

Introduction
Venous thromboembolism (VTE) is a common source of morbidity and mortality in the setting of malignancy, with the potential to present at the time of diagnosis, throughout treatment, and ultimately as a frequent cause of death.1 It has been well established that the diagnosis of malignancy itself is a potent risk factor for the development of deep vein thromboses/pulmonary emboli with several malignancies including, but not limited to, pancreatic, gastroesophageal, lung, and brain cancers demonstrating a particularly increased risk. 2,3 The risk for VTEs in patients with cancer has been estimated at 4-7.5 times greater than that for the general population. 4
Several contemporary studies have investigated independent predictors for VTEs in patients actively receiving chemotherapy leading to the development of risk models for identifying patients at highest risk. 5 The Khorana model is a validated scoring system that utilizes specific patient characteristics and laboratory values to stratify patients into low, intermediate, or high risk for venous thromboembolism; this model was developed in a study population of 4,066 cancer patients that were initiated on chemotherapy. The patients were observed for a median period of 2.5 months. In this model, five variables including site of primary cancer, prechemotherapy platelet count greater than 350 × 109/l, prechemotherapy leukocyte count greater than 11 × 109/l, hemoglobin less than 10g/dl, and BMI of 35kg/m2 were identified as quantifiable risk factors increasing the likelihood of developing symptomatic VTEs.6 Each variable was assigned a numerical value ranging from 0-2 and patients were stratified into three categories based on the total score obtained from the variables. The conclusion from this study was that patient in the low (score = 0) and intermediate (score = 1-2) risk group had a low incidence of VTE and would most likely not benefit from thromboprophylaxis. In contrast, Khorana et al observed that patients identified in the high risk group (score ≥ 3) had a higher risk of VTE and hence would most likely benefit from initiation of thromboprophylactic therapy. It is important to note that this group of patient (i.e. high risk scores) were a minority of the patients studied. This study mainly included patients with good performance status and did not adequately represent certain malignancies that are associated with a higher risk for VTE e.g. central nervous system malignancies.7
Subsequent models such as the Vienna VTE Risk Assessment Score have proposed that the inclusion of other biomarkers such as the D-dimer and the cell adhesion molecule soluble P-selectin could further enhance one's ability to predict thrombosis risk. 7,8 In fact, a host of other potential biomarkers for thrombosis risk have been investigated with preliminary data suggesting that elevated clotting factors, markers of inflammation, and procoagulant tissue factor associated microparticles (derived from the endothelium or cancer cells themselves) may all contribute to the underlying pathogenesis of cancer related VTEs; these biomarkers and their potential role in predicting risk of thrombosis in the cancer patient have been recently reviewed in detail elsewhere.9
Despite the fact that evidence has supported a causal relationship between chemotherapy and thrombosis for over three decades, it remains an underappreciated risk that has not been routinely incorporated into thrombosis risk assessment models.10 By the early 1980s, studies in women with breast cancer had demonstrated this increased risk of thrombosis in both the adjuvant setting and in metastatic disease. 11-12 In patients undergoing multidrug therapy for metastatic breast cancer (cyclophosphamide, methotrexate, 5-fluorouracil, vincristine, and prednisone), 17.6% developed thrombosis while on treatment, a majority of these being VTE, compared to just over 2% while receiving no therapy.10
Below we review specific anti-neoplastic drugs, both cytotoxic and targeted agents, that have been associated with an increased thrombotic risk and the proposed mechanisms for thrombosis. We conclude with a discussion of the implications for VTE prophylaxis and future considerations to reduce the risk of DVTs/PEs in the cancer population.
Cytotoxic Chemotherapy
Cisplatin
Cisplatin is a fairly ubiquitous chemotherapeutic agent, used in various combinations to treat a wide variety of malignancies. An appreciation of the increased vascular toxicity and thrombotic potential of cisplatin based therapies was noted not long after its FDA approval in 1978 for the treatment of testicular and ovarian cancers. 13 Increased indications for cisplatin-based treatments were associated with a concomitant rise in thrombotic events (both arterial and venous) that occurred in patients exposed to this chemotherapeutic agent. 14,15 Perhaps most telling is the marked increased in thrombotic events in patients treated with cisplatin compared to patients treated with other platinum based regimens. In the REAL-2 trial, 15.1% of patients treated with ECF (epirubicin-cisplatin-5FU) experienced some form of thromboembolic event during treatment, compared to 7.6% in patients randomized to EOX (epirubicin-oxaliplatin-capecitabine). 16
Such observations led to a large, single institution retrospective investigation of all thromboembolic events (TEE) in patients treated with a cisplatin-based regimen. This study demonstrated a TEE in 18.1% of patients either actively receiving cisplatin or having had completed cisplatin based therapy within the preceding four weeks, with over 90% related to venous thromboembolism.17 A subsequent meta-analysis of 38 phase II and III trials comparing cisplatin verse non-cisplatin treatments also demonstrated a significantly increased risk of VTEs in the cisplatin treated groups (RR=1.67, P=0.01). Notably, there was a marked variability in the incidence of VTEs among the trials included in this meta-analysis ranging from 0 to 17% in patients treated with cisplatin. 18
The mechanism of cisplatin induced hypercoagulability has not been precisely defined. In a small prospective study of thirteen patients treated with cisplatin, three developed arterial thrombosis. All of these three patients had elevated von Willebrand factor (vWF) levels prior to initiating cisplatin; vWF levels increased even further after cisplatin therapy to greater than 600% of normal.19 In addition, Lechner et al demonstrated in vitro that cisplatin-induced endothelial cell apoptosis results in the release of procoagulant endothelial microparticles that are able to generate thrombin through tissue factor independent pathways. 20
L-asparaginase
L-asparaginase is incorporated into induction regimens for the treatment of pediatric and adult acute lymphoblastic leukemia (ALL). L-asparaginase depletes intracellular asparagine and leads to decreased protein synthesis and subsequent cellular apoptosis. Early case reports documented an increased incidence of both thrombotic and hemorrhagic complications in the setting of L-asparaginase treatment. In a multicenter review of pediatric patients treated with asparaginase containing regimens between 1976-1980, 18 children of the 1547 studied developed a ‘severe’ thrombotic or hemorrhagic complication; 14 of these involved CNS events: 5 intracranial thromboses, 5 intracranial hemorrhages, and 4 with intracranial thrombosis with hemorrhagic conversion. 21 The risk of thrombotic complications in the adult population treated with asparaginase is also markedly elevated; retrospective data has demonstrated a 4.2% incidence of thrombosis in adults with ALL during induction therapy. 22 While the quintessential TEE associated with asparaginase therapy is intracranial dural sinus thrombosis, there is also a marked increase risk of venous thrombosis of the extremities, in particular central venous catheter related clots. 23
The thrombotic tendency seen in patients treated with asparaginase appears to be related to depletion of key proteins in the regulation of the coagulation pathway. In a series following daily plasma levels of protein C and S after initiating asparaginase therapy, protein C levels dropped to 30% of normal by days 6-10 of therapy, and protein S dropped to 41% by days 11-12. 24 Even more fundamental to the prothrombotic state, the synthesis of plasminogen and antithrombin (AT) is markedly impaired with asparaginase based therapy.25,26 The net effect is increased thrombin generation with impaired thrombin inhibition.
Fluoropyrimidines
5-Fluorouracil (5FU) features prominently into the treatment of gastrointestinal malignancies and has demonstrated activity against several other cancers, including breast and head and neck. The most feared cardiovascular complications of 5FU and its oral prodrug capecitabine are related to direct cardiac toxicity which can manifest as angina or even true myocardial infarction. The incidence of fluoropyrimidine induced cardiac toxicity has been estimated between 4.2 to 19% with a noted increased risk in the setting of higher doses and continuous infusions; while the effects are usually reversible after cessation of the drug, there have been case reports of fatal outcomes. 27,28 It is worth noting that fluoropyrimidine induced angina or ischemia is not due to coronary artery thrombosis, but is more likely related to arterial vasospasm or direct myocardial toxicity from metabolites of 5FU. 28,29
There is only limited evidence that 5FU increases the risk of VTE. Retrospective data in patients treated with 5FU and leucovorin for colon cancer demonstrated an incidence of VTE as high as 15%. 9 In a phase I clinical trial, 5FU in combination with G-CSF reached VTE rates of 29%.30 However, these rates are significantly higher than reported in most other trials. For example, Tournigand et al randomized 220 patients to sequential FOLFOX (5FU, leucovorin, oxaliplatin) followed by FOLFIRI (5FU, leucovorin, irinotecan) or the reverse sequence, and only 2 patients (one in each arm) developed symptomatic VTE in the form of PEs. 31
The degree to which 5FU itself causes VTE therefore remains somewhat uncertain; laboratory data would at least suggest that 5FU does contribute to a potentially prothrombotic environment through the depletion of protein C and increased thrombin activity. 32,33 Furthermore, animal models and human endothelial cell cultures exposed to 5FU demonstrated endothelial cell damage with the potential to promote thrombus formation. 34,35
Targeted Agents
Tamoxifen and Aromatase Inhibitors
The connection between tamoxifen and increased risk of thrombosis has been recognized since the 1970s and the start of its use in breast cancer treatment.36,37 Data from the Fisher trials for adjuvant treatment of local breast cancer identifies a relative risk (RR) of 4.0-6.0 in the 5-year setting and 3.25 in post 5-year setting38-40. Saphner evaluated data from 7 ECOG trials from 1977-1987 to further emphasize tamoxifen's relation to thrombosis. Premenopausal women that received tamoxifen and chemotherapy had an increased risk of venous and arterial thrombosis versus chemotherapy alone (2.8% vs 0.8%, 1.6% vs 0.0% respectively) 41. Comparisons looking at race show no difference in RR and mirror data from prior studies; RR of 2.17 and 3.19 in African American and White women treated with tamoxifen, with chemotherapy plus tamoxifen increasing to 10.70 and 15.49 respectively42. Interestingly, a Danish trial looked at the time course for the occurrence of these events in the adjuvant setting. As described in their analysis, the highest risk is during the first two years (adjusted RR of 3.8) with a non-significant increased risk in years 3-5 of therapy (adjusted RR of 1.8)43.
In contrast to tamoxifen, aromatase inhibitors (AI) have not been shown to increase the risk of thrombosis, although data in the immediate adjuvant setting is limited to comparisons between tamoxifen and AIs. The ATAC trial data showed a RR of 2.0 with tamoxifen and anastrazole versus anastrazole alone and a RR of 1.7 with tamoxifen versus anastrazole44, indicating that the thrombosis risk was most likely related to tamoxifen use. When looking at the “switch” trials in which patients were started on tamoxifen then changed to AI versus placebo, no increased risk of thrombosis was noted while patients were on AIs45,46 to further suggest that AIs are not associated with an increased risk for thrombosis.
Over the past two decades, there has been several options added to the oncologist's armamentarium, each with their unique adverse effects. Specifically when looking at venous thrombosis, of these new agents, two classes come to the forefront: VEGF inhibitors and immunomodulatory drugs.
Antiangiogenic Agents
The first approved by the FDA was bevacizumab in use for colon cancer, glioblastoma, and non-small cell lung cancer (NSCLC). This agent is a monoclonal antibody directed at vascular endothelial growth factor A (VEGF-A), which is released by malignant cells to activate the endothelium. Once activated, matrix metalloproteinases break down the extracellular matrix and allow for new vessel growth and subsequently continued tumor growth47.
Since its release, there have been discrepancies in the toxicities of this agent, with respect to thrombosis. Kabbinavar reported 23% in the treatment arm vs 6% in his control arm in the use of bevacizumab with 5-FU and leucovorin in metastatic colon cancer48. Contrary to this, Hurwitz in 2004 and Kabbinavar in 2005 showed no difference between the treatment arm and control arm in patients with metastatic colon cancer49,50. Subsequent meta-analyses have also drawn different conclusions about bevacizumab and risk of venous thromboembolism. Nalluri, et al looked at 7956 patients from 15 trials and reported a RR of 1.33 in both all-grade (11.9%) and high-grade VTE (6.3%)51. Another meta-analysis pooled 1745 patients from three trials showed no difference in overall rate of VTE (HR 0.89) but an increased risk of arterial thrombosis (HR 2.0)52.
The first of the oral tyrosine kinase inhibitors (TKI) that targeted VEGF, semaxanib, was never brought to market due to its vascular toxicity profile. In a Phase I trial with gemcitabine and paclitaxel, 8 of the first 19 patients suffered from a VTE event53. When evaluated in vitro, studies showed increased thrombin potential, E-selectin, von Willebrand factor, and soluble tissue factor. When combined with gemcitabine and cisplatin, this led to a concomitant activation of the coagulation cascade. They hypothesized that the endothelium, becoming starved of VEGF, activates and upregulates these molecules. In this state, it also becomes susceptible to the endothelial damage induced by cytotoxic therapies, leading to thrombosis54. Additionally, studies have shown an elevation in VEGF during thrombus resolution55. Over expression in the presence of a thrombus leads to increased neovascularization and recanalization56; two actions intrinsic to thrombus resolution. This suggests that VEGF targeted therapy is also associated with decreased thrombus resolution.
Other oral VEGF inhibitors are now available: sorafenib, sunitinib, pazopanib, vandetanib, and axitinib. These agents are broader in their targets than bevacizumab. Sunitinib for example affects VEGF receptor s-1, -2, -3, cKIT, Fms-like tyrosine kinase 3 (FLT-3), colony stimulating factor 1 receptor (CSF-1R), and RET. Sorafenib has similar targets but includes platelet derived growth factor receptor beta (PDGFR-β). In contrast to semaxanib, these agents have not been associated with increased risk of VTE. Two meta-analyses recently published cite a relative risk of 0.91 and 1.1 in comparison to non-TKI arms57,58. These agents, however, have been used as monotherapy for the time being in malignancies traditionally not known for thrombosis risk. In the future, combination therapy may reveal an increased thrombosis risk, much like thalidomide and other immunomodulatory agents, where the cytotoxic therapy induces thrombosis which is propagated by VEGF blockade. For example, Evans specifically looked at axitinib and its role in inhibiting thrombus resolution. They showed that the agent had no effect on initial thrombus generation as no difference in early fibrillar collagen content (a marker of thrombus organization), thrombus volume, neutrophil content, or recanalization was evident against a control. By day 17, there was significantly less of the aforementioned except for neutrophil count in the axitinib treated model. The natural progression of macrophage accumulation in thrombus was significantly impaired, which follows from VEGFR1 inhibition on macrophages59.
Immunomodulatory Agents
It is well known that multiple myeloma (MM), along with MGUS, is a prothrombotic state. Interestingly enough, IgA and IgG MGUS carries an increased thrombosis risk but IgM does not60. Multiple myeloma creates an inflammatory cytokine milieu with elevated levels of TNF, CRP, and IL-6 61. IL-6 in particular has been shown in vitro to trigger the coagulation cascade62. Other pathways have been presented as possible etiologies for this prothrombotic state, including a transitory acquired activated protein C resistance outside of Factor V Leiden related to disease activity63.
As a single agent, the risk for thrombosis with thalidomide in the treatment of multiple myeloma is not clearly increased, roughly 3-4%64. The use of thalidomide saw an increase in the incidence of VTE only once combined with other agents. Zangari did an up-front randomization to thalidomide or no thalidomide with anthracycline-based induction chemotherapy in newly diagnosed MM, citing a 28% versus 4% incidence of VTE65. In another multi-center trial looking at melphalan, prednisone, and thalidomide versus melphalan and prednisone alone in an older patient population, the incidence was 17% vs 2%66. This increased risk also occurs in patients receiving only steroids and no cytotoxic agents, as described by Rajkumar where the incidence was 17% with thalidomide/dexamethasone and 3% with dexamethasone alone67.
These effects are shared with the second generation agent lenalidomide. Similarly to thalidomide, when used as monotherapy the rates of thrombosis are not elevated (1% when used in MDS68, 4% when used in MM without thromboprophylaxis69). When used in conjunction with dexamethasone, this rate rises to approximately 11-14% prior to the use aspirin thrombophrophylaxis70,71. After the utilization of thromboprophylaxis, this rate fell to 1-3%72. Additionally, the dose schedule of the dexamethasone with the lenalidomide changes the endothelial manifestations with pulse steroids leading to greater variation in fibrinogen, P-selectin, and VEGF than weekly dexamethasone73.
Limited data exists on pomalidomide and thrombosis. In one Phase I trial of pomalidomide monotherapy not using thromboprophylaxis, 4 out of 32 patients developed VTE74. While this number is greater than that cited with other agents, the number of patients limits interpretation. A subsequent trial using pomalidomide with or without dexamethasone had rates of 2% and 3% respectively with all patients receiving thromboprophylaxis75.
This requirement of a second agent is similar to the anti-VEGF agents. Thalidomide and its brethren likely precipitate platelet adhesion and thrombosis by maintaining endothelium in a VEGF-starved state, unable to recuperate after cytotoxic chemotherapy induced vascular injury76. Other theories have been proposed. Kaushal et al proposed that thalidomide increases the expression of protease-activated receptor 177. PAR-1 is expressed on both platelets and endothelium. It presents its own ligand, which is irreversibly unmasked by thrombin. This leads to platelet activation, granule secretion, and aggregation. On the endothelium, PAR-1 facilitates platelet and neutrophil rolling and adhesion. Therefore, PAR-1 may serve as the connection between injury and the coagulation response78. Abdullah et al noted that thalidomide leads to conformation changes in GPIIb/IIIa via increased target for PAC-1, indicating platelet activation79, and further reinforcing the importance of platelet activity in thalidomide related thrombosis.
Supportive Agents
Corticosteroids
Steroids play a fundamental role in the supportive care of the cancer patient and are regularly incorporated into treatment protocols for hematologic malignancies (i.e. lymphoma, multiple myeloma). An association between corticosteroid excess and increased risk of thrombosis was noted outside the field of oncology over sixty years ago; natural history studies of patients with Cushing's Syndrome noted a relatively high rate of pulmonary emboli in a series of patients published in 1951.80 Increased risk of VTE secondary to exogenous steroid use was recently confirmed in a large case control study which included patients with malignancy; this study demonstrated an incidence rate ratio of 2.31 for DVT/PE in patients actively taking corticosteroids compared to controls.81As noted above, steroids in combination with immunomodulatory derivatives in the treatment of myeloma result in profoundly increased risks for VTEs. In addition, the use of high dose steroids (>/= 80mg dexamethasone per treatment cycle) for antiemetic purposes resulted in a significantly increased rate of VTEs (OR = 3.47, 95% CI: 1.2-10.3). 82
Mechanistically, corticosteroid use has been shown to increase circulating levels of clotting factors VII, VIII, XI and fibrinogen in healthy volunteers. 83 Further, studies of patients with Cushing's syndrome demonstrated increased clotting factors as well as evidence of decreased thrombolysis associated with increased plasminogen and alpha-2 antiplasmin levels. 84
Hematopoietic Growth Factors
Erythropoietin stimulating agents (ESAs) are used to increase hemoglobin levels and decrease the total number of transfused red blood cells in cancer patients undergoing myelosuppressive chemotherapy. ASCO guidelines recommend discussion of the risks and benefits of ESAs when hemoglobin levels drop below 10g/dl in this setting. 85 Multiple studies have demonstrated an increased risk of VTE, and a recent Cochrane review demonstrated that ESAs resulted in a relative risk of 1.52 for the development of VTEs (CI 1.34-1.74). 86 There has been some suggestion that this risk is reduced when ESAs are held until hemoglobin is less than 10g/dl, but this data is weak and not reproduced in other studies. 86,87 The prothrombotic nature of ESAs in the oncology patient is not likely due to elevated hemoglobin levels but is rather multifactorial; erythropoietin has been shown to decrease proteins C and S, increase PAI-1 production, and increase platelet activation. 88
Granulocyte colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) are used to aid granulocyte cell recovery after myelosuppressive chemotherapy. Prospective data has demonstrated that the use of white cell growth factor is associated with a significant risk of VTE (OR = 2.09, CI: 1.2-3.6) 5 In healthy patients treated with G-CSF prior to stem cell collection, markers for increased clotting activity (prothrombin fragment F1+2, thrombin-antithrombin complex, and D-dimer) were all elevated after exposure to G-CSF. In addition, G-CSF use resulted in some evidence of endothelial cell damage/activation with increased serum levels of thrombomodulin and von Willebrand factor. 89
Prophylactic therapy
Several studies have investigated the role of VTE prophylaxis in the ambulatory oncology patient. Two large clinical trials, the PROTECHT and the SAVE-ONCO trials, randomized patients with solid tumors to prophylactic dose low molecular weight heparins (LMWH) versus placebo. The PROTECHT study included 1150 patients actively receiving chemotherapy for lung, gastrointestinal, pancreatic, breast, ovarian, or head and neck cancers with the treatment group receiving the LMWH nadroparin; prophylactic nadroparin decreased the incidence of symptomatic arterial or venous thrombosis from 3.9% in the placebo group to 2.0% in the treatment arm (p = 0.02). 90 At the same time, the risk for minor bleeds was similar in both treatment and placebo groups with only a slightly increased risk for major bleeds (0.7% versus 0 in the placebo group). Similarly, the SAVE-ONCO trial included 1608 patients with locally advanced or metastatic solid tumors undergoing chemotherapy with patients randomized to the LMWH semuloparin vs placebo; again, the treatment arm demonstrated a statistically significant decrease in the primary end point of VTE/VTE related death (3.4% v. 1.2%; p <0.001). 91 Several other smaller trials investigating VTE prophylaxis in patients undergoing treatment for specific malignancies (pancreas, lung, breast, glioma) have been performed with mixed results. 92-94 While the SAVE-ONCO and PROTECHT studies were statistically positive, the results must be interpreted with caution. First, the rate of thrombosis in the control arm was fairly low in each trial (<5%), which suggests that the percentage of patients in these largely unselected populations that could benefit from prophylactic therapy is quite small. Second, one must appreciate that there was no suggestion that prophylaxis in the treatment groups had any impact on overall survival and that the only major bleeding events occurred in the treatment arms. Again the risk of major bleeding was quite small and not statistically significant, but does serve as a reminder that any intervention comes with potential costs. It is worth emphasizing that the FDA has not approved any drug with the indication of VTE prophylaxis in solid tumor patients undergoing systemic chemotherapy. ASCO guidelines have been recently updated regarding VTE prophylaxis in patients with solid tumors undergoing treatment; based on available data, the expert panel did not recommend routine thromboprophylaxis in cancer outpatients, but stated that it could be considered in a ‘highly selected’ population. 95
Exceptions to this recommendation are patients with multiple myeloma undergoing thalidomide or lenalidomide-based treatments. Current ASCO guidelines do recommend thromboprophylaxis with aspirin or LMWH in this clinical situation. As previously noted, patients with myeloma treated with a thalidomide/lenalidomide regimen are at an exceptionally increased risk for VTEs. Early studies demonstrated that prophylactic doses of LMWH significantly reduced the incidence of VTEs.96 The choice of prophylaxis has been based on two randomized clinical trials. Palumbo et al randomized patients treated with thalidomide to daily 100mg aspirin, 1.25mg warfarin, or 40mg enoxaparin; they reported a statistically insignificant trend towards a lower incidence of serious thrombotic events in the LWMH arm (5.0%) compared to warfarin (8.2%) or aspirin (6.4%).97 Of further interest, this study also identified several other characteristics that placed thalidomide treated patients at higher risk for DVTs: age >60, multiple comorbidities, poorer performance status, and patients not treated with bortezomib. Similarly, Larocca's group randomized patients undergoing treatment with lenolidomide to either aspirin or low dose enoxaparin; again, there was a statistically insignificant trend towards lower incidence of VTE in the LMWH group (1.2%) compared to the aspirin group (2.7%).98 The choice of prophylactic therapy is based on the expected risk of VTE which is dependent on the anti-myeloma therapy used and patient characteristics. Although aspirin is more appealing as an oral agent, LMWH is more effective in situations where the thrombosis risk is high such as in patient treated with the immunomodulatory agents thalidomide or lenalidomide as well as in patients who have additional risk factors for VTE. Most VTEs in myeloma patients occur within the first 6 months after initiation of therapy. 66,96 Although prophylactic therapy is usually provided for at least this duration of time, longer periods of therapy may be considered based on therapy- or patient-related risks factors. 99
In sharp contrast to the association of increased VTE risk with most anti-myeloma therapies, treatment of MM with proteasome inhibitor bortezomib is associated with a significantly decreased risk for VTEs. Importantly, this thromboprotective benefit holds true even when bortezomib is combined with anti-myeloma agents that are usually associated prothrombotic outcomes such a thalidomide or lenalidomide. 100,101 The molecular mechanism involved in orchestrating this thromboprotective effect is dependent of modulation of trascription factor Kruppel-like factor 2 (KLF2). As elucidated by Nayak et al, bortezomib therapy is associated with a prolonged time to thrombosis in a mouse model that is dependent upon transcription factor KLF2, a zinc-finger transcription factor with known vasculoprotective benefits. 102
Conclusion
Cancer patients undergoing systemic treatment for their malignancy are among the highest risk populations for thromboembolic complications; often, the treatment itself contributes to this risk. Recognition of the antineoplastic agents most likely to cause thrombosis can help raise provider awareness and lead to earlier diagnosis and treatment. Studies completed to date do not identify a definitive role for routine thromboprophylactic therapy to all patients with a diagnosis of malignancy who are undergoing therapy. Current recommendations however suggest that antithrombotic treatment be strongly considered in patients with an especially high risk of VTE based on the diagnosis, therapy, and other patient-related VTE-risk factors.
Figure 1.
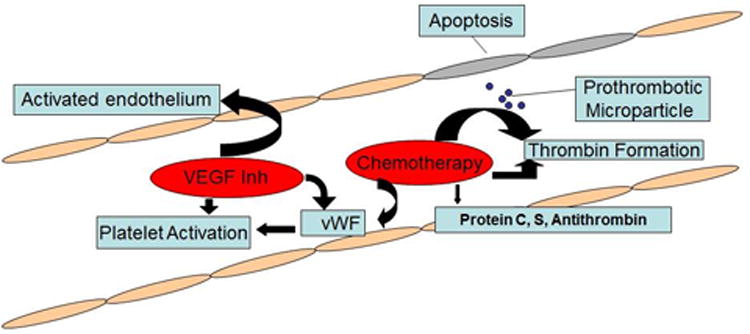
Cytotoxic chemotherapy has a multifactorial contribution to the risk of thrombosis. It induces vascular injury through apoptosis. In the case of cisplatin, this leads to release of prothrombotic particles that trigger thrombin generation via tissue factor independent mechanisms along with drastically increased vWF activity. Other agents, like 5-FU, also drive thrombin formation in combination with depleted protein C activity. L-asparaginase administration is tied to drastically decreased protein C, protein S, and antithrombin levels, creating a prothrombotic milieu through loss of anticoagulant factors. VEGF inhibition does not directly lead to thrombosis, but instead ‘primes’ the endothelium through a VEGF starved state to be more susceptible to injury. Additionally, platelet activation through PAR-1 and increased Gp llb/llla activity in the case of immunomodulatory agents or increased vWF among others in the case of small molecule inhibitors contributes to this ‘primed’ state.
Contributor Information
Peter Oppelt, Case Western Reserve University.
Anthony Betbadal, Case Western Reserve University.
Lalitha Nayak, Case Western Reserve University.
Bibliography
- 1.Khorana A, Francis CW, Culakova E, Kuderer NM, Lyman GH. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost. 2007;5(3):632–4. doi: 10.1111/j.1538-7836.2007.02374.x. [DOI] [PubMed] [Google Scholar]
- 2.Chew HK, Wun T, Harvey D, Zhou H, White RH. Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch Intern Med. 2006;166(4):458–64. doi: 10.1001/archinte.166.4.458. [DOI] [PubMed] [Google Scholar]
- 3.Cronin-Fenton DP, Søndergaard F, Pedersen LA, et al. Hospitalisation for venous thromboembolism in cancer patients and the general population: a population-based cohort study in Denmark, 1997-2006. Br J Cancer. 2010;103(7):947–53. doi: 10.1038/sj.bjc.6605883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blom JW, Doggen CJM. Malignancies, Prothrombotic Mutations, and the Risk of Venous Thrombosis. JAMA. 2014;293(6):715–722. doi: 10.1001/jama.293.6.715. [DOI] [PubMed] [Google Scholar]
- 5.Khorana AA, Francis CW, Culakova E, Lyman GH. Risk factors for chemotherapy-associated venous thromboembolism in a prospective observational study. Cancer. 2005;104(12):2822–2829. doi: 10.1002/cncr.21496. [DOI] [PubMed] [Google Scholar]
- 6.Khorana AA, Kuderer NM, Culakova E, et al. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood. 2008;111:4902–4907. doi: 10.1182/blood-2007-10-116327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thaler J, Cihan A, Pabinger I. Venous thromboembolism in cancer patients – Risk score and recent randomized controlled trials. Thrombosis and Hemostasis. 2013;108:1042–1048. doi: 10.1160/TH12-04-0241. [DOI] [PubMed] [Google Scholar]
- 8.Cihan A, Dunkler D, Marosi C, et al. Prediction of venous thromboembolism in cancer patients. Blood. 2010;116:5377–5382. doi: 10.1182/blood-2010-02-270116. [DOI] [PubMed] [Google Scholar]
- 9.Pabinger I, Thaler J, Cihan A. Biomakers for prediction of venous thromboembolism in cancer. Blood. 2013;122:2011–2018. doi: 10.1182/blood-2013-04-460147. [DOI] [PubMed] [Google Scholar]
- 10.Otten HM, Mathijssen J, Cate H, et al. Symptomatic Venous Thromboembolism in Cancer Patients Treated With Chemotherapy: An Underestimated Phenomenon. Arch Intern Med. 2004;164(190) doi: 10.1001/archinte.164.2.190. [DOI] [PubMed] [Google Scholar]
- 11.Goodnough LT, Saito H, Manni A, Jones PK, Pearson OH. Increased incidence of thromboembolism in stage IV breast cancer patients treated with a five-drug chemotherapy regimen. A study of 159 patients. Cancer. 1984;54(7):1264–8. doi: 10.1002/1097-0142(19841001)54:7<1264::aid-cncr2820540706>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 12.Levine MN, Gent M, Hirsh J, et al. The thrombogenic effect of anticancer drug therapy in women with stage II breast cancer. N Engl J Med. 1988;318(7):404–7. doi: 10.1056/NEJM198802183180703. [DOI] [PubMed] [Google Scholar]
- 13.Doll DC, List AF, Greco FA, Hainsworth JD, Hande KR, Johnson DH. Acute vascular ischemic events after cisplatin-based combination chemotherapy for germ-cell tumors of the testis. Ann Intern Med. 1986;105(1):48–51. doi: 10.7326/0003-4819-105-1-48. [DOI] [PubMed] [Google Scholar]
- 14.Numico G, Garrone O, Dongiovanni V, et al. Prospective evaluation of major vascular events in patients with nonsmall cell lung carcinoma treated with cisplatin and gemcitabine. Cancer. 2005;103(5):994–9. doi: 10.1002/cncr.20893.. [DOI] [PubMed] [Google Scholar]
- 15.Czaykowski PM, Moore MJ, Tannock IF. High risk of vascular events in patients with urothelial transitional cell carcinoma treated with cisplatin based chemotherapy. J Urol. 1998;160(6 Pt 1):2021–4. doi: 10.1097/00005392-199812010-00022. [DOI] [PubMed] [Google Scholar]
- 16.Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358(1):36–46. doi: 10.1056/NEJMoa073149. [DOI] [PubMed] [Google Scholar]
- 17.Moore Ra, Adel N, Riedel E, et al. High incidence of thromboembolic events in patients treated with cisplatin-based chemotherapy: a large retrospective analysis. J Clin Oncol. 2011;29(25):3466–73. doi: 10.1200/JCO.2011.35.5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seng S, Liu Z, Chiu SK, et al. Risk of venous thromboembolism in patients with cancer treated with Cisplatin: a systematic review and meta-analysis. J Clin Oncol. 2012;30(35):4416–26. doi: 10.1200/JCO.2012.42.4358. [DOI] [PubMed] [Google Scholar]
- 19.Licciardello JT, Moake JL, Rudy CK, Karp DD, Hong WK. Elevated plasma von Willebrand factor levels and arterial occlusive complications associated with cisplatin-based chemotherapy. Oncology. 1985;42(5):296–300. doi: 10.1159/000226049. [DOI] [PubMed] [Google Scholar]
- 20.Lechner D, Kollars M, Gleiss A, Kyrle PA, Weltermann A. Chemotherapy-induced thrombin generation via procoagulant endothelial microparticles is independent of tissue factor activity. J Thromb Haemost. 2007;5(12):2445–52. doi: 10.1111/j.1538-7836.2007.02788.x. [DOI] [PubMed] [Google Scholar]
- 21.Priest JR, Ramsay NK, Steinherz PG, et al. A syndrome of thrombosis and hemorrhage complicating L-asparaginase therapy for childhood acute lymphoblastic leukemia. J Pediatr. 1982;100(6):984–9. doi: 10.1016/s0022-3476(82)80535-0. [DOI] [PubMed] [Google Scholar]
- 22.Gugliotta L, Mazzucconi MG, Leone G, et al. Incidence of thrombotic complications in adult patients with acute lymphoblastic leukaemia receiving L-asparaginase during induction therapy: a retrospective study. The GIMEMA Group. Eur J Haematol. 1992;49(2):63–6. doi: 10.1111/j.1600-0609.1992.tb00032.x. [DOI] [PubMed] [Google Scholar]
- 23.Caruso V, Iacoviello L, Di Castelnuovo A, et al. Thrombotic complications in childhood acute lymphoblastic leukemia: a meta-analysis of 17 prospective studies comprising 1752 pediatric patients. Blood. 2006;108(7):2216–22. doi: 10.1182/blood-2006-04-015511. [DOI] [PubMed] [Google Scholar]
- 24.Bezeaud A, Drouet L, Leverger G, Griffin JH, Guillin MC. Effect of L-asparaginase therapy for acute lymphoblastic leukemia on plasma vitamin K-dependent coagulation factors and inhibitors. J Pediatr. 1986;108(5 Pt 1):698–701. doi: 10.1016/s0022-3476(86)81044-7. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell LG, Halton JM, Vegh PA, et al. Effect of disease and chemotherapy on hemostasis in children with acute lymphoid leukemia. Am J Pediatr Hematol Oncol. 1994;16(2):120–6. [PubMed] [Google Scholar]
- 26.Mitchell L, Hoogendoorn H, Giles AR, Vegh P, Andrew M. Increased endogenous thrombin generation in children with acute lymphoblastic leukemia: risk of thrombotic complications in L'Asparaginase-induced antithrombin III deficiency. Blood. 1994;83(2):386–91. [PubMed] [Google Scholar]
- 27.Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487–93. doi: 10.1007/s00280-005-0178-1. [DOI] [PubMed] [Google Scholar]
- 28.Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108–12. doi: 10.1159/000072334. [DOI] [PubMed] [Google Scholar]
- 29.Ang C, Kornbluth M, Thirlwell MP, Rajan RD. Capecitabine-induced cardiotoxicity: case report and review of the literature. Curr Oncol. 2010;17(1):59–63. doi: 10.3747/co.v17i1.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grem JL, McAtee N, Murphy RF, et al. Phase I and pharmacokinetic study of recombinant human granulocyte-macrophage colony-stimulating factor given in combination with fluorouracil plus calcium leucovorin in metastatic gastrointestinal adenocarcinoma. J Clin Oncol. 1994;12(3):560–8. doi: 10.1200/JCO.1994.12.3.560. [DOI] [PubMed] [Google Scholar]
- 31.Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22(2):229–37. doi: 10.1200/JCO.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 32.Feffer SE, Carmosino LS, Fox RL. Acquired protein C deficiency in patients with breast cancer receiving cyclophosphamide, methotrexate, and 5-fluorouracil. Cancer. 1989;63(7):1303–7. doi: 10.1002/1097-0142(19890401)63:7<1303::aid-cncr2820630713>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 33.Edwards RL, Klaus M, Matthews E, McCullen C, Bona RD, Rickles FR. Heparin abolishes the chemotherapy-induced increase in plasma fibrinopeptide A levels. Am J Med. 1990;89(1):25–8. doi: 10.1016/0002-9343(90)90093-s. [DOI] [PubMed] [Google Scholar]
- 34.Cwikiel M, Eskilsson J, Albertsson M, Stavenow L. The influence of 5-fluorouracil and methotrexate on vascular endothelium. An experimental study using endothelial cells in the culture. Ann Oncol. 1996;7(7):731–7. doi: 10.1093/oxfordjournals.annonc.a010723. [DOI] [PubMed] [Google Scholar]
- 35.Cwikiel M, Eskilsson J, Wieslander JB, Stjernquist U, Albertsson M. The appearance of endothelium in small arteries after treatment with 5-fluorouracil. An electron microscopic study of late effects in rabbits. Scanning Microsc. 1996;10(3):805–18. discussion 819. [PubMed] [Google Scholar]
- 36.Nevasaari K, Heikkinen M, Taskinen PJ. Tamoxifen and thrombosis. Lancet. 1978;2(8096):946–7. doi: 10.1016/s0140-6736(78)91668-9. [DOI] [PubMed] [Google Scholar]
- 37.Jungi WF, Alberto P, Wagenknecht L, Cavalli F, Martz G, Brunner KW. Antiestrogens: a new endocrine treatment possibility in metastasizing breast neoplasms. Experiences of the Swiss Cooperative Cancer Study Group with tamoxifen. Schweiz Med Wochenschr. 1978;108(34):1317–21. [PubMed] [Google Scholar]
- 38.Fisher B, Dignam J, Wolmark N, et al. Tamoxifen in treatment of intraductal breast cancer: National Surgical Adjuvant Breast and Bowel Project B-24 randomised controlled trial. Lancet. 1999;353(9169):1993–2000. doi: 10.1016/S0140-6736(99)05036-9. [DOI] [PubMed] [Google Scholar]
- 39.Fisher B, Dignam J, Bryant J, et al. Five versus more than five years of tamoxifen therapy for breast cancer patients with negative lymph nodes and estrogen receptor-positive tumors. J Natl Cancer Inst. 1996;88(21):1529–42. doi: 10.1093/jnci/88.21.1529. [DOI] [PubMed] [Google Scholar]
- 40.Fisher B, Costantino J, Redmond C, et al. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N Engl J Med. 1989;320(8):479–84. doi: 10.1056/NEJM198902233200802. [DOI] [PubMed] [Google Scholar]
- 41.Saphner T, Tormey DC, Gray R. Venous and arterial thrombosis in patients who received adjuvant therapy for breast cancer. J Clin Oncol. 1991;9(2):286–94. doi: 10.1200/JCO.1991.9.2.286. [DOI] [PubMed] [Google Scholar]
- 42.McCaskill-Stevens W, Wilson J, Bryant J, et al. Contralateral breast cancer and thromboembolic events in African American women treated with tamoxifen. J Natl Cancer Inst. 2004;96(23):1762–9. doi: 10.1093/jnci/djh321. [DOI] [PubMed] [Google Scholar]
- 43.Hernandez RK, Sørensen HT, Pedersen L, Jacobsen J, Lash TL. Tamoxifen treatment and risk of deep venous thrombosis and pulmonary embolism: a Danish population-based cohort study. Cancer. 2009;115(19):4442–9. doi: 10.1002/cncr.24508. [DOI] [PubMed] [Google Scholar]
- 44.Baum M, Budzar AU, Cuzick J, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet. 2002;359(9324):2131–9. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 45.Goss PE, Ingle JN, Martino S, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Cancer Inst. 2005;97(17):1262–71. doi: 10.1093/jnci/dji250. [DOI] [PubMed] [Google Scholar]
- 46.Jakesz R, Jonat W, Gnant M, et al. Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years' adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial. Lancet. 2005;366(9484):455–62. doi: 10.1016/S0140-6736(05)67059-6. [DOI] [PubMed] [Google Scholar]
- 47.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 48.Kabbinavar F, Hurwitz HI, Fehrenbacher L, et al. Phase II, Randomized Trial Comparing Bevacizumab plus Fluorouracil (FU)/leucovorin (LV) with FU/LV Alone in Patients with. Metastatic Colorectal Cancer. 2003:60–65. doi: 10.1200/JCO.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 49.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N Engl J Med. 2004:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 50.Kabbinavar FF, Hambleton J, Mass RD, Hurwitz HI, Bergsland E, Sarkar S. Combined Analysis of Efficacy: The Addition of Bevacizumab to Fluorouracil/leucovorin Improves Survival for Patients with. Metastatic Colorectal Cancer. 2005:3706–3712. doi: 10.1200/JCO.2005.00.232. [DOI] [PubMed] [Google Scholar]
- 51.Nalluri SR, Chu D, Keresztes R, Zhu X, Wu S. Risk of venous thromboembolism with the angiogenesis inhibitor bevacizumab in cancer patients: a meta-analysis. JAMA. 2008;300:2277–2285. doi: 10.1001/jama.2008.656. [DOI] [PubMed] [Google Scholar]
- 52.Scappaticci FA, Skillings JR, Holden SN, et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J Natl Cancer Inst. 2007;99:1232–1239. doi: 10.1093/jnci/djm086. [DOI] [PubMed] [Google Scholar]
- 53.Zangari M, Fink LM, Elice F, Zhan F, Adcock DM, Tricot GJ. Thrombotic events in patients with cancer receiving antiangiogenesis agents. J Clin Oncol. 2009;27:4865–4873. doi: 10.1200/JCO.2009.22.3875. [DOI] [PubMed] [Google Scholar]
- 54.Kuenen BC. Analysis of Coagulation Cascade and Endothelial Cell Activation During Inhibition of Vascular Endothelial Growth Factor/Vascular Endothelial Growth Factor Receptor Pathway in Cancer Patients. Arterioscler Thromb Vasc Biol. 2002;22(9):1500–1505. doi: 10.1161/01.atv.0000030186.66672.36. [DOI] [PubMed] [Google Scholar]
- 55.Waltham M, Burnand KG, Collins M, Smith A. Vascular endothelial growth factor and basic fibroblast growth factor are found in resolving venous thrombi. J Vasc Surg. 2000;32(5):988–96. doi: 10.1067/mva.2000.110882. [DOI] [PubMed] [Google Scholar]
- 56.Waltham M, Burnand KG, Collins M, McGuinness CL, Singh I, Smith A. Vascular endothelial growth factor enhances venous thrombus recanalisation and organisation. Thromb Haemost. 2003;89(1):169–76. [PubMed] [Google Scholar]
- 57.Qi WX, Min DL, Shen Z, et al. Risk of venous thromboembolic events associated with VEGFR-TKIs: a systematic review and meta-analysis. Int J Cancer. 2013;132(12):2967–74. doi: 10.1002/ijc.27979. [DOI] [PubMed] [Google Scholar]
- 58.Sonpavde G, Je Y, Schutz F, et al. Venous thromboembolic events with vascular endothelial growth factor receptor tyrosine kinase inhibitors: a systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol. 2013;87(1):80–9. doi: 10.1016/j.critrevonc.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 59.Evans CE, Grover SP, Humphries J, et al. Antiangiogenic therapy inhibits venous thrombus resolution. Arterioscler Thromb Vasc Biol. 2014;34:565–570. doi: 10.1161/ATVBAHA.113.302998. [DOI] [PubMed] [Google Scholar]
- 60.Kristinsson SY, Pfeiffer RM, Björkholm M, et al. Arterial and venous thrombosis in monoclonal gammopathy of undetermined significance and multiple myeloma: A population-based study. Blood. 2010;115:4991–4998. doi: 10.1182/blood-2009-11-252072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tricot GJ. New insights into role of microenvironment in multiple myeloma. Int J Hematol. 2002;76 Suppl 1:334–336. doi: 10.1007/BF03165279. [DOI] [PubMed] [Google Scholar]
- 62.Amrani DL. Regulation of fibrinogen biosynthesis: glucocorticoid and interleukin-6 control. Blood Coagul Fibrinolysis. 1990;1:443–446. [PubMed] [Google Scholar]
- 63.Elice F, Fink L, Tricot G, Barlogie B, Zangari M. Acquired resistance to activated protein C (aAPCR) in multiple myeloma is a transitory abnormality associated with an increased risk of venous thromboembolism. Br J Haematol. 2006;134:399–405. doi: 10.1111/j.1365-2141.2006.06208.x. [DOI] [PubMed] [Google Scholar]
- 64.Weber D, Rankin K, Gavino M, Delasalle K, Alexanian R. Thalidomide Alone or with Dexamethasone for Previously Untreated Multiple Myeloma. 2003:16–19. doi: 10.1200/JCO.2003.03.139. [DOI] [PubMed] [Google Scholar]
- 65.Zangari M, Anaissie E, Barlogie B, et al. Increased risk of deep-vein thrombosis in patients with multiple myeloma receiving thalidomide and chemotherapy. Blood. 2001;98:1614–1615. doi: 10.1182/blood.v98.5.1614. [DOI] [PubMed] [Google Scholar]
- 66.Palumbo A, Bringhen S, Caravita T, et al. Oral melphalan and prednisone chemotherapy plus thalidomide compared with melphalan and prednisone alone in elderly patients with multiple myeloma: Randomised controlled trial. Lancet. 2006;367:825–831. doi: 10.1016/S0140-6736(06)68338-4. [DOI] [PubMed] [Google Scholar]
- 67.Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III Clinical Trial of Thalidomide plus Dexamethasone Compared with Dexamethasone Alone in Newly Diagnosed Multiple Myeloma: A Clinical Trial Coordinated by the Eastern Cooperative. doi: 10.1200/JCO.2005.03.0221. [DOI] [PubMed] [Google Scholar]
- 68.Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1-risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111:86–93. doi: 10.1182/blood-2007-01-068833. [DOI] [PubMed] [Google Scholar]
- 69.Richardson P, Jagannath S, Hussein M, et al. Safety and efficacy of single-agent lenalidomide in patients with relapsed and refractory multiple myeloma. Blood. 2009;114(4):772–8. doi: 10.1182/blood-2008-12-196238. [DOI] [PubMed] [Google Scholar]
- 70.Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus. Dexamethasone for Relapsed Multiple Myeloma in North America. 2007:2133–2142. doi: 10.1056/NEJMoa070596. [DOI] [PubMed] [Google Scholar]
- 71.Dimopoulos M, Spencer A. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123–2132. doi: 10.1056/NEJMoa070594. [DOI] [PubMed] [Google Scholar]
- 72.De Stefano V, Za T, Rossi E. Venous thromboembolism in multiple myeloma. Semin Thromb Hemost. 2014;40(3):338–47. doi: 10.1055/s-0034-1370793. [DOI] [PubMed] [Google Scholar]
- 73.Rosovsky R, Hong F, Tocco D, et al. Endothelial stress products and coagulation markers in patients with multiple myeloma treated with lenalidomide plus dexamethasone: An observational study. Br J Haematol. 2013;160:351–358. doi: 10.1111/bjh.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schey SA, Fields P, Bartlett JB, et al. Phase I study of an immunomodulatory thalidomide analog, CC-4047, in relapsed or refractory multiple myeloma. J Clin Oncol. 2004;22(16):3269–76. doi: 10.1200/JCO.2004.10.052. [DOI] [PubMed] [Google Scholar]
- 75.Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123(12):1826–32. doi: 10.1182/blood-2013-11-538835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuenen BC. Analysis of prothrombotic mechanisms and endothelial perturbation during treatment with angiogenesis inhibitors. Pathophysiol Haemost Thromb. 2003;33 Suppl 1(suppl 1):13–4. doi: 10.1159/000073281. [DOI] [PubMed] [Google Scholar]
- 77.Kaushal V, Kaushal GP, Melkaveri SN, Mehta P. Thalidomide protects endothelial cells from doxorubicin-induced apoptosis but alters cell morphology. J Thromb Haemost. 2004;2:327–334. doi: 10.1046/j.1538-7933.2003.00573.x. [DOI] [PubMed] [Google Scholar]
- 78.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–64. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 79.Abdullah WZ, Roshan TM, Hussin A, Zain WSWM, Abdullah D. Increased PAC-1 expression among patients with multiple myeloma on concurrent thalidomide and warfarin. Blood Coagul Fibrinolysis. 2013;24(8):893–5. doi: 10.1097/MBC.0b013e3283642ee2. [DOI] [PubMed] [Google Scholar]
- 80.Plotz CM, Knowlton AI, Ragan C. The natural history of Cushing's syndrome. Am J Med. 1952;13(5):597–614. doi: 10.1016/0002-9343(52)90027-2. [DOI] [PubMed] [Google Scholar]
- 81.Johannesdottir SA, Horváth-Puhó E, Dekkers OM, et al. Use of glucocorticoids and risk of venous thromboembolism: a nationwide population-based case-control study. JAMA Intern Med. 2013;173(9):743–52. doi: 10.1001/jamainternmed.2013.122. [DOI] [PubMed] [Google Scholar]
- 82.Weijl NI, Rutten MF, Zwinderman AH, et al. Thromboembolic events during chemotherapy for germ cell cancer: a cohort study and review of the literature. J Clin Oncol. 2000;18(10):2169–78. doi: 10.1200/JCO.2000.18.10.2169. [DOI] [PubMed] [Google Scholar]
- 83.Brotman DJ, Girod JP, Posch A, et al. Effects of short-term glucocorticoids on hemostatic factors in healthy volunteers. Thromb Res. 2006;118(2):247–52. doi: 10.1016/j.thromres.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 84.Patrassi GM, Dal Bo Zanon R, Boscaro M, Martinelli S, Girolami A. Further studies on the hypercoagulable state of patients with Cushing's syndrome. Thromb Haemost. 1985;54(2):518–20. [PubMed] [Google Scholar]
- 85.Rizzo JD, Brouwers M, Hurley P, et al. American Society of Clinical Oncology/American Society of Hematology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. J Clin Oncol. 2010;28(33):4996–5010. doi: 10.1200/JCO.2010.29.2201. [DOI] [PubMed] [Google Scholar]
- 86.Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane database Syst Rev. 2012;12:CD003407. doi: 10.1002/14651858.CD003407.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grant MD, Piper M, Bohlius J, et al. Epoetin and Darbepoetin for Managing Anemia in Patients Undergoing Cancer Treatment : Comparative Effectiveness. Comp Eff Updat [Internet] Rockv Agency Healthc Res Qual. 2013 Available from: http://www.ncbi.nlm.nih.gov/books/ [PubMed]
- 88.Smith KJ, Bleyer AJ, Little WC, Sane DC. The cardiovascular effects of erythropoietin. Cardiovasc Res. 2003;59(3):538–48. doi: 10.1016/s0008-6363(03)00468-1. [DOI] [PubMed] [Google Scholar]
- 89.Falanga A, Marchetti M, Evangelista V, et al. Neutrophil activation and hemostatic changes in healthy donors receiving granulocyte colony-stimulating factor. Blood. 1999;93(8):2506–14. [PubMed] [Google Scholar]
- 90.Agnelli G, Gussoni G, Bianchini C, et al. Nadroparin for the prevention of thromboembolic events in ambulatory patients with metastatic or locally advanced solid cancer receiving chemotherapy: a randomised, placebo-controlled, double-blind study. Lancet Oncol. 2009;10(10):943–9. doi: 10.1016/S1470-2045(09)70232-3. [DOI] [PubMed] [Google Scholar]
- 91.Agnelli G, George DJ, Kakkar AK, et al. Semuloparin for thromboprophylaxis in patients receiving chemotherapy for cancer. N Engl J Med. 2012;366(7):601–9. doi: 10.1056/NEJMoa1108898. [DOI] [PubMed] [Google Scholar]
- 92.Haas SK, Freund M, Heigener D, et al. Low-molecular-weight heparin versus placebo for the prevention of venous thromboembolism in metastatic breast cancer or stage III/IV lung cancer. Clin Appl Thromb Hemost. 18(2):159–65. doi: 10.1177/1076029611433769. [DOI] [PubMed] [Google Scholar]
- 93.Perry JR, Julian JA, Laperriere NJ, et al. PRODIGE: a randomized placebo-controlled trial of dalteparin low-molecular-weight heparin thromboprophylaxis in patients with newly diagnosed malignant glioma. J Thromb Haemost. 2010;8(9):1959–65. doi: 10.1111/j.1538-7836.2010.03973.x. [DOI] [PubMed] [Google Scholar]
- 94.Maraveyas A, Waters J, Roy R, et al. Gemcitabine versus gemcitabine plus dalteparin thromboprophylaxis in pancreatic cancer. Eur J Cancer. 2012;48(9):1283–92. doi: 10.1016/j.ejca.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 95.Lyman GH, Khorana AA, Kuderer NM, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2013;31(17):2189–204. doi: 10.1200/JCO.2013.49.1118. [DOI] [PubMed] [Google Scholar]
- 96.Zangari M, Barlogie B, Anaissie E, et al. Deep vein thrombosis in patients with multiple myeloma treated with thalidomide and chemotherapy: effects of prophylactic and therapeutic anticoagulation. Br J Haematol. 2004;126(5):715–21. doi: 10.1111/j.1365-2141.2004.05078.x. [DOI] [PubMed] [Google Scholar]
- 97.Palumbo A, Cavo M, Bringhen S, et al. Aspirin, warfarin, or enoxaparin thromboprophylaxis in patients with multiple myeloma treated with thalidomide: a phase III, open-label, randomized trial. J Clin Oncol. 2011;29(8):986–93. doi: 10.1200/JCO.2010.31.6844. [DOI] [PubMed] [Google Scholar]
- 98.Larocca A, Cavallo F, Bringhen S, et al. Aspirin or enoxaparin thromboprophylaxis for patients with newly diagnosed multiple myeloma treated with lenalidomide. Blood. 2012;119(4):933–9. doi: 10.1182/blood-2011-03-344333. [DOI] [PubMed] [Google Scholar]
- 99.Palumbo A, Rajkumar SV, Dimopoulos MA, et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia. 2008;22(2):413–22. doi: 10.1038/sj.leu.2405062. [DOI] [PubMed] [Google Scholar]
- 100.Zamagni E, Brioli I, Tacchetti P, et al. Multiple myeloma, venous thromboembolism, and treatment-related risk of thrombosis. Semin Thromb Hemost. 2011;37(3):209–219. doi: 10.1055/s-0031-1273085. [DOI] [PubMed] [Google Scholar]
- 101.Zangari M, Fink L, Zhan F, et al. Low venous thromboembolic risk with bortezomib in multiple myeloma and potential protective effect with thalidomide/lenalidomide-based therapy: review of data from phase 3 trials and studies of novel combination regimens. Clin Lymphoma Myeloma Leuk. 2011;11(2):228–236. doi: 10.1016/j.clml.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 102.Nayak L, Shi H, Atkins GB, et al. The thromboprotective effect of bortezomib is dependent on the transcription factor Kruppel-like factor 2 (KLF2) Blood. 2014;123(24):3828–31. doi: 10.1182/blood-2014-01-547448. [DOI] [PMC free article] [PubMed] [Google Scholar]