

Abstract
NF-κB is one of the best-characterized transcription factors, providing the link between early membrane-proximal signaling events and changes in many inflammatory genes. MicroRNAs are small non-coding RNAs that regulate gene expression at the post-transcriptional level. Here, we evaluated the role of miR-26b in lipopolysaccharide (LPS)-induced inflammatory response in bovine alveolar macrophages (bAMs). LPS stimulation of bAMs upregulated miR-26b at 1 h, and down-regulated miR-26b at 6 and 36 h. Overexpression of miR-26b in bAMs enhanced the LPS-induced mRNA expression of proinflammatory cytokines and chemokines including TNF-α, IL-1β, IL-8, , IL-10, but directly inhibited that of IL-6. The similar trend was observed for the release of these cytokines and chemokines from bAMs. miR-26b directly bound the 3’-untranslated region of PTEN, leading to the reduction of PTEN protein in bAMs. miR-26b also enhanced the LPS-induced NF-κB signaling pathway as revealed by increased NFκB transcriptional activity and phosphorylation of p65, IκBα, Ikk and Akt. Moreover, PTEN silencing increased the LPS-induced mRNA expression of TNF-α, IL-1β, IL-6, IL-8, and IL-10, and up-regulated the NF-κB pathway. Taken together, we conclude that miR-26b participates in the inflammatory response of LPS-stimulated bAMs by modulating the NF-κB pathway through targeting PTEN.
INTRODUCTION
Bovine respiratory disease (BRD) complex, also known as shipping fever, is the biggest health obstacle for the cattle industry and causes great economic losses to the global cattle industry (1). Stressful management practices, environmental factors, and a variety of microorganisms are believed to contribute to the pathogenesis of BRD. Mannheimia haemolytica (formally known as Pasteurella haemolytica) is the primary bacterial agent responsible for the clinical disease (2). M. haemolytica produces various potential virulence factors including the endotoxin, lipopolysaccharide (LPS), which causes acute inflammation, hemorrhage, edema, and hypoxemia (3).
LPS is a component of the outer membrane of Gram-negative bacteria such as M. haemolytica and Escherichia coli. LPS initiates the innate immune responses via the Toll-like receptor 4 (TLR4) that works as a primary sensor to detect various microbial components (4). The stimulation of TLR4 activates the IκB kinase (Ikk) complex and leads to the phosphorylation of IκBα, resulting in its ubiquitin-induced degradation by the 26S proteasome (5). NF-κB heterodimers, p50/p65 are then released and translocated to the nucleus, where they bind to κB DNA sequence motifs and induce the expression of important immune-regulatory genes (5, 6), including inflammatory cytokines such as TNF-α and IL-6 (4, 7) and nitric oxide production (8). Thus, modulating NF-κB activity is a logical therapeutic approach for the treatment of inflammatory-mediated diseases.
The phosphatase and tensin homolog on chromosome ten (PTEN) gene was initially identified as a tumor suppressor (9, 10). PTEN plays an important role in multiple cellular events including proliferation and apoptosis (11). PTEN is a phosphoinositide phosphatase that dephosphorylates phosphatidylinositol 3, 4, 5-trisphosphate (PIP3) to PIP2 (12). PTEN acts as an natural inhibitor of the PI3K/Akt pathway. Dysfunction of PTEN results in PIP3 accumulation in cells, followed by the hyperactivation of the PI3K/Akt downstream signaling (11), leading to increased cell proliferation and survival. Down-regulation of PTEN also activates the NF-κB pathway signaling by increasing p65 subunit nuclear translocation in mouse mesangial cells (13). Thus, reduction of PTEN protein levels induces gene expression related to diverse cellular processes including inflammation (14-16).
MicroRNAs (miRNAs) are non-coding small RNAs (~21–23 nucleotides) that regulate many biological processes and contribute to various disorders including inflammatory diseases (17-19). miRNAs inhibit the expression of target genes at the post-transcriptional level (20, 21). The binding of a miRNA in the form of the RNA-induced silencing complex with 3’-untranslationed region (3’-UTR) of its target mRNA represses protein synthesis by translational inhibition and mRNA cleavge or decay (21). In rare cases, miRNAs can activate translation (22, 23). miRNAs are potential regulators of inflammatory responses in many immunoactive cells, including alveolar macrophages (24-26). Additionally, miRNAs are known to regulate the NF-κB pathway (27). miR-146a inhibits NF-κB activity by targeting TRAF6 (28). In contrast, miR-21 enhances NF-κB activity through repression of PTEN expression, forming a positive feedback loop (29, 30). miR-301a elevates NF-κB activity by down-regulating NF-κB repressing factor (NKRF) levels, which in turn further promotes miR-301a transcription (31). Multiple miRNAs including miR-21 (32-34), miR-26a (35) and miR-17-92 (36) have been shown to target PTEN.
Very little is known regarding the role of miRNAs in the progression of BRD. In addition, the biological function of miRNAs in modulating NF-κB signaling in bovine alveolar macrophages (bAMs) and their key target genes are still poorly understood. In this study, we identified miR-26b as an LPS-induced inflammatory responsive miRNA in bAMs. Further investigation revealed that miR-26b increased NF-κB activity and thus cytokine expression in bAMs challenged with LPS by negatively regulating PTEN. Our studies suggest that tight regulation of miR-26b is critical for the inflammatory response through NF-κB signaling in bAMs and may provide a new therapeutic target for BRD.
MATERIALS AND METHODS
Bovine alveolar macrophage isolation and LPS stimulation
Bovine lungs (Ralph’s Packing Company, Perkins, Oklahoma) were infused by gravity (30 cm height) with cold Hanks’ balanced salt solution (pH 7.4) without Ca2+ or Mg2+ (HBSS−). The infused fluid was gauze-filtered. bAMs were collected by centrifugation (200 xg, 10 min at 4°C). After being washed twice in HBSS−, bAMs were resuspended in Dulbecco’s Modified Eagle’s medium (DMEM ) (ATCC, Manassas, VA) with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Atlanta, GA) and seeded into 6-well cell culture plates at a density of 4 million cells per well. bAMs were allowed to adhere for 4 h and then stimulated with 1 μg/ml of Escherichia coli LPS (055:B5; Sigma-Aldrich, St Louis, MO) for 1 - 48 h. The cell viability was determined by trypan blue dye exclusion assay. The purity of bAMs was determined by immunocytochemistry with monoclonal anti-CD68 (EBM11). bAMs were maintained in DMEM containing 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin. We used E. coli LPS in the current study because a relatively pure preparation of E. coli LPS is commercially available. A side-by-side comparison study of LPS from E. Coli and M. haemolytica did reveal any obvious differences on bovine lung cells (37). Furthermore, the pattern of cytokine expression in bovine alveolar macrophages stimulated by LPS from M. haemolytica (38, 39) and from E. Coli (the current study) appears to be similar although the peak expression of cytokines showed a slight difference for some cytokines.
Immunocytochemistry
Freshly isolated bAMs were seeded in 24-well cell culture plates and allowed to adhere for 4 h. Then, cells were washed twice with phosphate buffered saline (PBS) (pH 7.4) and fixed with 4% paraformaldehyde in PBS for 30 min, followed by permeabilization with 0.1% Triton X-100 for 15 min and blocking with 1% bovine serum albumin (BSA) for 1 h. Cells were incubated with mouse monoclonal anti-CD68 (EBM11) (1:100) (Dako, Carpenteria, CA) at 4°C overnight. Cells were then washed and incubated with Alexa-Fluor-488-conjugated anti-(mouse IgG) secondary antibodies (1:500) (Bio-Rad, Hercules, CA). The negative controls were treated as described above except the omission of the primary antibody. 4', 6-Diamidino-2-phenylindole dihydrochloride (DAPI) (2.5 mg/ml) (Sigma-Aldrich, St Louis, MO) staining was performed for counting cells. Cells were viewed on a Nikon Eclipse TE 2000 U inverted fluorescence microscope or Nikon Eclipse E600 fluorescence microscope.
RNA isolation and quantitative real-time PCR
Total RNAs were extracted from bAMs using TRI Reagent (Molecular Research Center, Inc., Cincinnati, OH). Residual DNA was removed using DNA-free DNase (Ambion, Austin, TX). RNAs were reverse-transcribed into cDNA using M-MLV reverse transcriptase (Invitrogen, Carslbad, CA). Quantitative RT-PCR (qRT-PCR) was performed in triplicate on an ABI 7500 system (Applied Biosystems, Foster City, CA) using SYBR Green I detection and gene-specific primers (Table 1). The fluorescent signal was only detected in the amplification step for each cycle. A melt curve was generated to check the specificity of the amplification at the dissociation stage. 18S rRNA was used as an internal control. The relative amount of each mRNA to 18S rRNA was calculated with the equation 2− (CtmRNA −Ct18S).
Table 1.
Primers designed for PCR amplification
| Forward primer | Reverse primer | |
|---|---|---|
| 18s | CTGTTGCTCTCTTGGCAGCTT | GGTGGAAAGGTGTGGAATGTG |
| TNF α | TGATGCTGATTTGGTGACTGATT | TTATTTCTCGCCACTGACCAGTAG |
| IL 6 | CCAGAGAAAACCGAAGCTCTCA | TCCTTGCTGCTTTCACACTCA |
| IL 8 | CTGTTGCTCTCTTGGCAGCTT | GGTGGAAAGGTGTGGAATGTG |
| IL 10 | TGCATAGCTCAGCACTACTCTGTTG | GCTGGTTGGCAAGTGGATACA |
| IL 1β | TGAGCTGTTATTTGAGGCTGATG | TGAGAAATCTGCAGCTGGATGT |
| miR-26b | ACCCAGTTCAAGTAATTCAGGA | GCGAGCACAGAATTAATACGACTCACTATAGGTTTTTTTTTTTTAACCT |
| U6 | AGGCTCTGAAAGACCGAGTG | GCGAGCACAGAATTAATACGACTCACTATAGGTTTTTTTTTTTTVN |
| PTEN1 3’-UTR- WT (PTEN1-WT) |
CATGCTAGCCACCACTGACTCTGATCCAGAG | TCCGTCGACCATATGCAGTCTGGGCATATCA |
| PTEN1 3’-UTR- Mutant 1 (PTEN1-M1) |
ACACCATGAAAACAACTATCTATAAACTGAA | TTCAGTTTATTCAAGTTTGTTTTCATGGTGT |
| PTEN1 3’-UTR- Mutant 2 (PTEN1-M2) |
TAACTGTTAGGGAATTTTCTATCTATATTGAA TACATAT |
ATATGTATTCAATATAGATAGAAAATTCCCTAACAGTTA |
| PTEN2 3’-UTR- WT (PTEN2-WT) |
TAAGCTAGCGGAATGTGAAGGTCTGAATGA | ATGTCGACGCAACCACAGCCATCGTTAT |
| PTEN2 3’-UTR- Mutant 3 (PTEN2-M3) |
CTTACTTGTCTGAAGTTCGTAGACGGCATCA CT |
AGTGATGCCGTCTACGAACTTCAGACAAGTAAG |
| PTEN shRNA | GATCCGCTGAAAGACATTATGATACCTTCAA GAGAGGTATCATAATGTCTTTCAGCTTTTTG |
AATTCAAAAAGCTGAAAGACATTATGATACCTCTCTTGAAGGTATCAT AATGTCTTTCAGCG |
For quantification of miRNAs, 5 μg of total RNAs were poly (A)-tailed using A-Plus™ Poly (A) Polymerase tailing kit (Epicentre Biotechnologies, Madison, WI). 1.5 μg of poly (A)-tailed RNAs were reversed-transcribed into cDNA using oligo dT primers and M-MLV reverse transcriptase. qRT-PCR was performed using the target mature miRNA sequence as the forward PCR primer (Table 1) and a universal primer (5’- GCGAGCACAGAATTAATACGACTCAC-3’) as the reverse primer. U6 snRNA was used as an internal control. The relative amount of each miRNA to U6 snRNA was calculated by the comparative Ct method using the equation 2− (CtmiRNA −CtU6).
Cytokine measurement in culture medium
Immediately after collection, culture medium was centrifuged at 1,000 rpm for 5 min at 4°C to remove residual cells. The supernatant was harvested and stored at −80°C for subsequent analysis. Concentrations of TNF-α, IL-1β, IL-6, IL-8, and IL-10 in culture media were assayed using bovine ELISA kits: TNF-α (Raybiotech, Norcross, GA), IL-1β (Thermo Scientific, Waltham, MA), IL-6 (Thermo Scientific), IL-8 (MyBiosource, San Diego, CA), and IL-10 (MyBiosource) following manufacturers' instructions. All samples were assayed in duplicate. Cytokine concentrations were expressed as pg/ml based on relevant standard curves.
Target gene and pathway analyses of miR-26b
Web-based software of Pictar (URL: http://pictar.mdc-berlin.de/) and TargetScan (URL: http://www.targetscan.org/mmu_50/) were used to predict the pathways and targets of miR-26b. Commonly used algorithms give variable weight to: (i) complementarity to the miRNA seed region; (ii) evolutionary conservation of the miRNA recognition elements (MREs); (iii) free energy of the miRNA-mRNA heteroduplex; and (iv) mRNA sequence features outside the target site. TargetScan and PicTar focus on the seed region in miRNA targeting (40, 41). TargetScan requires an exact match to ≥7 bases of the seed sequence (40). PicTar imposes a stringent free energy cutoff for imperfect matches (41). Both TargetScan and PicTar improve their predictions because of evolutionary conservation. Furthermore, TargetScan improves predictions for non-conserved sequences and adds a ‘context score’ considering features in the surrounding mRNA which include local A-U content and location (near either end of the 3′-UTR is preferred) (42). mRNAs that have a high context score or multiple predicted MREs are more likely to be true targets.
Construction of lentiviral vectors
We constructed a lentiviral miR-26b expression vector as previously described (43). The lentiviral control vector (miR-con) contains a non-relevant sequence as a negative control. For PTEN silencing lentiviral vector, shRNA of PTEN (Table 1) was cloned into pSIH1-H1-copGFP Vector (System Biosciences, Mountain View, California, USA). The control shRNA vector was purchased from System Biosciences. The lentiviruses were generated in HEK 293T cells using Lenti-X HTX Packaging Mix (Clontech, Mountain View, CA), then titered and stored at −80°C until use.
Construction of 3’-UTR luciferase reporter vectors
We divided the three binding sites of PTEN into 2 segments for cloning due to the length of its 3’-UTR. PTEN1 contained two binding sites (43-49 and 1278-1285), and PTEN2 contained one binding site (2618-2625). PTEN1 and PTEN2 were PCR-amplified from bovine genomic DNA and cloned into the pmirGLO vector (Promega, Madison, WI) downstream of a firefly luciferase reporter gene using the Nhel I and Sal I restriction sites. We used over-lapping PCR to construct the luciferase reporters with mutant PTEN 3’-UTR of miR-26b binding sites. We first amplified the PTEN1 or PTEN2 as two fragments with overlapping mutation sites using two primer sets. The annealing two fragments were then used as a template to amplify the mutant PTEN1 or 2. The primer sequences are listed in Table 1.
Dual luciferase reporter assay
Raw 264.7 cells were seeded in a 96-well plate (2 ×104 cells/well). After being in culture for 24 h, the cells were co-transfected with 5 ng of wild-type or mutant PTEN 3’-UTR luciferase reporter construct and 100 ng of miR-26b expression plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The empty pmirGLO vector was used as a control. 24 h after transfection, cells were harvested and dual luciferase activities were measured with Dual Luciferase Reporter Assay System (Promega, Madison, WI). Data were normalized by dividing firefly luciferase activity by Renilla luciferase activity.
For the pathway activity, Raw 264.7 cells were seeded in a 96 well plate at a density of 2 ×104 cells/well. After being in culture for 24 h, the cells were transfected with 50 ng of miR-26b and 100 ng of NF-κB pathway-luciferase-reporter construct (SABiosciences, Frederick, MD). miR-con was used as a control. 24 h after transfection, LPS (1 μg/ ml) was added. After 18 h stimulation, the cells were harvested. NF-κB transcriptional activity was measured with Dual Luciferase Reporter Assay System. Data was expressed as a ratio of firefly luciferase activity to Renilla luciferase activity.
Western blotting
Cells were lysed in lysis buffer (Thermo Scientific, Rockford, IL) and protein concentrations were determined using DC protein assay kit (Bio-Rad, Hercules, CA). Twenty μg of protein extracts were separated by 10% SDS-PAGE and then transferred onto nitrocellulose membranes. Proper transfer was ensured by staining the membrane with Ponceau S. Membranes were blocked overnight with 5% dried skimmed milk powder in 100 mM TBST. Then, the membranes were incubated at 4°C overnight with primary antibodies: anti-PTEN (1:500, Bioss, Woburn, MA), anti-Akt (1:500, Bioss), anti-phospho-Akt (1:1,000, Millipore, Bellerica, MA), anti-Ikkβ (1:1,000, Cell Signaling Technologies, Beverly, MA), anti-Ikkα (1:1,000, Cell Signaling Technologies), anti-phospho-Ikkα/β (1:1,000, Cell Signaling Technologies, Beverly, MA), anti-IκBα (1:1,000, Cell Signaling Technologies, Beverly, MA), anti-phospho-IκBα (1:1,000, Cell Signaling Technologies, Beverly, MA), anti-NF-κB p65 (1:1,000, Abcam, Cambridge, MA), anti-phospho-NF-κB p65 (1:500 dilution, Bioss) and anti–GAPDH (1:500, Sigma-Aldrich) and anti-β-actin (1:500, Sigma-Aldrich). After being washed with TBST three times, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad, Hercules, CA) for 1 h. Blots were washed again and target proteins were visualized using the enhanced chemiluminescence detection system. All the experiments were repeated three times. All the results are from separate blots to avoid possible problems related to incomplete stripping.
Nitric oxide measurement
The level of nitric oxide production was monitored by measuring nitrite and nitrate concentration in the cultured medium. bAM culture media were collected and centrifuged at 4°C for 5 min to remove the residual cells. The amount of nitric oxide in the supernatant was measured by mixing the medium with the same volume of Griess reagent (100 μl) in a 96-well plate. The plate was incubated for 15 min at room temperature and read at 570 nm by a spectrophotometer (44).
RESULTS
Viability and purity of bovine macrophages
The viabilities of the isolated bAMs were 72 ± 1.8 % and 92 ± 0.5% (means ± SE, n=9) before and after attachment as revealed by trypan blue staining. The purity of the attached bAMs was determined by Wright’s Giemsa staining and immunostaining for the surface antigen CD68, which is a cell surface marker of macrophages (Supplementary Fig. 1). The immunostaining showed that 96.1 ± 0.5% (means ± SE, n=9) cells were positive for CD68. Only cell preparations containing bAMs with > 95% purity were used for our studies.
LPS induces a time-dependent cytokine mRNA expression and nitric oxide production in bAMs
LPS induces rapid production and release of inflammatory cytokines and chemokines known to be involved in lung inflammation and acute lung injury (45). We incubated bAMs with LPS for various time periods and determined cytokine and chemokine expression. TNF-α and IL-6 mRNA expression peaked at 1 h after LPS stimulation of bAMs (Fig. 1A, B) while IL-1β and IL-10 mRNA expression peaked at 6 and 36 h post-LPS exposure, respectively (Fig. 1C, D).
Fig. 1. LPS induces mRNA expression of cytokines and NO production from bAMs.
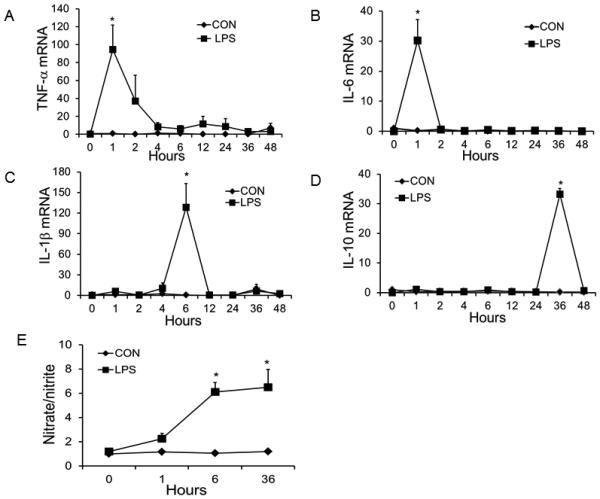
bAMs were stimulated with LPS (1 μg/ml) for various times. Controls were bAMs without LPS stimulation. The mRNA expression of TNF-α (A), IL-6 (B), IL-1β (C), and IL-10 (D) was measured by real-time PCR using 18S rRNA as an endogenous control. (E) NO production in the medium was determined using the Griess reagent in triplicate. Data was expressed as a fold change to 0 h control. Results are shown as means ± SE. *P<0.05 vs. 0 h (n=3 cell preparations).
It is known that nitric oxide generated from LPS-stimulated bAMs causes cytotoxic injury to pulmonary cells (46). NO also plays an important role in numerous pathophysiological conditions including inflammation, and is induced in a nuclear factor (NF)-κB-dependent manner (47-49). LPS enhanced NO production in bAMs as revealed by increased nitrate and nitrite levels in culture media, reaching a maximum at 6 h post-LPS stimulation (Fig. 1E). Based on cytokine expression and NO production, we chose 1, 6 and 36 h for further studies.
miR-26b is dynamically changed in bAMs after LPS stimulation
Based on our preliminary microRNA microarray studies, we found that miR-26b is a LPS-responsive miRNA in bAMs. Real-time PCR analysis revealed that miR-26b expression in bAMs was increased in the early phase, but decreased in the later stages after LPS stimulation (Fig. 2).
Fig. 2. miR-26b expression in LPS-stimulated bAMs.
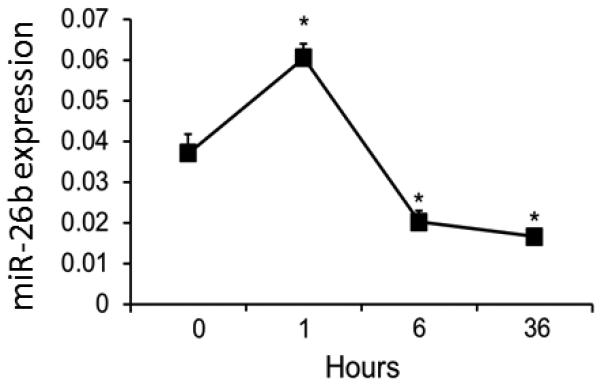
bAMs were treated with LPS (1 μg/ ml) for 0, 1, 6 and 36 h. miR-26b expression level was determined by real-time PCR. Data was normalized to U6 snRNA. Results are expressed as means ± SE. *P<0.05 vs. 0 h (n=3 cell preparations).
miR-26b enhances LPS-induced mRNA expression of TNF-α, IL-1β, IL-8, and IL-10 but represses that of IL-6
We further explored whether miR-26b regulates inflammatory cytokine and chemokine mRNA levels in LPS-stimulated bAMs. We first determined miR-26b expression levels after infecting bAMs with different MOIs of miR-26b lentivirus or lentiviral control (VC). Maximal expression of miR-26b was achieved at a MOI of 50 (Fig. 3A). We then infected bAMs with miR-26b lentivirus or VC at a MOI of 50 and challenged them with LPS for 0, 1, 6 and 36 h. The blank control group (BC) was bAMs without virus infection. As seen in Fig. 3B-F, the combination of miR-26b and LPS greatly augmented TNF-α, IL-8, IL-1β, and IL-10 mRNA expression. However, miR-26b markedly reduced the level of IL-6 mRNA, consistent with the report that IL-6 is a direct target of miR-26b (50).
Fig. 3. miR-26b enhances LPS-induced mRNA and protein expression of TNF-α, IL-8, IL-1β, and IL-10, but represses that of IL-6.
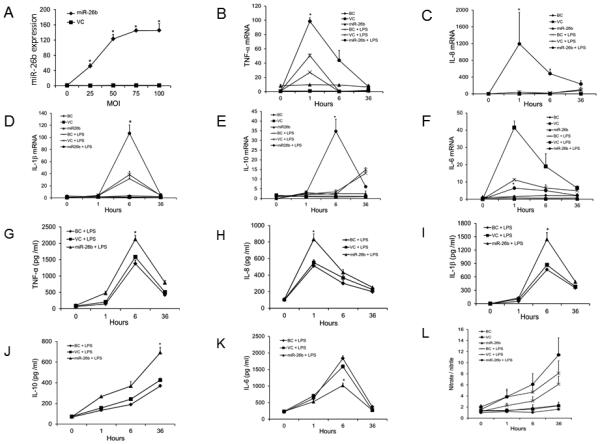
(A) bAMs were infected with various multiplicity of infection (MOI) of miR-26b lentivirus or lentiviral control vector (VC). miR-26b expression level was determined by real-time PCR. Data was expressed as a fold change to 0 h VC. Results are expressed as means ± SE (n=3 cell preparations). (B-L) bAMs were infected with lentiviruses expressing miR-26b or VC at a MOI of 50. bAMs that were not infected with lentiviruses are blank control (BC). Then, bAMs were stimulated with or without LPS (1 μg/ml) for 1, 6 and 36 h. The mRNA expression of TNF-α (B), IL-8 (C), IL-1β (D), IL-10 (E) and IL-6 (F) at indicated time points was analyzed by real-time PCR. The expression of each gene was normalized to the average of BC at 0 h. The protein levels of TNF-α (G), IL-8 (H), IL-1β (I), IL-10 (J), and IL-6 (K), as well as NO production (L) in the culture media were assayed with a specific ELISA or Griess reagent. Results are expressed as means ± SE. *P<0.05 vs. VC+LPS (n=3 cell preparations).
Cytokine protein production in culture media of bAMs was quantified by ELISA. In the LPS-challenged miR-26b-infected bAMs, we observed a significant increase in TNF-α, IL-8, IL-1β, and IL-10 protein and a decrease in IL-6 protein in the culture media as compared to the levels of these cytokines in the VC group treated with LPS (Fig. 3G-K).
miR-26b promotes LPS-induced NO production
Since NO is up-regulated by LPS and proinflammatory cytokines, we next examined NO production in miR-26b-challenged bAMs. NO production was increased after 6 h of LPS stimulation in lentiviral miR-26b-infected bAMs compared to VC-infected bAMs but did not reach the significant level (Fig. 3L). These results suggest that miR-26b and LPS synergistically enhance TNF-α, IL-1β, IL-8, and IL-10 but repress IL-6 expression at the mRNA level, resulting in enhanced TNF-α, IL-1β, IL-8, IL-10 protein and NO production but decreased IL-6 protein levels in the culture media.
PTEN is repressed by miR-26b
To decipher how miR-26b regulates LPS-induced cytokine expression, we used the bioinformatics tool, Targetscan, to predict target genes of miR-26b. Among these potential targets, there are 3 binding sites in the 3’-UTR of PTEN, which is highly conserved in mammals including cows, humans, mice and rats. Furthermore, PTEN inhibition activates Akt, which consequently activates Ikk, leading to degradation of IκB and nuclear translocation of the transcription factor NF-κB (51). Therefore, we chose PTEN for further analysis.
To verify whether PTEN is a potential target of miR-26b, Raw 264.7 cells were co-transfected with the PTEN 3’-UTR firefly luciferase reporter vector and miR-26b or the control vector, miR-Con. Due to the length of the PTEN 3’-UTR, we cloned binding sites 1 and 2 into one luciferase reporter vector (PTEN1-WT), and cloned binding site 3 into a second reporter vector (PTEN2-WT) (Fig. 4A). We found that miR-26b repressed activities of both PTEN1 and PTEN2 (Fig. 4B). When we mutated the binding site 1 or 2 in PTEN1 (PTEN1-M1 or PTEN1-M2), the inhibition of the reporter activities by miR-26b was less compared to PTEN1-WT. Furthermore, the mutation of both binding sites (PTEN1-M1+M2) completely abolished the miR-26b inhibition. Similarly, the mutation of the binding site 3 in PTEN2 (PTEN2-M3) also resulted in no inhibition of the reporter activity by miR-26b. These results suggest that three binding sites in 3’-UTR of PTEN are all involved in the binding of miR-26b.
Fig. 4. PTEN is a target of miR-26b.
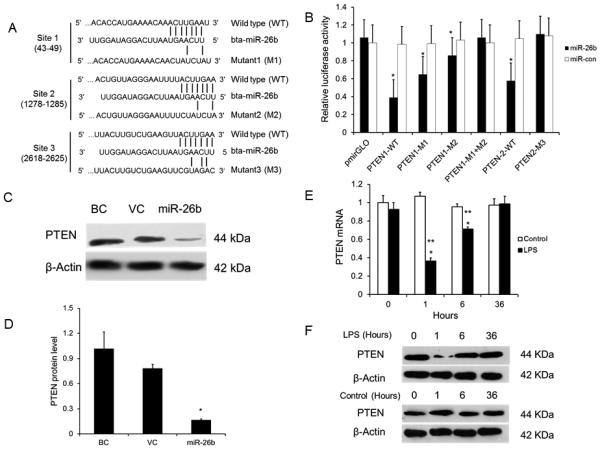
(A) The potential interaction between bta-miR-26b and the putative binding sites in the 3’-UTR of the bovine PTEN gene. Mutations of bta-miR-26b binding sites in the 3’-UTR of bovine PTEN are indicated. (B) 3’-UTR luciferase reporter assays. HEK293T cells were co-transfected with wild-type (WT) PTEN1, PTEN2 or their mutants (M1, M2, M1+M2 or M3) and miR-26b or miR-con for 24 h. Firefly luciferase activity was normalized to Renilla luciferase activity. The results were expressed a ratio of empty pmirGLO vector + miR-con (means ± SE). *P<0.05 vs. miR-con (n=3 cell preparation). (C) Western blotting for PTEN in the bAMs without lentivirus (BC) or infected with lentivirus expressing miR-26b or the lentiviral control vector (VC). (D) The blotting bands were quantified by ImageJ. The relative PTEN expression levels were normalized to β-actin. Results are expressed as means ± SE. *P<0.05 vs. BC (n=3 cell preparation). (E-F) bAMs were stimulated with LPS (1 μg/ml) for various times. Controls were bAMs without LPS stimulation. The mRNA levels of PTEN were measured by real-time PCR using 18S rRNA as an endogenous control (E). *p<0.05 vs. miR-con at the same time point; **P<0.05 vs. miR-26b at 0 h stimulation (n=3 cell preparation). The protein level of PTEN was measured by western blotting (F).
To further confirm the results, western blotting was used to determine the endougenous PTEN protein in miR-26b-overexpressing bAMs. The result showed that PTEN protein level was reduced by miR-26b overexpression (Fig. 4C, D).
miR-26b expression was increased at 1 h and then declined at 6 h and 36 h in bAMs stimulated with LPS (Fig. 2). We determined PTEN expression in LPS-stimulated bAMs as used in Fig. 2 to see whether miR-26b is inversely correlated to PTEN expression. The results showed that mRNA and protein levels of PTEN were reduced at 1 h and then gradually recoverd at 6 h and 36 h after LPS stimulation (Fig. 4E, F). Taken together, we concluded that PTEN is a target of miR-26b in bAMs.
miR-26b enhances LPS-induced NF-κB signaling pathway
PTEN is known to inhibit Akt phosphorylation and thus the NF-κB pathway (52, 53). Therefore, we next investigated whether miR-26b can activate NF-κB signaling. We first used a dual luciferase reporter assay to assess the effect of miR-26b on the NF-κB pathway. We found that miR-26b up-regulated the activity of the NF-κB luciferase reporter construct after LPS stimulation compared with the control vector, miR-Con (Fig. 5A). Then, we determined the effect of miR-26b on LPS-induced phosphorylation of Akt, Ikk, IκBα and p65. miR-26b greatly enhanced the LPS-stimulated phosphorylation of Akt, Ikk, IκBα and P65 at the early time point (15 min) (Fig. 5B). The phosphorylation of these proteins sustained up to 60 min, the longest time point measured in the current study. Thus, these results show that miR-26b and LPS cooperatively up-regulate the NF-κB singling pathway.
Fig. 5. miR-26b activates NF-κB signaling.
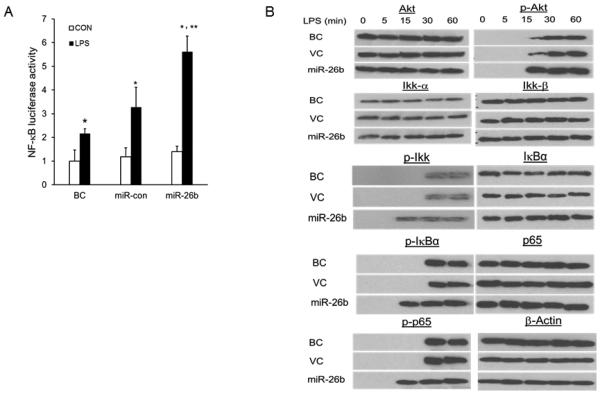
(A) Raw 264.7 cells were co-transfected with a NF-κB luciferase-reporter construct and miR-26b or miR-Con. After 24 h culture, cells were stimulated with or without LPS (CON). Blank Control (BC): Raw 264.7 cells transfected with only NF-κB luciferase-reporter construct. The firefly luciferase activity was normalized to Renilla luciferase activity. Results are expressed as fold change over blank control (BC) without LPS stimulation. Results are expressed as means ± SE. **P<0.05 vs. miR-Con with LPS, *P<0.05 vs. CON (n=3 cell preparation). (B) Western blot analysis of phosphorylated Akt, Ikk, IκBα and p65 in lysates from bAMs infected with miR-26b or control lentivirus (VC) at a MOI of 50 for 16 h, and then treated for 0 – 60 min (above lanes) with LPS. Blank control (BC): bAMs without virus infection.
PTEN silencing enhances LPS-induced cytokine expression and activates the NF-κB signaling pathway
In order to assess the role of PTEN in the LPS-induced NF-κB signaling pathway and in inflammatory responses in bAMs, we silenced PTEN using a PTEN shRNA lentivirus to see whether the expression of cytokines and chemokines and NF-κB signaling pathway were affected. PTEN shRNA significantly reduced the expression of PTEN protein in bAMs (Fig. 6A). To determine whether silencing PTEN causes the same cytokine and chemokine responses in LPS-induced bAMs as miR-26b, bAMs were infected with PTEN shRNA lentivirus or the control lentivirus (Con-shRNA) at a MOI of 50 for 24 h and then treated with LPS for 0, 1, 6 and 36 h. Similar to miR-26b, PTEN silencing markedly increased the mRNA expression of TNF-α and IL-1β at 1 and 6 h (Fig. 6B, D). However, PTEN silencing enhanced IL-8 and IL-10 mRNA expression at 36 h rather than at 1 or 6 h as for the case of miR-26b (Fig. 6D, E). In contrast to miR-26b, which decreased the mRNA expression of IL-6 (Fig. 3F), PTEN silencing increased IL-6 mRNA level (Fig. 6F). This is consistent with the observation that IL-6 is a direct target of miR-26b (50).
Fig. 6. PTEN silencing promotes LPS-induced TNF-α, IL-8, IL-1β, IL-10 and IL-6 mRNA expression.
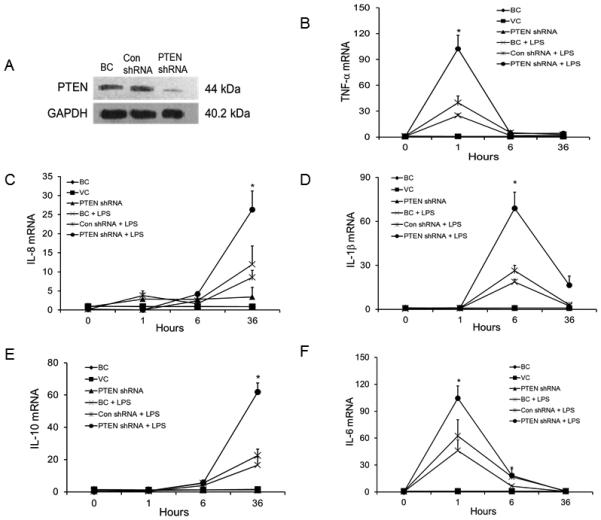
bAMs were infected with PTEN silencing lentivirus (PTEN shRNA) or virus control (Con shRNA) and cultured for 24 h. Blank control (BC): bAMs without virus infection. (A) Western blot analysis was performed to determine PTEN expression. (B-F) infected or un-infected bAMs were stimulated with or without LPS (1 μg/ml) for 1, 6 and 36 h. The mRNA expression of TNF-α (B), ), IL-8 (C), IL-1β (D), IL-10 (E) and IL-6 (F) at indicated time points was analyzed by real-time PCR. The expression of each gene was normalized to the average of BC control samples at 0 h. Results are expressed as means ± SE. *P<0.05 vs. Con shRNA + LPS (n=3 cell preparations).
To investigate the molecular mechanism by which PTEN silencing influences the LPS-induced NF-κB pathway in bAMs, we determined the phosphorylation of the components involved in the pathway by western blotting (Fig. 7). The phosphorylation of Akt, Ikk, IκBα, and p65 were enhanced by PTEN silencing after 15 min of LPS stimulation in bAMs, which is the same pattern as for miR-26b (Fig. 5B).
Fig. 7. PTEN silencing activates NF-κB signaling in bAMs.
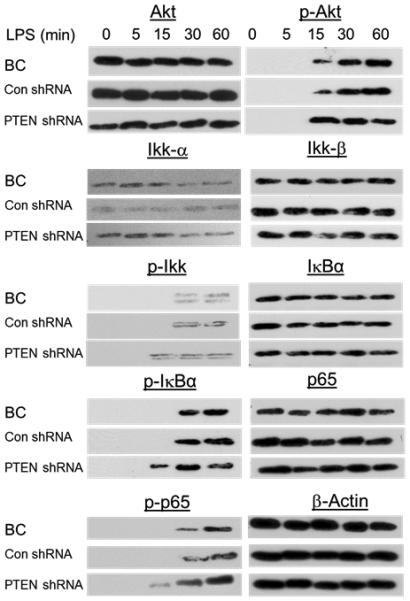
Western blot analysis of phosphorylated Akt, Ikk, IκBα and p65 in lysates of bAMs infected with lenti-viruses expressing PTEN shRNA or vector control (Con shRNA) of MOI 50 for 16 h, and then treated for 0 – 60 min (above lanes) with LPS. Blank control (BC): bAMs without virus infection.
DISCUSSION
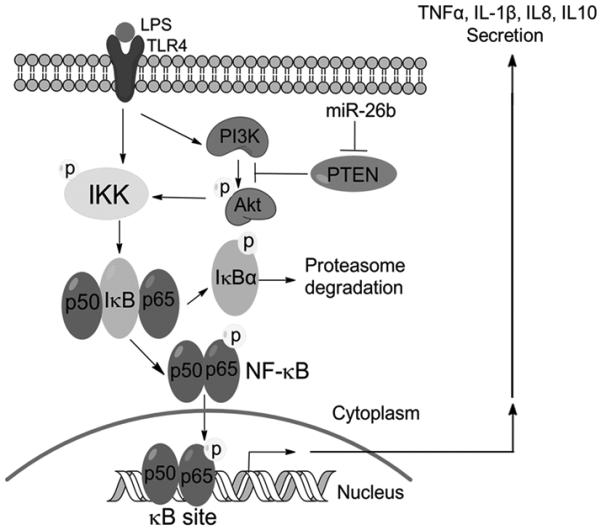
The expression and functions of miRNAs are cell- and tissue-specific. The role of miRNAs in the inflammatory response of bovine alveolar macrophages to LPS challenge has not been investigated in detail. In this study, miR-26b was found to be a LPS-responsive miRNA in bAMs. miR-26b increased the expression of inflammatory cytokines and chemokines, and production of nitric oxide. miR-26b also enhanced the NF-κB pathway by targeting PTEN, therefore acting upstream of the NF-κB pathway. Taken together, these data reveal that miR-26b promotes inflammatory responses in LPS-stimulated bAMs through the activation of the NF-κB pathway by inhibiting PTEN (Fig. 8). It raises the possibility for the development of novel therapeutic strategies for decreasing the morbidity associated with BRD by regulating miR-26b expression.
Fig. 8. Proposed mechanism of miR-26b enhancement of LPS-stimulated NF-κB pathway through down-regulation of PTEN.
Alterations in gene expression including miRNA expression can regulate cellular responses to exogenous stimulation. LPS is one of the potential virulence factors and is known as an inducer of inflammation playing a significant role in the lesion severity in lung tissue (54, 55). Since BRD is a multi-factorial complex phenotype, dissecting molecular networks involved is essential for understanding the pathogenesis of BRD. It has been demonstrated that miRNAs play important roles in bovine monocyte inflammatory and metabolic networks (56). Many LPS-responsive miRNAs also have a role in proinflammatory cytokine production in other species rather than bovine (57-59). miR-223, for example, is down-regulated in RAW264.7 cells challenged with LPS, which leads to the up-regulation of signal transducer and activator of transcription 3 (STAT3) and promotes pro-inflammatory IL-6 and IL-1β transcription (57). It is unknown if miR-223 has a similar role in alveolar macrophages. This is of interest since these cells play a critical role in the innate immunity against infections of BRD. In our study, miR-26b was identified as one of the LPS responsive miRNAs in bAMs. To our knowledge, this is the first report to study the function of LPS-responsive miRNAs in bovine alveolar macrophages and the first to report that miR-26b is an important regulator of proinflammatory cytokines and chemokines production in these cells.
miR-26 is involved in many biological processes. It has been suggested that miR-26 may play a significant role in tumor formation, since miR-26 expression is frequently abnormal in tumors (60). miR-26b also enhances human pituitary tumor cell behavior through the direct regulation of PTEN (61). Estrogen-repressed miR-26a regulates numerous genes associated with cell growth and proliferation in breast tumors (62). miR-26b is down-regulated in carcinoma-associated fibroblasts from estrogen receptor-positive breast cancers , which results in enhanced cell migration and invasion (63). miR-26b may regulates obesity-related insulin sensitivity and inflammatory responses (64). In spite of all these studies, there are very few functional studies of miR-26b in bovine lungs associated with inflammation. We demonstrated that miR-26b promotes pulmonary inflammation by enhancing mRNA expression levels of TNF-α, IL-1β, IL-8, and IL-10 but not IL-6, supporting a previous finding that IL-6 is a direct target of miR-26b (50). Furthermore, secreted cytokine protein levels mirrored those found in the mRNA, establishing that these changes are functionally significant. In all, these findings add to the growing body of cell- and tissue-specific functions of miR-26b.
NF-κB is a transcription factor that controls a variety of important genes encoding transcriptional factors, adhesion molecules, cytokines and growth factors involved in inflammation and the immune response. LPS is a potent activator of NF-κB in alveolar macrophages (8). Because of its importance to many biological processes, NF-κB activation and activity are tightly regulated by a battery of endogenous mechanisms. The excessive and prolonged production of pro-inflammatory mediators triggered by NF-κB pathway, such as TNF-α and IL-6 may result in tissue damage (7). Evidence is growing rapidly that establishes an important etiologic role of miRNAs in the initiation and progression of pathological inflammation and immune responses. miRNA control of NF-κB is emerging as a significant mechanism in disease and normal homeostasis (27).
Several studies have reported the involvement of miRNAs in the regulation of NF-κB signaling. Estradiol inhibits LPS-stimulated NF-κB pathway through correlated regulation of miR-125b and let-7a in primary human macrophages (65). miR-155 reduces the production of proinflammatory cytokines through the activation of NF-κB by targeting IKKε in epithelial cell line (66). Little is known concerning the function of miR-26b in the regulation of NF-κB signaling in bovine alveolar macrophages. In this study, we proved that miR-26b up-regulates the LPS-stimulated NF-κB signaling pathway by suppressing PTEN in bAMs, providing new insight into the regulatory role that miR-26b plays in the NF-κB pathway in alveolar macrophages.
PTEN is well known as a tumor suppressor gene and frequently deleted or mutated in a wide variety of human cancers including lung cancers (67). PTEN encodes a protein that has sequence homology with phosphatases that dephosphorylate phosphates of serine/threonine and tyrosine phosphates on protein (9, 10). PTEN also dephosphorylates PIP3 and PIP2. Akt has been demonstrated as one of the important downstream targets of PI3K (68). Ikk is one of the downstream targets of activated Akt (52). In addition, Ikk is an upstream gene in the NF-κB pathway. Akt-mediated effects on target gene transcription lead to various cellular responses of cancers (67). However, it is unknown whether miRNAs acting through PTEN play any role in bovine lung inflammation and whether such an effect is mediated through the NF-κB pathway. In this study, we demonstrated that PTEN is a target of miR-26b. Additionally, we found that NF-κB signaling is up-regulated by PTEN silencing, which results in the enhancement of inflammatory cytokine mRNA expression levels including IL-6. However, under PTEN inhibition, the peak times of IL-8 and IL-10 shifted to 36 h compared with 1 and 6 h with miR-26b overexpression. This leaves us an interesting direction to study in the future.
In summary, miR-26b enhances inflammatory responses in vitro through the promotion of the NF-κB pathway by directly targeting PTEN (Fig. 8).Consequently, miR-26b has a significant mechanistic role in NF-κB activation in bAMs and may be responsible for the inflammatory reaction that is important in the development of bovine lung diseases caused by gram-negative bacteria. Therefore, strategies aimed at mitigating miR-26b expression to reduce NF-κB activation in bAMs may represent a potential therapy for ameliorating disease in BRD.
Supplementary Material
Acknowlegements
We thank Ralph’s Packing Company for providing the bovine lungs.
Footnotes
This work was supported by the USDA National Institute of Food and Agriculture under Award Number 09PRE2300211 and the National Heart, Lung and Blood Institute under Award Numbers R01HL116876 (to L.L.), and we used the Molecular Biology Core facility supported by the National Institute of General Medical Sciences under Award number P20GM103648. The content is solely the responsibility of the authors and does not necessarily represent the official views of the USDA or National Institutes of Health.
References
- 1.Griffin D, Chengappa MM, Kuszak J, McVey DS. Bacterial pathogens of the bovine respiratory disease complex. Vet Clin North Am Food Anim Pract. 2010;26:381–394. doi: 10.1016/j.cvfa.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Rehmtulla AJ, Thomson RG. A review of the lesions in shipping fever of cattle. Can Vet J. 1981;22:1–8. [PMC free article] [PubMed] [Google Scholar]
- 3.Czuprynski CJ, Leite F, Sylte M, Kuckleburg C, Schultz R, Inzana T, Behling-Kelly E, Corbeil L. Complexities of the pathogenesis of Mannheimia haemolytica and Haemophilus somnus infections: challenges and potential opportunities for prevention? Anim Health Res Rev. 2004;5:277–282. doi: 10.1079/ahr200483. [DOI] [PubMed] [Google Scholar]
- 4.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 6.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 8.Li YH, Yan ZQ, Brauner A, Tullus K. Activation of macrophage nuclear factor-kappa B and induct ion of inducible nitric oxide synthase by LPS. Respir Res. 2002;3:23. doi: 10.1186/rr173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 11.Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. PTEN function: the long and the short of it. Trends Biochem Sci. 2014;39:183–190. doi: 10.1016/j.tibs.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 13.Feng XJ, Liu SX, Wu C, Kang PP, Liu Q, Hao J, Li HB, Li F, Zhang Y, Fu XH, Zhang SB, Zuo LF. The PTEN/PI3K/Akt signalling pathway mediates HMGB1-induced cell proliferation by regulating the NF-kappaB/cyclin D1 pathway in mouse mesangial cells. Am J Physiol Cell Physiol. 2014;306:C1119–C1128. doi: 10.1152/ajpcell.00385.2013. [DOI] [PubMed] [Google Scholar]
- 14.Singh AP, Arora S, Bhardwaj A, Srivastava SK, Kadakia MP, Wang B, Grizzle WE, Owen LB, Singh S. CXCL12/CXCR4 Protein Signaling Axis Induces Sonic Hedgehog Expression in Pancreatic Cancer Cells via Extracellular Regulated Kinase- and Akt Kinase-mediated Activation of Nuclear Factor kappaB: IMPLICATIONS FOR BIDIRECTIONAL TUMOR-STROMAL INTERACTIONS. J Biol Chem. 2012;287:39115–39124. doi: 10.1074/jbc.M112.409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39:493–506. doi: 10.1016/j.molcel.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kansal S, Bhatnagar A, Agnihotri N. Fish oil suppresses cell growth and metastatic potential by regulating PTEN and NF-kappaB signaling in colorectal cancer. PLoS One. 2014;9:e84627. doi: 10.1371/journal.pone.0084627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y, Schetter AJ, Yang GB, Nguyen G, Mathe EA, Li P, Cai H, Yu L, Liu F, Hang D, Yang H, Wang XW, Ke Y, Harris CC. microRNA and inflammatory gene expression as prognostic marker for overall survival in esophageal squamous cell carcinoma. Int J Cancer. 2013;132:2901–2909. doi: 10.1002/ijc.27954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 19.Foster PS, Plank M, Collison A, Tay HL, Kaiko GE, Li J, Johnston SL, Hansbro PM, Kumar RK, Yang M, Mattes J. The emerging role of microRNAs in regulating immune and inflammatory responses in the lung. Immunol Rev. 2013;253:198–215. doi: 10.1111/imr.12058. [DOI] [PubMed] [Google Scholar]
- 20.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 21.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 22.Vasudevan S, Tong YC, Steitz JA. Switching from repression to activation: MicroRNAs can up-regulate translation. Science. 2007;318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 23.Wu L, Belasco JG. Let me count the ways: mechanisms of gene regulation by miRNAs and siRNAs. Mol Cell. 2008;29:1–7. doi: 10.1016/j.molcel.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 24.Coutinho LL, Matukumalli LK, Sonstegard TS, Van Tassell CP, Gasbarre LC, Capuco AV, Smith TPL. Discovery and profiling of bovine microRNAs from immune-related and embryonic tissues. Physiol Genomics. 2007;29:35–43. doi: 10.1152/physiolgenomics.00081.2006. [DOI] [PubMed] [Google Scholar]
- 25.Graff JW, Powers LS, Dickson AM, Kim J, Reisetter AC, Hassan IH, Kremens K, Gross TJ, Wilson ME, Monick MM. Cigarette smoking decreases global microRNA expression in human alveolar macrophages. PLoS One. 2012;7:e44066. doi: 10.1371/journal.pone.0044066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie T, Liang J, Liu N, Wang Q, Li Y, Noble PW, Jiang D. MicroRNA-127 inhibits lung inflammation by targeting IgG Fcgamma receptor I. J Immunol. 2012;188:2437–2444. doi: 10.4049/jimmunol.1101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-kappaB signaling. J Mol Cell Biol. 2011;3:159–166. doi: 10.1093/jmcb/mjr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paik JH, Jang JY, Jeon YK, Kim WY, Kim TM, Heo DS, Kim CW. MicroRNA-146a downregulates NFkappaB activity via targeting TRAF6 and functions as a tumor suppressor having strong prognostic implications in NK/T cell lymphoma. Clin Cancer Res. 2011;17:4761–4771. doi: 10.1158/1078-0432.CCR-11-0494. [DOI] [PubMed] [Google Scholar]
- 29.Ruan Q, Wang T, Kameswaran V, Wei Q, Johnson DS, Matschinsky F, Shi W, Chen YH. The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic beta cell death. Proc Natl Acad Sci U S A. 2011;108:12030–12035. doi: 10.1073/pnas.1101450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marquez RT, Wendlandt E, Galle CS, Keck K, McCaffrey AP. MicroRNA-21 is upregulated during the proliferative phase of liver regeneration, targets Pellino-1, and inhibits NF-kappaB signaling. Am J Physiol Gastrointest Liver Physiol. 2010;298:G535–541. doi: 10.1152/ajpgi.00338.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Z, Li Y, Takwi A, Li B, Zhang J, Conklin DJ, Young KH, Martin R, Li Y. miR-301a as an NF-kappaB activator in pancreatic cancer cells. EMBO J. 2011;30:57–67. doi: 10.1038/emboj.2010.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pezzolesi MG, Platzer P, Waite KA, Eng C. Differential expression of PTEN-targeting microRNAs miR-19a and miR-21 in Cowden syndrome. Am J Hum Genet. 2008;82:1141–1149. doi: 10.1016/j.ajhg.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H, Kong W, He L, Zhao JJ, O'Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, Cheng JQ. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68:425–433. doi: 10.1158/0008-5472.CAN-07-2488. [DOI] [PubMed] [Google Scholar]
- 35.Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH, Sohn-Lee C, le Sage C, Agami R, Tuschl T, Holland EC. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327–1337. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao CC, Srinivasan L, Calado DP, Patterson HC, Zhang BC, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McClenahan D, Hellenbrand K, Atapattu D, Aulik N, Carlton D, Kapur A, Czuprynski C. Effects of lipopolysaccharide and Mannheimia haemolytica leukotoxin on bovine lung microvascular endothelial cells and alveolar epithelial cells. Clin Vaccine Immunol. 2008;15:338–347. doi: 10.1128/CVI.00344-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoo HS, Maheswaran SK, Lin G, Townsend EL, Ames TR. Induction of inflammatory cytokines in bovine alveolar macrophages following stimulation with Pasteurella haemolytica lipopolysaccharide. Infect Immun. 1995;63:381–388. doi: 10.1128/iai.63.2.381-388.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lafleur RL, Malazdrewich C, Jeyaseelan S, Bleifield E, Abrahamsen MS, Maheswaran SK. Lipopolysaccharide enhances cytolysis and inflammatory cytokine induction in bovine alveolar macrophages exposed to Pasteurella (Mannheimia) haemolytica leukotoxin. Microb Pathog. 2001;30:347–357. doi: 10.1006/mpat.2000.0438. [DOI] [PubMed] [Google Scholar]
- 40.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 42.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao C, Huang C, Weng T, Xiao X, Ma H, Liu L. Computational prediction of MicroRNAs targeting GABA receptors and experimental verification of miR-181, miR-216 and miR-203 targets in GABA-A receptor. BMC Res Notes. 2012;5:91. doi: 10.1186/1756-0500-5-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai SH, Lin-Shiau SY, Lin JK. Suppression of nitric oxide synthase and the down-regulation of the activation of NFkappaB in macrophages by resveratrol. Br J Pharmacol. 1999;126:673–680. doi: 10.1038/sj.bjp.0702357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeyaseelan S, Chu HW, Young SK, Worthen GS. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect Immun. 2004;72:7247–7256. doi: 10.1128/IAI.72.12.7247-7256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoo HS, Rutherford MS, Maheswaran SK, Srinand S, Ames TR. Induction of nitric oxide production by bovine alveolar macrophages in response to Pasteurella haemolytica A1. Microb Pathog. 1996;20:361–375. doi: 10.1006/mpat.1996.0034. [DOI] [PubMed] [Google Scholar]
- 47.Chakravortty D, Hensel M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect. 2003;5:621–627. doi: 10.1016/s1286-4579(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 48.Baker BJ, Park KW, Qin H, Ma X, Benveniste EN. IL-27 inhibits OSM-mediated TNF-alpha and iNOS gene expression in microglia. Glia. 2010;58:1082–1093. doi: 10.1002/glia.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimizu M, Ogura K, Mizoguchi I, Chiba Y, Higuchi K, Ohtsuka H, Mizuguchi J, Yoshimoto T. IL-27 promotes nitric oxide production induced by LPS through STAT1, NF-kappaB and MAPKs. Immunobiology. 2013;218:628–634. doi: 10.1016/j.imbio.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 50.Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, Ivanov AR, Wolf DA, Mizgerd JP. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat Cell Biol. 2009;11:1157–1163. doi: 10.1038/ncb1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 52.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wan X, Helman LJ. Levels of PTEN protein modulate Akt phosphorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene. 2003;22:8205–8211. doi: 10.1038/sj.onc.1206878. [DOI] [PubMed] [Google Scholar]
- 54.Confer AW. Update on bacterial pathogenesis in BRD. Anim Health Res Rev. 2009;10:145–148. doi: 10.1017/S1466252309990193. [DOI] [PubMed] [Google Scholar]
- 55.Wessely-Szponder J, Urban-Chmiel R, Wernicki A, Bobowiec R. Effect of leukotoxin of Mannheimia haemolytica and LPS of E. coli on secretory response of bovine neutrophils in vitro. Pol J Vet Sci. 2005;8:99–105. [PubMed] [Google Scholar]
- 56.Lawless N, Reinhardt TA, Bryan K, Baker M, Pesch B, Zimmerman D, Zuelke K, Sonstegard T, O'Farrelly C, Lippolis JD, Lynn DJ. MicroRNA regulation of bovine monocyte inflammatory and metabolic networks in an in vivo infection model. G3 (Bethesda) 2014;4:957–971. doi: 10.1534/g3.113.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, Cao X, Wang Q. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1beta production in macrophages by targeting STAT3. PLoS One. 2012;7:e42971. doi: 10.1371/journal.pone.0042971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O'Leary JJ, Ruan Q, Johnson DS, Chen Y, O'Neill LA. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 2010;11:141–147. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- 60.Gao J, Liu QG. The role of miR-26 in tumors and normal tissues (Review) Oncol Lett. 2011;2:1019–1023. doi: 10.3892/ol.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palumbo T, Faucz FR, Azevedo M, Xekouki P, Iliopoulos D, Stratakis CA. Functional screen analysis reveals miR-26b and miR-128 as central regulators of pituitary somatomammotrophic tumor growth through activation of the PTEN-AKT pathway. Oncogene. 2013;32:1651–1659. doi: 10.1038/onc.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maillot G, Lacroix-Triki M, Pierredon S, Gratadou L, Schmidt S, Benes V, Roche H, Dalenc F, Auboeuf D, Millevoi S, Vagner S. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009;69:8332–8340. doi: 10.1158/0008-5472.CAN-09-2206. [DOI] [PubMed] [Google Scholar]
- 63.Verghese ET, Drury R, Green CA, Holliday DL, Lu X, Nash C, Speirs V, Thorne JL, Thygesen HH, Zougman A, Hull MA, Hanby AM, Hughes TA. MiR-26b is down-regulated in carcinoma-associated fibroblasts from ER-positive breast cancers leading to enhanced cell migration and invasion. J Pathol. 2013;231:388–399. doi: 10.1002/path.4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu G, Ji C, Shi C, Fu H, Zhu L, Zhu L, Xu L, Chen L, Feng Y, Zhao Y, Guo X. Modulation of hsa-miR-26b levels following adipokine stimulation. Mol Biol Rep. 2013;40:3577–3582. doi: 10.1007/s11033-012-2431-0. [DOI] [PubMed] [Google Scholar]
- 65.Murphy AJ, Guyre PM, Pioli PA. Estradiol suppresses NF-kappa B activation through coordinated regulation of let-7a and miR-125b in primary human macrophages. J Immunol. 2010;184:5029–5037. doi: 10.4049/jimmunol.0903463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao B, Liu Z, Li BS, Tang B, Li W, Guo G, Shi Y, Wang F, Wu Y, Tong WD, Guo H, Mao XH, Zou QM. Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J Infect Dis. 2009;200:916–925. doi: 10.1086/605443. [DOI] [PubMed] [Google Scholar]
- 67.Akca H, Demiray A, Tokgun O, Yokota J. Invasiveness and anchorage independent growth ability augmented by PTEN inactivation through the PI3K/AKT/NFkB pathway in lung cancer cells. Lung Cancer. 2011;73:302–309. doi: 10.1016/j.lungcan.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 68.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.