

Abstract
The use of ion/ion reactions to effect gas-phase alkylation is demonstrated. Commonly used fixed-charge “onium” cations are well-suited for ion/ion reactions with multiply deprotonated analytes because of their tendency to form long-lived electrostatic complexes. Activation of these complexes results in an SN2 reaction that yields an alkylated anion with the loss of a neutral remnant of the reagent. This alkylation process forms the basis of a general method for alkylation of deprotonated analytes generated via electrospray, and is demonstrated on a variety of anionic sites. SN2 reactions of this nature are demonstrated empirically and characterized using density functional theory (DFT). This method for modification in the gas phase is extended to the transfer of larger and more complex R groups that can be used in later gas-phase synthesis steps. For example, N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide (CMC) is used to transfer a carbodiimide functionality to a peptide anion containing a carboxylic acid. Subsequent activation yields a selective reaction between the transferred carbodiimide group and a carboxylic acid, suggesting the carbodiimide functionality is retained through the transfer process. Many different R groups are transferable using this method, allowing for new possibilities for charge manipulation and derivatization in the gas phase.
Keywords: Ion/ion reactions, “Onium” cations, Alkyl ion transfer, Esterification
Introduction
G as-phase alkylation through intracluster SN2 reactions has been observed for over a decade. These observations were made by using electrospray ionization (ESI) with high salt concentrations, which allows for the formation of an anion/cation salt complex bound together by electrostatic interactions. In many cases, a tetraalkylammonium reagent makes up the cation component of the complex and, upon collisional activation, transfers an alkyl cation to its counterion through an SN2 reaction. This transfer results in both the alkylation of the anionic reagent and formation of a neutral amine as the remainder of the reagent [1]. Collisional activation of the cluster ion can also result in an elimination pathway (E2), wherein a proton, rather than a charged alkyl group, is transferred from the cation to the anion counterpart. The competition between these two pathways has been subjected to in-depth scrutiny by Gronert [2]. Gas-phase alkylation in this manner has been observed with a variety of analytes, such as oligonucleotides [3], peptides [4, 5], lipids [6–9], and polyoxometalates [10–12], showing that the process can occur with a variety of analyte systems.
The use of relatively high concentrations of salts in sample solutions needed to generate the cluster ions in solution, however, can lead to experimental complications. Even millimolar concentrations of salts can be deleterious to ESI efficiency, leading to signal suppression [13, 14] and peak broadening [15]. In some cases, high salt concentration leads to frequent clogging of electrospray emitters (especially in the case of nano-ESI). Mass spectra obtained under such conditions are also often complicated by the appearance of prominent signals attributable to cluster ions unrelated to the analyte. Altering the bulk sample for ESI does not allow for fine control of which anions and cations are paired together in a cluster. In addition, the degree of analyte adduction (the number of cations attached to an anion) in a cluster is a difficult parameter to tune with ESI solution conditions. These issues related to altering bulk solution conditions can be avoided via the use of pulsed dual nESI [16] to carry out ion/ion reactions in the gas phase. Pulsed dual nESI provides facile control of the ratio of cations to anions to maximize the desired complex abundance in the gas phase. Adding the ability to isolate the desired reactants after pulsed dual nESI also removes much of the background chemical noise associated with bulk solution with high salt concentrations, simplifying ion/ion reaction spectra.
Ion/ion reactions can occur either via charged particle transfer at a crossing on the energy hypersurface or via a long-lived complex. The former pathway is most likely for small particles, such as an electron or a proton, and need not involve the formation of a long-lived complex. Small charged particle transfer, however, can also proceed via a complex. The reactions of multiply charged proteins of opposite polarity provide a case in which both proton transfer at a crossing point and complex formation are observed [17]. Reactions other than simple proton or electron transfer, on the other hand, generally require the formation of a long-lived complex intermediate. For example, sulfo-N-hydroxysuccinimide (sulfo-NHS)-containing reagents have been shown to form a complex with analytes containing primary amines or guanidino groups in the positive mode [18–20]. Activation of these complexes yields a signature loss of sulfo-NHS, indicating that a primary amine or guanidine within the analyte initiated a nucleophilic attack on the carbonyl carbon of the NHS reagent, ultimately resulting in stable amide bond formation [18]. This NHS reactivity in the gas phase allows for the modification of primary amine- or guanidine-containing peptides, as well as protein cross-linking in the gas phase [21]. Amide bond formation in the gas phase has also been reported using carbodiimide reagents to react with carboxylic acid-containing analytes in the negative mode [22].
The complexes involved in alkylation of an anion with tetraalkylammonium cations are inherently well-suited for investigation via ion/ion chemistry. In order to form a complex and subsequently effect a covalent modification in the gas phase, the participating cation and anion must satisfy two criteria. First, both ions must have “sticky” charge-bearing moieties that are likely to lead to strong electrostatic interactions in the ion/ion complex without low energy proton transfer channels. This condition maximinzes the possibility for the generation of a long-lived complex, first, allowing time for covalent chemistry to occur. Examples of “sticky” charge-bearing moieties include “onium” cations and sulf(on)ate and phosph(on)ate anions. Second, the reactants must contain reactive groups that will interact with each other to undergo the desired chemistry in a solvent-free environment. Tetraalkylammonium cations contain a fixed charge that is less prone to proton transfer reactions, while they also possess the alkyl chain necessary for gas-phase alkylation reactions. Similarly, anionic sites, including carboxylates, phosphates, and sulfonates in particular, are capable of forming a complex through ion/ion reactions. Once the complex is formed and activated, these anionic sites participate in the alkylation reaction by receiving a charged alkyl group.
Gas-phase alkyl ion transfer has been used within the context of lipid analysis by adducting a zwitterionic lipid to a singly charged anion such as formate, acetate, or chloride in the solution phase and subjecting the complexes generated in solution to collision-induced dissociation (CID) [23, 24]. This process leads to alkyl ion transfer to the counterion (i.e., formate, acetate, or chloride) leading to an analyte lipid anion that generates useful structural information upon subsequent activation. In a recent publication, Stutzman et al. demonstrated alkyl transfer from phosphatidyl choline cations with simultaneous proton transfer to doubly deprotonated dicarboxylic acid reagents to generate a charge inverted species. Collisional activation of the resultant lipid anion yielded more informative fragmentation compared with the singly charged cationic phosphatidyl choline species [25]. Gronert has utilized adducts between tetraalkylammonium cations and dianions generated in solution to study the competition between elimination (proton transfer) and substitution (alkylation) pathways in the gas phase [2], as well as demonstrate a degree of stereoselectivity in the alkylation process [4]. Herein, we demonstrate alkylation of anions in the gas phase via ion/ion reactions and compare this process to previously reported alkylation methods. DFT calculations are also performed to characterize the energy surface of both the elimination and substitution pathways in multiple reactions. Additionally, we demonstrate gas-phase alkylation as a means to functionalize anions. While previous studies have focused on the transfer of simple alkyl chains to anionic sites, we investigate using this gas-phase SN2 reaction to add new functional groups to analytes of interest through ion/ion reactions. Tandem MS studies are used to show that the reactive functionalities are maintained following transfer from an ammonium reagent to an anion.
Experimental
Materials
Tetramethylammonium acetate, tetraethylammonium chloride, trimethylsulfonium iodide, tetramethylphosphonium bromide, benzyltriethylammonium chloride, (2 -aminoethyl)trimethylammonium chloride hydrochloride, N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMC), 2,6-naphthalenedisulfonic acid (NDSA), adenosine 5′-triphosphate (ATP), and ethylenedi-aminetetraacetic acid (EDTA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The model peptides DGAI LDGAILD and DGAIL were synthesized by NeoBioSci (Cambridge, MA, USA). Methanol was purchased from Mallinckrodt (Phillipsburg, NJ, USA). Ammonium, phosphonium, and sulfonium reagents, peptides, EDTA, ATP, NDSA, and CMC were prepared as 0.1 mg/mL solutions in water and methanol in a 1:1 ratio.
Mass Spectrometry
All experiments were performed using a QTRAP 4000 hybrid triple quadrupole/linear ion trap (AB SCIEX, Concord, ON, Canada) that has been previously modified for ion/ion reactions using alternately pulsed dual nESI. Typical experiments for this work consisted of injection of anions (doubly deprotonated), followed by injection of cations (“onium” reagent) into the q2 reaction cell modified to allow for mutual storage of both ion polarities [26]. Reactants of both polarities that are injected into this reaction cell undergo rf/DC isolation as they pass through the preceding quadrupole cell, Q1. Once in Q2, the selected ions are allowed to react for a defined period of time (on the order of hundreds of milliseconds to a second). Reaction products are then transferred to Q3, where subsequent MSn experiments are performed, followed by the use of mass-selective axial ejection for mass analysis [27].
Density Functional Theory Calculations
DFT calculations were used to map out the reaction pathways of the various reagents complexed with carboxylate. All structure optimizations and energies were calculated using the Gaussian 09 package at the M06/6-311G++(2d,p) [28]. Transition states were searched for using QST2 and QST3 options and confirmed with intrinsic coordinate reaction (IRC) calculations.
Results and Discussion
Alkylation Via Ion/Ion Reactions
Long-lived complexes must retain an absolute charge greater than zero for subsequent activation and mass analysis following the initial ion/ion reaction. When using a fixed charge tetraalkylammonium monocation as the alkylating reagent, the analyte must be at least doubly deprotonated to preserve charge upon formation of a stable electrostatic complex. Ethylenediaminetetraacetic acid (EDTA) is a simple analyte that can be used to demonstrate gas-phase alkylation. EDTA contains four carboxylic acid functionalities, allowing for facile generation of the doubly deprotonated ion using nESI, as demonstrated in Figure 1a. In the case of alkylation of doubly deprotonated EDTA by fixed-charge tetraethylammonium cations, carboxylate sites serve both as anchors to form an electrostatic interaction and as the reactive sites for alkylation. Mutual storage of isolated [EDTA − 2H]2− and tetraethylammonium leads to the postion/ion reaction spectrum of Figure 1b. Proton transfer to yield the [EDTA − H]− is clearly apparent and, in principle, can arise from proton transfer at a crossing point on the ion/ion energy surface, ion/molecule proton transfer from adventitious neutral species in the vacuum system, and via a long-lived complex. Formation of a stable long-lived complex is also observed along with a product consistent with ethyl cation transfer and a nominal [EDTA − H −H2O]− ion. Isolation and activation of the complex via CID generates the product ion spectrum of Figure 1c. This spectrum shows the product consistent with ethyl cation transfer and concomitant loss of triethylamine, deprotonated EDTA from proton transfer, and the peak at m/z 273 (indicated by ⋄) consistent with [EDTA − H − H2O]−. This result shows that ethyl cation transfer and proton transfer are competitive channels in CID of the complex. Although water loss is a common occurrence in negative mode CID of some carboxylate-containing ions [29–31], the alkyl esterified EDTA anions generated either in solution or via ion/ion reactions all show dominant alcohol loss upon CID (data not shown). For example, ion trap collisional activation of the ethyl transfer product in Figure 1) shows exclusive loss of ethanol to yield the nominal [EDTA − H − H2O]− product. We therefore expect that the [EDTA − H − H2O]− product ion population can arise from sequential fragmentation in both the ethyl cation and proton transfer channels.
Figure 1.

A step-by-step demonstration of the alkylation of EDTA in the gas phase: (a) isolation of doubly deprotonated EDTA, (b) product ion spectrum following the ion/ion reaction of the anionic analyte with tetraethylammonium, and (c) collisional activation of the isolated electrostatic complex (the precursor ion is indicated with the lightning bolt image). Nominal water loss from the proton transfer peak as well as ethanol loss from the ethylated product, both m/z 273, are represented with an open diamond symbol (◇)
Performance of Alternate ‘Onium’ Reagents
Although quaternary ammonium-containing reagents have been shown to be effective alkylating reagents in the gas phase, sulfonium and phosphonium reagents were also investigated as alkylation reagents. Trimethylsulfonium has previously been shown to be a superior methylating reagent relative to tetramethylammonium in the sense that the transition state barrier and reaction enthalpy of methyl transfer from trimethylsulfonium are both lower than the respective values for methylation using tetramethylammonium [5]. Figure 2 displays the CID spectra of complexes consisting of doubly deprotonated EDTA with tetramethylammonium (Figure 2a), tetramethylphosphonium (Figure 2b), and trimethylsulfonium (Figure 2c). Activation of the complex between [EDTA − 2H]2− and tetramethylammonium exclusively yields methyl transfer, along with some consecutive methanol loss (Figure 2a). In contrast with the tetraethylammonium cation experiment summarized in Figure 1, the proton transfer channel is not present with tetramethylammonium. Conversely, activation of the analogous complex with tetramethylphosphonium displays only proton transfer (Figure 2b). It should be noted that phosphonium-centered reagents with longer alkyl chains also yield only proton transfer. Trimethylsulfonium, however, is capable of transferring either a proton or a methyl group, as observed in Figure 2c.
Figure 2.

Fragmentation spectra of [EDTA − 2H]2− complexed with (a) tetramethylammonium, (b) tetramethylphosphonium, and (c) trimethylsulfonium. The precursor ion being subjected to CID is indicated with the lightning bolt image. Nominal water loss from the proton transfer peak as well as methanol loss from the methylated product, both m/z 273, are represented with an open diamond symbol (◇)
In order to shed light on why these three reagents display different reactivities and to investigate the difference in reactivity between tetramethylammonium and tetraethylammonium, DFT calculations were performed using the Gaussian 09 package [32] to map the reaction pathway of both alkylation and proton transfer channels. In the latter case, comparing tetramethylammonium and tetraethylammonium, it is shown above that while tetramethylammonium solely follows the methyl transfer pathway (Figure 2a), tetraethylammonium can follow either pathway (Figure 1c). The energies of the transition states and products, determined at the M06/6-311++G(2d,p) level of theory and shown in Figure 3, are consistent with proton transfer and ethyl transfer being competitive processes. In these calculations, acetate is used as the model anion to which a proton or alkyl group is transferred. Figure 3 shows that the transition state energy for proton transfer (0.97 eV, blue) is close to but somewhat greater than that of ethyl transfer (0.91 eV, red). Both reactions are observed in Figure 2c with alkyl transfer being the more dominant process. Similar calculations were carried out for the three methyl ‘onium’ reagents (see below).
Figure 3.

A reaction coordinate displaying transition states and products of both the proton transfer (blue) and ethyl transfer (red) pathways from collisionally activating a complex between [EDTA − 2H]2− and tetraethylammonium. Coordinates for structures in the calculations are available as Supplemental Information
Table 1 lists the zero-point energies of products and transition states for the methyl cation transfer pathways and the zero-point energies of products for the proton transfer pathways relative to the complex for the three methyl “onium” reagents (ammonium, phosphonium, and sulfonium). Transition states could not be found for the proton transfer pathways for these three reagents. Nevertheless, some insights can be drawn from the calculated values. As seen in Figure 2a, activation of the complex of doubly deprotonated EDTA with tetramethylammonium apparently results exclusively in methyl cation transfer. With the absence of any evidence for [EDTA − H]−, the [EDTA − H − H2O]− ion is likely to arise from methyl loss from the alkyl cation transfer product. The calculations indicate that the transition state barrier for methyl transfer to acetate is 0.94 eV relative to the beginning complex, whereas the proton transfer products are at 1.79 eV relative to the complex. The products from methyl cation transfer, on the other hand, are calculated to be −0.49 eV relative to the complex. The highly endothermic nature of the proton transfer process alone can account for the lack of proton transfer from tetramethylammonium. The tetramethylphosphonium zero-point energy values show a far lower disparity in the product energies for proton transfer (1.20 eV) and methyl cation transfer (0.38 eV) than the tetramethylammonium case. However, perhaps more significant is the substantially higher transition state energy for methyl cation transfer (1.87 eV) from the tetramethylphosphonium cation compared with the tetramethylammonium case. The high barrier calculated for this process likely underlies the absence of alkyl ion transfer from tetraalkylphosphonium cations. The only case in the experiments of Figure 2, in which both methyl cation transfer and proton transfer occur, is with trimethylsulfonium as the cationic reactant (Figure 2c). The alkyl cation transfer process is similarly favorable thermodynamically (−0.91 eV) much like the tetramethylammonium cation (−0.49 eV), and the barrier for methyl cation transfer is lower at 0.75 eV from trimethylsulfonium. The fact that proton transfer is observed at all is likely related to the fact that the proton transfer channel is not as strongly disfavored (0.96 eV) as it is for the tetramethylammonium cation (1.79 eV).
Table 1.
The Calculated Zero-Point Energies of the Products and Transition States for Methyl Cation Transfer in the Dissociation of Acetate Complexes with Tetramethylammonium, Tetramethylphosphonium, and Trimethylsulfonium as well as the Zero-Point Energies of the Products from Proton Transfer from Dissociation of the Corresponding Complexes. All Calculations Were Performed Using M06/6-311++G(2d,p). Coordinates for Structures in the Calculations are Available as Supplemental Information
| Reagent | Proton transfer | Methyl transfer | |
|---|---|---|---|
|
| |||
| Products (eV) | TS (eV) | Products (eV) | |
| (CH3)4N+ | 1.79 | 0.94 | −0.49 |
| (CH3)4P+ | 1.20 | 1.87 | 0.38 |
| (CH3)3S+ | 0.96 | 0.75 | −0.91 |
Alkylation of Alternative Anionic Sites
Tetraalkylammonium reagents have also been found to alkylate a variety of anionic sites beyond carboxylates [3, 6–12]. Ethylation of sulfonate and phosphate anions is shown in Figure 4. Doubly deprotonated 2,6-naphthalenedisulfonic acid (NDSA) is used as a model sulfonate anion for alkylation in Figure 4a. As is the case for carboxylates, ethylation and proton transfer are both prevalent pathways upon CID of the reaction complex between tetraethylammonium and doubly deprotonated NDSA. The common neutral loss of SO3 [33] after proton transfer is also observed as a secondary fragment in lower abundance. Ethylation was demonstrated on other sulfonate-containing small organic molecules as well, yielding results similar to what was observed in experiments with NDSA.
Figure 4.

CID of the reaction complex between tetraethylammonium and (a) doubly deprotonated (NDSA) or (b) doubly deprotonated ATP. The precursor ion being subjected to CID is indicated with the lightning bolt image
Figure 4b displays the ethylation of a phosphate anionic site within adenosine 5′-triphosphate (ATP). CID of the complex between doubly deprotonated ATP and tetraethylammonium largely yields ethylated ATP along with common H3PO4 neutral losses. The proton transfer pathway is present, yet the singly deprotonated ATP abundance is minimal compared with that of ethylated ATP. The ion/ion method presented here appears to have the same diversity characterized by previous work [3–12] in the sense that many anionic sites can be alkylated with fixed-charged “onium” reagents regardless of whether the complex is generated in solution or via ion/ion reactions in the gas phase.
Alkylation Via Ion/Ion Reactions for Functionalizing Peptides
Most gas-phase alkylation studies have used solution-generated clusters to effect alkylation with small alkyl chains from tetraalkylammonium reagents. However, this method can also be used to modify anionic sites with either larger alkyl chains or alkyl groups with varying functional groups as shown in Figure 5a and b. For all sections of Figure 5, the doubly deprotonated model peptide, DGAIL, undergoes an ion/ion reaction with a quaternary ammonium reagent, forming complexes that are then isolated and subjected to CID. In Figure 5a, [DGAIL −2H]2− forms a complex with (2 -aminoethyl)trimethylammonium. When this complex is activated, both the alkylation and proton transfer pathways are observed. The most abundant product from the alkylation pathway is methylation of DGAIL, which is expected given that there are three methyl groups and only one 2-aminoethyl group. It is highly likely that the proton transfer from this CID experiment originates from the 2-aminoethyl group, as ammonium compounds with methyl groups have been shown to effect methylation exclusively (Figure 2a). Nonetheless, there remains a significant degree of 2-aminoethyl transfer, amounting to approximately one-third of the abundance of the methylation peak. In this case, it does not appear that one group (methyl versus 2-aminoethyl) is favored over the other for alkylation. The possibility of transferring a more bulky functional group is demonstrated in Figure 5b. Benzyltriethylammonium was reacted with doubly deprotonated DGAIL to form a stable reaction complex that, upon CID, proceeds through both alkylation and proton transfer pathways. In this case, however, the benzyl transfer pathway is by far the most abundant peak in the spectrum, despite being outnumbered 3:1 by ethyl groups, implying that it is much more favorable to transfer a benzyl group than an ethyl group. This observation agrees with an experiment performed by Gronert et al. [2] in which benzyl transfer within complex generated in the solution phase was the most favorable pathway, followed by proton transfer and ethyl transfer. Figure 5c demonstrates a case in which functional groups cannot be transferred with this alkylation process. CID of the reaction complex between doubly deprotonated DGAIL and (3-carboxypropyl)trimethylammonium results in only proton transfer. Even with three methyl groups on the ammonium reagent (bearing in mind that methyl groups do not display the capacity for proton transfer), there is no alkylation observed. This is likely due to the presence of the acidic proton located at the end of the carboxypropyl functionality. It should also be noted that during the ion/ion reaction, very little complex was observed. It would appear that transferring functional groups containing an acidic proton is a limitation of the process of alkylation through ion/ion reactions in the gas-phase.
Figure 5.

Demonstration of ‘alkylation’ via ion/ion reactions to functionalize the model peptide, DGAIL using (a) (2-aminoethyl)trimethylammonium, (b) benzyltriethylammonium, and (c) (3-carboxypropyl)trimethylammonium. The precursor ion being subjected to CID is indicated with the lightning bolt image. Nominal water losses are represented with a degree symbol (°) whereas ammonia loss is designated with an asterisk (*). Alcohol losses are again displayed with the open diamond symbol (◇). Nominal water loss from the proton transfer peak, as well as respective alcohol losses from the alkylated product, both appearing at m/z 273, are represented with an open diamond symbol (◇)
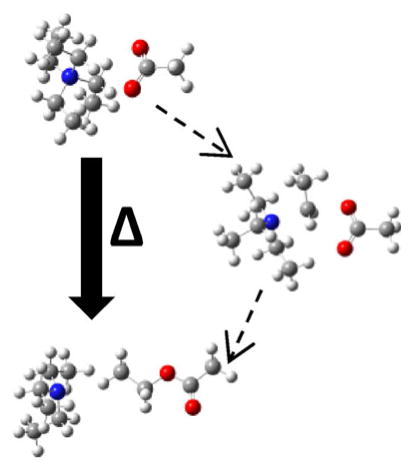
To demonstrate the retention of functional integrity of R groups transferred to anionic analytes, a reagent with the capacity to alkylate anions and react selectively with other moieties was used. N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide (CMC, Figure 6) is a reagent that has been previously studied in the gas phase. Briefly, CMC has a fixed positive charge on a quaternary ammonium with a carbodiimide functionality (see structure in Figure 6). In an ion/ion reaction, CMC forms an electrostatic complex with multiply charged anions. Upon CID of this complex, CMC selectively reacts with a carboxylic acid to form a stable amide bond, displaying a signature loss of an isocyanate derivative (125 Da neutral loss) in this covalent modification [22]. The carbodiimide functionality is located along a chain that stems from the quaternary ammonium moiety, making it a transferrable group using the methods described above. Doubly deprotonated DGAILDGAILD was used to demonstrate the transfer and utilization of a functional group using gas-phase alkylation. In this experiment, doubly deprotonated DGAILDGAILD reacts with CMC to form a long-lived complex. Upon isolation and CID of the complex, one pathway, as seen previously, is loss of 125 Da, which indicates amide bond formation via carbodiimide reactivity. Another pathway, however, is the transfer of the entire carbodiimide functionality from the quaternary ammonium to the peptide, adding 151 Da to DGAILDGAILD. This ion is isolated and subjected to further activation, resulting in the spectrum shown in Figure 6. Although the spectrum is dominated by various water losses that are characteristic of aspartic acid residues, the 125 Da loss (along with a water loss from the 125 Da loss) is present, indicating amide bond formation, and suggesting that the carbodiimide functionality has been preserved after the carbodiimide group is transferred to the peptide.
Figure 6.

CID of the alkylation product from reacting doubly deprotonated DGAILDGAILD with CMC. The precursor ion being subjected to CID is indicated with the lightning bolt image. The nominal water loss from the charge-reduced peak and the alcohol loss from the alkylated product are represented with an open diamond symbol (◇). Sequence fragments containing the amide bound modification from the selective reactivity of CMC are indicated by the double-dagger symbol (‡)
Conclusions
This work demonstrates the gas-phase alkylation of various anionic sites via ion/ion reactions with “onium” reagents. Tetraalkylammonium reagents are inherently well-suited as reactants in ion/ion reactions as they are “sticky” enough to form long-lived electrostatic complexes while also containing the desired reactive moieties. Sulfonium centers are also presented as a viable alkylation reagent, whereas phosphonium centers do not display any alkylation reactivity, apparently because of a high transition state barrier compared with the proton transfer pathway. Alkylation through ion/ion reactions can be used as a tool for functionalizing analytes in the gas-phase. As demonstrated in this and previous works, alkylation with “onium” reagents may be a general technique for O-alkylation. Although transfer using ion/ion reactions has been demonstrated with simple R groups, this technique also has the capability to transfer more reactive functionalities, such as carbodiimides. In the case of carbodiimide reagents, the selective reactivity towards carboxylic acids is preserved during the substitution reaction. However, functional groups containing an acidic proton, such as the 3-carboxypropyl group, seem to favor the proton transfer pathway more so than the alkylation pathway. The work presented here adds another tool for covalent modification in the gas phase. This method can be used to introduce new functional groups to a reagent as a gas-phase synthesis process.
Supplementary Material
Acknowledgments
James Riedeman, Brian Finney, and the Zwier Laboratory at Purdue are acknowledged for critical discussions that moved this work forward. This work was supported by the National Institutes of Health under grant GM 45372.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-015-1077-2) contains supplementary material, which is available to authorized users.
References
- 1.Gross DS, Williams ER. On the dissociation and conformation of gas-phase methonium ions. Int J Mass Spectrom. 1996;157:305–318. doi: 10.1016/S0168-1176(96)04407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gronert S. Gas phase studies of the competition between substitution and elimination reactions. Acc Chem Res. 2003;36:848–857. doi: 10.1021/ar020042n. [DOI] [PubMed] [Google Scholar]
- 3.Hodyss R, Cox HA, Beauchamp JL. Cluster phase reactions: alkylation of triphosphate and DNA anions with alkylammonium cations. J Phys Chem A. 2004;108:10030–10034. [Google Scholar]
- 4.Gronert S, Fagin AE, Okamoto K. Steroselectivity in the collision-activated reactions of gas phase salt complexes. J Am Soc Mass Spectrom. 2004;15:1509–1516. doi: 10.1016/j.jasms.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Yoo EJH, Feketeová L, Khairallah GN, O’Hair RAJ. Intercluster reactions show that (CH3)2S+CH2CO2H is a better methyl cation donor than (CH3)3N+CH2CO2H. Eur J Mass Spectrom. 2011;17:159–166. doi: 10.1255/ejms.1115. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X, Reid GE. Multistage tandem mass spectrometry of anionic phosphatidylcholine lipid adducts reveals novel dissociation pathways. Int J Mass Spectrom. 2006;242:242–255. [Google Scholar]
- 7.Lehmann WD, Koester M, Erben G, Keppler D. Characterization and quantification of rat bile phosphatidylcholine by electrospray-tandem mass spectrometry. Anal Biochem. 1997;246:102–110. doi: 10.1006/abio.1996.9941. [DOI] [PubMed] [Google Scholar]
- 8.Khaselev N, Murphy RC. Structural characterization of oxidized phospholipid products derived from arachidonate-containing plasmenyl glycerophosphocholine. J Lipid Res. 2000;41:564–572. [PubMed] [Google Scholar]
- 9.Pacetti D, Malavolta M, Bocci F, Boselli E, Frega NG. High-performance liquid chromatography/electrospray ionization ion-trap tandem mass spectrometric analysis and quantification of phosphatidylcholine molecular species in the serum of cystic fibrosis subjects supplemented with docosahexaenoic acid. Rapid Commun Mass Spectrom. 2004;18:2395–2400. doi: 10.1002/rcm.1639. [DOI] [PubMed] [Google Scholar]
- 10.Johnson GE, Hasan NM, Laskin J. Influence of heteroanion and ammonium cation size on the composition and gas-phase fragmentation of polyoxovanadates. Int J Mass Spectrom. 2013;354:333–341. [Google Scholar]
- 11.Cao J, Li C, Zhang Z, Xu C, Yan J, Cui F, Hu C. Intriguing role of a quaternary ammonium cation in the dissociation chemistry of Keggin polyoxometalate anions. J Am Mass Spectrom. 2012;23:366–374. doi: 10.1007/s13361-011-0296-4. [DOI] [PubMed] [Google Scholar]
- 12.Cao J, Xu C, Fan Y, Zhang X, Hu C. Selective production of electrostatically-bound adducts of alkyl cations/polyoxanions by the collision-induced fragmentations of their quaternary ammonium counterparts. J Am Soc Mass Spectrom. 2013;24:884–894. doi: 10.1007/s13361-013-0598-9. [DOI] [PubMed] [Google Scholar]
- 13.Constantopoulos TL, Jackson GS, Enke CG. Effects of salt concentration on analyte response using electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 1999;10:625–634. doi: 10.1016/S1044-0305(99)00031-8. [DOI] [PubMed] [Google Scholar]
- 14.Sojo LE, Lum G, Chee P. Internal standard signal suppression by coeluting analyte in isotope dilution LC-ESI-MS. Analyst. 2003;128:51–54. doi: 10.1039/b209521c. [DOI] [PubMed] [Google Scholar]
- 15.Nettleton EJ, Sunde M, Lai Z, Kelly JW, Dobson CM, Robinson CV. Protein subunit interactions and structural integrity of amyloidogenic transthyretins: evidence from electrospray mass spectrometry. J Mol Biol. 1998;281:553–564. doi: 10.1006/jmbi.1998.1937. [DOI] [PubMed] [Google Scholar]
- 16.Xia Y, Liang X, McLuckey SA. Pulsed dual electrospray ionization for ion/ion reactions. J Am Soc Mass Spectrom. 2005;16:1750–1756. doi: 10.1016/j.jasms.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Wells JM, Chrisman PA, McLuckey SA. Formation and characterization of protein–protein complexes in vacuo. J Am Chem Soc. 2003;125:7238–7249. doi: 10.1021/ja035051l. [DOI] [PubMed] [Google Scholar]
- 18.Mentinova M, McLuckey SA. Covalent modification of gaseous peptide ions with N-hydroxysuccinimide ester reagent ions. J Am Chem Soc. 2010;132:18248–18257. doi: 10.1021/ja107286p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mentinova M, McLuckey SA. Intra- and inter-molecular cross-linking of peptide ions in the gas phase: reagents and conditions. J Am Soc Mass Spectrom. 2011;22:912–921. doi: 10.1007/s13361-011-0103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mentinova M, Barefoot NZ, McLuckey SA. Solution versus gas-phase modification of peptide cations with NHS-ester reagents. J Am Soc Mass Spectrom. 2012;23:282–289. doi: 10.1007/s13361-011-0291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webb IK, Mentinova M, McGee WM, McLuckey SA. Gas-phase intramolecular protein crosslinking via ion/ion reactions: ubiquitin and a homobifunctional sulfo-NHS ester. J Am Soc Mass Spectrom. 2013;24:733–743. doi: 10.1007/s13361-013-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prentice BM, Gilbert JD, Stutzman JR, Forrest WP, McLuckey SA. Gas-phase reactivity of carboxylic acid functional groups with carbodiimides. J Am Soc Mass Spectrom. 2013;24:30–37. doi: 10.1007/s13361-012-0506-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pulfer M, Murphy RC. Electrospray mass spectrometry of phospholipids. Mass Spectrom Rev. 2003;22:332–364. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- 24.Han X, Gross RW. Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev. 2005;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 25.Stutzman JR, Blanksby SJ, McLuckey SA. Gas-phase transformation of phosphatidylcholine cations to structurally informative anions via ion/ion chemistry. Anal Chem. 2013;85:3752–3757. doi: 10.1021/ac400190k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia Y, Wu J, McLuckey SA, Londry FA, Hager JW. Mutual storage mode ion/ion reactions in a hybrid linear ion trap. J Am Soc Mass Spectrom. 2004;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 27.Londry FA, Hager JW. Mass selective axial ion ejection from a linear quadrupole ion trap. J Am Soc Mass Spectrom. 2003;14:1130–1147. doi: 10.1016/S1044-0305(03)00446-X. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account. 2008;120:215–241. [Google Scholar]
- 29.Bowie JH, Brinkworth CS, Dua S. Collision-induced fragmentations of the (M – H)− parent ions of underivatized peptides: an aid to structure determination and some unusual negative ion cleavages. Mass Spectrom Rev. 2002;21:87–107. doi: 10.1002/mas.10022. [DOI] [PubMed] [Google Scholar]
- 30.Brinkworth CS, Dua S, McAnoy AM, Bowie JH. Negative ion fragmentations of deprotonated peptides: backbone cleavages directed through both Asp and Glu. Rapid Commun Mass Spectrom. 2001;15:1965–1973. doi: 10.1002/rcm.457. [DOI] [PubMed] [Google Scholar]
- 31.Waugh RJ, Bowie JH, Hayes RN. Collision-induced dissociations of deprotonated peptides dipeptides containing aspartic or glutamic acids. J Mass Spectrom. 1991;26:250–256. [Google Scholar]
- 32.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C.01. Guassian, Inc; Walingford, CT, USA: 2010. [Google Scholar]
- 33.Attygalle AB, Garcia-Rubio S, Ta J, Meinwald J. Collisionally-induced dissociation mass spectra of organic sulfate anions. J Chem Soc, Perkin Trans. 2001;2:498–506. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.