

Abstract
Chronic traumatic encephalopathy (CTE) is a progressive neurodegenerative disorder linked to repetitive traumatic brain injury (TBI) and characterized by deposition of hyperphosphorylated tau at the depths of sulci. We sought to determine the presence of chronic traumatic encephalopathy (CTE) pathology in a brain bank for neurodegenerative disorders for individuals with and without a history of contact sports participation. Available medical records of 1,721 men were reviewed for evidence of past history of injury or participation in contact sports. Subsequently, cerebral cortical samples were processed for tau immunohistochemistry in cases with a documented history of sports exposure as well as age- and disease-matched men and women without such exposure. For cases with available frozen tissue, genetic analysis was performed for variants in APOE, MAPT, and TMEM106B. Immunohistochemistry revealed 21 of 66 former athletes had cortical tau pathology consistent with CTE. CTE pathology was not detected in 198 individuals without exposure to contact sports, including 33 individuals with documented single-incident TBI sustained from falls, motor vehicle accidents, domestic violence, or assaults. Among those exposed to contact sports, those with CTE pathology did not differ from those without CTE pathology with respect to noted clinicopathologic features. There were no significant differences in genetic variants for those with CTE pathology, but we observed a slight increase in MAPT H1 haplotype, and there tended to be fewer homozygous carriers of the protective TMEM106B rs3173615 minor allele in those with sports exposure and CTE pathology compared to those without CTE pathology. In conclusion, this study has identified a small, yet significant, subset of individuals with neurodegenerative disorders and concomitant CTE pathology. CTE pathology was only detected in individuals with documented participation in contact sports. Exposure to contact sports was the greatest risk factor for CTE pathology. Future studies addressing clinical correlates of CTE pathology are needed.
Keywords: chronic traumatic encephalopathy, traumatic brain injury, sports, microtubule-associated protein tau, brain bank
Introduction
Chronic traumatic encephalopathy (CTE) is a progressive neurodegenerative disorder linked to repetitive traumatic brain injury [5, 8, 9, 35]. First described in boxers as “dementia pugilistica” [4], CTE is now used more widely to describe a neurodegenerative disorder associated with a variety of contact sports, as well as traumatic brain injury sustained in military training and combat [6, 11, 23]. Clinical symptoms of CTE usually occur 8-10 years after exposure to repetitive brain injury, and can progress from headaches, loss of attention and short-term memory loss, to behavioral changes, rage dyscontrol, depression, suicidality, dementia, and gait abnormalities [20]. Two major clinical variants of CTE have been described, with younger patients exhibiting more pronounced behavioral or mood disturbances and older patients exhibiting cognitive impairment that overlaps with other age associated cognitive disorders, such as Alzheimer's disease (AD) [33].
Neuropathologically, CTE is defined by deposition of hyperphosphorylated tau in neurofibrillary tangles, threads and astrocytes, as well as variable axonal and transactive response DNA-binding protein 43 (TDP-43) pathology [20, 22]. Tau deposition is typically focal and characteristically most severe at the depths of cerebral sulci, often surrounding penetrating cortical blood vessels. Cross sectional studies in experienced athletes suggest CTE pathology progresses from focal deposits (“epicenters”) in the neocortex to multifocal cortical lesions and to later involvement of medial temporal lobe, basal ganglia, diencephalon and brainstem. A staging scheme has been proposed, with cortical pathology in Stages I and II, and more extensive subcortical pathology in Stages III and IV [23].
CTE pathology can be accompanied by other neurodegenerative processes. Subsets of patients with CTE have amyloid-β, TDP-43 or α-synuclein pathology [16, 23, 31, 32]. CTE pathology has also been reported in patients with amyotrophic lateral sclerosis (ALS) [7]. TDP-43 pathology is detected in up to 80% of CTE cases, and pathologically confirmed ALS is reported in approximately 12% of CTE cases [15, 22, 23].
Between 1982 and 2008, over 160 million men and women participated in sports in the USA at either the high school or collegiate level [3]. The proportion of this cohort at risk for developing CTE is unknown. While current studies have focused on characterizing CTE in defined cohorts of mostly professional athletes and military veterans, there are no studies that have examined the frequency of CTE in unselected individuals from the general population. As an initial effort to address this problem, we screened for CTE pathology in brains of individuals with exposure to contact sports and matched controls without such exposure, all derived from a brain bank for neurodegenerative disorders.
Methods
Case material
The Mayo Clinic brain bank in Jacksonville, FL is an IRB-approved brain bank that holds brains of individuals with a range of neurodegenerative disorders, as well as a smaller number of neurologically normal controls. In addition to formalin-fixed, paraffin-embedded and frozen brain tissue, variable medical records are available for study. Between 1997 and 2014, 4,711 cases were acquired by the brain bank. All autopsies were performed after consent from the legal next-of-kin or an individual holding power-of-attorney. The inclusion criteria for this study were presence of paraffin-embedded tissue, at least minimal medical documentation, and male sex. The major exclusion criteria were a neuropathologic diagnosis of progressive supranuclear palsy, corticobasal degeneration, Pick's disease, and cases with a mutation in the microtubule associated protein tau gene (MAPT). Atypical tauopathies, including primary age-related tauopathy, which did not meet diagnostic criteria for progressive supranuclear palsy, corticobasal degeneration, or Pick's disease and lacked a known MAPT mutation, were included in this study (FTLD-tau). The resulting cohort consisted of 1,721 cases, of which available medical records were queried for information regarding participation in contact sports. These cases were received from the following sources: State of Florida Alzheimer's Disease Initiative (N=611), Mayo Clinic Morris K. Udall Center of Excellence for Parkinson's Disease (N=345), Mayo Clinic Jacksonville Alzheimer's Disease Research Center (N=191), CurePSP: Society for PSP | CBD and Related Disorders (N=177), consultation cases (N=161), Mayo Clinic Jacksonville hospital autopsy cases (N=139), Mayo Clinic Jacksonville ALS Clinic (N=39), Einstein Aging Study (N=35), Mayo Clinic Rochester Alzheimer's Disease Patient Registry (N=12), and Mayo Clinic Alzheimer's Disease Research Center (N=11). The depth of the clinical information on head trauma (and subsequently contact sports exposure) varied between these sources.
Pertinent clinical information derived from the available records included gender, age at symptom onset, symptom duration, age at death, height, education, family history of dementia or parkinsonism, alcohol use (including amount), tobacco use (including amount), military service, contact sports participation, the level of sports participation, and documented head trauma associated with sports participation. Exposure to non-sports related head trauma, including falls with head injury, motor vehicle accidents, violent assaults, and domestic abuse was recorded as well. Contact sports were defined as organized athletics with the potential for repetitive traumatic brain injury to the participants, including American football, boxing, wrestling, rugby, soccer, ice hockey, basketball, baseball, martial arts, and rodeo. Non-contact sports that were excluded from the contact sports exposure group included track and field, cross country, swimming, tennis, and golf. Online searches of obituaries were conducted to validate and supplement clinical records for individuals with contact sports exposure and controls using full name, date of birth and date of death.
Neuropathologic controls included men without documented exposure to contact sports selected from the 1,721 men identified in the screen of medical records. They were matched on a two-to-one ratio to those with exposure to contact sports with respect to age at death and comorbid pathological processes. Sixty-six women without documented sports exposure were similarly matched. Clinical controls (n=408) for genetic analysis were evaluated by a neurologist at Mayo Clinic Florida and had no neurological disorder or family history of neurological disease. All were Caucasian males and the median age at the time of blood draw was 68 years.
Neuropathologic assessment
Most of the 1,721 brains were received with the left hemibrain fixed in 10% formalin and the right hemibrain frozen (N=1,257). Formalin-fixed brains underwent systematic and standardized sampling, and neuropathologic evaluation by a single neuropathologist (DWD). Regions sampled on all cases processed by the Mayo Clinic brain bank included neocortex (6×; primary and association cortices), hippocampus (2×), basal forebrain, striatum, thalamus, midbrain, pons, medulla, and cerebellum (2×). Neuropathologic evaluation included review of histologic stains (hematoxylin and eosin), thioflavin S fluorescent microscopy, and select immunohistochemistry at Dr. Dickson's discretion (i.e., α-synuclein and TDP-43). Braak Neurofibrillary Tangle Stage and Thal Phase were assigned using thioflavin S slides and published criteria (Supplemental Table 1) [2, 34].
To assess CTE pathology, 5 μm thick sections from convexity cerebral cortices [dorsolateral middle frontal gyrus (Brodmann area 46/9) and inferior parietal lobule (Brodmann area 39/40)] were processed for immunohistochemistry. Following deparaffinization in xylene and reagent alcohol, antigen retrieval was performed by steaming slides in distilled water for 30 minutes. All immunohistochemistry was conducted using a DAKO Autostainer and DAKO EnVision™+ reagents. The tau antibody used was CP13 (mouse monoclonal; 1:1,000; a gift from Dr. Peter Davies, Feinstein Institute for Medical Research). The TDP-43 antibody pS409/410 (mouse monoclonal; 1:5,000; CosmoBio USA, Inc.) was used for immunohistochemistry in the posterior hippocampus. Immunostained slides were counterstained with hematoxylin and coverslipped. For cases suspected of having CTE pathology based on screening of standard (1.5 × 5.5-cm) histologic slides, coronal slabs of formalin-fixed tissue at the level of the infundibulum were sectioned at 50 μm thickness on a sliding microtome and immunostained as free floating sections with CP13 [23]. Cases with CTE pathology were staged using whole mount coronal slides according to the McKee staging scheme (Table 1).
Table 1. Clinical and demographic features of men with contact sports exposure.
| Case # | CTE | Sport | Sport Level | Veteran | Education | FHx | Clinical Dx | Path Dx | Onset | Death | Duration | Alcohol | Tobacco |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | I | Bk | HS | – | ? | – | ALS | ALS | 45 | 50 | 5 | + | – |
| 2 | I | F | HS | – | 18 | + | FTD | FTLD-FUS | 37 | 41 | 4 | + | – |
| 3 | I | F | HS | + | ? | + | ALS | ALS | 45 | 47 | 2 | ? | ? |
| 4 | I | F | HS | + | 16 | – | PDD | AD+LBD | 65 | 79 | 14 | + | + |
| 5 | I | F | HS, Co | – | 16 | – | DLB | LBD | 66 | 71 | 5 | + | + |
| 6 | I | F/Bk/Bb | HS, Co[Bb], sPro[Bb] | + | 16 | – | MSA | AD+LBD | 63 | 74 | 11 | ? | ? |
| 7 | I | F/W | HS, Co, Pro[W] | + | 16 | + | FTD/CVA | AD | ? | 80 | ? | + | + |
| 8 | II | ? | ? | + | 16 | + | AD | AD | 83 | 87 | 4 | – | – |
| 9 | II | F | ? | – | ? | – | ALS | ALS | 30 | 45 | 15 | + | + |
| 10 | II | Bb | sPro | + | 12 | – | NCI | AD | NA | 87 | NA | + | + |
| 11 | II | Bo | Am | – | 5 | + | AD | AD | 75 | 87 | 12 | + | – |
| 12 | II | F | HS, Co | – | 18 | – | NCI | AD | NA | 68 | NA | + | + |
| 13 | II | F | HS, Co | – | 16 | + | NCI | normal | NA | 51 | NA | – | + |
| 14 | II | F | HS, Co | + | 15 | – | PSP | LBD | 73 | 79 | 6 | – | – |
| 15 | III | F | HS | – | 17 | + | AD/PD | AD+LBD | 53 | 75 | 22 | + | + |
| 16 | III | F | HS | + | 14 | + | DLB | AD+LBD | 70 | 78 | 8 | + | + |
| 17 | III | F | HS, Co, Pro | + | 16 | + | VaD | FTLD-tau | 67 | 70 | 3 | – | – |
| 18 | III | F/Ru | ? | + | 16 | + | PDD | LBD | 65 | 81 | 16 | + | + |
| 19 | III | F/Bb | Co | – | 16 | – | AD | FTLD-tau | 75 | 82 | 7 | + | – |
| 20 | IV | Bo | Pro | + | 5 | – | AD v DLB | AD | 77 | 93 | 16 | – | – |
| 21 | IV | F/Bk/Bb | HS, Co, sPro[Bb] | + | 16 | + | PSP | FTLD-tau | 60 | 81 | 21 | ? | ? |
| 22 | – | Bb | Co, sPro | – | 18 | – | FTD-MND | FTLD-TDP | ? | 71 | ? | – | – |
| 23 | – | Bb | Co, sPro | + | 16 | + | DLB | AD+LBD | 62 | 76 | 14 | – | – |
| 24 | – | Bb | HS | – | 16 | – | PD | LBD | 45 | 73 | 28 | ? | ? |
| 25 | – | Bb | HS | – | 12 | + | AD/aphasia | AD | 76 | 86 | 10 | + | + |
| 26 | – | Bb | HS | + | 18 | – | CBS v FTD | AD+LBD | 56 | 65 | 9 | + | + |
| 27 | – | Bb | HS, Co | + | 16 | + | FTD v DLB | VaD | 67 | 76 | 9 | + | + |
| 28 | – | Bo | Am | – | 14 | + | AD | AD | 55 | 71 | 16 | + | + |
| 29 | – | Bo | Am | + | 12 | + | AD | AD | 63 | 74 | 11 | – | – |
| 30 | – | Bo | Am | + | 12 | + | AD v DLB | AD | 70 | 80 | 10 | – | + |
| 31 | – | Bo | Am | + | 20 | + | AD | AD | 67 | 73 | 6 | + | + |
| 32 | – | Bo | Am | + | 14 | – | AD | AD | 62 | 68 | 6 | ? | ? |
| 33 | – | Bo | ? | + | ? | – | AD v DLB | AD+LBD | 63 | 70 | 7 | – | + |
| 34 | – | F | Co | – | 16 | + | AD v FTD | AD | 57 | 65 | 8 | + | + |
| 35 | – | F | Co | + | 16 | + | PDD | AD+LBD | 63 | 74 | 11 | – | – |
| 36 | – | F | HS | – | 20 | – | CBS | AD | 59 | 67 | 8 | + | – |
| 37 | – | F | HS | – | 18 | – | FTD | AD | 69 | 78 | 9 | ? | ? |
| 38 | – | F | HS | – | 16 | – | AD v DLB | AD | 56 | 64 | 8 | + | + |
| 39 | – | F | HS | – | 20 | + | AD | AD | 53 | 61 | 8 | + | + |
| 40 | – | F | HS | – | ? | – | AD | AD+LBD | 74 | 78 | 4 | + | – |
| 41 | – | F | HS | – | 18 | – | AD v DLB | AD+LBD | 66 | 77 | 11 | + | – |
| 42 | – | F | HS | + | 12 | – | AD v DLB | AD+LBD | 78 | 87 | 9 | + | + |
| 43 | – | F | HS | + | ? | – | ALS | ALS | 72 | 74 | 2 | – | ? |
| 44 | – | F | HS | + | 16 | – | AD | FTLD-TDP | 48 | 56 | 8 | – | – |
| 45 | – | F | HS | + | 18 | + | AD/VaD | AD | 70 | 83 | 13 | + | + |
| 46 | – | F | HS | + | 14 | + | AD | AD | 60 | 81 | 21 | + | + |
| 47 | – | F | HS | + | 21 | + | AD | AD | 70 | 80 | 10 | + | + |
| 48 | – | F | HS, Co | – | 22 | – | FTD | AD | 69 | 73 | 4 | + | – |
| 49 | – | F | HS, Co | – | 16 | + | AD/VaD | AD+LBD | 52 | 60 | 8 | + | + |
| 50 | – | F | HS, Co | + | 18 | – | DLB | LBD | 85 | 90 | 5 | ? | ? |
| 51 | – | F | HS, Co | + | 16 | + | AD | FTLD-TDP | 80 | 89 | 9 | + | – |
| 52 | – | F | HS, Co | + | 16 | – | AD | AD | 55 | 59 | 4 | + | – |
| 53 | – | F | ? | – | ? | – | AD | AD | 73 | 78 | 5 | + | + |
| 54 | – | F | ? | – | ? | – | PDD | LBD | 73 | 93 | 20 | + | + |
| 55 | – | F | ? | – | 16 | – | MSA | MSA | 69 | 76 | 7 | – | – |
| 56 | – | F | ? | + | 16 | – | NCI | ATP | NA | 64 | NA | + | – |
| 57 | – | F/Bk | HS | – | 16 | + | PDD | AD+LBD | 49 | 57 | 8 | – | – |
| 58 | – | F/Bk | HS, Co[Bk] | – | 18 | – | AD | AD | 63 | 73 | 10 | + | + |
| 59 | – | F/Bo | ? | + | 14 | – | AD | AD+LBD | 69 | 79 | 10 | + | + |
| 60 | – | F/S | HS | + | 20 | + | AD | AD | 70 | 79 | 9 | – | + |
| 61 | – | H | ? | + | ? | + | AD v DLB | AD+LBD | 68 | 78 | 10 | + | + |
| 62 | – | MA | ? | + | 22 | + | AD | AD | 76 | 80 | 4 | + | – |
| 63 | – | Ro | ? | – | 12 | – | AD | AD+LBD | 64 | 70 | 6 | + | – |
| 64 | – | S | Am | – | ? | – | AD | AD | 66 | 73 | 7 | + | – |
| 65 | – | ? | ? | – | 18 | – | AD/PD | AD+LBD | 72 | 79 | 7 | + | + |
| 66 | – | ? | ? | – | 14 | – | MSA v PDD | LBD | 71 | 79 | 8 | + | + |
AD: Alzheimer's disease, ALS: amyotrophic lateral sclerosis, Am: amateur, ATP: Alzheimer-type pathology (does not meet criteria for AD), Bb: baseball, Bk: basketball, Bo: boxing, Co: college, CBS: corticobasal syndrome, CTE: CTE Stage (-, no CTE), CVA: cerebrovascular accident, DLB: dementia with Lewy bodies, Dx: diagnosis, F: football, FHx: family history of dementia or parkinsonism, FTD: frontotemporal dementia, FTLD-FUS: frontotemporal lobar degeneration with fused in sarcoma protein, FTLD-tau: frontotemporal lobar degeneration with atypical tau protein, FTLD-TDP: frontotemporal lobar degeneration with TDP-43 protein, H: hockey, HS: high school, LBD: Lewy body disease, MA: martial arts, MND: motor neuron disease, MSA: multiple systems atrophy, NA: not applicable, NCl: no cognitive impairment, PD: Parkinson's disease, PDD: Parkinson's disease dementia, Pro: professional, PSP: progressive supranuclear palsy, Ro: rodeo, Ru: rugby, S: soccer, sPro: semi-professional, VaD: vascular dementia, v: versus, W: wrestling, ?: unknown
Genetic analysis
Genomic DNA was extracted from cerebellum of frozen brains of men with exposure to contact sports or from blood of neurologically normal men. Genotyping was performed with TaqMan Allelic Discrimination Assay on an ABI 7900HT Fast Real-Time PCR system (Applied Bio-systems). A single SNP (rs1052553) was used to determine MAPT haplotype and TMEM106B (rs3173615) and two SNPs (rs7412 and rs429358) were used to determine APOE genotype. Five μL reactions consisting of 20 ng of DNA, 1× TaqMan genotyping master mix and 1× TaqMan SNP genotyping assay with appropriate genomic controls were used to genotype the four variants. Thermal cycling conditions consisted of one enzyme activation step at 95°C for 10 minutes followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Genotype calls were performed with SDS 2.2.2 software.
Statistical methods
Statistical analysis was conducted using SigmaPlot software (version 11.0, Systat Software Inc.). Rank sum test was performed when comparing continuous quantitative variables, while the chi-square test was performed when comparing categorical variables between cases and controls, and between cases with and without CTE pathology. Fisher's exact test was used to compare presence or absence of CTE pathology in former football players due to small sample size. A statistically significant difference was considered for two-sided P-values < 0.05.
Results
Retrospective review of available records of 1,721 men identified 66 patients with history of exposure to contact sports (Figure 1). Demographic and clinical features of this cohort are summarized in Table 1. Most (54 men) participated in a single sport (34 American football, 8 boxers, 7 baseball, 1 basketball, 1 ice hockey, 1 soccer, 1 rodeo and 1 martial artist), but nine participated in multiple sports [football plus: baseball (1), basketball (2), basketball and baseball (2), boxing (1), wrestling (1), rugby (1), and soccer (1)]. Three subjects had comments in medical records (“possible concussion while playing sports as a youngster,” “three concussions with being not fully conscious while doing sports,” and “brief loss of consciousness playing sports”) suggestive of contact sports, but without details on the specific sport.
Figure 1.
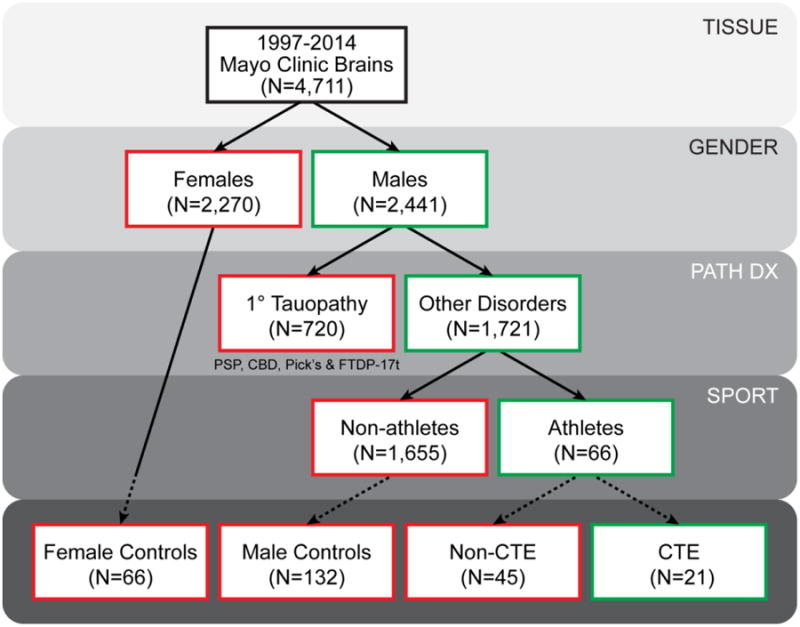
Selection criteria for Mayo Clinic Florida cohort. There are 4,711 brains with available formalin-fixed tissue recruited between the years 1997 and 2014. Of these 4,711 brains, 2,441 were male and 1,721 males did not have a prior neuropathologic diagnosis (Path Dx) of a primary tauopathy, namely progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick's disease (Pick's), or frontotemporal lobar degeneration with parkinsonism with a known MAPT mutation (FTDP-17t). Available clinical records of these 1,721 males were queried for any description of involvement in a contact sport, and 66 individuals did have notation regarding former athletics participation. Screening cortical tissue via tau immunohistochemistry (dashed lines) identified 21 cases with CTE pathology and 45 cases that failed to meet study standards of CTE pathology. In addition, tau immunohistochemistry was also performed on 66 female and 132 male age- and disease-matched (Path Dx) controls without documentation of sports involvement.
Sixty-five of the 66 men were Caucasian (one Hispanic). The average level of education (n=56) was 15.9 years. The average height (n=35) was 176 cm. Alcohol usage (n=59) was endorsed by 43 individuals and denied by 16 individuals. Of the 43 former athletes that endorsed alcohol use, 11 individuals were noted to have consumed alcohol minimally or periodically, 24 individuals on average consumed less than 2 alcoholic drinks per day, and 8 individuals were described as ‘heavy’ drinkers at some point throughout their documented clinical course or consumed 2 drinks or more per day. Tobacco usage (n= 58) was endorsed by 33 individuals and denied by 25 individuals. Of the 33 former athletes that endorsed tobacco use, 5 smoked for 0-10 pack years, 17 smoked for 11-30 pack years, and 11 smoked for 30 or more pack years. Additionally, 35 men served in the United States Armed Forces. A control cohort of 198 disease-matched individuals (132 men and 66 women) was selected based upon age at death. None of these individuals had documented involvement in contact sports; however, 33 had history of head trauma, including falls (n=14), motor vehicle accidents (n=10), violent assaults (n=4), domestic abuse (n=3), and 2 unspecified head trauma.
Tau immunohistochemistry was performed on sections from the dorsolateral frontal and parietal lobes. Of the 66 men with exposure to contact sports, 21 (32%) had tau pathology consistent with CTE (Figure 2). This was characterized by subpial thorn-shaped astrocytes (TSA) at the depths of sulci; neurofibrillary tangles (NFT), pretangles and threads in superficial cortical layers; as well as perivascular epicenters in the cortical ribbon at the depths of sulci composed of TSA, NFT and threads.[17] Tau immunohistochemistry uncovered select characteristic features, including pretangles and TSA, which were not observed on thioflavin-S microscopy. The overall pattern of tau pathology was patchy and best demonstrated on the whole hemisphere coronal slides (Figure 3). If Alzheimer-type pathological change was present, it was distinguished by more uniform involvement of lamina 3 and 5 of the cortical ribbon with no overt predilection for depths of sulci (Figure 4). CTE tau pathology was observed in both frontal and parietal cortices at a similar frequency in 17 cases, only in the parietal cortex in 2 cases, and only in the frontal cortex in 2 cases. Regarding CTE pathologic distribution, 7 cases were classified as CTE Stage I, 7 CTE Stage II, 5 CTE Stage III, and 2 CTE Stage IV. Tau immunohistochemistry was also performed on frontal and parietal cortical tissue of 198 disease- and age-matched controls. No CTE pathology was observed in any control case, including 33 controls with documented head trauma (unrelated to contact sports).
Figure 2.
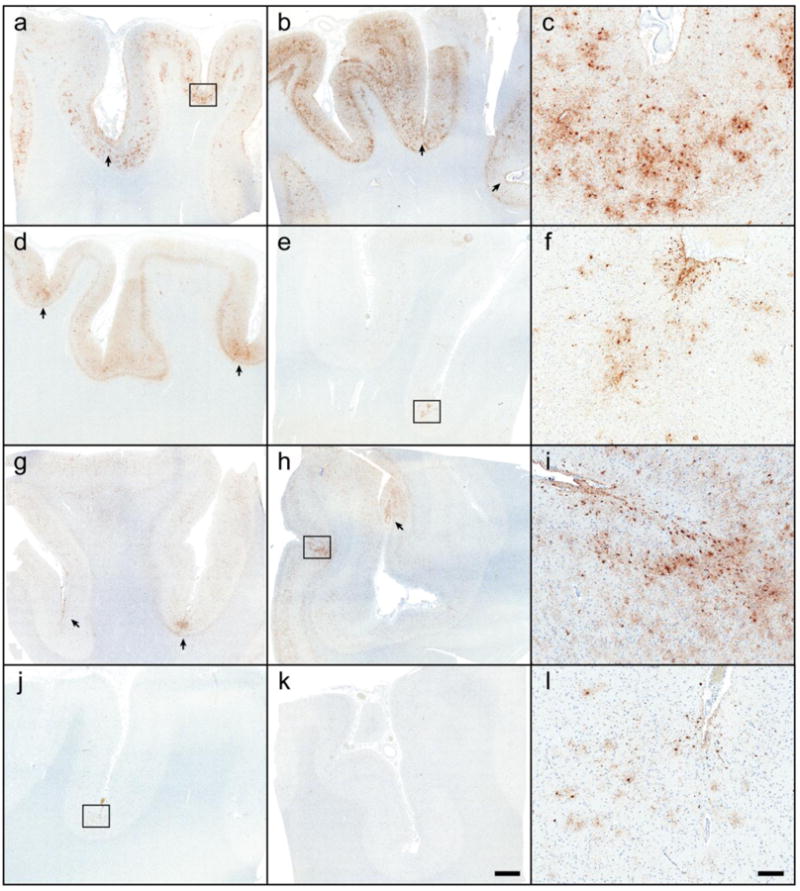
Chronic traumatic encephalopathy in the Mayo Clinic Florida cohort. Tau immunohistochemistry on tissue from the frontal cortex (a, d, g, j) and parietal cortex (b, e, h, k) in 66 athletes and 198 controls revealed CTE pathology in 21 cases, including a former boxer (a-c: Case #20), a former high school and collegiate American football player (d-f: Case #12), and a multi-sport athlete (football, basketball and baseball)(g-i: Case #21). One case initially without clinical documentation of contact sports and CTE pathology was subsequently discovered to be a former high school football player (j-l: Case #4). Microscopically, CTE foci (arrows and boxes; c, f, i, l) were characterized by clustered glial and neuronal lesions primarily at the depths of sulci and surrounding vessels. Bar: 2 mm on large sections, 20 μm on insets.
Figure 3.
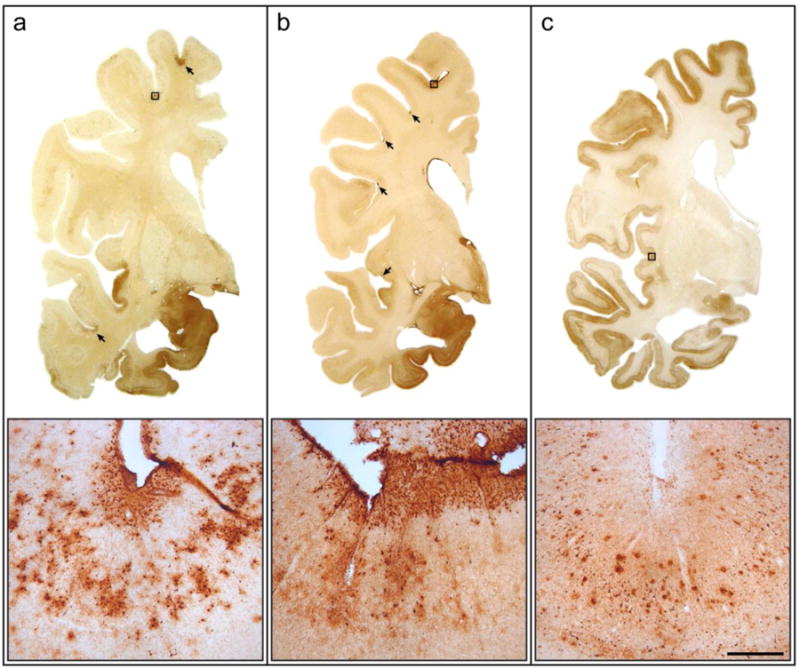
Distribution of chronic traumatic encephalopathy foci in mixed neurodegenerative cases. In 3 former football players (ages 78, 79 and 81), cortical foci of CTE pathology are observed in the frontal and temporal cortices of two individuals, one with pathologic Alzheimer's disease and Lewy body disease (a: Case #16) and one with pathologic Lewy body disease (b: Case #14), but not in a third individual with pathologic Alzheimer's disease (c: Case #46). Distributed prominently at the depths of sulci, some foci are large and span the entirety of the cortical gray matter (a) while others lesions are discrete, concentrated at the subpial surface (b). Bar: 15 mm coronal images, 200 μm inset.
Figure 4.
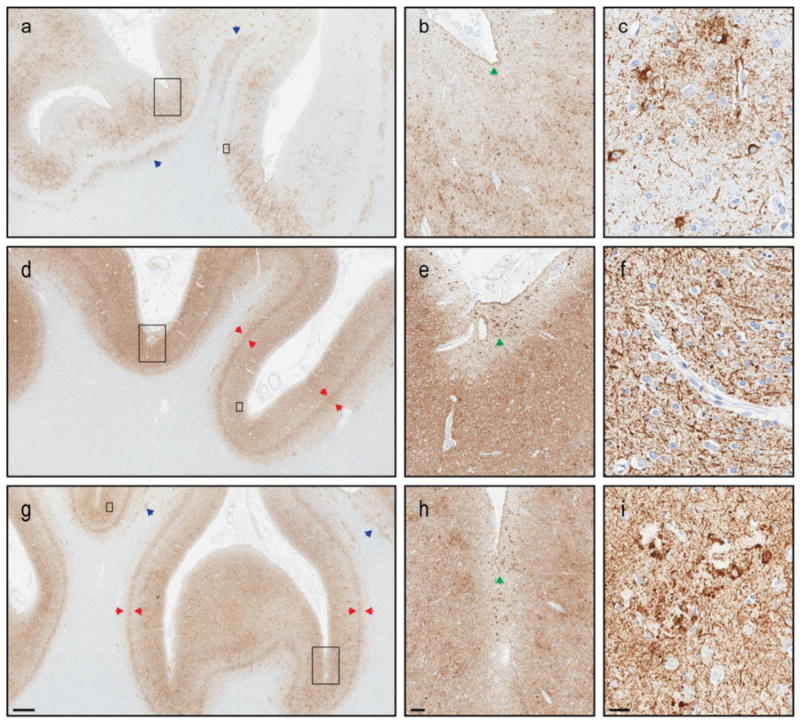
Neuropathologic differentiation of chronic traumatic encephalopathy and Alzheimer's disease. Tau pathology in the frontal cortex of 3 male amateur boxers, 1 with CTE (Case #11: a-c) and 2 without CTE (Case #29: d-f; Case #32: g-i). All 3 cases have concomitant Alzheimer's disease (Braak Stage 6, Thal Phase 5). Co-morbid CTE pathology is patchy (a) with marked prominence at the depth of cerebral sulci (b). CTE pathognomonic lesions include focal neurofibrillary tangles and astrocytic tau pathology adjacent to penetrating blood vessels (c). This contrasts the more uniform laminar distribution of Alzheimer tau pathology (d, g), particularly in layer 5 (red arrows). Similar to Case #11, Case #29 and #32 both show subpial thorn-shaped astrocytes (b, e, h; green arrows) at the depths of sulci. However, this often associated CTE feature is non-specific without additional perivascular pathology (f) or neuritic tau pathology associated with cerebral amyloid angiopathy (i). Tau-immunoreactive white matter astrocytes (blue arrows) are another pathologic feature commonly observed in CTE (a) which lack disease specificity (g). Ultimately, confluent tau pathology in cases with a high burden of Alzheimer pathology may prohibit a definitive CTE diagnosis. Large inset: b, e, h; small inset: c, f, i. Measure bar: 1mm a, d, g; 100 μm b, e, h; 25 μm c, f, i.
Of the 21 patients with exposure to contact sports and CTE pathology, 11 played football, 5 multiple sports [football plus: baseball (1), basketball and baseball (2), rugby (1), and wrestling (1)], 2 boxing, 1 baseball, 1 basketball, and 1 unspecified involvement with loss of consciousness. Of the 43 patients with exposure to contact sports through football, 16 (37%) had CTE pathology, and of these, 6 played up to high school level, 7 played to college, and 1 played professional football. In 27 former football players without evidence of CTE pathology, 15 played up to the high school, 7 played up to college, and none played professional football. There were no significant differences in the proportion of football players with CTE pathology with respect to level of involvement (p=0.175).
The previously defined neurodegenerative conditions in 20 of the 21 patients with CTE pathology included AD (n=6), mixed AD and Lewy body disease (LBD; n=4) (Figure 3a), pure LBD (n=3) (Figure 3b), ALS (n=3), and frontotemporal lobar degeneration (FTLD) with fused in sarcoma protein (n=1) or atypical tau protein (n=3). Hippocampal TDP-43 pathology was observed at a similar frequency in former athletes with (7/21; 33%) and without (14/45; 31%) CTE pathology (Supplemental Table 1). Comparing the patients with exposure to contact sports and CTE pathology to those without CTE pathology, there were no significant differences in age at symptom onset, symptom duration, age at death, height, education, history of alcohol or tobacco use, or prior military service (Table 2). There was no increase in frequency of family history of dementia, parkinsonism, or ALS in patients with CTE pathology compared to those without CTE pathology.
Table 2. Comparison of clinical features between former athletes with and without chronic traumatic encephalopathy.
| Clinical feature (median value) | CTE (#) | non-CTE (#) | p-value |
|---|---|---|---|
| Age at symptom onset (age) | 65 (17) | 67 (43) | 0.646 |
| Symptom duration (number of years) | 8 (17) | 8 (43) | 0.941 |
| Age at death (age) | 78 (21) | 74 (45) | 0.736 |
| Height (cm) | 175 (10) | 175 (25) | 0.686 |
| Education (years) | 16 (18) | 16 (38) | 0.146 |
| History of alcohol use | 13Y | 5N | 30Y | 11N | 1.000 |
| none to rare [no, minimal, periodically] | 7 | 20 | – |
| low to moderate [<2 drinks/day] | 10 | 14 | – |
| high [≥2 drinks/day; heavy] | 1 | 7 | – |
| History of tobacco use | 10Y | 8N | 23Y | 17N | 0.882 |
| none to rare [no, 0-10 pack years] | 10 | 20 | – |
| low to moderate [11-30 pack years] | 7 | 10 | – |
| high [30+ pack years] | 1 | 10 | – |
| Military service | 12Y | 9N | 23Y | 22N | 0.847 |
| Clinically documented combat | 3 | 3 | – |
| Family history of neurodegeneration | 11Y | 10N | 19Y | 25N | 0.667 |
CTE: chronic traumatic encephalopathy, N: no, Y: yes, Italicized text: comment in clinical notes, #: number of cases
Genetic variants associated with neurodegeneration, including MAPT, APOE and TMEM106B, were assessed. Given the small sample size of CTE cases (n=17 with frozen tissue) these results were considered exploratory, and no statistically significant differences were observed (Table 3; Supplemental Table 1). APOE ε4 allele tended to be higher in those with exposure to contact sports with (26.5%) or without (38.4%) CTE pathology compared to clinical controls (13.4%). There was a slight increase in the frequency of MAPT H1/H1 genotype in men with exposure to contact sports and CTE pathology (70.6%) compared to those without CTE pathology (62.8%) and to clinical controls (55.1 %). The H1 haplotype frequency was similar between former athletes with (82.4%) and without (79.1%) CTE pathology, both higher than clinical controls (74.4%). MAPT subhaplotyping results are displayed in Supplemental Table 2. There was a decreased frequency of TMEM106B homozygous minor allele in patients with exposure to contact sports and CTE pathology (5.9%) compared to those without CTE pathology (23.3%) and to clinical controls (17.2%).
Table 3. APOE, MAPT, and TMEM106B genotypes in men with exposure to contact sports.
| Variant/genotype | CTE pathology (n=17) | No pathology (n=43) | Clinical controls (n=408) |
|---|---|---|---|
| APOE | |||
| ε2/ε2 | 0 (0.0%) | 0 (0.0%) | 1 (0.2%) |
| ε2/ε3 | 1 (5.9%) | 2 (4.7%) | 60 (14.7%) |
| ε2/ε4 | 0 (0.0%) | 2 (4.7%) | 7 (1.7%) |
| ε3/ε3 | 8 (47.1%) | 15 (34.9%) | 245 (60.0%) |
| ε3/ε4 | 7 (41.2%) | 17 (39.5%) | 88 (21.6%) |
| ε4/ε4 | 1 (5.9%) | 7 (16.3%) | 7 (1.7%) |
| ε2 | 1 (2.9%) | 4 (4.7%) | 69 (8.5%) |
| ε3 | 24 (70.6%) | 49 (57.0%) | 638 (78.2%) |
| ε4 | 9 (26.5%) | 33 (38.4%) | 109 (13.4%) |
| MAPT | |||
| H1/H1 | 12 (70.6%) | 27 (62.8%) | 225 (55.1%) |
| H1/H2 | 4 (23.5%) | 14 (32.6%) | 157 (38.5%) |
| H2/H2 | 1 (5.9%) | 2 (4.7%) | 26 (6.4%) |
| H1 | 28 (82.4%) | 68 (79.1%) | 607 (74.4%) |
| H2 | 6 (17.6%) | 18 (20.9%) | 209 (25.6%) |
| TMEM106B rs3173615 | |||
| CC | 7 (41.2%) | 7 (16.3%) | 137 (33.6%) |
| CG | 9 (52.9%) | 26 (60.5%) | 201 (49.3%) |
| GG | 1 (5.9%) | 10 (23.3%) | 70 (17.2%) |
| C | 23 (67.6%) | 40 (46.5%) | 475 (58.2%) |
| G | 11 (32.4%) | 46 (53.5%) | 341 (41.8%) |
Discussion
Repetitive brain injury sustained during participation in contact sports is considered one of the etiologies of CTE pathology [21]. Contact sports in which CTE pathology has been reported include American football [26, 27], baseball [21], boxing [4], ice hockey [23], rugby [10], soccer [21], and wrestling [28] while other athletic activities including basketball, lacrosse, and martial arts also pose a threat of possible brain injury. Screening for history of exposure to these sports in 1,721 men in the Mayo Clinic brain bank for neurodegenerative disorders identified 66 individuals with exposure to contact sports. Football was the most common (n=43), followed by baseball (n=10), boxing (n=9), and basketball (n=5). Of the 66 individuals, 23 played one or more sports during high school, 14 played one or more sports during college, and 15 played their sport beyond college at the amateur (n=7), semi-professional (n=5), or professional level (n=3).
Of the 66 individuals with exposure to contact sports, 21 had pathology consistent with CTE. All had the pathognomonic lesions of CTE pathology [17], namely foci of cortical tau pathology at the depths of sulci and characterized by tau immunoreactive TSA in subpial and perivascular spaces, as well as perivascular NFT and threads (Figure 2). Two-thirds of the cases displayed mild to moderate CTE pathology (CTE Stages I and II) whereas one-third of the cases harbored more severe CTE pathology (CTE Stages III and IV) [23]. Tau immunohistochemistry demonstrated greater sensitivity to the pathognomonic lesions of CTE than thioflavin-S. This finding is reminiscent of thiazin red labeling of other primary tauopathies reported by Uchihara and colleagues [36]. With 153 unique cases of CTE in the literature as of August 2013 [19], addition of our 21 cases increases this reported frequency by 14%.
While some of the cases had concomitant Alzheimer-type pathological change, it could be distinguished from CTE pathology (Figure 4). Whole mount sections were useful to adjudicate difficult cases as the focal nature of epicenters could be more easily appreciated (Figure 3). It is remotely possible that high Alzheimer-type pathology (Supplemental Table 1) masked subtle CTE pathology. If this is the case, then our results are a conservative estimate of the frequency of CTE pathology. Research from Stein and colleagues has also noted differences in the accumulation of Aβ plaques in AD and CTE, but suggests select, focal Aβ pathology might be accelerated in CTE cases [32]. The role of Aβ pathology in CTE has been and continues to be debated and warrants further research emphasis.
Cortical lesions were most specific for CTE, but underlying white matter astrocytic pathology was also frequent. In this series, white matter astrocytic tau pathology was most marked in two boxers. Tau-immunoreactive astrogliopathy, including subependymal, subpial, gray matter, and white matter astrocytes, is an increasingly recognized neuropathology in elderly brains [18] and not restrictive to CTE. Astrocytic pathology in CTE is often focal and at the depths of sulci, whereas tau astrogliopathy in the elderly can be widespread and has no known predilection for sulcal depths. However, tau astrogliopathy must be considered in the differential diagnosis of CTE, especially those with advanced age.
No statistically significant differences were found between men with exposure to contact sports with CTE pathology and those without CTE pathology for any clinical or demographic feature, including age at symptom onset, symptom duration, age at death, height, and education. Some studies have noted substance abuse is frequent in CTE [23, 25], however, this relationship was not observed in our cohort. Moderate to high alcohol and tobacco usage did not appear to be more prevalent amongst CTE cases compared to former athletes without CTE pathology. CTE pathology has been described in military veterans who have sustained blast injuries [11]. In our cohort of 35 patients who served in the US Armed Forces (6 in a combat role), CTE pathology was not more frequent in veteran athletes than non-veteran athletes.
The genetic underpinnings of CTE have yet to be elucidated. Genetic research conducted to date has focused on APOE, a well-known risk factor in AD. While several studies with smaller cohorts have described a higher frequency of the APOE ε4 allele and ε4/ε4 genotype in CTE [13, 20, 33], larger studies have failed to replicate this finding [19, 23]. In the present study, no association was found for APOE ε4 and CTE pathology. The significance of the increased frequency of APOE ε4 allele observed in those exposed to contact sports, regardless of presence of CTE pathology, is likely coincidental. Since APOE is a driving factor for amyloid pathology [14] and CTE is primarily a tauopathy, it was of interest to know whether variants in MAPT would be associated with CTE pathology, as has been shown for progressive supranuclear palsy [29], corticobasal degeneration [12] and tangle-predominant dementia [30]. No significant differences were found regarding the frequency of the H1 haplotype, although it did appear higher in the CTE cases compared to controls. It is estimated that more than 50% of CTE cases have concomitant TDP-43 pathology [22]. The strongest genetic risk factor currently known for TDP-43 pathology in a range of disorders, including FTLD [37], AD [24, 38], and LBD [1], is a common variant in the gene for an uncharacterized lysosomal transmembrane protein 106b (TMEM106B). We observed a trend for decrease in homozygous carriers of the protective minor allele of rs3173615 in individuals with exposure to contact sports (Table 3).
In summary, CTE pathology was found in at least 1.2% of men in a neurodegenerative brain bank exclusive of progressive supranuclear palsy, corticobasal degeneration, Pick's disease, or a known MAPT mutation; however, the frequency was much higher (32%) in patients with documented participation in contact sports. The results of our screen suggest that individuals with documented participation in contact sports are at an increased risk for CTE pathology compared to those with no such exposure or for whom exposure to contact sports is unknown. The findings have important implications for public health given the number of individuals with past exposure to contact sports. While our brain bank cohort is predominantly Caucasian and enriched in individuals residing in Southeastern United States, the generalizability of our findings is probably greater than that from series of selectively recruited professional athletes [21]. Assessment of frequency or severity of head trauma in affected individuals and the possible clinical correlates of CTE pathology could not be evaluated given the retrospective nature of this study. Our findings should be considered provisional, and it is imperative to screen other brain bank cohorts for CTE pathology, particularly cohorts with minimal concomitant neurodegenerative pathology that can make recognition of the pathognomonic lesion problematic, to determine the frequency of CTE pathology in those with exposure to contact sports. Large cohorts also need to be studied to confirm potential genetic risk for CTE pathology due to MAPT and TMEM106B, including cohorts prospectively following individuals who do and do not participate in contact sports.
Supplementary Material
Acknowledgments
We would like to thank the patients and their families. Without their generous donation, these studies would not be possible. We would also like to thank Monica Castanedes-Casey, Virginia Philips, and Linda Rousseau for their neurohistologic assistance, Michael DeTure for brain banking, and Deann Gibson for coordination of brain donations. This study was supported by the National Institute of Health (NIH) P50 NS072187 (OAR, ZKW, RR, DWD), R01 NS076471 (RR), R01 NS078086 (OAR), UO1 NS086659-01 (ACM), P30 AG13846 (ACM; supplement 0572063345-5). Other support included an unrestricted gift from Carl Edward Bolch Jr. and Susan Bass Bolch (ZKW), Department of Veterans Affairs (ACM), Veterans Affairs Biorepository CSP 501 (ACM), Sports Legacy Institute (ACM), National Operating Committee on Standards for Athletic Equipment (ACM), the National Football League (ACM), the Andlinger Foundation (ACM), and the WWE (ACM).
References
- 1.Aoki N, Murray ME, Ogaki K, et al. Hippocampal sclerosis in Lewy body disease is a TDP-43 proteinopathy similar to FTLD-TDP Type A. Acta Neuropathol. 2015;129:53–64. doi: 10.1007/s00401-014-1358-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 3.Cantu RC, Mueller FO. The prevention of catastrophic head and spine injuries in high school and college sports. Br J Sports Med. 2009;43:981–986. doi: 10.1136/bjsm.2009.067728. [DOI] [PubMed] [Google Scholar]
- 4.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychological Medicine. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 5.DeKosky ST, Blennow K, Ikonomovic MD, Gandy S. Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nature Reviews Neurology. 2013;9:192–200. doi: 10.1038/nrneurol.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeKosky ST, Ikonomovic MD, Gandy S. Traumatic brain injury--football, warfare, and long-term effects. N Engl J Med. 2010;363:1293–1296. doi: 10.1056/NEJMp1007051. [DOI] [PubMed] [Google Scholar]
- 7.Fournier CN, Gearing M, Upadhyayula SR, Klein M, Glass JD. Head injury does not alter disease progression or neuropathologic outcomes in ALS. Neurology. 2015;84:1788–1795. doi: 10.1212/WNL.0000000000001522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gavett BE, Stern RA, Cantu RC, Nowinski CJ, McKee AC. Mild traumatic brain injury: a risk factor for neurodegeneration. Alzheimers Res Ther. 2010;2:18. doi: 10.1186/alzrt42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavett BE, Stern RA, McKee AC. Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med. 2011;30:179–188, xi. doi: 10.1016/j.csm.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geddes JF, Vowles GH, Nicoll JA, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathologica. 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein LE, Fisher AM, Tagge CA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra160. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houlden H, Baker M, Morris HR, et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56:1702–1706. doi: 10.1212/wnl.56.12.1702. [DOI] [PubMed] [Google Scholar]
- 13.Jordan BD, Kanik AB, Horwich MS, et al. Apolipoprotein E epsilon 4 and fatal cerebral amyloid angiopathy associated with dementia pugilistica. Annals of Neurology. 1995;38:698–699. doi: 10.1002/ana.410380429. [DOI] [PubMed] [Google Scholar]
- 14.Josephs KA, Tsuboi Y, Cookson N, Watt H, Dickson DW. Apolipoprotein E epsilon 4 is a determinant for Alzheimer-type pathologic features in tauopathies, synucleinopathies, and frontotemporal degeneration. Arch Neurol. 2004;61:1579–1584. doi: 10.1001/archneur.61.10.1579. [DOI] [PubMed] [Google Scholar]
- 15.King A, Sweeney F, Bodi I, et al. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer's disease. Neuropathology. 2010;30:408–419. doi: 10.1111/j.1440-1789.2009.01085.x. [DOI] [PubMed] [Google Scholar]
- 16.Kokjohn TA, Maarouf CL, Daugs ID, et al. Neurochemical profile of dementia pugilistica. J Neurotrauma. 2013;30:981–997. doi: 10.1089/neu.2012.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koroshetz W. Report from the First NIH Consensus Conference to Define the Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. 2015 http://www.ninds.nih.gov/research/tbi/ReportFirstNIHConsensusConference.htm.
- 18.Kovacs GG, Milenkovic I, Wohrer A, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathologica. 2013;126:365–384. doi: 10.1007/s00401-013-1157-y. [DOI] [PubMed] [Google Scholar]
- 19.Maroon JC, Winkelman R, Bost J, et al. (2015) Chronic Traumatic Encephalopathy in Contact Sports: A Systematic Review of All Reported Pathological Cases. PLoS One. 2015;10:e0117338. doi: 10.1371/journal.pone.0117338. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKee AC, Daneshvar DH, Alvarez VE, Stein TD. The neuropathology of sport. Acta Neuropathologica. 2014;127:29–51. doi: 10.1007/s00401-013-1230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray ME, Cannon A, Graff-Radford NR, et al. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol. 2014;128:411–421. doi: 10.1007/s00401-014-1302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Omalu B, Bailes J, Hamilton RL, et al. Emerging Histomorphologic Phenotypes of Chronic Traumatic Encephalopathy in American Athletes. Neurosurgery. 2011;69:173–183. doi: 10.1227/NEU.0b013e318212bc7b. [DOI] [PubMed] [Google Scholar]
- 26.Omalu BI, DeKosky ST, Hamilton RL, et al. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery. 2006;59:1086–1092. doi: 10.1227/01.NEU.0000245601.69451.27. discussion 1092-1083. [DOI] [PubMed] [Google Scholar]
- 27.Omalu BI, DeKosky ST, Minster RL, et al. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–134. doi: 10.1227/01.neu.0000163407.92769.ed. discussion 128-134. [DOI] [PubMed] [Google Scholar]
- 28.Omalu BI, Fitzsimmons RP, Hammers J, Bailes J. Chronic traumatic encephalopathy in a professional American wrestler. J Forensic Nurs. 2010;6:130–136. doi: 10.1111/j.1939-3938.2010.01078.x. [DOI] [PubMed] [Google Scholar]
- 29.Rademakers R, Melquist S, Cruts M, et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Human Molecular Genetics. 2005;14:3281–3292. doi: 10.1093/hmg/ddi361. [DOI] [PubMed] [Google Scholar]
- 30.Santa-Maria I, Haggiagi A, Liu XM, et al. The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathologica. 2012;124:693–704. doi: 10.1007/s00401-012-1017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nature Reviews Neurology. 2013;9:211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein TD, Montenigro PH, Alvarez VE, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathologica. 2015;130:21–34. doi: 10.1007/s00401-015-1435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81:1122–1129. doi: 10.1212/WNL.0b013e3182a55f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 35.Turner RC, Lucke-Wold BP, Robson MJ, et al. Repetitive traumatic brain injury and development of chronic traumatic encephalopathy: a potential role for biomarkers in diagnosis, prognosis, and treatment? Front Neurol. 2012;3:186. doi: 10.3389/fneur.2012.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uchihara T, Nakamura A, Yamazaki M, et al. Different conformation of neuronal tau deposits distinguished by double immunofluorescence with AT8 and thiazin red combined with Gallyas method. Acta Neuropathologica. 2001;102:462–466. doi: 10.1007/s004010100401. [DOI] [PubMed] [Google Scholar]
- 37.Van Deerlin VM, Sleiman PMA, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP43 inclusions. Nature Genetics. 2010;42:234–U234. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu L, De Jager PL, Yang J, et al. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology. 2015;84:927–934. doi: 10.1212/WNL.0000000000001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.