

Summary
Mutations in the ADAR gene that encodes the ADAR1 RNA editing enzyme cause Aicardi-Goutières Syndrome (AGS), a severe autoimmune disease associated with an aberrant type I interferon response. How ADAR1 prevents autoimmunity remains incompletely defined. Here, we demonstrate that ADAR1 is a specific and essential negative regulator of the MDA5-MAVS RNA sensing pathway. Moreover, we uncovered a MDA5-MAVS-independent function for ADAR1 in the development of multiple organs. We showed that the p150 isoform of ADAR1 uniquely regulated the MDA5 pathway, whereas both the p150 and p110 isoforms contributed to development. Abrupt deletion of ADAR1 in adult mice revealed that both of these functions were required throughout life. Our findings delineate genetically separable roles for both ADAR1 isoforms in vivo, with implications for the human diseases caused by ADAR mutations.
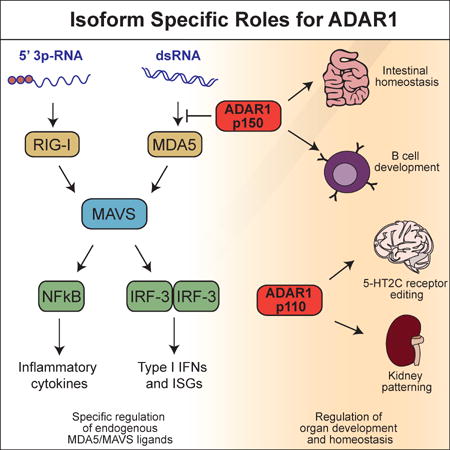
Graphical Abstract
Introduction
Intracellular detection of foreign nucleic acids initiates the production of type I interferons (IFNs) and is essential for host defense against virus infection (Goubau et al., 2013). Characterization of the RIG-I-like receptor-MAVS RNA sensing pathway and the cGAS-STING DNA sensing pathway has illuminated the earliest events of virus detection in molecular detail (Wu and Chen, 2014). Given the millions of molecules of RNA and the billions of base pairs of genomic DNA present in all nucleated cells, negative regulation of these pathways has emerged as a key mechanism to prevent autoreactivity. Much of our understanding of this regulation comes from the genetic dissection of a severe human autoimmune disease called Aicardi-Goutières Syndrome (AGS), first described over 30 years ago as a monogenic disorder associated with the aberrant production of type I IFNs (Aicardi and Goutieres, 1984; Lebon et al., 1988). Crow and colleagues have identified seven human genes that are mutated in AGS, providing a framework for studying the mechanisms that limit activation of intracellular nucleic acid sensors: TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1 (MDA5) (Crow et al., 2015; Crow and Manel, 2015).
Among the best characterized of the AGS enzymes is Trex1, a cytosolic DNA exonuclease. Trex1-deficient mice develop lethal autoimmune disease that is entirely dependent on cGAS, STING, the IRF3 transcription factor, the type I IFN receptor, and lymphocytes, thus defining Trex1 as a specific and essential negative regulator of the intracellular DNA sensing pathway (Ablasser et al., 2014; Gall et al., 2012; Gao et al., 2015; Gray et al., 2015; Stetson et al., 2008). Similarly, SAMHD1 is a dNTP phosphohydrolase that prevents cDNA synthesis by retroviral reverse transcriptase enzymes, strongly suggesting that the cGAS-STING pathway also drives autoimmunity in AGS caused by SAMHD1 mutations (Goldstone et al., 2011; Hrecka et al., 2011; Laguette et al., 2011; Rehwinkel et al., 2013). Interestingly, identification of the innate immune pathway regulated by RNase H2 is complicated by the early embryonic lethality of RNase H2-deficient mice, caused by accumulation of ribonucleotides in genomic DNA and massive genome instability (Hiller et al., 2012; Reijns et al., 2012).
The identification of AGS mutations in the ADAR gene that encodes the ADAR1 enzyme revealed a disease mechanism that does not fit into the cGAS-STING pathway (Rice et al., 2012). The ADAR gene in mammals encodes two open reading frames that give rise to two protein isoforms: a p110 isoform that is constitutively and ubiquitously expressed; and a larger p150 isoform that is IFN-inducible and contains two complete zDNA-binding domains that are not found in p110 (Bass, 2002; Schwartz et al., 1999). ADAR1 is a deaminase that converts adenosine to inosine (A to I) within double-stranded regions of RNA (Bass, 2002). In coding regions, inosine is decoded as guanosine (G), leading to amino acid changes or stop codon read-through at edited sites. In introns of mRNAs, editing can create or destroy splice acceptor sites. In double-stranded regions of RNA, the I-U base pair is unstable compared to the parental A-U base pair, leading to changes in secondary structure of RNA that can result in degradation (Wang et al., 2013). In mammals, ADAR1 edits coding RNAs (Hartner et al., 2004), microRNAs (Yang et al., 2006), and the RNA transcripts of the prolific SINE retroelements (Osenberg et al., 2010). Interestingly, ADAR1 deficiency in mice results in embryonic lethality, accompanied by massive overproduction of type I IFNs and hematopoietic failure (Hartner et al., 2004; Hartner et al., 2009; Wang et al., 2000). A number of mechanisms have been proposed to explain the dramatic phenotype of Adar−/− mice, including roles for ADAR1 in modulation of microRNA biogenesis (Ota et al., 2013), control of the length of mRNA 3′ untranslated regions (UTRs; Bahn et al., 2015), physical sequestration of RNAs from RIG-I (Yang et al., 2014), and the direct suppression of RIG-I and MDA5 activity by RNAs that contain inosine (Vitali and Scadden, 2010). In addition, a recent study identified live births of Adar−/−Mavs−/− mice (Mannion et al., 2014). However, the precise innate immune pathways regulated by ADAR1 and the contributions of these pathways to the phenotypes of Adar−/− mice remain undefined.
In this study, we define ADAR1 as a specific negative regulator of the MDA5-MAVS pathway. Moreover, we reveal an essential role for ADAR1 in multi-organ development and homeostasis that is independent of the MDA5-MAVS pathway. We show that the two isoforms of ADAR1 contribute independently to each of these functions.
Results
ADAR1 is a specific negative regulator of the MDA5-MAVS pathway
To define the innate immune receptor(s) and signaling pathways responsible for the embryonic lethality of Adar−/− mice, we analyzed over 700 live births of Adar+/− intercrosses, alone and on four additional genetic backgrounds: Tmem173 (Sting)−/−, Mavs−/−, Ifih1 (Mda5)−/−, and Ddx58 (Rigi)+/−. Consistent with previous reports defining fully penetrant embryonic lethality of Adar−/− mice (Hartner et al., 2004; Wang et al., 2000), we found no live births of Adar−/− mice (Figure 1A). Similarly, no Adar−/−Tmem173−/− mice were born, and the frequencies of live Adar+/+Tmem173−/− and Adar+/−Tmem173−/− mice were indistinguishable from mice born from the Adar+/− intercross, formally demonstrating that the STING pathway plays no role in the phenotypes associated with ADAR1 deficiency (Figure 1B). In contrast, and consistent with a recent report (Mannion et al., 2014), Adar−/−Mavs−/− mice were born at frequencies that were statistically indistinguishable from expected Mendelian ratios (Figure 1C). Thus, the MAVS pathway, and not the STING pathway, drives the embryonic lethality of Adar−/− mice.
Figure 1. Rescue of Adar−/− mice to birth by Ifih1/Mda5 and MAVS deficiency.

(A) Live births from Adar+/− intercross.
(B) Live births from Adar+/−Tmem173−/− intercross.
(C) Live births from Adar+/−Mavs−/− intercross.
(D) Live births from Adar+/−Ifih1−/− intercross.
(E) Live births from Adar+/−Ddx58+/− intercross. Percent rescue p>0.05 by Chi Square goodness-of-fit for Adar+/−Mavs−/− intercross and Adar+/−Ifih1−/− intercross.
We next evaluated intercrosses of Adar+/− mice with mice lacking the two principal RNA sensors upstream of MAVS: MDA5 (Ifih1) and RIG-I (Ddx58; Kato et al., 2006). Remarkably, Adar−/−Ifih1−/− mice were born at expected Mendelian frequencies (Figure 1D). The Adar-Ddx58 cross was complicated by the embryonic lethality of most Ddx58−/− mice (Kato et al., 2005), which occurs for reasons that remain poorly defined. Therefore, we bred Adar+/− mice to Ddx58+/− mice and analyzed over 300 live births. We recovered only two Ddx58−/− single mutant mice and no Adar−/−Ddx58−/− mice (Figure 1E).
We performed timed matings of Adar+/− mice on the four backgrounds described above, harvested embryos at day 11.5 of development (E11.5, before the onset of lethality in Adar−/− mice), and assessed the innate immune response using quantitative RT-PCR analysis of six interferon stimulated genes (ISGs). We found that MAVS deficiency completely reversed the elevated ISG signature of Adar−/− embryos, but STING deficiency had no effect (Figure 2A). Moreover, and identical to Adar−/−Mavs−/− embryos, we found that Adar−/−Ifih1−/− embryos also lacked the increased ISG expression seen in Adar−/− embryos. Importantly, the single Adar−/−Ddx58−/− embryo that we recovered had elevated ISG expression that was identical to control Adar−/−Ddx58+/+ embryos (Figure 2B). Taken together, these data define MDA5-MAVS, and not RIG-I-MAVS, as the specific innate immune pathway responsible for both the dysregulated ISG expression and the embryonic lethality of Adar−/− mice.
Figure 2. MDA5 and MAVS deficiency reverse the IFN signature in Adar−/− embryos.

Quantitative RT-PCR on a panel of 6 ISGs from whole E11.5 embryos.
(A) Adar+/+ (white, n=3), Adar−/− (grey, n=3), Adar−/−Sting−/− (blue, n=4), Adar−/−Mavs−/− (red, n=5). Interferon signature assessed by Wilcoxon Signed Rank test compared to Adar+/+: Adar−/− p=0.03, Adar−/−Sting−/− p=0.03, Adar−/−Mavs−/− p=0.31.
(B) Adar+/+Ifih1−/− (white, n=3), Adar−/−Ifih1−/− (red, n=3), Adar−+/+Ddx58+/+ (white stripes, n=3), Adar−−/−Ddx58+/+ (grey stripes, n=2), Adar+/+Ddx58−/− (light blue stripes, n=2), Adar−−/−Ddx58−/− (blue stripes, n=1). Interferon signature assessed by Wilcoxon Signed Rank test compare to Adar+/+controls, Adar−/−Ifih1−/− p=0.56, Adar−−/−Ddx58+/+ p=0.03, Adar+/+Ddx58−/− p=0.69, Adar−−/−Ddx58−/− p=0.03.
To extend our findings to human cells, we used a lenti-CRISPR approach to disrupt the endogenous ADAR gene in HEK 293T cells. We designed a guide RNA to target an exon shared by both isoforms of ADAR1, transduced and selected targeted cells, and then derived a clonal line of ADAR-null cells with frameshift mutations in all three alleles of ADAR (Figure 3A; HEK 293T cells are triploid for chromosome 3). We confirmed by western blot that these ADAR-null cells lacked expression of both isoforms of ADAR1 protein (Figure 3B). We then introduced expression vectors for either RIG-I or MDA5 into these cells or control cells, and measured the IFN response using an interferon-stimulated response element (ISRE)-luciferase reporter. We found that RIG-I responded identically in control and ADAR-null cells at each concentration of transfected expression vector (Figure 3C). Remarkably, the MDA5-activated IFN response was enhanced in ADAR-null HEK 293T cells compared to control cells at all concentrations of plasmid (Figure 3C). These findings, together with our analysis of Adar−/− mice and embryos, strongly suggest that ADAR1 deficiency results in the accumulation of endogenous, immunostimulatory RNAs that are specifically detected by MDA5 and not by RIG-I.
Figure 3. ADAR1 specifically regulates the MDA5 pathway in human cells.

(A) Generation of ADAR-null HEK 293T cells by lentiCRISPR targeting. The CRISPR target site in exon 4 of the ADAR gene is indicated in blue, and the protospacer adjacent motif (PAM) is shown in green. The Cas9 cleavage site is indicated with an arrow. Deletions in the three ADAR alleles, each of which results in a frameshift, are shown by the red dashes.
(B) Western blot of ADAR1 protein using lysates from control HEK 293T cells and an ADAR-lentiCRISPR-targeted clone of HEK 293T cells.
(C) The indicated HEK 293T cells were transfected with 25ng ISRE-luciferase reporter plasmid, with or without the indicated amounts of plasmids encoding RIG-I or MDA5. Cells were analyzed for relative luciferase units 24 hours after transfection. Mean±SD; ***: p<0.0001 in Two way ANOVA test with Tukey’s multiple comparison. Data are representative of 4 independent experiments.
MAVS-dependent and MAVS-independent gene expression changes in Adar−/− embryos
To assess global changes in gene expression caused by ADAR1 deficiency beyond the specific ISGs analyzed in Figure 2, we performed RNA-Seq analysis of gene expression in whole E11.5 embryos, comparing Adar−/− to Adar+/+ embryos, as well as Adar−/−Mavs−/− to Adar+/+Mavs−/− embryos (Figure 4 and Supplementary Tables 1 and 2). In both cases, the great majority of dysregulated genes exhibited increased expression in the Adar−/− embryos compared to respective controls, suggesting that ADAR1 is primarily a negative regulator of gene expression (Figure 4A, 4B). We found over 900 genes with significantly different expression levels in Adar−/− versus Adar+/+ embryos, 180 of which were ISGs (Figure 4A). As expected, the increased expression of nearly all of these ISGs was restored to normal levels in the Adar−/−Mavs−/− embryos (Figure 4A, 4B, red dots). However, and unexpectedly, we identified over 200 genes with similarly dysregulated expression in both Adar−/− and Adar−/−Mavs−/− embryos compared to their respective controls (Figure 4A–4C, blue dots). Bioinformatics analysis of these genes revealed highly enriched biological processes and transcription factor networks controlled by ADAR1 in a MAVS-independent fashion. These included genes involved in lipid metabolism and transport, associated with up-regulation of PPAR-γ transcriptional targets in Adar−/− embryos (Figure 4D, 4E). Moreover, we identified targets of the TCF1, TCF14, TCF3, and HOXA13 transcription factors that control cell fate specification during development (Figure 4D, 4E). Importantly, we found no evidence for residual inflammatory gene expression in Adar−/−Mavs−/− embryos (Fig. 4D, 4E), suggesting that the entire dysregulated innate immune response caused by ADAR1 deficiency depends on MAVS signaling. Thus, ADAR1 controls expression of two classes of genes: the first class is innate immune response genes driven by MAVS signaling, whereas the second class is MAVS-independent and implicated in development and metabolism.
Figure 4. MAVS-dependent and MAVS-independent gene expression in Adar−/− embryos.

RNA-Seq was performed on rRNA-depleted RNA from whole E11.5 embryos of the indicated genotypes.
(A) Comparison of gene expression between Adar−/− (n=3) and Adar+/+ (n=3) embryos. Data are plotted as log2 fold change in gene expression on the y-axis, with normalized log2 counts per million (CPM) on the x-axis. Grey dots denote genes with insignificant differences in expression. Blue dots denote non-ISGs with differential expression (p≤0.01). Red dots indicate ISGs with differential expression (p≤0.01).
(B) Comparison of gene expression between Adar−/−Mavs−/− (n=3) and Mavs−/− (n=3) embryos, using the same criteria as in (A).
(C) Genes with differential expression (p≤0.01) in either pairwise comparison were plotted, with Adar−/− vs. Adar+/+ on the y-axis, and Adar−/−Mavs−/− vs. Mavs−/− on the x-axis. Blue and red genes are the same as (A) and (B).
(D) Biological pathways enriched among the genes with dysregulated expression in both Adar−/− embryos and Adar−/−Mavs−/−embryos identified in (C).
(E) Transcription factor binding sites enriched among the MAVS-independent differentially expressed genes from (C).
For D and E, fold enrichment relative to the representation of these pathways in the genome is shown on the left y-axis and the blue bars. Significance of enrichment is indicated by hyper geometric p-value on the right y-axis and the black symnbols/line. Analysis was performed using FunRich software.
MDA5-MAVS-independent control of multi-organ development by ADAR1
Consistent with the identification of a substantial set of MAVS-independent genes regulated by ADAR1 (Figure 4), we observed a fully penetrant postnatal mortality in both Adar−/−Mavs−/− mice and Adar−/−Ifih1−/− mice, with the majority of neonates dying by two days of age (Figure 5A, 5B). We analyzed the phenotypes of the small number of Adar−/−Mavs−/− mice that survived past one week of age, comparing them to Adar+/+Mavs−/− littermates. The double knockout mice were severely runted compared to their Mavs−/− littermates, but they appeared to be feeding, as evidenced by the presence of milk in their stomachs (data not shown). However, the Adar−/−Mavs−/− mice were largely unresponsive and immobile, with a “trembling” phenotype (Supplementary Video 1). We performed a thorough histological analysis of tissues and organs in three mice of each genotype, revealing a number of novel, MAVS-independent developmental phenotypes caused by ADAR1 deficiency. First, the kidneys of Adar−/−Mavs−/− mice had a profound change in architecture, with multiple discrete lobes of the outer medulla and marked disorganization of the tubules in the corticomedullary junction (Figure 5C). This contrasts with the single-lobed outer medulla of normal rodent kidneys, revealing an unexpected role for ADAR1 in regulation of kidney patterning during development. Second, we observed a dramatic dysregulation of gastrointestinal homeostasis in Adar−/−Mavs−/− mice (Figure 5C). This phenotype was apparent throughout the intestine, although most pronounced in the small intestine. Intestinal lesions included moderate superficial enterocyte vacuolation, mild to moderate villar fusion, and extensive apoptosis of enterocytes that was especially pronounced in the crypts, with mild proliferation and mild mixed inflammation (enteritis; Figure 5C). Third, we found a near complete lack of organized lymphoid follicles in both lymph nodes and spleens of Adar−/−Mavs−/− mice, on both histology (Figure 5C) and immunofluorescence microscopy performed to localize T cells and B cells (Figure 5D). In the spleens of Adar−/−Mavs−/− mice, there was extramedullary hematopoiesis of both myeloid and erythroid lineages (Figure 5C). Fourth, flow cytometry analysis of splenocytes revealed a dramatic reduction in the frequency of mature B cells, but relatively normal numbers of T cells, as well as increased frequencies of CD11c+ dendritic cells (Figure 5E). These phenotypes of Adar−/−Mavs−/− mice reveal an essential role for ADAR1 in the development and homeostasis of multiple organs, independent of its role as a negative regulator of the MDA5-MAVS-mediated antiviral response. Importantly, we demonstrate that the failure of hematopoiesis in Adar−/− mice is an indirect consequence of the aberrant, MDA5-MAVS-dependent antiviral response, revealing instead a selective, MDA5-MAVS-independent role for ADAR1 in development of B cells.
Figure 5. Postnatal mortality and severe developmental defects in Adar−/−Mavs−/− mice.

(A) Postnatal survival curves for Adar−/−Mavs−/− mice.
(B) Postnatal survival curves for Adar−/−Ifih1−/− mice.
(C) Adar−/−; Mavs−/− mice have developmental defects of the kidney (top row; papillae marked with black asterisks), small intestine (second row), lymph node (third row; follicles marked with white asterisks), and spleen (fourth row; lymphoid regions enclosed by dashed circle). Images are hematoxylin and eosin with magnification indicated.
(D) Immunofluorescence microscopy of splenic sections with anti-B220 (green), anti-CD8 and anti-CD4 (red), and DAPI (blue).
(E) Analysis of splenocytes by flow cytometry shows severe B cell deficiency in Adar−/−; Mavs−/− mice. Mice in C-D were 20 days old. Mice in E were 15 days old. Mice in F were 13 days old.
Independent contributions of ADAR1 isoforms to regulation of MDA5-MAVS and development
We evaluated the relative in vivo contributions of the p150 and p110 isoforms of ADAR1 to regulation of MDA5-MAVS-dependent autoimmunity versus MDA5-MAVS-independent organ development. To do this, we studied Adar p150−/− mice, which were generated by targeted disruption of the unique promoter and first exon that encodes the amino terminus of the p150 isoform (Ward et al., 2011). Adar p150−/− mice are embryonic lethal, similar to Adar−/− mice that lack both the p150 and p110 isoforms of ADAR1 (Ward et al., 2011). Given that the embryonic lethality of Adar−/− mice is entirely MDA5- and MAVS-dependent (Figure 1), we hypothesized that the ADAR1 p150 isoform might specifically regulate the MDA5-MAVS pathway. We performed western blot analysis of Adar p150+/− and Adar p150−/− MEF extracts and confirmed the absence of p150 protein in these cells, but normal levels of the p110 isoform (Figure 6A). Next, we intercrossed Adar p150+/−Mavs−/− mice and observed complete rescue of the Adar p150−/−Mavs−/− mice to birth, demonstrating that the p150 isoform of ADAR1 is the unique and essential negative regulator of the MAVS pathway (Figure 6B). Remarkably, and unlike the Adar−/−Mavs−/− mice that died shortly after birth (Figure 5A), most of the Adar p150−/−Mavs−/− mice survived to weaning (Figure 6C). Moreover, these mice were active and mobile, ate solid food, and did not tremble, although they were smaller than their control littermates (Supplementary video 1). We performed a histological analysis of Adar p150−/−Mavs−/− mice, comparing them to Adar−/−Mavs−/− mice (Figure 5) and to controls. We found that the kidney abnormalities that were present in the Adar−/−Mavs−/− mice were largely absent in Adar p150−/−Mavs−/− mice, demonstrating that p110 is a specific regulator of kidney development (Figure 6D). However, the dysregulated intestinal homeostasis was similar between Adar p150−/−Mavs−/− mice compared to the Adar−/−Mavs−/− mice (Figure 6D). Moreover, the lymph nodes and spleens of Adar p150−/−Mavs−/− mice were still devoid of lymphoid follicles (Figure 6D). Consistent with this, we observed reduced numbers of peripheral B cells and increased numbers of CD11c+ myeloid cells in Adar p150−/−Mavs−/− mice (Figure 6E), identical to Adar−/−Mavs−/− mice (Figure 5E). Thus, expression of the p110 isoform of ADAR1 in Adar p150−/−Mavs−/− mice is sufficient to restore kidney development, but not intestinal homeostasis or B cell development.
Figure 6. Independent roles for ADAR1 isoforms in regulation of MDA5-MAVS and development.

(A) Western blot of ADAR1 protein expression in p150+/− or p150−/− MEFs.
(B) Live births for mice from the p150+/−Mavs−/− intercross.
(C) Postnatal survival curves for mice from the p150+/−Mavs−/− intercross.
(D) Hematoxylin and eosin-stained tissue sections of the indicated organs of p150−/−; Mavs−/− mice and controls are shown, with magnification indicated.
(E) Analysis of splenocytes by flow cytometry shows severe B cell deficiency and an increase in CD11c+ myeloid cells in p150−/−Mavs−/− mice. Two sample t-test for p values p150+/+Mavs−/− n=3, p150−/−Mavs−/− n=3 *** p<0.002, **p=0.004.
(F) Representative chromatograms of 2-HT2C receptor transcript editing in brains of 15–21 day old mice of the indicated genotypes. Mice in E and F were 21 days old. Mice in D were 15 days old.
We next examined editing of the mRNA encoding the 5-HT2C serotonin receptor that mediates numerous actions of serotonin in the central nervous system, including feeding and sleep behavior, as well as inhibition of neuronal excitability (Frank et al., 2002; Tecott et al., 1995). A prior study identified ADAR1-dependent editing at two specific adenosines within the 5-HT2C open reading frame in cultured embryonic neurons, termed the “A” and “B” sites (Hartner et al., 2004). We therefore measured editing of these sites in cDNAs prepared from brains of ~20 day old Adar−/−Mavs−/− and Adar p150−/−Mavs−/− mice. Consistent with the prior study, we found that both the “A” and “B” sites of 5-HT2C mRNAs were extensively edited in control Mavs−/− mice, and that this editing was absent in Adar−/−Mavs−/− mice (Figure 6F). Interestingly, the Adar p150−/−Mavs−/− mice had normal levels of editing at these sites, demonstrating that ADAR1 p110 is both necessary and sufficient for editing of these sites in vivo (Figure 6F). This finding reveals a specific editing event mediated by p110 and not by p150. These, as well as other potential p110-specific editing events that remain to be identified, may contribute to the phenotypic differences between Adar−/−Mavs−/− mice and Adar p150−/−Mavs−/− mice. Together, these data demonstrate independent roles for ADAR1 isoforms: the p150 isoform regulates the MDA5-MAVS pathway, and both isoforms contribute to development.
ADAR1 controls both innate immunity and homeostasis in adult mice
Our findings demonstrating roles for ADAR1 in controlling both the MDA5-MAVS pathway and multi-organ development led us to test whether these functions are similarly required in adult mice. We crossed mice with a floxed Adar allele (Adarfl/fl; (Hartner et al., 2009) to mice expressing a tamoxifen-inducible Ert2-Cre transgene under the control of the broadly expressed UBC promoter (Ruzankina et al., 2007), on both Mavs+/+ and Mavs−/− backgrounds. We treated these mice daily for three days by intraperitoneal injection of tamoxifen, which resulted in widespread deletion of the floxed Adar alleles in Ert2-Cre-expressing mice as measured by genotyping of ear tissue (Figure 7A). We found that the tamoxifen injections resulted in a drop in body temperature in all treated mice, regardless of Cre expression or Adar genotype (Figure 7B). One day after the third tamoxifen treatment, and only four days after the initial treatment, we found that the Adarfl/flMavs+/+Ert2-Cre-expressing mice had an exacerbated reduction in body temperature (Figure 7B), were hunched and unresponsive, and required immediate euthanasia. We measured the abundance of 32 serum cytokines and chemokines in these mice and controls and found dramatically elevated levels of many of them, including TNFα, CXCL10, and IL-10, thus revealing a profound systemic inflammatory response instigated by abrupt Adar deletion on a Mavs+/+ background (Figure 7C). In contrast, the Adarfl/flMavs−/−Ert2-Cre-expressing mice recovered body temperature after cessation of tamoxifen treatments and appeared healthy for five more days (Figure 7B). At day 8 post treatment, we noted that these mice were abnormal compared to controls, so we prepared these mice for serum cytokine analysis and histological evaluation. We found that the systemic cytokine response was absent in the Adarfl/flMavs−/−Ert2-Cre-expressing mice, demonstrating that MAVS controls the entire inflammatory response caused by Adar deletion in adult mice (Figure 7C). However, we found that the small intestines of mice with Adar deletion on a Mavs−/− background were shortened relative to Cre-expressing control mice, and their colons were thickened and devoid of formed stools (Figure 7D). Histological analysis revealed a disruption of intestinal homeostasis in these mice (Figure 7E, 7F), reminiscent of the intestinal phenotype of Adar−/−Mavs−/− and Adar p150−/−Mavs−/− mice. Together, these data demonstrate that the requirement for ADAR1 in regulation of both the MDA5-MAVS pathway and tissue homeostasis is maintained in adult mice.
Figure 7. ADAR1 regulates both the MDA5-MAVS pathway and tissue homeostasis in adults.

Mice were treated with 2 mg tamoxifen i.p. daily for 3 days and observed for gross pathology.
(A) Samples of the ear were genotyped for Adar deletion following tamoxifen treatment.
(B) Surface body temperate readings for tamoxifen-treated mice from 20–120 hours after initial tamoxifen injection.
(C) Serum cytokine levels determined from mice at Day 3 or Day 8.
(D) Ex vivo dissection of the gastrointestinal tract at Day 8; Stomach (S), small intestine (SI), cecum (Ce) and large intestine (LI) are indicated.
(E) Representative hematoxylin and eosin-stained ileum sections of tamoxifen-treated mice of the indicated genotypes at Day 8. Original magnification is indicated in the bottom right.
(F) Histological scoring of inflammation in the gut at Day 8. SI=small intestine; CE=cecum; Prox=Proximal colon; Mid=Mid-colon. Mean inflammation score of Adarfl/flMavs−/− UBC:Ert2Cre+ intestines compared to either control is significant by one-way ANOVA, p=0.002.
Discussion
Our findings reveal two important roles for ADAR1. First, ADAR1 is a specific and essential negative regulator of the MDA5- and MAVS-dependent antiviral response. Second, ADAR1 is a key regulator of multi-organ development and homeostasis, independent of the MDA5-MAVS pathway. These roles are both genetically and temporally separable, with the MDA5-MAVS pathway entirely responsible for the embryonic lethality, and the MDA5-MAVS-independent pathway responsible for the postnatal mortality of Adar−/− mice. Moreover, we demonstrate that ADAR1 isoforms independently contribute to these two functions, with the p150 isoform essential for regulation of the MDA5-MAVS pathway, and the p110 isoform contributing to development. Finally, we demonstrate that ADAR1 regulates both innate immunity and tissue homeostasis in adult mice.
Our data provide insight into the ADAR gene mutations in humans that cause AGS (Crow et al., 2015; Rice et al., 2012). The regulation of the MDA5-MAVS pathway by ADAR1 p150, and not the RIG-I-MAVS pathway, strongly suggests that ADAR1 p150 modifies a discrete pool of RNAs to prevent their specific detection by MDA5. Moreover, our findings reveal a clear genetic pathway linking the ADAR and IFIH1 mutations found in AGS (Rice et al., 2014; Rice et al., 2012), a rationale for the ADAR AGS mutations that affect only the p150 isoform of ADAR1 (Crow et al., 2015; Rice et al., 2012), and a biological framework for understanding the numerous IFIH1 gene polymorphisms in humans that are associated with type I diabetes, systemic lupus erythematosus, and Graves disease (Gateva et al., 2009; Nejentsev et al., 2009; Smyth et al., 2006; Sutherland et al., 2007). Given the embryonic lethality and robust IFN signature of Adar−/− mice, as well as the rapid MAVS-dependent inflammatory response that arises after abrupt Adar deletion in adults, we propose that the ADAR1-regulated endogenous MDA5 RNA ligands are broadly expressed, highly immunostimulatory, or both. Definitive identification of these RNAs will provide insight into the elusive ligand specificity of MDA5 (Wu et al., 2013), with implications for the underlying mechanisms of self/non-self discrimination by intracellular nucleic acid sensors.
Analysis of Adar−/− and Adar p150−/− mice on a Mavs−/− background revealed isoform-specific contributions of ADAR1 to kidney development, intestinal homeostasis, B cell development, and 5-HT2C serotonin receptor editing. Further work will be required to determine whether these functions are mediated by “precision editing” of adenosines in mRNA open reading frames that result in new coding potential (as is the case for 5-HT2C), or whether some of these functions require previously described roles for ADAR1 in control of micro-RNA processing, mRNA stability, or mRNA 3′-UTR length (Ota et al., 2013; Bahn et al., 2015).
Importantly, no AGS patients are homozygous for null alleles of ADAR, and most AGS-mutant ADAR1 enzymes are competent for RNA editing in vitro (Rice et al., 2012). Distinct ADAR mutations in humans also cause dyschromatosis symmetrica hereditaria (DSH; OMIM 127400; (Hayashi and Suzuki, 2013) and bilateral striatal necrosis (BSN; OMIM 271930; (Livingston et al., 2014). Some DSH and BSN cases are associated with a mild IFN signature that is less pronounced compared to AGS with ADAR mutations, but the clinical presentations of DSH and BSN differ significantly from classical AGS (Livingston et al., 2014; Rice et al., 2012). Our identification of an MDA5-MAVS-independent role for ADAR1 in development and tissue homeostasis may shed light on the various human phenotypes associated with ADAR mutations. We propose that the more than 150 known mutations in ADAR represent a spectrum of effects on ADAR1 regulation of the MDA5-MAVS pathway versus ADAR1 control of tissue homeostasis. Specific disease presentations may reflect the extent to which each pathway is compromised by a particular mutation. For example, AGS mutations likely impact primarily the MDA5-MAVS response controlled by the p150 isoform, leaving the developmental roles of ADAR intact.
A very recent study by Walkley and colleagues described the phenotype of knockin mice with a point mutation in the Adar gene that disrupts catalytic activity (E861A; (Liddicoat et al., 2015). Consistent with our findings, they identified two individual AdarE861A/E861AIfih1−/− mice that were rescued to birth. Interestingly, and in contrast to our data demonstrating fully penetrant postnatal mortality in Adar−/−Ifih1−/− mice, the rescued AdarE861A/E861AIfih1−/− mice were largely normal in appearance, with no evident gross lesions. It is tempting to speculate that the differences in postnatal phenotypes revealed in our study and theirs may reflect an important distinction between the catalytic mutant and the null allele of Adar. A direct comparison of the null allele and the catalytic mutant allele of Adar on an Ifih1−/− or Mavs−/− background would resolve this issue.
In summary, we have identified independent roles for ADAR1 isoforms in regulation of the antiviral response and control of tissue development and homeostasis, with implications for the human diseases caused by ADAR mutations.
Experimental Procedures
Mice
Adarfl/fl mice were kindly provided by Dr. Stuart Orkin (Hartner et al., 2009) and were bred to B6.129S4-Meox2tm1(cre)Sor mice to delete the Adar allele in the germline, and to B6;129S-Tg(UBC-cre/ERT2)1Ejb to allow for tamoxifen-induced widespread deletion of Adar in adult mice. Both Cre-expressing mouse lines were purchased from the Jackson Laboratory (stock numbers 003755 and 008085). The Adar+/− mice resulting from the B6.129S4-Meox2tm1(cre)Sor cross were subsequently bred to Tmem173−/− (Sting−/−) mice (Ishikawa et al., 2009), Mavs−/− mice (Gall et al., 2012), Ddx58−/− (Rigi−/−) mice (Kato et al., 2005), or Ifih1−/− (Mda5−/−) mice (Gitlin et al., 2006). Adar p150+/− gametes were generously provided by Dr. M. B. A. Oldstone (Ward et al., 2011). Sentinel mice (Crl:CD1[ICR]; Charles River, Wilmington, MA) were tested quarterly for endo- and ectoparasites, mouse hepatitis virus, mouse parvovirus, and rotavirus; and tested annually for Mycoplasma pulmonis, pneumonia virus of mice, reovirus 3, Sendai virus, and Theiler murine encephalomyelitis virus. All experiments were done in accordance with the Institutional Animal Care and Use Committee guidelines of the University of Washington.
Histology
Tissues were fixed in 10% neutral buffered formalin, paraffin embedded, cut into 4-5μm sections and routinely stained with hematoxylin and eosin. All tissues were coded to remove genotype identification. Tissues evaluated included lung, heart, esophagus, kidney, ureter, bladder, liver, pancreas, spleen, lymph nodes, salivary glands, stomach, small intestine, large intestine, and reproductive tract. Additionally, for the Adar−/−Mavs−/− mice, decalcified cross sectional images of the skull and brain were also evaluated.
Flow cytometry and Cytokine Measurements
Single cell suspensions from spleen, bone marrow, thymus or blood were isolated and stained with antibodies for CD3 (145-2C11), CD4 (RM4-5), CD8β (H35-17.2), B220 (RA3-6B2), CD11c (N418), CD11b (M1/70), CD45.2 (104), and/or Ter119; Cells were analyzed with a FACSCanto (BD Biosciences) and analyzed with FlowJo software (TreeStar). Measurement of serum cytokines was performed using the Milliplex-70K-PX32 mouse cytokine/chemokine magnetic bead panel (Millipore), according to the manufacturers instructions.
Immunofluorescence microscopy
Spleens were frozen in Optimal Cutting Temperature (OCT) media (Sakura). Tissues were cut into 7-μm sections and treated with ice-cold acetone. Sections were stained with directly conjugated antibodies: CD8α (53–6.7, eBioscience), CD4 (RM4-5, eBioscience), and B220 (RA3-6B2, eBioscience). Nuclei were stained with 1 ug/mL DAPI. Stained slides were mounted with Prolong Gold antifade reagent (Life Technologies), imaged using a Nikon Eclipse 90i microscope and analyzed using Adobe Photoshop software.
Quantitative RT-PCR
Embryos were harvested into TRIzol (Life Technologies), homogenized through an 18-G needle attached to a 3 mL syringe, followed by RNA extraction according to manufacturer’s instructions. RNA was treated with DNase (Ambion) and 1 μg was reverse-transcribed using RNA to cDNA EcoDry Premix (Double Primed) (Clontech). cDNA was used for PCR with EVA Green reagents (Bio-Rad Laboratories) on a Bio-Rad CFX96 Real-Time System. The abundance of each interferon-stimulated gene mRNA was normalized to that of HPRT mRNA and results were compared with genetic control embryos for calculation of relative induction.
The primers used were:
Cxcl10 Fwd:AAGTGCTGCCGTCATTTTCTGCCTC, Cxcl10 Rev:CTTGATGGTCTTAGATTCCGGATTC;
Isg15 Fwd:GGTGTCCGTGACTAACTCCAT, Isg15 Rev:TGGAAAGGGTAAGACCGTCCT;
Ifit1 Fwd:GCCATTCAACTGTCTCCTG, Ifit1 Rev:GCTCTGTCTGTGTCATATACC;
Ifit2 Fwd:AGTACAACGAGTAAGGAGTCACT, Ifit2 Rev:AGGCCAGTATGTTGCACATGG (Primer bank ID: 6680363a1);
Mx1 Fwd:GACCATAGGGGTCTTGACCAA, Mx1 Rev:AGACTTGCTCTTTCTGAAAAGCC (Primer bank ID: 6996930a1);
Rsad2 Fwd:TGCTGGCTGAGAATAGCATTAGG, Rsad2 Rev:GCTGAGTGCTGTTCCCATCT (Primer bank ID: 31543946a1);
Hprt Fwd:GTTGGATACAGGCCAGACTTTGTTG, Hprt Rev:GAGGGTAGGCTGGCCTATAGGCT (Spandidos et al., 2010);
Htr2c (5ht2c) Fwd:TGTCCCTAGCCATTGCTGATATG, Htr2c Rev:TGTCAACGGGATGAAGAATGCC.
Deletion of ADAR1 in adult mice
Tamoxifen-induced deletion was performed according to the protocol available from the Jackson Laboratory. In brief, tamoxifen (Sigma-Aldrich) was dissolved in corn oil (Sigma-Aldrich) at 20 mg/mL overnight at 37°C, filtered through a 0.22 μm Millex GP PES membrane and stored at 4°C. Mice were administered 100 μL of tamoxifen via i.p. injection once a day for three days. On day four, Adarfl/flMavs+/+ Cre-positive mice were moribund, so no further injections were given. Mice were sacrificed and analyzed by histology and flow cytometry.
RNA-Seq library preparation
Total RNA was harvest from embryos as described above. Ribosomal RNA was depleted using the RiboZero™ Magnetic Kit (Human/Mouse/Rat) from Illumina. Libraries were prepared from the Ribo-depleted RNA using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (New England BioLabs) with the following modifications: Ribo-depleted RNA was fragmented for 5 min at 94°C to obtain >300bp fragments and first strand synthesis reaction was incubated for 50 min at 42°C. PCR Library enrichment was performed using the KAPA HiFi DNA Polymerase (KAPA Biosystems) with Illumina barcoded sequencing primers. Library size distribution was confirmed on an Agilent 2100 Bioanalyzer with a High Sensitivity DNA Chip (Agilent Technologies). Barcoded libraries were combined and sequenced with an Illumina NextSeq, resulting in approximately 60 million paired-end 150-base pair sequencing reads per embryo.
Alignment and analysis of RNA-Seq data
Fastq files were aligned using STAR 2.4.2a (Dobin et al., 2013), with the following parameters for the indices: STAR--runMode genomeGenerate--genomeDir /mnt/indices/--genomeFastaFiles /mnt/genome/Mus_musculus.GRCm38.dna.SORTED.fa--sjdbGTFfile /mnt/transcriptome/Mus_musculus.GRCm38.81.gtf--runThreadN 32 . Alignment was performed with the following parameters: STAR--genomeDir /mnt/indices/--readFilesIn /mnt2/data/run2/${i}_R1.fastq.gz /mnt2/data/run2/${i}_R2.fastq.gz--readFilesCommand zcat--runThreadN 32--outSAMtype BAM SortedByCoordinate--quantMode GeneCounts--outFilterMismatchNmax 30--outFileNamePrefix /mnt/results/run2/${i}.star.--outFilterMultimapNmax 20--outSAMattributes All.
All alignments were performed on Amazon EC2 c3.8xlarge instance using a Ubuntu13.04 base AMI. The read count files produced by STAR (unstranded) were used for differential expression analysis using the Bioconductor package edgeR (Robinson et al., 2010). Following calculation of the normalization factors, low-expressing genes were discarded. Counts were then subject to the estimateGLMCommonDisp function followed by glmFit. Differential expression for comparison between phenotypes was then performed using the glmLRT function. ISGs were annotated using the Interferome web tool (http://www.interferome.org/interferome/search/showSearch.jspx), defined as all Type I IFN-regulated genes in all tissues of Mus musculus with a cutoff of 3-fold expression. Read count and fastq files are available at SRA. For bioinformatics analysis, we used FunRich software to determine the enrichment of specific biological pathways and transcription factor networks among genes with dysregulated expression in both Adar−/− and Adar−/−Mavs−/− embryos, relative to the representation of these pathways in the genome.
CRISPR targeting of ADAR1 in human cells
For targeting of ADAR with CRISPR-Cas9, we used a lentiviral vector in which an RNA polymerase III promoter–driven guide RNA and an RNA polymerase II promoter–driven Cas9-T2A cassette (including sequence encoding a protein for resistance to puromycin) were constitutively expressed from a single, self-inactivating lentivirus upon integration into the host cell genome. Lentivirus pseudotyped with vesicular stomatitis virus envelope glycoprotein was produced by transfection of 2.5 × 106 HEK 293T cells for 48 h in 10-cm plates with 10 μg of the CRISPR-Cas9 ADAR targeting construct, 9 μg psPAX-2 (a lentiviral packaging plasmid) and 1 μg pVSV-G (plasmid encoding vesicular stomatitis virus envelope glycoprotein). 2.5 × 106 HEK 293T cells were transduced with the viral supernatants on day 3 after harvest, then were selected for 3 d with 5 μg/ml puromycin (Life Technologies). Subsequent single-cell cloning was performed by serial dilution. Targeting of the ADAR locus via CRISPR was evaluated by restriction fragment length polymorphism with an ApoI (New England Biolabs) restriction site that overlapped the CRISPR targeting site. Products were separated by electrophoresis through a 3% MetaPhor agarose gel (Lonza). ADAR mutations were identified by PCR amplification of the surrounding sequence, cloning into pCDNA3, and sequencing of nine independent plasmids. ADAR1 protein loss was confirmed by immunoblot analysis of whole-cell extracts with or without 24 hours of human IFNβ treatment (100 U/mL, R&D Systems) with rabbit polyclonal anti-ADAR1 (12317; Cell Signaling Technologies) and mouse monoclonal anti-β actin (AC-74; Sigma). The sequences of the guide RNA target sequence is (sense), 5′-GGACAGGAGACGGAATTCGC-3′.
ISRE-luciferase reporter assays
1 × 105 HEK 293T cells with or without ADAR1 expression in 24 well plates were transfected with 25 ng ISRE-luciferase reporter plasmid (Takara Bio Inc.) with 0, 12.5, 25, 50, 100, or 200 ng of pCDNA.3-expressing human RLRs using Lipofectamine® 2000 (Life Technologies) and then incubated for 24 h. Cells were lysed in Passive Lysis Buffer (Promega) and luciferase activity was assessed using the Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions and read using a Centro LB 960 Luminometer (Berthold Technologies).
Statistical Analysis
Statistical significance of difference between groups was assessed using Wilcoxon Rank Sum, Chi Square Goodness-of-fit or two-way ANOVA with Tukey’s multiple comparison test, as indicated in the figure legends. Values of p<0.05 were considered statistically significant. All analyses were performed using Prism v6.0 software (GraphPad).
Supplementary Material
Acknowledgments
We are grateful to Stuart Orkin for providing mice with the Adar conditional allele; to Ming R. Loo and Michael Gale, Jr for providing Mavs−/−, Ifih1−/−, and Ddx58−/− mice; to Shizuo Akira for the Ddx58−/− mice; to Marco Colonna for the Ifih1−/− mice; to Glen Barber for Tmem173−/− mice; to M. B. A. Oldstone for Adar p150+/− gametes; to Yanick Crow for helpful insights; to Stephanie Cambier for bioinformatics analysis; to Brian Johnson, Kerrie Allen, and the staff of UW Histology and Imaging Core for their technical expertise; and to members of the Stetson and Bevan labs for helpful discussions. DBS is a scholar of the Rita Allen Foundation and a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease. This work was supported by grants from the NIH (AI084914-DBS and NDP; T32GM007270-KP), and the Lupus Research Institute (DBS).
Footnotes
Author Contributions
K.P. and D.B.S. conceived of the project and wrote the manuscript. K.P. performed the experiments. C.C.F. and N.D.P. analyzed RNA-Seq data. J.M.S. and P.M.T. performed the histological analyses. All authors edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol. 2014;192:5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. doi: 10.1002/ana.410150109. [DOI] [PubMed] [Google Scholar]
- Bahn JH, Ahn J, Lin X, Zhang Q, Lee JH, Civelek M, Xiao X. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nature communications. 2015;6:6355. doi: 10.1038/ncomms7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. American journal of medical genetics Part A. 2015;167A:296–312. doi: 10.1002/ajmg.a.36887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Stryker MP, Tecott LH. Sleep and sleep homeostasis in mice lacking the 5-HT2c receptor. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2002;27:869–873. doi: 10.1016/S0893-133X(02)00353-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr, Barber GN, Stetson DB. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, Chen ZJ. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1516465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- Goubau D, Deddouche S, Reis ESC. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Treuting PM, Woodward JJ, Stetson DB. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutieres Syndrome. J Immunol. 2015;195:1939–1943. doi: 10.4049/jimmunol.1500969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Suzuki T. Dyschromatosis symmetrica hereditaria. The Journal of dermatology. 2013;40:336–343. doi: 10.1111/j.1346-8138.2012.01661.x. [DOI] [PubMed] [Google Scholar]
- Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med. 2012;209:1419–1426. doi: 10.1084/jem.20120876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci. 1988;84:201–208. doi: 10.1016/0022-510x(88)90125-6. [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A, et al. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2014;51:76–82. doi: 10.1136/jmedgenet-2013-102038. [DOI] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell reports. 2014;9:1482–1494. doi: 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare Variants of IFIH1, a Gene Implicated in Antiviral Responses, Protect Against Type 1 Diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenberg S, Paz Yaacov N, Safran M, Moshkovitz S, Shtrichman R, Sherf O, Jacob-Hirsch J, Keshet G, Amariglio N, Itskovitz-Eldor J, Rechavi G. Alu sequences in undifferentiated human embryonic stem cells display high levels of A-to-I RNA editing. PLoS One. 2010;5:e11173. doi: 10.1371/journal.pone.0011173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153:575–589. doi: 10.1016/j.cell.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Maelfait J, Bridgeman A, Rigby R, Hayward B, Liberatore RA, Bieniasz PD, Towers GJ, Moita LF, Crow YJ, et al. SAMHD1-dependent retroviral control and escape in mice. Embo J. 2013;32:2454–2462. doi: 10.1038/emboj.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell stem cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T, Rould MA, Lowenhaupt K, Herbert A, Rich A. Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science. 1999;284:1841–1845. doi: 10.1126/science.284.5421.1841. [DOI] [PubMed] [Google Scholar]
- Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland A, Davies J, Owen CJ, Vaikkakara S, Walker C, Cheetham TD, James RA, Perros P, Donaldson PT, Cordell HJ, et al. Genomic polymorphism at the interferon-induced helicase (IFIH1) locus contributes to Graves’ disease susceptibility. The Journal of clinical endocrinology and metabolism. 2007;92:3338–3341. doi: 10.1210/jc.2007-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, Julius D. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- Vitali P, Scadden AD. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat Struct Mol Biol. 2010;17:1043–1050. doi: 10.1038/nsmb.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IX, So E, Devlin JL, Zhao Y, Wu M, Cheung VG. ADAR regulates RNA editing, transcript stability, and gene expression. Cell reports. 2013;5:849–860. doi: 10.1016/j.celrep.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Khillan J, Gadue P, Nishikura K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science. 2000;290:1765–1768. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- Ward SV, George CX, Welch MJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MB. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci U S A. 2011;108:331–336. doi: 10.1073/pnas.1017241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- Yang S, Deng P, Zhu Z, Zhu J, Wang G, Zhang L, Chen AF, Wang T, Sarkar SN, Billiar TR, Wang Q. Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J Immunol. 2014;193:3436–3445. doi: 10.4049/jimmunol.1401136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.