbiprofen and extend its time in the joint, leading to extended release locally for more than 24 hours. [111] Gelatin has also been used to deliver many nonsteroidal anti-inflammatory drugs and proteins such as anti-TNF to reduce inflammation. [112, 113]
Synthetic polymeric particles are one of the most commonly utilized delivery technologies due to their lack of immunogenicity, tunability, and ability to synthesize them reproducibly. The most widely used synthetic polymer is poly(lactic-co-glycolic acid) (PLGA). PLGA has been used to fabricate particles for delivery of many types of drugs. PLGA has a history of use in FDA-approved systems because it degrades into naturally existing metabolites (lactic and glycolic acid) and is fully resorbable, although lactic acid can be toxic at high levels. One such technology is Lupron®, which is used currently used to deliver drugs such as testosterone, clarithromycin, lovastatin, and progesterone long term, up to 6 months, to treat prostate cancer, endometriosis, fibroids, and central precocious puberty. Flexion Therapeutics is another company that specialized in the use of PLGA microspheres for OA therapy. They have developed three formulations of PLGA microspheres for OA treatment and therapy, FX006, FX007, and FX005. All formulations are intended for IA injection or local delivery. FX006 is a sustained release steroid injectable in phase 3 development for patients with severe OA pain.[114] FX007 is a PLGA formulation for local administration of TrkA receptor antagonist intended for post-operative pain. FX005, like FX006 is intended for late or end stage OA patients. This formulation is an IA, sustained release particle that delivers p38 MAP kinase inhibitor. This company has seen very promising pre-clinical and clinical results from these formulations for extended pain relief. FX006 (0.28mg) was able to almost completely eliminate painful gait in a rat arthritis model out to day 32. The free drug (Kenalog-40®) reduced painful gait, but not to the extent of the PLGA formulation and at day 32, the gait analysis score for the free drug was ~3x higher than FX006.
Almost all PLGA particles experience an initial burst release of the drug, which is an important dosing consideration for sustained release formulations in order to be sure local drug toxicity does not occur. Other synthetic polymers have also been extensively tested for sustained and targeted drug release. By tailoring the polymer composition, microspheres have been developed that are responsive to a variety of stimuli. For example, polypropylene sulfide (PPS) is responsive to reactive oxygen species (ROS) (undergoes a phase change from hydrophobic to hydrophilic) and was recently utilized for the first time to form microparticles that enabled “on demand” release of antioxidants. [78] OA is a disease associated with inflammation. Reactive oxygen species are major mediators of the inflammatory cycle. By introducing stimuli responsive particles, such as PPS polymeric particles, “environmentally responsive” drug release can be achieved. PPS has a unique ability to become hydrophilic when exposed to ROS such as hydrogen peroxide, triggering the release of hydrophobic encapsulated drugs. Since ROS triggers the release of the drug, these particles provide local, sustained therapy which is activated “on-demand” during cycles of oxidative stress.
Polymer microparticles have been utilized to deliver a variety of therapeutic cargo relevant to reduction of inflammation in the setting of OA. Interluekin-1 (IL-1) is a positive regulator of inflammation and can be inhibited to stop or slow the progression of OA symptoms. Interleukin-1 receptor antagonist (IL-1Ra) is a natural protein inhibitor of IL-1 and has been on of the most thoroughly investigated biologic drugs for treatment of OA. One model of arthritis, discussed above, involves the spontaneous onset of arthritic symptoms in mice lacking the IL-1Ra gene. Because this gene is an important regulator of RA and OA, IL-1Ra is a widely studied therapeutic for arthritis. Whitmire et al. used self-assembling nanoparticles to deliver IL-1Ra to rat knee joints and showed prolonged retention over free IL-1Ra. The nanoparticles are formed from a block co-polymer consisting of a hydrophobic block (cyclohexyl methacrylate, CHM) and a hydrophilic block (tetraethylene glycol methacrylate, TEGM) with a tethering moiety, paranitrophenol (pNP) used to attached the IL-1Ra protein. The nanoparticle delivery system retained 20% of the delivered IL-1Ra at day 10 which is significantly higher than the free IL-1Ra that remained in the joint at day 10. The half-life of the IL-1Ra nanoparticles was significantly higher at 3.01 +/- 0.09 days compared to the soluble IL-1Ra at only 0.96 +/- 0.08 days. [115] This study highlights the ability of polymeric particles to supply a local depot of drug that is less readily cleared from arthritic joints.
While larger particles have been used to enhance pharmacokinetics through physical size, more convention nanoparticle sizes (i.e., 100-200 nm) have also shown promise, especially when used in conjunction with targeting ligands that improve binding and retention within OA joints. In a study conducted by Rothenfluh at el., bio-functional polymeric nanoparticles were used to target arthritis and increase retention in cartilage. [90] In this study, PPS nanoparticles are synthesized using PEG-PPS-PEG block copolymers. These nanoparticles were surface functionalized with a peptide (WYRGRL) that targets collagen 1. In healthy mice, fluorescent nanoparticles were injected IA. The particles were tracked via fluorescence to monitor cell invasion and ability to infiltrate the cartilage. The WYRGRL-PPS nanoparticles were able to infiltrate both the cartilage and chondrocytes. The WYRGRL-PPS particles compared to a control peptide were also retained within the ECM at higher concentrations than the non-targeted PPS particles. A 44.8 fold increase and a 71.7 fold increase at 24 and 48hrs respectively was seen in the ratio of ECM to intracellular fluorescence of the targeted particles compared to the non-targeted particles.[90] This study specifically highlights the functional benefits of incorporating appropriate targeting ligands into polymer drug delivery systems.
There is an abundance of studies that have used polymeric microparticles for arthritis drug delivery. Rather than summarize every study in text, the following table highlights some of the more recent studies of polymer-drug systems and their outcomes in various arthritis models.
Future Perspectives and Conclusions
As surveyed herein, particle based drug delivery systems show promise for improving the pharmacokinetic and pharmacodynamics of OA drugs, including providing means for sustained therapeutic action with fewer side effects and longer-term benefits. Materials that have made it to clinical trial are of particular interest for future studies. We have highlighted many uses of liposomes for human application in other fields such as cancer treatment.[62] Due to their high biocompatibility, development of liposomes for OA applications looks very promising not only for drug delivery but also for OA detection and targeting.[2, 3] Polymeric nano- and microparticles have high potential for development for use in OA applications. Several of the common polymers used for the production of these particles are FDA approved for other biomedical applications, so the potential for these particles to progress into clinical trial is high. Polymeric delivery systems have the capacity to provide much needed extension for targeted delivery and prolonged release of drugs for OA prevention, treatment, and detection. However, significant challenges remain for clinical translation of these polymeric delivery vehicles within the pharmaceutical industry. Many of the drug delivery systems pair drugs that are already on the market or in use for treatment of OA with new polymers to improve either route of delivery or reduce off target side effects. Marketing these polymeric drugs delivery systems remains a challenge for the OA industry. Continued evaluation of advanced, targeted polymeric drug delivery systems through robust preclinical studies will be necessary to optimizing their ability to improveme pharmacokinetics and reduce side effects. These studies will be key for justifying the progression of these technologies from pre-clinical testing to clinical trials and bringing them to market.
Figure 5.
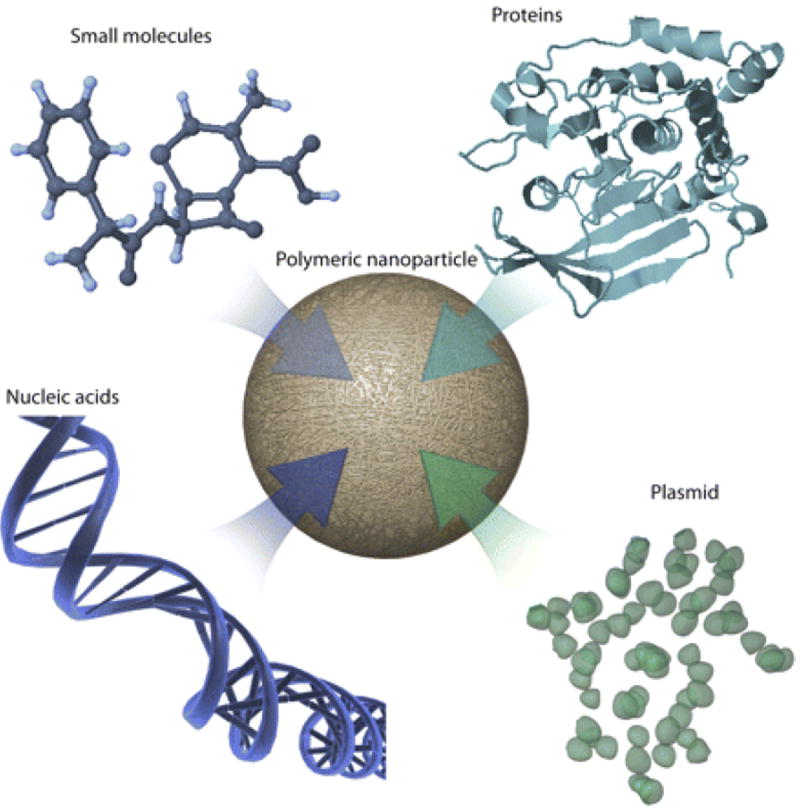
Graphical representation of polymeric nano- and microparticles, which can deliver both hydrophobic and hydrophilic compounds. [1] Figure reproduced with permission from ASPET.
Table 1.
This table highlights some of the polymer-drug combinations as they have been applied to OA and their results.
| Polymer Type | Drug | Model/Route of Delivery | Outcome | Reference |
|---|---|---|---|---|
| Poly(L-lactic acid) (PLA) | Paclotaxel | carrageenan induced rabbit model of arthritis/IA Injections | 20% paclitaxel loaded PLA microspheres in the 35-100 um size range delivered intra-articularly reduced all measure of inflammation | [116] |
| PLA | Methotrexate (MTX) | Rabbit induced arthritis model/IA Injections | 10 fold increase in MTX retention in joint compared to free MTX after intra articular injection | [117] |
| PLGA-PEG | methacrylic derivative of ibuprofen | Ex vivo sheep joints | Decreased burst release of drug and prolonged sustained release for up to 3 months | [118] |
| PLGA | Clonidine | In vitro drug release | Achieves controlled release for up to 30 days of hydrophilic drug | [119] |
| PLGA | Lornoxicam | Rat induced arthritis model/IA Injections | Reduced drug plasma levels compared to free drug, retention time increased after intra-articular injections | [120, 121] |
| PLGA | Naproxen Sodium | Rabbit induced arthritis model via intra-articular injection of ovalbumin and Freud’s Complete Adjuvant/IA Injection | Improved cure of articular arthritis when treated with PLGA loaded particles compared to BSA microspheres. | [122] |
| PLGA | ibuprofen/Labrafil | In vitro drug release | Prolonged drug release with addition of labrafil | [123] |
| PLGA | Methylpredniso lone (MP) | Rat induced arthritis model/IA Injection | Rapid increase in MP concentration in plasma at 30 minutes compared to MP suspension | [124] |
| PLGA | Dexamethasone | mouse dorsal air pouch model/local Injection | Similar DXM release with varying polymer molecular weights | [100] |
| PLGA | Betamethasone sodium phosphate (BSP) | rat air-pouch model/antigen-induced arthritic rabbit model/local or IA Injection | joint swelling significantly decreased at 21 days with administration of PLGA drug loaded particles | [125] |
| PLA and PLGA | hyaluronate | Arthritis and osteoarthritis rat models/IA Injection | administration of particles did not worsen already altered articular tissues and did not cause inflammation in healthy rat knees | [126] |
| PLGA | PTH(1-34) | Papain-induced OA rat model/IA Injection | Effect of PTH/PLGA microspheres on suppressing the OA progression was similar to that of a once-every-three-day injections | [127] |
| PLGA | Sulforaphane (SFN) | Surgically induced OA (ACL transection) in rats/local delivery (or injection) | Treatment with SFN-PLGA microspheres inhibited the mRNA and protein expression of COX-2, ADAMTS-5 and MMP-2 induced by LPS in articular chondrocytes. Intra-articular SFN-PLGA microspheres delayed the progression of surgically induced OA in rat. | [128] |
| PLGA | siRNA against TNF-a | preclinical model of RA/local injection | PLGA microspheres slowly released siRNAs effectively inhibited the expression of TNF-a in arthritic joints | [129] |
| Chitosan | Flurbiprofin | Rat knee joints/IA Injection | significant extended release of flurbiprofen from microspheres in comparison with its solution | [110] |
Footnotes
Conflict of Interest:
There are no conflicts of interests for the production of this review article.
References
- 1.Morachis JM, Mahmoud EA, Almutairi A. Physical and chemical strategies for therapeutic delivery by using polymeric nanoparticles. Pharmacological reviews. 2012;64(3):505–519. doi: 10.1124/pr.111.005363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho H, et al. Detection of early cartilage damage using targeted nanosomes in a posttraumatic osteoarthritis mouse model. Nanomedicine: Nanotechnology, Biology and Medicine. 2015 doi: 10.1016/j.nano.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 3.Cho H, et al. Theranostic immunoliposomes for osteoarthritis. Nanomedicine. 2014;10(3):619–27. doi: 10.1016/j.nano.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dufès C, I, Uchegbu F, Schätzlein AG. Dendrimers in gene delivery. Advanced Drug Delivery Reviews. 2005;57(15):2177–2202. doi: 10.1016/j.addr.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 5.Oerlemans C, et al. Polymeric Micelles in Anticancer Therapy: Targeting, Imaging and Triggered Release. Pharmaceutical Research. 2010;27(12):2569–2589. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerwin N, Hops C, Lucke A. Intraarticular drug delivery in osteoarthritis. Adv Drug Deliv Rev. 2006;58(2):226–42. doi: 10.1016/j.addr.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 7.Loeser RF. Osteoarthritis year in review 2013: biology. Osteoarthritis and Cartilage. 2013;21(10):1436–1442. doi: 10.1016/j.joca.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crielaard BJ, et al. Glucocorticoid-Loaded Core-Cross-Linked Polymeric Micelles with Tailorable Release Kinetics for Targeted Therapy of Rheumatoid Arthritis. Angewandte Chemie International Edition. 2012;51(29):7254–7258. doi: 10.1002/anie.201202713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valdes AM, Spector TD. Genetic epidemiology of hip and knee osteoarthritis. Nat Rev Rheumatol. 2011;7(1):23–32. doi: 10.1038/nrrheum.2010.191. [DOI] [PubMed] [Google Scholar]
- 10.Issa S, Sharma L. Epidemiology of osteoarthritis: An update. Current Rheumatology Reports. 2006;8(1):7–15. doi: 10.1007/s11926-006-0019-1. [DOI] [PubMed] [Google Scholar]
- 11.Richette P, et al. Benefits of massive weight loss on symptoms, systemic inflammation and cartilage turnover in obese patients with knee osteoarthritis. Annals of the Rheumatic Diseases. 2011;70(1):139–144. doi: 10.1136/ard.2010.134015. [DOI] [PubMed] [Google Scholar]
- 12.Tanamas S, et al. Does knee malalignment increase the risk of development and progression of knee osteoarthritis? A systematic review. Arthritis Care & Research. 2009;61(4):459–467. doi: 10.1002/art.24336. [DOI] [PubMed] [Google Scholar]
- 13.Buckwalter JA, Brown TD. Joint injury, repair, and remodeling: roles in post-traumatic osteoarthritis. Clin Orthop Relat Res. 2004;(423):7–16. [PubMed] [Google Scholar]
- 14.Lee AS, et al. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene. 2013;527(2):440–447. doi: 10.1016/j.gene.2013.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Jordan JM. Epidemiology of Osteoarthritis. Clinics in geriatric medicine. 2010;26(3):355–369. doi: 10.1016/j.cger.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turkiewicz A, et al. Current and future impact of osteoarthritis on health care: a populationbased study with projections to year 2032. Osteoarthritis and Cartilage. 22(11):1826–1832. doi: 10.1016/j.joca.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 17.Kotlarz H, et al. Insurer and out-of-pocket costs of osteoarthritis in the US: Evidence from national survey data. Arthritis & Rheumatism. 2009;60(12):3546–3553. doi: 10.1002/art.24984. [DOI] [PubMed] [Google Scholar]
- 18.Conaghan PG, et al. Impact and therapy of osteoarthritis: the Arthritis Care OA Nation 2012 survey. Clin Rheumatol. 2014 doi: 10.1007/s10067-014-2692-1. [DOI] [PubMed] [Google Scholar]
- 19.Fortin PR, et al. Timing of total joint replacement affects clinical outcomes among patients with osteoarthritis of the hip or knee. Arthritis & Rheumatism. 2002;46(12):3327–3330. doi: 10.1002/art.10631. [DOI] [PubMed] [Google Scholar]
- 20.Bijlsma JW, Berenbaum F, Lafeber FP. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377(9783):2115–26. doi: 10.1016/S0140-6736(11)60243-2. [DOI] [PubMed] [Google Scholar]
- 21.Bradley JD, et al. Comparison of an antiinflammatory dose of ibuprofen, an analgesic dose of ibuprofen, and acetaminophen in the treatment of patients with osteoarthritis of the knee. N Engl J Med. 1991;325(2):87–91. doi: 10.1056/NEJM199107113250203. [DOI] [PubMed] [Google Scholar]
- 22.Kirwan JR. The effect of glucocorticoids on joint destruction in rheumatoid arthritis. The Arthritis and Rheumatism Council Low-Dose Glucocorticoid Study Group. N Engl J Med. 1995;333(3):142–6. doi: 10.1056/NEJM199507203330302. [DOI] [PubMed] [Google Scholar]
- 23.Kirwan JR, Rankin E. 8 Intra-articular therapy in osteoarthritis. Baillière’s Clinical Rheumatology. 1997;11(4):769–794. doi: 10.1016/s0950-3579(97)80009-x. [DOI] [PubMed] [Google Scholar]
- 24.Derendorf H, et al. Pharmacokinetics and pharmacodynamics of glucocorticoid suspensions after intra-articular administration. Clin Pharm Ther. 1986;39(3):313–317. doi: 10.1038/clpt.1986.45. [DOI] [PubMed] [Google Scholar]
- 25.Larsen C, et al. Intra-articular depot formulation principles: Role in the management of postoperative pain and arthritic disorders. Journal of pharmaceutical sciences. 2008;97(11):4622–4654. doi: 10.1002/jps.21346. [DOI] [PubMed] [Google Scholar]
- 26.Nanomaterials for the Local and Targeted Delivery of Osteoarthritis Drugs. Journal of Nanomaterials. 2012;2012:13. [Google Scholar]
- 27.Bertrand N, et al. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Advanced Drug Delivery Reviews. 2014;66(0):2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2(5):347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 29.Peppas NA, et al. Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Advanced Materials. 2006;18(11):1345–1360. [Google Scholar]
- 30.Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Advanced Drug Delivery Reviews. 2001;47(1):113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 31.Maibaum L, Dinner AR, Chandler D. Micelle Formation and the Hydrophobic Effect†. The Journal of Physical Chemistry B. 2004;108(21):6778–6781. [Google Scholar]
- 32.Eetezadi S, Ekdawi SN, Allen C. The challenges facing block copolymer micelles for cancer therapy: In vivo barriers and clinical translation. Adv Drug Deliv Rev. 2014 doi: 10.1016/j.addr.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 33.O’Reilly RK, Hawker CJ, Wooley KL. Cross-linked block copolymer micelles: functional nanostructures of great potential and versatility. Chemical Society Reviews. 2006;35(11):1068–1083. doi: 10.1039/b514858h. [DOI] [PubMed] [Google Scholar]
- 34.Oe Y, et al. Actively-targeted polyion complex micelles stabilized by cholesterol and disulfide cross-linking for systemic delivery of siRNA to solid tumors. Biomaterials. 2014;35(27):7887–7895. doi: 10.1016/j.biomaterials.2014.05.041. [DOI] [PubMed] [Google Scholar]
- 35.Kazunori K, et al. Block copolymer micelles as vehicles for drug delivery. Journal of Controlled Release. 1993;24(1–3):119–132. [Google Scholar]
- 36.Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–9. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 37.Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13(11):813–827. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi HS, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165–70. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto Y, et al. Long-circulating poly(ethylene glycol)-poly(D,L-lactide) block copolymer micelles with modulated surface charge. J Control Release. 2001;77(1-2):27–38. doi: 10.1016/s0168-3659(01)00451-5. [DOI] [PubMed] [Google Scholar]
- 40.Papahadjopoulos D, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 1991;88(24):11460–4. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao W, et al. In situ growth of a stoichiometric PEG-like conjugate at a protein’s N-terminus with significantly improved pharmacokinetics. Proc Natl Acad Sci U S A. 2009;106(36):15231–6. doi: 10.1073/pnas.0904378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, et al. Matrix Metalloproteinase Responsive, Proximity-Activated Polymeric Nanoparticles for siRNA Delivery. Advanced Functional Materials. 2013;23(24):3040–3052. doi: 10.1002/adfm.201202215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta MK, et al. Poly(PS-b-DMA) micelles for reactive oxygen species triggered drug release. Journal of Controlled Release. 2012;162(3):591–598. doi: 10.1016/j.jconrel.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, et al. Dual MMP7-Proximity-Activated and Folate Receptor-Targeted Nanoparticles for siRNA Delivery. Biomacromolecules. 2014;16(1):192–201. doi: 10.1021/bm501394m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ponta A, Bae Y. PEG-poly (amino acid) block copolymer micelles for tunable drug release. Pharmaceutical research. 2010;27(11):2330–2342. doi: 10.1007/s11095-010-0120-z. [DOI] [PubMed] [Google Scholar]
- 46.Wu C, et al. Fabrication of complex micelles with tunable shell for application in controlled drug release. Macromolecular bioscience. 2009;9(12):1185–1193. doi: 10.1002/mabi.200900232. [DOI] [PubMed] [Google Scholar]
- 47.Li J, et al. A Reduction and pH Dual-Sensitive Polymeric Vector for Long-Circulating and Tumor- Targeted siRNA Delivery. Advanced Materials. 2014;26(48):8217–8224. doi: 10.1002/adma.201403877. [DOI] [PubMed] [Google Scholar]
- 48.Dahlman JE, et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nano. 2014;9(8):648–655. doi: 10.1038/nnano.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Convertine AJ, et al. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J Control Release. 2009;133(3):221–9. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Convertine AJ, et al. pH-Responsive Polymeric Micelle Carriers for siRNA Drugs. Biomacromolecules. 2010;11(11):2904–2911. doi: 10.1021/bm100652w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson CE, et al. Balancing Cationic and Hydrophobic Content of PEGylated siRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity in Vivo. ACS Nano. 2013;7(10):8870–8880. doi: 10.1021/nn403325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miteva M, et al. Tuning PEGylation of mixed micelles to overcome intracellular and systemic siRNA delivery barriers. Biomaterials. 2015;38(0):97–107. doi: 10.1016/j.biomaterials.2014.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pittella F, et al. Enhanced endosomal escape of siRNA-incorporating hybrid nanoparticles from calcium phosphate and PEG-block charge-conversional polymer for efficient gene knockdown with negligible cytotoxicity. Biomaterials. 2011;32(11):3106–3114. doi: 10.1016/j.biomaterials.2010.12.057. [DOI] [PubMed] [Google Scholar]
- 54.PANYAM J, et al. Rapid endo-lysosomal escape of poly(dl-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. The FASEB Journal. 2002;16(10):1217–1226. doi: 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]
- 55.Safinya CR, Ewert KK. Materials chemistry: Liposomes derived from molecular vases. Nature. 2012;489(7416):372–374. doi: 10.1038/489372b. [DOI] [PubMed] [Google Scholar]
- 56.Allen TM, Chonn A. Large unilamellar liposomes with low uptake into the reticuloendothelial system. FEBS Letters. 1987;223(1):42–46. doi: 10.1016/0014-5793(87)80506-9. [DOI] [PubMed] [Google Scholar]
- 57.Allen TM, Cullis PR. Liposomal drug delivery systems: From concept to clinical applications. Advanced Drug Delivery Reviews. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 58.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 59.Gubernator J. Active methods of drug loading into liposomes: recent strategies for stable drug entrapment and increased in vivo activity. Expert Opinion on Drug Delivery. 2011;8(5):565–580. doi: 10.1517/17425247.2011.566552. [DOI] [PubMed] [Google Scholar]
- 60.Akbarzadeh A, et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):102. doi: 10.1186/1556-276X-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abraham SA, et al. The liposomal formulation of doxorubicin. Methods in enzymology. 2005;391:71–97. doi: 10.1016/S0076-6879(05)91004-5. [DOI] [PubMed] [Google Scholar]
- 62.Slingerland M, Guchelaar HJ, Gelderblom H. Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov Today. 2012;17(3-4):160–6. doi: 10.1016/j.drudis.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 63.Mulder WJM, et al. Lipid-based nanoparticles for contrast-enhanced MRI and molecular imaging. NMR in Biomedicine. 2006;19(1):142–164. doi: 10.1002/nbm.1011. [DOI] [PubMed] [Google Scholar]
- 64.van den Hoven JM, et al. Liposomal drug formulations in the treatment of rheumatoid arthritis. Molecular pharmaceutics. 2011;8(4):1002–1015. doi: 10.1021/mp2000742. [DOI] [PubMed] [Google Scholar]
- 65.Zimmerman SC, Lawless LJ. Dendrimers IV. Springer; 2001. Supramolecular chemistry of dendrimers; pp. 95–120. [Google Scholar]
- 66.Gillies ER, Fréchet JMJ. Dendrimers and dendritic polymers in drug delivery. Drug Discovery Today. 2005;10(1):35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- 67.Patil ML, et al. Surface-Modified and Internally Cationic Polyamidoamine Dendrimers for Efficient siRNA Delivery. Bioconjugate Chemistry. 2008;19(7):1396–1403. doi: 10.1021/bc8000722. [DOI] [PubMed] [Google Scholar]
- 68.Lee CC, et al. Designing dendrimers for biological applications. Nat Biotech. 2005;23(12):1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 69.Li Y, et al. A smart and versatile theranostic nanomedicine platform based on nanoporphyrin. Nat Commun. 2014;5 doi: 10.1038/ncomms5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller TM, et al. Synthesis and characterization of a series of monodisperse, 1,3,5-phenylenebased hydrocarbon dendrimers including C276H186 and their fluorinated analogs. Journal of the American Chemical Society. 1992;114(3):1018–1025. [Google Scholar]
- 71.Tyssen D, et al. Structure Activity Relationship of Dendrimer Microbicides with Dual Action Antiviral Activity. PLoS ONE. 2010;5(8):e12309. doi: 10.1371/journal.pone.0012309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patri AK, Kukowska-Latallo JF, Baker JR., Jr Targeted drug delivery with dendrimers: Comparison of the release kinetics of covalently conjugated drug and non-covalent drug inclusion complex. Advanced Drug Delivery Reviews. 2005;57(15):2203–2214. doi: 10.1016/j.addr.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 73.Svenson S. Dendrimers as versatile platform in drug delivery applications. European Journal of Pharmaceutics and Biopharmaceutics. 2009;71(3):445–462. doi: 10.1016/j.ejpb.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 74.Midoux P, et al. Polymer-Based Gene Delivery: A Current Review on the Uptake and Intracellular Trafficking of Polyplexes. Current Gene Therapy. 2008;8(5):335–352. doi: 10.2174/156652308786071014. [DOI] [PubMed] [Google Scholar]
- 75.Gajbhiye V, et al. Dendrimers as therapeutic agents: a systematic review. Journal of Pharmacy and Pharmacology. 2009;61(8):989–1003. doi: 10.1211/jpp/61.08.0002. [DOI] [PubMed] [Google Scholar]
- 76.Joshi N, Grinstaff M. Applications of dendrimers in tissue engineering. Current topics in medicinal chemistry. 2008;8(14):1225–1236. doi: 10.2174/156802608785849067. [DOI] [PubMed] [Google Scholar]
- 77.Napoli A, et al. Oxidation-responsive polymeric vesicles. Nat Mater. 2004;3(3):183–9. doi: 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]
- 78.Poole KM, et al. ROS-responsive microspheres for on demand antioxidant therapy in a model of diabetic peripheral arterial disease. Biomaterials. 2015;41(0):166–175. doi: 10.1016/j.biomaterials.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Joshi RV, et al. Dual pH- and temperature-responsive microparticles for protein delivery to ischemic tissues. Acta Biomaterialia. 2013;9(5):6526–6534. doi: 10.1016/j.actbio.2013.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schubert S, Delaney JT, Jr, Schubert US. Nanoprecipitation and nanoformulation of polymers: from history to powerful possibilities beyond poly (lactic acid) Soft Matter. 2011;7(5):1581–1588. [Google Scholar]
- 81.Hornig S, et al. Synthetic polymeric nanoparticles by nanoprecipitation. Journal of Materials Chemistry. 2009;19(23):3838–3840. [Google Scholar]
- 82.Tseng CHT, et al. Continuous precipitation of ceria nanoparticles from a continuous flow micromixer. The International Journal of Advanced Manufacturing Technology. 2013;64(1-4):579–586. [Google Scholar]
- 83.Zhu Z. Flash Nanoprecipitation: Prediction and Enhancement of Particle Stability via Drug Structure. Molecular pharmaceutics. 2014;11(3):776–786. doi: 10.1021/mp500025e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bensaid S, et al. Flow field simulation and mixing efficiency assessment of the multi-inlet vortex mixer for molybdenum sulfide nanoparticle precipitation. Chemical Engineering Journal. 2014;238:66–77. [Google Scholar]
- 85.Fang RH, et al. Large-scale synthesis of lipid–polymer hybrid nanoparticles using a multi-inlet vortex reactor. Langmuir. 2012;28(39):13824–13829. doi: 10.1021/la303012x. [DOI] [PubMed] [Google Scholar]
- 86.Capretto L, et al. Microfluidic and lab-on-a-chip preparation routes for organic nanoparticles and vesicular systems for nanomedicine applications. Advanced drug delivery reviews. 2013;65(11):1496–1532. doi: 10.1016/j.addr.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 87.Adolph EJ, et al. Enhanced Performance of Plasmid DNA Polyplexes Stabilized by a Combination of Core Hydrophobicity and Surface PEGylation. J Mater Chem B Mater Biol Med. 2014;2(46):8154–8164. doi: 10.1039/C4TB00352G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. International Journal of Nanomedicine. 2006;1(3):297–315. [PMC free article] [PubMed] [Google Scholar]
- 89.Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed Engl. 2006;45(8):1198–215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 90.Rothenfluh DA, et al. Biofunctional polymer nanoparticles for intra-articular targeting and retention in cartilage. Nat Mater. 2008;7(3):248–254. doi: 10.1038/nmat2116. [DOI] [PubMed] [Google Scholar]
- 91.Hayder M, et al. A Phosphorus-Based Dendrimer Targets Inflammation and Osteoclastogenesis in Experimental Arthritis. Science Translational Medicine. 2011;3(81):81ra35. doi: 10.1126/scitranslmed.3002212. [DOI] [PubMed] [Google Scholar]
- 92.Singer II, et al. VDIPEN, a metalloproteinase-generated neoepitope, is induced and immunolocalized in articular cartilage during inflammatory arthritis. The Journal of Clinical Investigation. 1995;95(5):2178–2186. doi: 10.1172/JCI117907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang J, et al. Local Delivery of Indomethacin to Arthritis-Bearing Rats through Polymeric Micelles Based on Amphiphilic Polyphosphazenes. Pharmaceutical Research. 2007;24(10):1944–1953. doi: 10.1007/s11095-007-9322-4. [DOI] [PubMed] [Google Scholar]
- 94.Koo O, Rubinstein I, Önyüksel H. Actively Targeted Low-Dose Camptothecin as a Safe, Long-Acting, Disease-Modifying Nanomedicine for Rheumatoid Arthritis. Pharmaceutical Research. 2011;28(4):776–787. doi: 10.1007/s11095-010-0330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Coimbra M, et al. Antitumor efficacy of dexamethasone-loaded core-crosslinked polymeric micelles. Journal of Controlled Release. 2012;163(3):361–367. doi: 10.1016/j.jconrel.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 96.Wilson DR, et al. Synthesis and evaluation of cyclosporine A-loaded polysialic acid– polycaprolactone micelles for rheumatoid arthritis. European Journal of Pharmaceutical Sciences. 2014;51(0):146–156. doi: 10.1016/j.ejps.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 97.Dagar S, et al. VIP grafted sterically stabilized liposomes for targeted imaging of breast cancer: in vivo studies. Journal of Controlled Release. 2003;91(1–2):123–133. doi: 10.1016/s0168-3659(03)00242-6. [DOI] [PubMed] [Google Scholar]
- 98.Sethi V, et al. Novel, biocompatible, and disease modifying VIP nanomedicine for rheumatoid arthritis. Mol Pharm. 2013;10(2):728–38. doi: 10.1021/mp300539f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Letchford K, Burt H. A review of the formation and classification of amphiphilic block copolymer nanoparticulate structures: micelles, nanospheres, nanocapsules and polymersomes. European Journal of Pharmaceutics and Biopharmaceutics. 2007;65(3):259–269. doi: 10.1016/j.ejpb.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 100.Butoescu N, et al. Dexamethasone-containing biodegradable superparamagnetic microparticles for intra-articular administration: physicochemical and magnetic properties, in vitro and in vivo drug release. European Journal of Pharmaceutics and Biopharmaceutics. 2009;72(3):529–538. doi: 10.1016/j.ejpb.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 101.Pradal J, Jordan O, Allémann E. Intra-articular drug delivery for arthritis diseases: the value of extended release and targeting strategies. Journal of Drug Delivery Science and Technology. 2012;22(5):409–419. [Google Scholar]
- 102.van den Hoven JM, et al. Liposomal drug formulations in the treatment of rheumatoid arthritis. Mol Pharm. 2011;8(4):1002–15. doi: 10.1021/mp2000742. [DOI] [PubMed] [Google Scholar]
- 103.Elron-Gross I, Glucksam Y, Margalit R. Liposomal dexamethasone–diclofenac combinations for local osteoarthritis treatment. International Journal of Pharmaceutics. 2009;376(1–2):84–91. doi: 10.1016/j.ijpharm.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 104.Hofkens W, et al. Liposomal targeting of prednisolone phosphate to synovial lining macrophages during experimental arthritis inhibits M1 activation but does not favor M2 differentiation. PLoS One. 2013;8(2):e54016. doi: 10.1371/journal.pone.0054016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dong J, et al. Intra-articular delivery of liposomal celecoxib–hyaluronate combination for the treatment of osteoarthritis in rabbit model. International journal of pharmaceutics. 2013;441(1):285–290. doi: 10.1016/j.ijpharm.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 106.Vanniasinghe AS, et al. Targeting fibroblast-like synovial cells at sites of inflammation with peptide targeted liposomes results in inhibition of experimental arthritis. Clinical Immunology. 2014;151(1):43–54. doi: 10.1016/j.clim.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 107.Hayder M, et al. Anti-inflammatory properties of dendrimers per se. ScientificWorldJournal. 2011;11:1367–82. doi: 10.1100/tsw.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singh A, et al. Nanoengineered Particles for Enhanced Intra-Articular Retention and Delivery of Proteins. Advanced Healthcare Materials. 2014;3(10):1562–1567. doi: 10.1002/adhm.201400051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grund S, Bauer M, Fischer D. Polymers in Drug Delivery—State of the Art and Future Trends. Advanced Engineering Materials. 2011;13(3):B61–B87. [Google Scholar]
- 110.Kawadkar J, Chauhan MK. Intra-articular delivery of genipin cross-linked chitosan microspheres of flurbiprofen: Preparation, characterization, in vitro and in vivo studies. European Journal of Pharmaceutics and Biopharmaceutics. 2012;81(3):563–572. doi: 10.1016/j.ejpb.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 111.Ryan SM, et al. An intra-articular salmon calcitonin-based nanocomplex reduces experimental inflammatory arthritis. Journal of Controlled Release. 2013;167(2):120–129. doi: 10.1016/j.jconrel.2013.01.027. [DOI] [PubMed] [Google Scholar]
- 112.Lu Y, et al. Preparation and evaluation of biodegradable flubiprofen gelatin micro-spheres for intra-articular administration. Journal of microencapsulation. 2007;24(6):515–524. doi: 10.1080/02652040701433479. [DOI] [PubMed] [Google Scholar]
- 113.Janssen M, et al. Drugs and Polymers for Delivery Systems in OA Joints: Clinical Needs and Opportunities. Polymers. 2014;6(3):799–819. [Google Scholar]
- 114.Kumar A, et al. Sustained efficacy of intra-articular FX006 in a rat model of osteoarthritis. Osteoarthritis and Cartilage. 2012;20:S289. doi: 10.1016/j.joca.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 115.Whitmire RE, et al. Self-assembling nanoparticles for intra-articular delivery of antiinflammatory proteins. Biomaterials. 2012;33(30):7665–7675. doi: 10.1016/j.biomaterials.2012.06.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liggins R, et al. Intra-articular treatment of arthritis with microsphere formulations of paclitaxel: biocompatibility and efficacy determinations in rabbits. Inflammation Research. 2004;53(8):363–372. doi: 10.1007/s00011-004-1273-1. [DOI] [PubMed] [Google Scholar]
- 117.Liang LS, et al. Methotrexate loaded poly (l-lactic acid) microspheres for intra-articular delivery of methotrexate to the joint. Journal of pharmaceutical sciences. 2004;93(4):943–956. doi: 10.1002/jps.20031. [DOI] [PubMed] [Google Scholar]
- 118.Bédouet L, et al. Synthesis of hydrophilic intra-articular microspheres conjugated to ibuprofen and evaluation of anti-inflammatory activity on articular explants. International Journal of Pharmaceutics. 2014;459(1–2):51–61. doi: 10.1016/j.ijpharm.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 119.Gaignaux A, et al. Development and evaluation of sustained-release clonidine-loaded PLGA microparticles. International journal of pharmaceutics. 2012;437(1):20–28. doi: 10.1016/j.ijpharm.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 120.Zhang Z, et al. Enhanced targeting efficiency of PLGA microspheres loaded with Lornoxicam for intra-articular administration. Drug delivery. 2011;18(7):536–544. doi: 10.3109/10717544.2011.596584. [DOI] [PubMed] [Google Scholar]
- 121.Zhang Z, Huang G. Intra-articular lornoxicam loaded PLGA microspheres: enhanced therapeutic efficiency and decreased systemic toxicity in the treatment of osteoarthritis. Drug delivery. 2012;19(5):255–263. doi: 10.3109/10717544.2012.700962. [DOI] [PubMed] [Google Scholar]
- 122.Bozdag S, et al. In vitro evaluation and intra-articular administration of biodegradable microspheres containing naproxen sodium. Journal of microencapsulation. 2001;18(4):443–456. doi: 10.1080/02652040010018641. [DOI] [PubMed] [Google Scholar]
- 123.Fernandez-Carballido A, et al. Sterilized ibuprofen-loaded poly (D, L-lactide-co-glycolide) microspheres for intra-articular administration: effect of γ-irradiation and storage. Journal of microencapsulation. 2004;21(6):653–665. doi: 10.1080/09687860400008437. [DOI] [PubMed] [Google Scholar]
- 124.Panusa A, et al. Methylprednisolone-loaded PLGA microspheres: A new formulation for sustained release via intra-articular administration. A comparison study with methylprednisolone acetate in rats. Journal of pharmaceutical sciences. 2011;100(11):4580–4586. doi: 10.1002/jps.22722. [DOI] [PubMed] [Google Scholar]
- 125.Horisawa E, et al. Prolonged anti-inflammatory action of DL-lactide/glycolide copolymer nanospheres containing betamethasone sodium phosphate for an intra-articular delivery system in antigen-induced arthritic rabbit. Pharmaceutical research. 2002;19(4):403–410. doi: 10.1023/a:1015123024113. [DOI] [PubMed] [Google Scholar]
- 126.Zille H, et al. Evaluation of intra-articular delivery of hyaluronic acid functionalized biopolymeric nanoparticles in healthy rat knees. Bio-medical materials and engineering. 2010;20(3):235–242. doi: 10.3233/BME-2010-0637. [DOI] [PubMed] [Google Scholar]
- 127.Eswaramoorthy R, et al. Sustained release of PTH(1-34) from PLGA microspheres suppresses osteoarthritis progression in rats. Acta Biomater. 2012;8(6):2254–62. doi: 10.1016/j.actbio.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 128.Ko J-Y, et al. Sulforaphane–PLGA microspheres for the intra-articular treatment of osteoarthritis. Biomaterials. 2013;34(21):5359–5368. doi: 10.1016/j.biomaterials.2013.03.066. [DOI] [PubMed] [Google Scholar]
- 129.Presumey J, et al. PLGA microspheres encapsulating siRNA anti-TNFalpha: efficient RNAimediated treatment of arthritic joints. European Journal of Pharmaceutics and Biopharmaceutics. 2012;82(3):457–464. doi: 10.1016/j.ejpb.2012.07.021. [DOI] [PubMed] [Google Scholar]