

Abstract
Recruitment of foamy monocytes to inflamed endothelium expressing vascular cell adhesion molecule-1 (VCAM-1) contributes to the development of plaque during atherogenesis. Foamy CD11c+ monocytes arise in the circulation during the onset of hypercholesterolemia and recruit to nascent plaque, but the mechanism of CD11c/CD18 and very late antigen-4 (VLA-4) activation and cooperation in shear resistant cell arrest on VCAM-1 is ill defined. Within one week of the onset of a Western high-fat diet (WD) in apoE-/- mice, an inflammatory subset of foamy monocytes emerged comprising ¼ of the circulating population. These cells expressed ∼3-fold more CD11c/CD18 and 50% higher chemokine receptors than non-foamy monocytes. Recruitment from blood to a VCAM-1 substrate under shear stress was assessed ex-vivo using a unique artery-chip microfluidic assay. It revealed that foamy monocytes from mice on a WD increased their adhesiveness over 5 weeks, rising to twice that of mice on a normal diet (ND) or CD11c-/- fed a WD. Shear resistant capture of foamy human or mouse monocytes was initiated by high affinity CD11c, which directly activated VLA-4 adhesion via phosphorylated spleen tyrosine kinase and paxillin within focal adhesion complexes. Lipid uptake and activation of CD11c are early and critical events in signaling VLA-4 adhesive function on foamy monocytes competent to recruit to VCAM-1 on inflamed arterial endothelium.
Keywords: Atherosclerosis, monocytes, CD11c, Ly6C, paxillin, Syk
Introduction
Monocyte recruitment from the circulation to inflamed endothelium expressing vascular cell adhesion molecule-1 (VCAM-1) is an obligate step during the formation of atherosclerotic lesions and represents a potential target for drug based therapies to ameliorate plaque formation.(1, 2) However, precise identification of the inflammatory monocytes that contribute to atherogenesis remains elusive. Mouse models of hypercholesterolemia such as apolipoprotein E deficient (apoE-/-) mice are commonly used to study atherogenesis.(3-5) Fed a Western high fat diet (WD) these mice circulate high levels of lipoproteins at concentrations that closely match that in hypertriglyceridemic humans.(6) With the onset of hyperlipidemia, monocytes are mobilized from bone marrow and splenic reservoirs to the circulation, whereupon a fraction accumulate lipid and traffic to inflamed endothelium expressing vascular cell adhesion molecule 1 (VCAM-1).(1, 4, 5) A critical step in atherogenesis is the early recruitment of inflammatory monocytes and their differentiation into foamy macrophages in nascent plaque, however, the mechanism through which lipid uptake initiates monocyte activation and integrin mediated shear resistant arrest is ill defined.(5)
Two major classes of monocytes increase their numbers in the circulation during hypercholesterolemia and recruit to inflamed endothelium: “classical” or inflammatory monocytes are characterized as Ly6ChighCCR2highCX3CR1low in mice and CD14++CD16− in humans. These subsets are thought to be the precursors of inflammatory foamy macrophages in established aortic lesions.(4) A second subset denoted as “patrolling” or anti-inflammatory monocytes that express Ly6ClowCCR2lowCX3CR1high in mice and CD14+CD16+ in humans, recruit to inflamed endothelium early in the process of lesion formation, give rise to CD11c+ foamy macrophages, and extravasate at the endothelial interface and accumulate in organs.(7, 8) The propensity for the trafficking and emigration of specific monocyte subsets is dictated by their relative expression of chemokine C-C motif receptor 2 (CCR2), C-C motif receptor 5 (CCR5), and chemokine C-X3-C motif receptor 1 (CX3CR1) that signal integrin activation and guide cell migration to arterial sites of nascent plaque formation.(7, 8) A direct comparison of inflammatory activation in the context of lipid mediated integrin-dependent recruitment on VCAM-1 between mouse and human monocyte subsets is lacking.
An early event in inflammation associated with hyperlipidemia is membrane upregulation of β2-integrin CD11c/CD18 on circulating monocytes. In contrast, β1-integrin very late antigen-4 (VLA-4) expression levels remain constant on circulating monocytes.(3, 5, 9, 10) Durable bond formation on VCAM-1 involves an upshift in VLA-4 affinity and subsequent receptor coalescence within focal clusters that promote high avidity binding and shear resistant adhesion on inflamed endothelium.(10-13) High affinity CD11c can also recognize VCAM-1 on mouse and human endothelium and in the high affinity conformation signals the activation of VLA-4, thereby enhancing recruitment of monocytes and exacerbating plaque formation.(3, 10, 14) In fact, the relative expression of CD11c on foamy monocytes increases with cardiovascular risk in subjects with metabolic syndrome, and receptor number increases as a function of the severity of myocardial injury in ischemic patients.(10) Its functional role in atherogenesis was recently demonstrated by genetic deletion in apoE-/-/CD11c-/- double knockout mice, which were protected from compartmentalization of monocytes in adipose tissue and in atherosclerotic plaque.(3, 15) Thus, the dynamics of CD11c expression during the onset of hypertriglyceridemia and its role in activation of VLA-4 may provide a means of predicting their participation in atherogenesis.
In the current study we measured the emergence of foamy monocytes within one week of the onset of hypercholesterolemia in mice and employed an ex-vivo model to produce TGRL loaded foamy monocytes from humans fed a high fat meal in order to better delve the mechanism by which CD11c activates VLA-4 during inflammation. Our experimental strategy was twofold: Phenotypic changes were tracked in monocytes sampled from the circulation of apoE-/- and CD11c-/-/apoE-/- mice fed a normal versus high fat diet for 5-weeks. We assessed ex-vivo the molecular pathway associated with CD11c and VLA-4 dependent capture of monocytes on VCAM-1 using a unique artery-chip microfluidic assay. Our data reveal that in hypertriglycerdemic mice a subset of lipid-laden monocytes expressing Ly6ClowCD11chigh emerge in the circulation and exhibit enhanced capacity to activate VLA-4. This subset is functionally similar to human CD14++CD16+ (Mon2) monocytes that take up TGRL and capture on inflamed aortic endothelium.(16) Conversion of CD11c from low to high affinity was sufficient to activate VLA-4 within focal clusters that redistribute to lipid rafts where spleen tyrosine kinase (Syk) and paxillin stabilize integrin mediated monocyte arrest. These data reveal the earliest phenotypic changes in monocyte activation during hyperlipidemia and indicate that CD11c expression and signaling on foamy monocytes provides a biomarker and potential target for intervention of atherogenesis.
Materials and Methods
Animals and Diet
Genetic deletion of CD11c bred with apoE-/- mice was achieved as described previously.(3) Both apoE-/- and CD11c-/-/apoE-/- mice were backcrossed for 12 generations onto a C57BL6 wild type stain. Male mice were used and fed a normal chow diet (ND, PicoLab Rodent Chow 5010; LabDiet, St. Louis, MO) until 8 weeks old and then switched to a Western high-fat diet (WD; 21% fat [w/w], 0.15% cholesterol [w/w]; DyetsInc, Bethlehem, Pa) and maintained on WD for 1-5 weeks. This has been shown to induce hypercholesterolemia in mice lacking the apoE gene. The α4(Y991A) C57BL6 strain were maintained on a normal chow diet and used at 10-12 weeks old.
Blood Collection
At predetermined time points mice were sacrificed using CO2 asphyxiation and blood was obtained via cardiac puncture using a 3ml syringe (BD San Diego) and 30-gauge × 1″ needle (BD San Diego) and injected into Vacutainer® collection tubes. For experiments studying primary human monocytes subjects were recruited from the general population near Davis, California. Blood was obtained by venipuncture into Vacutainer® collection tubes and maintained at room temperature. For flow cytometry and blood differential counts blood was drawn into tubes containing K2EDTA. For lab-on-a-chip adhesion assays blood was drawn into tubes containing sodium heparin. Blood differential counts were determined using a Coulter Act diff Hematology Analyzer (Beckman-Coulter)
Monoclonal Antibodies and Inhibitors
The following antibodies were purchased from Biolegend (San Diego, CA) and used for flow cytometry and immunofluorescence microscopy: anti-mouse CD11c (Alexa Fluor® 488 [AF488], clone N418), anti-mouse CD115 (phycoerythrin [PE]-conjugated, clone AFS98), anti-mouse CD49d (PE-conjugated or unconjugated, clone R-12), anti-mouse Ly6C (PerCp/Cy5.5-conjugated or AF647-conjugated, clone HK1.4), anti-human CD14 (AF488-conjugated or PE-conjugated, clone M5E2), anti-human CD16 (PE/Cy5-conjugated, clone 3G8), anti-human CD11c (PE-conjugated or unconjugated, clone BU15), and anti-human Syk (unconjugated, clone 4D10.2). The following antibodies were purchased from AbCam (Cambridge, MA) and used for IP, western blot, and immunofluorescence microscopy: anti-human paxillin (unconjugated, clone 5H11), anti-human CD11c (unconjugated, clone MM0422-3J16), and anti-human CD49d (clone EPR1355Y and clone HP2.1). The following secondary antibodies were purchased from Life Technologies (Green Island, NY): goat anti-mouse IgG (AF488-conjugated or HRP-conjugated), and goat anti-rabbit IgG (AF555-conjugated or HRP-conjugated). CD11c allosteric agonist 496B, which activates CD11c promoting ligand binding, and CD11c allosteric antagonist 496K, which inactivates CD11c inhibiting ligand binding, were generously provided by Eli Lilly Corp.(17) For flow cytometry, the stain indices for each antibody were calculated using the formula:
Where Xpos is the median fluorescence intensity of positive stained population, Xneg is the median fluorescence intensity of a population that does not express the target receptor, and SD is the standard deviation of the negative population. For function blocking studies antibodies were used at 10μg/ml. The paxillin inhibitor (6-B345TTQ, Sigma Aldrich) and pSyk inhibitor (622387-85-3, EMD Millipore) were used at a concentration of 50μM and 50nM respectively. Inhibitors were incubated with cells for 10 minutes at 37°C.
Flow Cytometry
Mouse whole blood obtained via cardiac puncture was drawn and immediately cooled to 4°C. After labeling with fluorescent antibodies for 30 minutes, red blood cells were removed with RBC Lysis buffer (Biolegend, San Diego, CA). Data were acquired on a BD FACScan cytometer within 2 hours of cardiac puncture. Cells were characterized as having positive expression of membrane adhesion molecules (CD11c, CD49d) and chemokine receptors (CCR2, CX3CR1, CCR5) based on a significant increase in mean fluorescence intensity over FMO controls. Foamy monocytes were defined as CD115 positive cells that had a side scatter (SSC) value greater than 2 standard deviations above the mean SSC of monocytes from ND fed mice. This criterion for defining foamy monocytes was assessed using the lipid dye BODIPY to stain intracellular lipid. More than 95% of SSC high monocytes stained positive for BODIPY.
Triglyceride-Rich Lipoprotein Isolation
Isolation of TGRL from hypertriglyceridemic subjects has been described previously.(10) Blood samples for lipoprotein isolation were collected into BD Vacutainer plasma separator tubes containing lithium heparin 3.5 h after consumption of the high-fat meal and spun at room temperature for 10 min at 1,300 g to remove cellular particulate. Following centrifugation, plasma was transferred to ultracentrifuge tubes (Beckman-Coulter) and centrifuged at 280,000 g at 14°C for 18 h. Triglyceride-rich lipoproteins (TGRLs) were obtained by aspirating the top layer of plasma (ρ< 1.0063 g/mL). The isolate's apolipoprotein B content was measured with a total human apoB ELISA (AlerCheck).
Mononuclear Cell Isolation
Human mononuclear cells were obtained as described previously.(18) Heparanized whole blood was diluted 1:1 with HBSS without Ca2+ and Mg2+ and then layered over mononuclear cell separation medium Lymphoprep® (ρ= 1.077g/ml)(Axis-Shield, Oslo Norway) and spun at 800 × g for 25 minutes at 25°C. The mononuclear band was isolated with a 16-gauge × 2.5″ safety cathlet, and then spun down at 200 × g for 10 minutes. Mononuclear cells were washed twice in HBSS without Ca2+ and Mg2+ and reconstituted in HBSS containing 1mM Ca2+ and 1mM Mg2+.
Vascular Cell Adhesion Molecule-1 coating on Glass Coverslips
We prepared 25mm diameter No. 1.5 glass coverslips in a 1:1 solution of H2S04 and H2O2, rinsed them with water, and then dipped them in a solution of acetone (Sigma Aldrich) and 4% (vol/vol) 3-aminopropyltriethoxysilane (Pierce). Goat F(ab')2 anti-human IgG Fc (Pierce) at 100μg/mL was incubated on the glass for 1 h at room temperature. Coverslips were then incubated with 5μg/mL of mouse recombinant vascular cell adhesion molecule-1 (mVCAM-1)–Fc chimera (R&D Systems) followed by 0.1% human serum albumin to block nonspecific binding of leukocytes. This method of linking VCAM-1 results in a density of ∼2000 molecules/μm2.
Artery-on-a-chip Microfluidic Assembly
The device was designed to allow utility of four independent flow channels with dimensions of 60μm×2mm×8mm (h×w×l). An interconnected spiderweb pattern surrounding the channels allowed the device to be vacuumed sealed over a glass coverslip. The microfluidic networks were designed in AutoCAD (Autodesk) and then printed as a photomask by CAD/Art services Inc. A master mold for the device was created by coating a 200mm diameter silicon wafer with SU-8 50 photoresist (MicroChem) to a height of 60μm and then exposing the wafer to UV light through the photomask containing the microfluidic channel pattern as described previously.(19) Microfluidic chambers were fabricated by pouring polydimethylisloxane (PDMS) Sylgard 184 prepolymer (Dow Corning) over the master silicon wafers. Reservoir and vacuum access holes were punched directly into the PDMS chambers using a needle.
Whole Blood Adhesion Assay
Heparinized mouse whole blood was added to the artery-chip chamber reservoir and withdrawn through a single channel using a syringe pump (Kent Scientific) at a flow rate resulting in a physiological shear stress of 2 dynes/cm2 at the glass fluid interface (a wall shear stress corresponding with athero-prone regions in the arterial vasculature).(20) Arrested monocytes were fixed with 1 × Lyse/Fix Buffer (Biolegend) and the coverslip was removed from the PDMS chamber and then incubated with hanks balanced salt solution (HBSS) containing 1% human serum albumin (HSA) for 30 minutes at room temperature. Coverslips were then washed twice with Dubelccos Phosphate Buffer Solution 1× stained with PE anti-CD115 and BODIPY.
Receptor Co-localization
Mouse blood was perfused through the microfluidic chamber for 4 minutes over a substrate of recombinant mouse VCAM-1. HBSS was then flowed through for 4 minutes to wash unbound cells and then Lyse/Fix Buffer 1× was perfused through for 2 minutes to fix arrested cells. The glass coverslip was removed from the chamber and placed in a dish containing HBSS + 1% HSA for 30 minutes at room temperature. For experiments imaging co-localization of VLA-4 with CD11c (or CD45) cells were stained with 5μg/ml PE anti-CD49d and 5μg/ml AF488 anti-CD11c (or 5μg/ml AF488 anti-CD45) for 30 minutes and then washed twice with DPBS. Coverslips were then mounted in Prolong® Gold Antifade Reagent with DAPI (Life Technologies) and imaged on a Nikon Eclipse TE2000-S microscope coupled with a Hamamatsu ORCA-ER CCD camera and a Nikon Apo TIRF 60× oil objective (N.A.= 1.49). Fluorophores were excited with a 2 mW HeNe 488 nm laser and an adjustable (1-20mW) 552nm OBIS laser diode (Coherent). For co-localization analysis, monocytes were identified using DAPI nuclear stain to visualize kidney bean shaped morphology specific to monocytes. ImageJ v1.46 was used to calculate the Pearson correlation to compare the overlap of CD49d (red) and CD11c (or CD45) (green) labeled receptors.
Fibronectin Coated Fluorescent Bead Assay
Fluorescent beads coated with ligand were used to probe VLA-4 receptor function on cells in suspension as described previously.(21) Carboxylated fluorescent latex beads (diameter = 2μm; Life Technologies) were washed three times in DPBS free of calcium or magnesium ions then incubated with 10μg/ml of recombinant human fibronectin for 1 hour at 37°C. Beads were then washed twice in DBPS, dispersed by sonication, and then counted before use. Monocytes (5 × 105) were mixed with 1 × 107 Fn coated beads in a 500μL volume of HBSS with calcium and magnesium ions in a mixing tube containing a small magnetic stir bar. The sample was then placed in a specially designed mixing mixing chamber to maintain the sample at 37°C and stir suspensions at 500 rpm with a magnetic bar, corresponding to between 1-2 dynes/cm2 which is equivalent to the hydrodynamic shear stress in the microfluidic channels.(22) The mixing chamber was positioned just upstream of the sample injection nozzle of a FACScan (Becton Dickinson) and the suspen was. Monocytes and discrete beads were discriminated based on forward scatter and side scatter parameters and a live gate was placed over the monocyte population in order to limit detection to monocytes and monocyte-bead events. 10,000 events were collected at a sampling rate of 10 ms over the course of over the 10 minutes total collection time. The mean number of beads per monocyte (NB) was calculated by summing the product of the fraction of monocytes (MONb) with the number of beads bound (b), and then dividing by the total number of monocytes with and without (MON0) Fn beads:
With this technique, the maximum number of beads per cell that could be resolved was six at each timepoint measured. Monocytes that bound more beads were summed at 6.
LDV-FITC Binding Assay
Monocytes maintained in HBSS + Ca/Mg + 0.1% HSA were stimulated with 496B (10μg/mL), TGRL (apoB 100μg/mL), MCP-1 (30nM), or Mn2+ (1mM) in the presence of 4nM of LDV-FITC with continuous mixing in a customized mixing device described previously. Binding of LDV-FITC to monocytes was constantly monitored using flow cytometry and reached maximum binding by 10 minutes of incubation.
Immunoprecipitation of CD11c
Human mononuclear cells were isolated from whole blood and suspended in HBSS with calcium and magnesium ions. We measured CD11c on lymphocytes in the mononuclear cell prep and confirmed that lymphocytes did not express CD11c (data not shown). Mononuclear cells were then incubated for 15 minutes at 37°C with allosteric antibodies against CD11c that activate (496B), inactivate (496K), or bind CD11c but not alter its conformation (BU15). Separately, mononuclear cells were incubated with TGRL at 100μg/ml of apoliprotein B or intralipid for 30 minutes at 37°C and then washed in HBSS containing 2mM EDTA to remove surface bound lipoproteins. Mononuclear cells were then lysed with IP wash/lysis buffer (Pierce) and protein was measured using bicinchoninic acid assay (Pierce). Samples were then diluted to 700μg/ml of protein using IP wash/lysis buffer and incubated with anti-CD11c (clone MM0422-3J16) for 4 hours at 4°C. Protein A/G agarose resin from Pierce classic IP kit was used to pull down CD11c, and the resin was then washed 4 times with IP wash/lysis buffer. Equal concentrations of protein sample were subjected to gel electrophoresis and then protein was transferred to a polyvinylidene membrane. The membranes were blocked with 5% milk for 1 hour and then primary antibodies were used to probe for CD49d, paxillin, Syk, and pSyk. An HRP-secondary was then used to stain the primary antibodies and membranes were developed using ECL (super signal west pico chemiluminescent substrate, Pierce). Data from blots was collected using a ChemiDoc™ MP system and protein density in each band was calculated using ImageJ v1.46.
Statistics
Data are presented as mean±SEM unless otherwise specified. Multiple groups were compared using one-way ANOVA with Tukey posttest. All analyses were carried out using Graph Pad Prism v6.0d for Mac (GraphPad Software, Inc, La Jolla, CA).
Study Approval
All protocols and procedures involving apoE-/-, CD11c-/-/apoE-/-, and α4(Y991A) mice were approved by the IACUC at the University of California Davis. Whole blood monocytes and TGRL were obtained from human subjects on the basis of an approved institutional review board protocol at the University of California, Davis. Written consent was obtained from human subjects prior to enrollment in the study.
Results
The onset of hypercholesterolemia and emergence of foamy Ly6Clow monocytes in blood is independent of CD11c expression
Hypercholesterolemia promotes monocyte uptake of circulating lipid and recruitment to arterial sites of nascent plaque development within weeks of initiating a WD in apoE-/- mice.(5, 23) This motivated assessment of the dynamics of emergence of foamy monocytes in the circulation of WD versus ND fed apoE-/- mice over a 5-week study interval. Blood monocytes were identified by flow cytometry based on positive expression of CD115 and were characterized as foamy based on elevated side scatter (SSC) and BODIPY fluorescence intensity above that on non-foamy controls (Figure 1A). Foamy monocytes increased in the circulation most rapidly within the first week of the onset of a WD and plateaued at ∼30% of total monocytes at 5 weeks (Figure 1B). Nonfat diet control mice exhibited no detectable change in lipid content of circulating monocytes, nor neutrophils or lymphocytes, after 1 week indicating that lipid uptake in the blood is specific to monocytes and the effect of a WD (Supplemental Figure 1A and 1B). The number of circulating monocytes that were foamy remained equivalent between apoE-/- and CD11c-/-/apoE-/- mice over 5 weeks of the WD (Figure 1B and Supplemental Figure 1C). Discrimination of monocytes based on Ly6C expression revealed the vast majority of foamy monocytes in blood of mice fed a WD were the Ly6Clow subset (∼80%), which out numbered Ly6Chigh by ∼4:1 and this ratio did not change significantly over 5 weeks regardless of CD11c expression (Figure 1C). We conclude that during the early onset of hypercholesterolemia in apoE-/- mice a subset of foamy Ly6Clow monocytes emerge, and CD11c-/- monocytes maintain the capacity to take up lipid in circulation.
Figure 1. Foamy monocytes are predominantly Ly6Clow and CD11chigh are prevalent in blood after 1 week of a high fat diet in apoE-/- mice independent of CD11c expression.
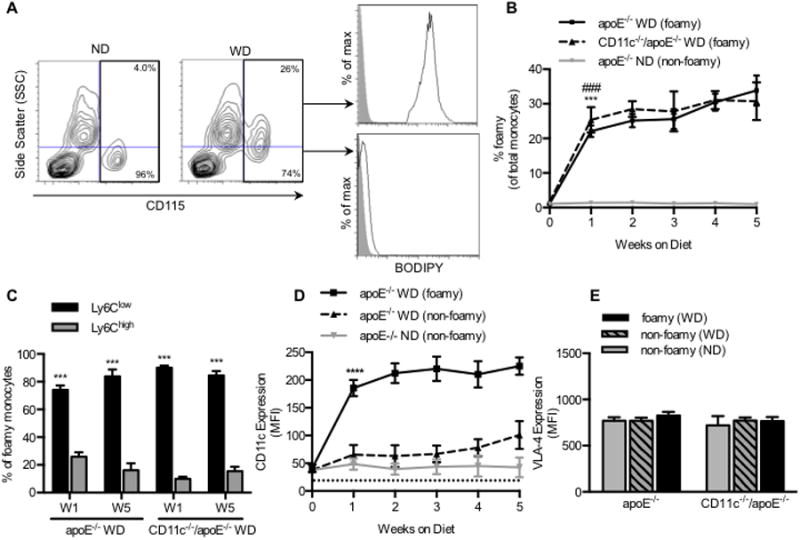
(A) Monocytes were identified based upon CD115 positivity. Monocytes that had taken up lipid and became foamy were discriminated by SSC intensity. BODIPY fluorescence (black line) was measured on both foamy (SSC high) and non-foamy (SSC low) populations and compared to unstained control (gray filled histogram). (B) Percent of foamy monocytes in apoE-/- mice consuming a ND (gray line) or a WD (solid black line) and in CD11c-/-/apoE-/- mice consuming a WD (dashed black line) (n= 4-5 mice per timepoint). Significance was determined by comparing apoE-/- WD, CD11c-/-/apoE-/- WD, and apoE-/- ND using a repeated measures ANOVA. #,*p<0.05,##,**p<0.01,###,***p<0.005 (C) Proportion of foamy monocytes that are Ly6Clow or Ly6Chigh. Significance comparing Ly6Clow versus Ly6Chigh was determined using paired Student's t-test. (D) CD11c expression measured on foamy (solid black line) and non-foamy (dashed black line) monocytes from apoE-/- mice fed a WD and non-foamy from apoE-/- fed a ND (gray line) (n = 4-5 mice per timepoint). Significance comparing foamy to non-foamy from WD was determined using one-way ANOVA with Tukey posttest. *p<0.05. (E) VLA-4 expression measured on monocytes from apoE-/- and CD11c-/-/apoE-/- fed a ND or WD for 1 week (n = 3 mice per group).
Foamy monocytes in the circulation of hypercholesterolemic mice upregulate membrane CD11c
It has been reported that CD11c-/-/apoE-/- mice develop smaller atherosclerotic lesions after 12 weeks on a WD and that monocytes from these mice exhibit a reduced capacity to arrest on VCAM-1 under hydrodynamic shear stress.(3) Furthermore, CD11c expression on circulating monocytes increases with the extent of hypertriglyceridemia in humans and hypercholesterolemia in mice.(3, 10, 15) This suggests that CD11c upregulation occurs as a consequence of metabolic and inflammatory signaling, but the dynamics of this process has not previously been examined. CD11c expression increased 3-fold after 1 week on foamy Ly6Clow monocytes, while no such increase was detected on non-foamy monocytes (Figure 1D). Lipid uptake also elicited CD11c upregulation on Ly6Chigh monocytes, but receptor numbers were 5-fold lower compared with Ly6Clow (Supplemental Figure 2A and 2B). Elevated expression of CD11c on all foamy monocytes was sustained out to 5 weeks, whereas VLA-4 was expressed at equivalent levels and remained constant on all subsets of circulating monocytes regardless of lipid uptake (Figure 1E). Moreover, no significant change in CD11c or VLA-4 expression on circulating neutrophils was detected (Supplemental Figure 2D and 2E). Thus, CD11c expression increased most rapidly early in the course of diet-induced hypercholesterolemia, mirroring the dynamics in emergence of foamy Ly6Clow monocytes in the circulation of apoE-/- mice.
Foamy monocytes upregulate atherogenic chemokine receptors and activate VLA-4 dependent adhesion within lipid rafts enriched in CD11c
Mice deficient in the MCP-1 receptor (CCR2), fractalkine receptor (CX3CR1), or RANTES receptor (Regulated on Activation normal T-cell Expressed and Secreted receptor, CCR5) exhibit impaired monocyte recruitment and development of mature plaque.(24) This observation prompted measurement of CCR2, CX3CR1, and CCR5 chemokine receptors and adhesion function of blood monocytes following chemokine stimulation (Supplemental Figure 2C). No significant differences in chemokine receptor expression was detected on non-foamy monocytes in blood from WD (Figure 2A) or ND fed mice (data not shown). In contrast, foamy monocytes from apoE-/- mice fed for 1 week on WD upregulated CCR2 and CX3CR1 (28% and 70% increase in median fluorescence intensity (MFI)) compared with non-foamy monocytes, respectively. In contrast, no significant change in CCR5 on foamy or non-foamy monocytes was detected. Monocyte adhesive capacity was assessed ex vivo by shearing cell suspensions with an excess of fluorescence beads coated with the VLA-4 ligand fibronectin (Fn) and measuring the kinetics of capture by flow cytometry (Figure 2B).(25) VLA-4 adhesion was low on non-foamy monocytes, which did not increase bead capture from the baseline following the onset of shear mixing. Foamy monocytes exhibited ∼1-fold greater binding avidity than non-foamy, which was abrogated by addition of anti-VLA-4 (Figure 2B). Bead capture increased ∼2-fold above baseline upon stimulation with MCP-1 (30nM) or fractalkine (10nM) (Figure 2C). In contrast, there was no significant change in VLA-4 avidity following addition of RANTES, which was consistent with the low expression level and lack of increase in CCR5 on foamy or non-foamy monocytes. These data indicate that foamy monocytes in the blood of hypercholesterolemic mice have increased baseline capacity to adhere via VLA-4 and are primed for heightened response to chemokine activation by MCP-1 and fractalkine within 1 week of the onset of a WD.
Figure 2. Foamy monocytes adopt an inflammatory phenotype associated with increased chemokine receptors, VLA-4 adhesion, and colocalization with CD11c within lipid rafts.
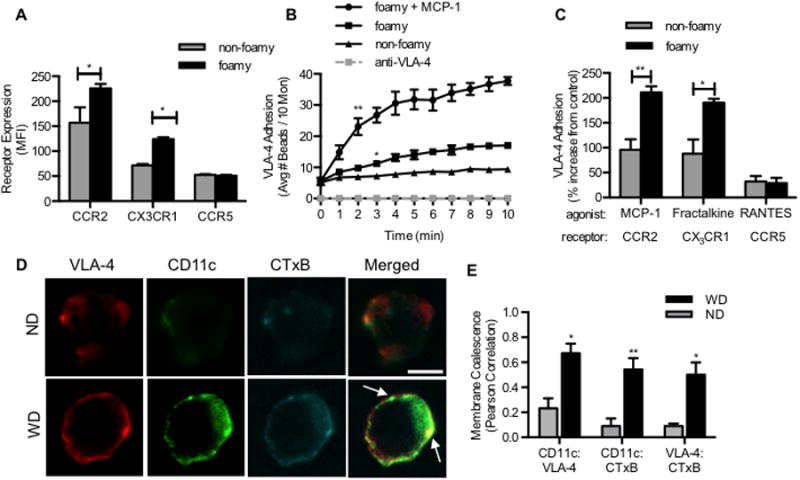
(A) Receptor expression of CCR2, CX3CR1, and CCR5 measured on foamy and non-foamy monocytes from apoE-/- mice fed a high fat diet for 1 week (n = 4-5 mice). Significance comparing foamy with non-foamy monocytes was determined using paired Student t-test. *p<0.05, **p<0.01, ***p<0.005. (B) A real-time readout of VLA-4 avidity based upon binding of fibronectin beads was performed by flow cytometry as described in the Methods. Capture of Fn beads sheared with foamy vs. non-foamy monocytes over 10 minutes in the presence of 30nM MCP-1 or anti-VLA-4 blocking antibody (n = 3-4 separate experiments) Significance comparing foamy and non-foamy was determined using multiple Student t-test. (C) Percent increase in VLA-4 adhesion over control (0.01% DMSO) following addition of MCP-1 (30nM), fractalkine (10nM), or RANTES (15nM)(n = 3 separate experiments). Significance comparing foamy and non-foamy was determined using paired Students t-test. (D) Monocytes from apoE-/- mice were labeled with antibodies against CD11c, VLA-4 and lipid rafts (CTxB). Confocal micrographs are representative of 30 monocytes observed from either a ND or HFD fed mouse (n = 3 mice per group). (E) Integrin coalescence in membrane lipid rafts was determined using Pearson correlation. Significance comparing ND and WD monocytes was determined using Student t-test.
To examine the mechanism underlying integrin mediated bead capture, we labeled and imaged lipid rafts on the membrane of foamy monocytes in suspension since they are known to play an important role in the coalescence and clustering of activated integrins and facilitate stable shear resistant adhesion.(26-28) Monocytes were isolated from mouse blood and labeled with fluorescent antibodies against VLA-4, CD11c, and fluorescent cholera toxin subunit B (CTxB), which preferentially binds to the ganglioside GM1 protein expressed in lipid rafts. Although CD11c and VLA-4 were diffusely expressed, these integrins were twice as likely to colocalize on the plasma membrane in general, and ∼5-fold more likely to coalesce within lipid raft domains, on monocytes from WD compared to ND controls (Figure 2D and 2E). These data indicate that the increase in VLA-4 adhesive function on freshly isolated foamy monocytes in suspension correlates with CD11c upregulation and integrin colocalization within lipid rafts.
Monocyte recruitment to VCAM-1 is dependent on CD11c, VLA-4, and lipid rafts
Monocytes in circulation take up lipid and are primed for recruitment on inflamed endothelium predominantly via VLA-4 binding to VCAM-1.(29, 30) We employed an artery-on-a-chip (A-chip) microfluidic device to quantify ex-vivo the propensity of foamy monocytes isolated from hyperlipidemic humans and mice to capture on VCAM-1.(10) Human aortic endothelial cells (HAEC) stimulated with the inflammatory cytokine tumor necrosis factor-α (TNF-α) provides a pro-atherogenic model to study integrin-mediated recruitment of monocytes.(29, 31) Mononuclear cells were isolated from hypertriglyceridemic subjects in the postprandial state and subsequently sheared over a substrate of inflamed HAEC in the A-chip microchannels (Supplementary Figure 3A and 3B). The prevalence of foamy monocyte capture was assessed by immunofluorescence labeling of recruited cells with the lipid partitioning dye BODIPY (Supplemental Figure 3A). Foamy monocytes sheared over inflamed HAEC captured twice as efficiently as non-foamy monocytes and adhesion was 30% dependent on CD11c and 70% dependent on VLA-4 (Supplemental Figure 3B). To directly compare human and mouse monocyte capture on VCAM-1, we next examined monocyte recruitment in a reduced system in which the glass substrate was coated with recombinant VCAM-1-IgG. Annealing VCAM-1 to the substrate at a site density equivalent to that on inflamed HAEC (data not shown), we detected a 2:1 recruitment efficiency of foamy to non-foamy monocytes, similar to capture on the HAEC monolayer. Further, monocyte capture was abrogated with an anti-VLA-4 blocking antibody yet the efficiency of capture remained partially CD11c dependent (Supplemental Figure 3B). This reduced A-chip system provided an ideal means of examining ex-vivo the dynamics of integrin mediated capture of circulating monocytes on VCAM-1 over the 5-week course of a WD versus ND in apoE-/- mice. Whole blood samples were collected and sheared over recombinant VCAM-1-IgG in the A-chip device at a wall shear stress of 2 dynes/cm2 and captured monocytes were fixed and H&E stained for identification based on characteristic morphology and expression of CD115 (Supplemental Figure 3C and 3D). Following 1 week of a WD, the capture efficiency of monocytes, defined as the number arrested normalized to the whole blood count, increased more than 2-fold from a baseline of ∼3 arrested up to 10 per 1000 infused monocytes (Figure 3A). Over the ensuing 4 weeks, monocytes from mice fed a WD increased by ∼4-fold over baseline. In comparison, blood monocytes from CD11c-/-/apoE-/- mice exhibited only a ∼1-fold increase, recruiting with an efficiency equivalent to that of mice fed a ND. By comparison the recruitment of neutrophils in blood on VCAM-1 arrested with 100-fold lower efficiency than monocytes on WD (Supplemental Figure 3E). These data indicate that monocytes from hypercholesterolemic mice markedly increase their capacity to recruit to VCAM-1 within 1 week of a WD. Moreover, the steady increase in the efficiency of monocyte recruitment observed up to 5 weeks of WD required CD11c expression.
Figure 3. Foamy monocytes recruit on a VCAM-1 substrate under shear stress in a lipid raft and VLA-4 dependent manner.
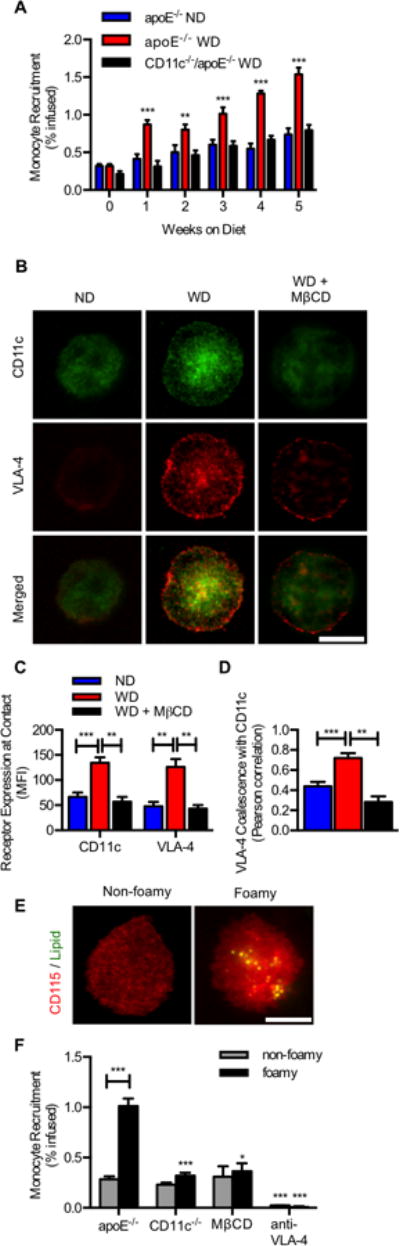
(A) Whole blood was sheared ex-vivo on recombinant VCAM-1 in a lab-chip microfluidic device. Recruitment efficiency of monocytes plotted for apoE-/- versus CD11c-/-/apoE-/- mice fed ND or WD for 1-5 weeks. Significance comparing WD with ND mice (n=4-5 per time point) was determined using one-way ANOVA with Tukeys post test. **p<0.01, ***p<0.005. (B) Focal adhesions enriched in integrins and lipid rafts support monocyte arrest on VCAM-1. Antibody tagged CD11c and VLA-4 show distribution within the contact region. Images are representative of 30-50 arrested cells per channel. (Scale bar = 10μm). (C) Integrin enrichment within the region of membrane contact on VCAM-1 as detected by TIRF microscopy for arrested monocytes from WD (n=5) versus ND (n=5) and disruption of lipid rafts after MβCD treatment. (D) VLA-4 coalescence with CD11c within focal adhesions quantified using Pearson coefficient. Significance was determined using one-way ANOVA with Tukey posttest. (E) Foamy monocytes following arrest were identified using CD115 (red) and lipid droplets were stained with BODIPY (green). Images are representative of 100-150 arrested cells per channel (Scale bar = 10μm). (F) Capture efficiency of foamy vs. non-foamy monocytes identified on chip following shearing of blood isolated from apoE-/- or CD11c-/-/apoE-/- mice on ND and 1 week WD. Significance comparing ND and 1 week WD was calculated using a Student t-test, *p<0.05 (n=5 separate experiments). Significance comparing apoE-/-, CD11c-/-/apoE-/-, while blocking VLA-4 was determined using one way ANOVA with Tukey posttest.
To assess the distribution of CD11c and VLA-4 receptors within the plane of adhesive contact of monocytes recruited on the VCAM-1 substrate, we employed total internal reflection fluorescence microscopy (TIRF) that restricts detection of fluorescent reporters to within ∼100 nm of the glass substrate where integrin/VCAM-1 bonds reside (Figure 3B). The number of CD11c and VLA-4 receptors in contact with VCAM-1 was ∼1-fold greater on arrested monocytes from WD versus ND mice (Figure 3C). Moreover, these integrins were twice as likely to coalesce in focal clusters on monocytes from blood of WD versus ND fed mice, such that 80% of VLA-4 was spatially co-located with CD11c following arrest (Figure 3D). Treatment of monocytes with the lipid raft inhibitor methyl-β-cyclodextrin (MβCD) reduced integrin coalescence within the region of contact with VCAM-1 to that observed with monocytes from ND mice (Figure 3C and 3D). This phenomenon was integrin specific, since coalescence between VLA-4 and CD45, a highly expressed membrane receptor on monocytes, remained at the baseline level of 20% that was equivalent between monocytes from ND and WD mice (Supplemental Figure 4A-C).
The prevalence of foamy monocytes captured on VCAM-1 was assessed on the A-chip by immunofluorescence labeling of membrane CD115 and intracellular lipid with BODIPY (Figure 3E).(10) Recruitment efficiency was equivalent between non-foamy monocytes obtained from ND and WD mice (data not shown). In contrast, foamy monocytes from apoE-/- mice fed a WD recruited ∼3-fold more efficiently than non-foamy monocytes and this increase was attributed to CD11c, since recruitment efficiency was equivalent between foamy and non-foamy monocytes in blood from apoE-/-/CD11c-/- mice (Figure 3F). Inhibition of lipid rafts with MβCD reduced recruitment of foamy monocytes to a level similar to non-foamy monocytes. In all cases, blocking VLA-4 binding to VCAM-1 by pretreatment with antibody abrogated recruitment. These data indicate that hypercholesterolemia amplifies the adhesive capacity of monocytes to capture on VCAM-1 in a manner dependent on spatial colocalization of CD11c and VLA-4 into bond clusters that coalesce within lipid raft microdomains. This integrin cooperativity was a function of lipid uptake to form foamy monocytes in the circulation regardless of subset since the relative fraction of foamy Ly6Clow to Ly6Chigh that arrested on VCAM-1 was maintained at 4:1, reflecting their respective numbers in the circulation of apoE-/- mice (data not shown).
CD11c expression and VLA-4 adhesion on foamy monocytes is upregulated following lipid uptake
We next addressed the mechanism by which monocyte uptake of lipoprotein in suspension elicits CD11c activation and VLA-4 dependent adhesion under shear stress. Mononuclear cells were isolated from blood collected from ND fed mice and incubated with triglyceride rich lipoproteins (TGRL). This TGRL was derived from human plasma and comprised of VLDL and chylomicron lipid particles(29), previously reported to elicit a pro-atherogenic response based on its capacity to upregulate VCAM-1 on HAEC and induce activation of VLA-4 adhesion following uptake by human monocytes.(9, 10) Mouse monocytes became foamy following in vitro exposure to TGRL as confirmed by an increase in SSC and BODIPY staining, similar to that observed ex vivo in circulating monocytes from apoE-/- mice fed a WD (Figure 4A). Monocyte activation following TGRL uptake was assessed based on upregulation of CD11c expression, an upshift in VLA-4 affinity, and VLA-4 dependent capture of Fn beads. VLA-4 affinity was detected as increased binding of the small fluorescently labeled peptide 4-[(N′-2-methylphenyl) ureido]-phenylacetyl-L-leucyl-L-α-aspartyl-L-valyl-L-prolyl-L-alanyl-L-alanyl-L-lysine - FITC (LDV-FITC) to monocytes incubated with TGRL.
Figure 4. Monocyte uptake of TGRL elicits increased CD11c expression and VLA-4 adhesion in lipid rafts dependent on paxillin binding and lipid rafts.
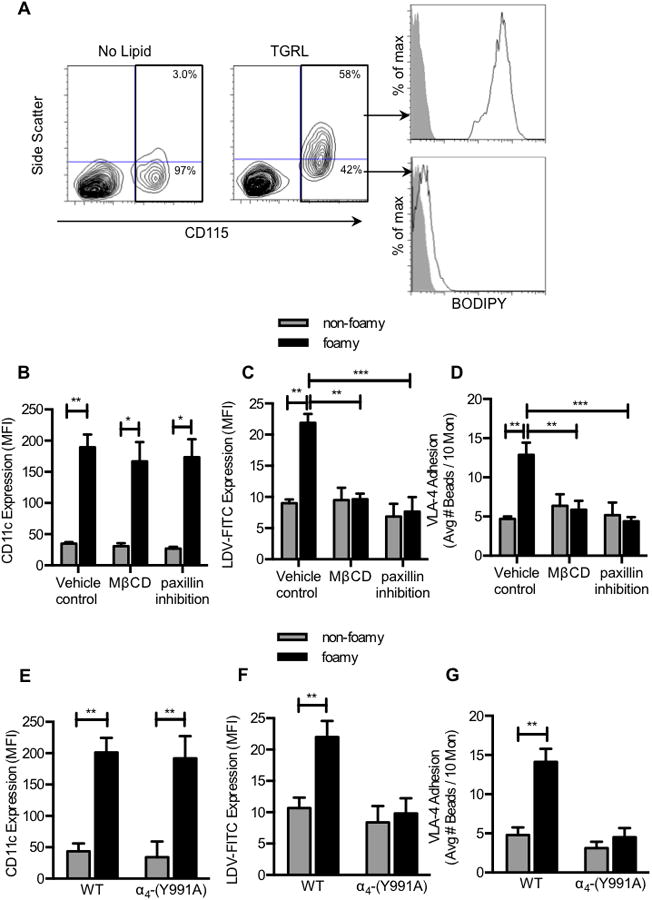
Mouse mononuclear cells were isolated from blood from apoE-/- mice maintained on a ND. Cells were incubated with human TGRL at 100μg/ml apoB for 30 minutes at 37°C. (A) FACS plots of foamy and non-foamy monocytes following uptake of TGRL. Monocytes containing lipid stained positive for BODIPY. Prior to incubation with TGRL, mouse mononuclear cells were treated with 0.01% DMSO (Vehicle Control), 10nM MβCD, or paxillin inhibitor 6-B345TTQ (50 μM). (B) CD11c expression on foamy vs. non-foamy monocytes. (C) VLA-4 affinity measured by LDV-FITC (30nM) binding to foamy vs. non-foamy monocytes at a time point of maximum adhesion (∼10 minutes). (D) VLA-4 adhesion measured by Fn bead binding in sheared monocyte suspensions. (n = 4 separate experiment utilizing TGRL isolated from 4 separate hypertriglyceridemic human subjects). Significance was determined by Student t-test. *p<0.05, **p<0.01. Mouse mononuclear cells were isolated from blood from α4-(Y991A) or wild type mice and incubated with human TGRL at 100μg/ml apoB for 30 minutes at 37°C. (E) CD11c expression on foamy vs. non-foamy monocytes. (F) VLA-4 affinity and (G) VLA-4 adhesion on foamy vs. non-foamy monocytes measured at a time point of maximum adhesion. (n = 5 separate experiments fed TGRL isolated from hypertriglyceridemic human subjects) Significance was determined by Student t-test. **p<0.01.
VLA-4 affinity and adhesion increased ∼1-fold and this correlated with a ∼7-fold upregulation of CD11c on foamy monocytes compared to non-foamy monocytes in the same suspensions (Figure 4B-D). Pretreatment with the lipid raft inhibitor MβCD abrogated the increase in VLA-4 affinity and avidity, but did not affect the upregulation of CD11c indicating that uptake of TGRL and CD11c upregulation was independent of lipid raft formation shown to be necessary for downstream VLA-4 activation.
Paxillin is a cytoplasmic adapter protein that binds to the α4 subunit of VLA-4 and stabilizes the high affinity conformation necessary for monocyte adhesion to β1-integrin ligands under shear.(32) Pretreatment with the paxillin antagonist 6-B345TTQ prevents its interaction with the cytoplasmic domain of α4 and is reported to reduce monocyte recruitment during inflammation.(33) Treatment with this antagonist did not diminish uptake of TGRL and subsequent upregulation of CD11c, but abrogated the increase in VLA-4 affinity and concomitant Fn bead capture (Figure 4B-D). To confirm that it was indeed paxillin association with the α4 cytoplasmic domain that stabilized high affinity VLA-4, we measured changes in VLA-4 affinity and Fn bead binding on monocytes from α4-(Y991A) mice, which lack the intracellular domain that supports paxillin binding to VLA-4. Uptake of TGRL by monocytes isolated from α4-(Y991A) mice induced foaminess and activated the increased expression of CD11c to a similar level as the control wild type C57BL6 mice (WT) (Figure 4E). However, activation of VLA-4 affinity and avidity in foamy monocytes from α4-(Y991A) mice was prevented (Figure 4F-G). These data indicate that paxillin binding at the Y991A domain is a critical step in TGRL mediated activation of VLA-4 to bind ligand. We conclude that monocyte uptake of lipid elicits upregulation of CD11c that in turn activates VLA-4 affinity and avidity preferentially on foamy monocytes in a lipid raft and paxillin-dependent manner.
Activation of high affinity CD11c directly activates VLA-4 adhesion by complexing with paxillin and phospho-spleen tyrosine kinase (Syk)
Previous studies in T-cells revealed that CCR2 ligation by MCP-1 signals activation of VLA-4 to form focal adhesions with VCAM-1 through cytosolic association with phospho-Syk and paxillin.(34, 35) We hypothesized that a similar mechanism governs activation of foamy monocyte induced via outside-in signaling by high affinity CD11c. Outside-in signaling of VLA-4 was compared with inside-out signaling via MCP-1 binding to CCR2, by measuring the level of LDV-FITC binding and the kinetics of Fn bead capture by human monocytes in suspension. The high affinity conformation of CD11c was detected with the antibody reporter mAb 3.9. Outside-in signaling was induced by the binding of 496B, a monoclonal antibody that recognizes the I-domain allosteric site of CD11c upshifting the receptor to a high affinity conformation.(10, 36) Titrating the activation of high affinity CD11c by addition of TGRL, MCP-1, or 496B, elicited a proportional increase in VLA-4 capture of Fn beads and binding of LDV-FITC (Figure 5A and 5B). Allosteric activation of CD11c with 496B elicited the steepest increase in VLA-4 adhesion to Fn beads, such that an equivalent amount of bead capture required activation of half the number of high affinity CD11c stimulated by MCP-1. Outside-in or inside-out signaling of VLA-4 activation was dependent on paxillin and Syk phosphorylation as shown by pretreatment with the respective inhibitors that lowered Fn bead binding to the baseline of untreated controls (Figure 5B and 5C). In contrast, activation of high affinity CD11c by all stimuli was unaffected by inhibition of paxillin and pSyk (Figure 5D). Thus, human and mouse monocytes exhibit a similar functional response of VLA-4 that correlates with the extent of outside-in signaling via high affinity CD11c.
Figure 5. High affinity CD11c activates VLA-4 adhesion within a macromolecular complex initiated by pSyk and dependent upon paxillin.
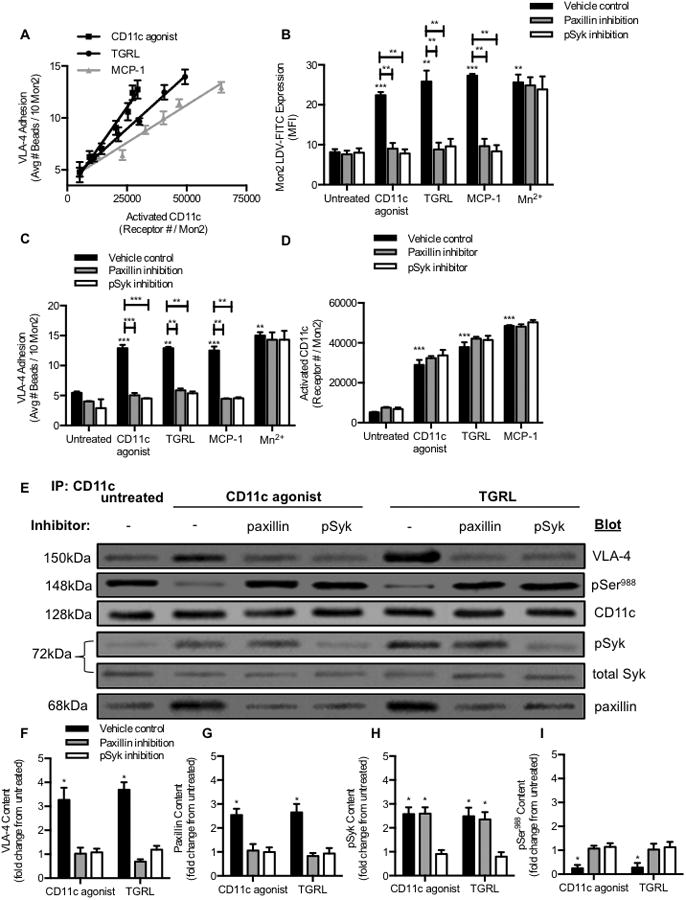
(A) Human mononuclear cells isolated from blood were treated with anti-CD11c agonist 496B at concentrations (0.5, 1, 2, 5, 10, 20 μg/mL), MCP-1 at concentrations (0.1, 1, 5, 10, 30, 100nM) or TGRL at concentrations (ApoB: 25, 50, 100 μg/mL). VLA-4 adhesion plotted vs. high affinity CD11c expression detected by anti-CD11c mAb 3.9. (B) VLA-4 binding to LDV FITC at time point of maximum adhesion, (C) VLA-4 adhesion, or (D) high affinity CD11c on mononuclear cells pretreated with 0.01% DMSO, 50μM paxillin inhibitor, or 3μM pSyk inhibitor for 15 minutes and then incubated with anti-CD11c antibody BU15 (control) (10μg/mL), anti-CD11c agonist 496B (10μg/mL), TGRL (100μg/ml apoB), MCP-1 (30nM), or 1mM Mn2+. (E) CD11c association with VLA-4 on human monocytes that were isolated and suspended for 15 min at 37°C with control anti-CD11c BU15, activated with TGRL (100μg/mL apoB) or allosteric anti-CD11c agonist 496B (10μg/mL). Western blot detection of CD11c, VLA-4, paxillin, pSyk, and pSer988 on monocytes that were lysed and immunoprecipitated by anti-CD11c coupled to protein-A/G agarose resin. (F-I) Protein content per band was normalized to CD11c density and the fold change for VLA-4 and paxillin following activation with anti-CD11c agonist 496B or TGRL was compared to that obtained by treatment with the control anti-CD11c BU15. Phosphorylated Syk was normalized to total Syk protein for each condition to establish pSyk/Syk ratio and its fold change was compared to the control anti-CD11c BU15. Significance comparing the results against the untreated sample was determined using multiple Mann-Whitney tests. *p<0.05 (n= 3 separate experiments).
To quantify the association between CD11c and VLA-4 integrins in response to monocyte activation in suspension, immunoprecipitation and western blot analysis was employed following allosteric activation of CD11c with 496B, or uptake of TGRL. Since phosphorylation of Syk and recruitment of paxillin to the α4-cytoplasmic domain are required for stabilizing high affinity VLA-4 integrins at sites of focal adhesion, we probed the complex pulled down by immunoprecipitation of CD11c with antibodies specific to these cytosolic molecules.(32, 37, 38) In response to activation with TGRL or 496B, there was a ∼3-fold increase in pSyk, paxillin, and VLA-4 (Figure 5E-I). Paxillin binding to the α4-cytoplasmic domain and a conformational shift of VLA-4 to high affinity requires dephosphorylation of α4 at the serine-988 region (Ser988).(39) To assess the phosphorylation state of Ser988 following activation of CD11c, we probed the immunoprecipiated complex using an antibody specific to pSer988. Activation of CD11c with 496B or TGRL resulted in a concomitant decrease in pSer988 to the baseline level detected in vehicle controls (Figure 5E and 5I). Thus, activated CD11c induces the dephosyphorylation of Ser988 that provides a substrate for the assembly of paxillin and pSyk to bind and stabilize high affinity VLA-4 at focal sites of adhesion.
To probe the sequence of events involved in the dephosphorylation of Ser988 and the association of pSyk and paxillin with the integrin complex, monocytes were pretreated with pSyk or paxillin inhibitors prior to activation of CD11c. Treatment with paxillin inhibitor reduced the amount of VLA-4 pulled down with CD11c to the baseline and prevented dephosphoylation of Ser988 at the α4-cytoplasmic domain. However, paxillin inhibition did not affect the binding of pSyk to activated CD11c (data not shown). Treatment with the pSyk inhibitor reduced both paxillin and VLA-4 association with CD11c and suppressed dephosphoylation of Ser988 (Figure 5F-I). This result suggests that phosphorylation of Syk by high affinity CD11c precedes its association with paxillin and VLA-4. Collectively these data indicate a sequence that is initiated by lipid uptake and an upshift in CD11c affinity that triggers the association with VLA-4, contemporaneous with activation of Syk, recruitment of paxillin, and dephosphorylation of Ser988 within a macromolecular membrane complex that supports stable bond formation with fibronectin or VCAM-1.
Activation of CD11c to high affinity is required for TGRL but not MCP-1 stimulated VLA-4 adhesion
To further examine how induction of the high affinity conformation of CD11c facilitates VLA-4 mediated adhesion within lipid rafts, we examined in real-time the kinetics of Fn bead capture and imaged integrin coalescence within the region of adhesive contact in fixed and stained samples. Human monocytes in suspension were stimulated with either MCP-1 or TGRL and CD11c was imaged with the non-blocking control antibody BU15, or locked into low affinity by pretreatment with the allosteric inhibitor antibody 496K, or treated with MβCD to inhibit lipid raft formation (Figure 6A-C). A similar rate of VLA-4 mediated capture of Fn beads was induced following stimulation with TGRL or MCP-1. However, locking CD11c in a low affinity conformation with 496K, or inhibition of lipid raft formation, blocked to baseline the Fn bead capture activated by TGRL uptake. In contrast, stimulation with MCP-1 elicited bead capture that was independent of high affinity CD11c or recruitment of VLA-4 into lipid rafts (Figure 6B and 6C). Confocal imaging of the region of adhesive contact revealed the coalescence of high affinity CD11c with VLA-4 within lipid rafts (e.g. Capture frequency: 1 in 5 monocytes with beads) (Figure 6D). The few monocytes with bound Fn beads (e.g. Capture frequency: <1 in 10 monocytes with beads) following TGRL activation in the presence of 496K or MβCD inhibitors revealed fewer focal clusters of VLA-4 within diffuse lipid rafts. MCP-1 activation resulted in uniformly distributed VLA-4, which did not necessarily redistribute to lipid raft domains. We conclude that uptake of lipoprotein and formation of foamy monocytes induces the activation of CD11c and its coalescence within lipid rafts, which in turn precipitates the focal clustering of high avidity VLA-4 competent for shear resistant ligand binding.
Figure 6. High affinity CD11c elicits VLA-4 coalescence in lipid rafts to activate adhesion under shear stress.
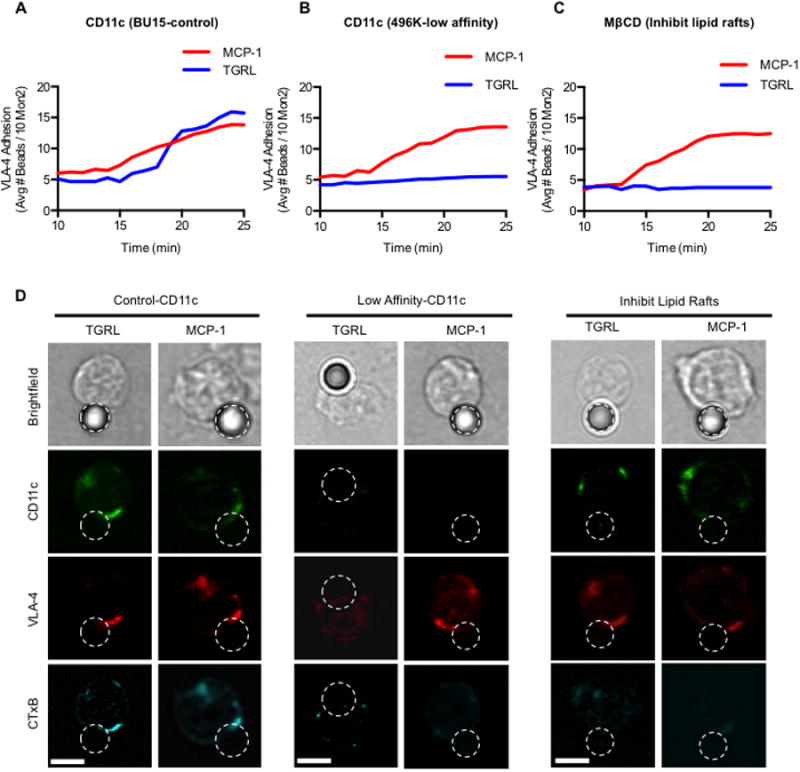
Human mononuclear cells isolated from blood were pretreated with anti-CD11c antibodies (A) BU15 or (B) 496K or (C) MβCD and VLA-4 bead adhesion was measured for 10 minutes to establish a baseline for each treatment. Activation was elicited by addition of TGRL (100μg/mL apoB) or MCP-1 (30nM) and kinetics of VLA-4 capture of Fn beads at ∼1 dyn/cm2 measured in real-time by FACS. (D) Confocal fluorescence imaging of captured Fn-beads by monocytes in shear. Brightfield image followed by antibodies recognizing high affinity-CD11c (green), total-VLA-4 (red), and lipid rafts-CTxB (cyan). (scale bar = 5μm).
Discussion
Identification of the inflammatory subset of circulating monocytes that recruit to nascent plaque during atherogenesis has been elusive. In the current study, we report that Ly6ClowCD11chigh constitute the major component (>80%) of foamy monocytes that emerge in the circulation within 1-week of the onset of hypercholesterolemia. This subset adopted a pro-inflammatory phenotype primed for enhanced VLA-4 adhesion in response to signaling via chemokine receptors (CCR2, CX3CR1) and uptake of lipids. Shear resistant monocyte arrest on VCAM-1 resulted from a redistribution of high affinity CD11c and VLA-4 into focal adhesions that were stabilized by paxillin within lipid raft enriched membrane microdomains. Monocyte uptake of native lipoprotein high in triglycerides was sufficient to activate CD11c from a low to high affinity state, which induced phosphorylation of Syk in a process that precipitated its physical coupling with VLA-4 and paxillin. These data reveal a novel mechanism by which human and mouse monocytes activate VLA-4 dependent recruitment on VCAM-1 that is distinct from chemokine signaling and regulated by the relative number of high affinity CD11c expressed on an inflammatory subset of foamy monocytes.
Of the two major monocyte subsets that circulate in murine blood, Ly6Chigh are typically associated with a pro-inflammatory phenotype,(40, 41) whereas Ly6Clow are often referred to as patrolling cells that monitor vascular integrity and function in angiogenesis.(42) However, Ly6Clow also recruit to sites of plaque formation and their numbers in circulation correlate with lesion size in apoE-/- mice, suggesting they also contribute to atherogenesis.(24) Supporting this is the recent study of Robbins et. al.,(5) who reported using an apoE-/- model that foamy macrophages in nascent plaque are primarily derived from monocyte subsets that recruit over the initial 4 weeks of the onset of hypercholesterolemia. We observed Ly6Clowand Ly6Chigh foamy monocytes in the circulation of apoE-/- mice within 1 week of a WD and their relative numbers were maintained at ∼4:1 ratio throughout the 5 weeks of observation. Importantly, both subsets of foamy monocytes increased their capacity to activate VLA-4 adhesion in a manner dependent upon the relative expression of CD11c. Using the identical mouse model, Xu et. al. reported that proliferating monocytes from the circulation of apoE-/- mice express CD11c and are present at sites of nascent plaque formation as early as 2 weeks following the onset of hypercholesterolemia.(43) These foamy monocytes in the plaque were shown to produce inflammatory cytokines such as TNFα and IL-1β. Likewise, we observed ex vivo that monocytes from the circulation when incubated with TGRL and captured on VCAM-1 produced TNFα and IL-1β (Supplemental Figure 4D). Thus, foamy monocytes (both Ly6Chigh and Ly6Clow) upregulate CD11c and increase their recruitment efficiency to VCAM-1 during the early stage of plaque formation.
CD11c expression is upregulated on the plasma membrane from a storage pool in cytosolic granules within minutes of lipid uptake.(44) Neither CD11c expression nor membrane upregulation was required for uptake and internalization of lipid as supported by the fact that equivalent numbers of foamy monocytes were detected in blood samples from apoE-/- and CD11c-/-/apoE-/- mice fed a WD. In this regard, Xu et. al., also found that CD11c was upregulated on foamy monocytes within hours of a bolus injection and uptake of isolated cholesterol ester-rich very low-density lipoproteins into the circulation.(43) Similarly, we show that exposure to pro-atherogenic TGRL also induces the formation of foamy monocytes, suggesting that lipoprotein uptake is the chief source of this mode of activation. Emergence of foamy monocytes and an increase in their capacity to recruit to VCAM-1 under shear stress rose most dramatically within the first week of the onset of a WD, at which time they adhered twice as efficiently as those from mice on a ND. The capacity for lipid uptake to activate CD11c and increase VLA-4 affinity and avidity on foamy monocytes was consistent between mouse and human models. Human Mon2 (CD14++CD16+) isolated postprandial from hypertriglyceridemic human subjects, or those activated ex-vivo following TGRL uptake, upregulated CD11c affinity that directly correlated with the extent of VLA-4 mediated adhesion.(10) The mechanism for CD11c mediated activation of VLA-4 adhesion to ligand is likely dependent on both an increase in its affinity for ligand and redistribution into focal clusters, given that the overall number of VLA-4 receptors on activated monocytes remains constant.(45) Examination of integrin distribution beneath arrested monocytes revealed twice as many VLA-4 and CD11c receptors colocalized in focal clusters within lipid raft microdomains adherent to VCAM-1, a process required for enhanced recruitment efficiency.
Cooperativity between β1- and β2-integrins appears to involve a cis-interaction between high affinity CD11c and VLA-4. This activation occurs downstream of Syk phosphorylation, which triggers the dephosphorylation of Ser988 on the α4 subunit, which leads to subsequent recruitment of paxillin(39, 46) and stabilization of high affinity VLA-4.(35, 47) Outside-in signaling was achieved using the CD11c activating antibody 496B to allosterically stabilize a high affinity conformation and VLA-4 mediated activation, which was functionally equivalent to monocyte uptake of TGRL. This approach established a causal link between the upshift in CD11c affinity state and an increase in VLA-4 affinity and adhesive capacity. A putative mechanism for VLA-4 activation via high affinity CD11c may involve their redistribution in membrane rafts proximal with Src-homology 2 (SH2) domains on SLP-76 or CrkL, adaptor proteins, both shown to trigger activation following interaction with integrin cytodomains. (48-50) A hierarchy in the level of VLA-4 activation was detected, with signaling via the allosteric agonist 496B most potent and TGRL and MCP-1 least potent. These data suggest that conversion of CD11c to high affinity is a direct regulator of VLA-4 adhesion and implicates pSyk activation of paxillin as downstream regulators in the signaling pathway. This conclusion is supported by previous reports that phosphorylation of Syk at the cyto-domain of CD18 activates binding of paxillin (51), which serves as a substrate in stabilizing α4β1 into a high affinity conformation.(32, 33) In regards to the sequence, we found that inhibiting paxillin binding or phosphorylation of Syk had no effect in conversion of CD11c to high affinity, but abrogated the downstream dephosphorylation of Ser988 on the α4 cytosolic subunit that is required for activation of VLA-4. Using the allosteric antagonist antibody 496K to maintain CD11c in a low affinity conformation, we found that the upshift in CD11c affinity was critical for TGRL, but not MCP-1 activation of VLA-4 adhesion. A similar level of inhibition was observed by depleting lipid rafts with MβCD, which interfered with VLA-4 and CD11c colocalization and consequently diminished the efficiency of monocyte Fn bead capture and arrest on VCAM-1. We conclude that lipid uptake induced high affinity CD11c functions as a direct regulator of VLA-4 avidity through activation of pSyk and dephosphorylation of Ser988 on the α4 subunit, both of which are associated with focal adhesion formation stabilized by paxilin within lipid rafts at sites of shear resistant adhesion on VCAM-1.
A question that remains unanswered is; what is the precise mechanism by which lipoprotein uptake by monocytes confers activation of CD11c? We recently published that uptake of TGRL via low-density lipoprotein receptor related protein-1 (LRP-1) mediates signaling through phospholipase C. (9, 10) In the current study, we show this pathway is necessary for CD11c activation as depicted in Figure 7A. Further, we propose that local release of intracellular calcium occurs downstream of lipid uptake, which is known to precipitate PIP3 and small-GTPases including Rap1 that can activate CD11c at its cytodomain. (52, 53) Uptake of oxidized low-density lipoprotein and recognition by toll-like receptor-4 (TLR4) signals through phosphorylation of Syk SH2 domains, which can in turn activate the immunoreceptor tyrosine-based activation motifs (ITAMs).(54) The mechanism by which CD11c becomes active and recruited within lipid rafts may occur in a manner similar to ITAM mediated outside-in activation of the β2-integrin LFA-1.(55) Upon redistribution with lipid rafts, active CD11c may associate with constitutively available signaling molecules such as ITAMs that facilitate phosphorylation and signaling via Syk as depicted in Figure 7B.(56) We showed that phosphorylation of Syk by high affinity CD11c triggered the dephosphorylation of Ser988 that provided a substrate for binding of paxillin to VLA-4 in a focal adhesion complex within lipid rafts (Figure 7C). In this manner, high affinity CD11c coupled in the membrane to pSyk could associate with paxillin to initiate conversion of VLA-4 to high affinity where they coalesce into focal clusters that facilitate bond formation with VCAM-1 (Figure 7D).
Figure 7. Signaling events following monocyte uptake of lipid and activation of CD11c.
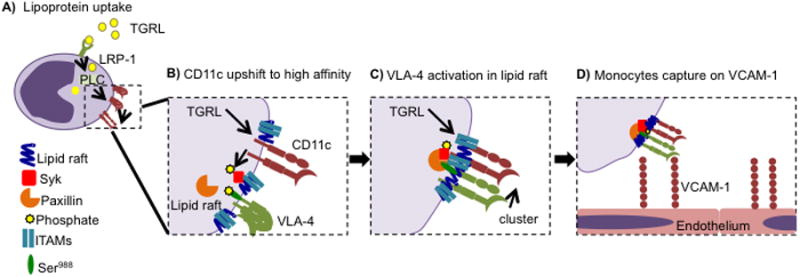
(A) Monocytes take up lipid in the blood via LRP-1, which results in upshift in affinity of CD11c via a PLC-dependent mechanism. (B) Activation of CD11c in response to TGRL uptake and PLC signaling results in coupling with ITAMs and phosphorylation of Syk within lipid rafts. (C) Phosphorylated Syk is associated with clustering of paxillin and VLA-4 with CD11c within focal adhesions. (D) Monocyte capture on VCAM-1 is mediated by high avidity focal adhesion domains within lipid rafts containing activated CD11c and VLA-4.
In conclusion, our data reveals that within a week of diet-induced hypercholesterolemia a subset of monocytes take up lipid and adopt an inflammatory phenotype associated with increased CD11c expression and shear resistant capture on VCAM-1. Human Mon2 inflammatory monocytes also activated VLA-4 mediated adhesion in response to uptake of TGRL, which elicited an upshift in CD11c affinity state that amplified monocyte recruitment to VCAM-1 under shear stress. We conclude that CD11c expression and activation may serve not only as a faithful biomarker of the pro-atherogenic state of monocytes in circulation, but also as a potential target for inhibiting their early recruitment into nascent plaque.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Britta Drier and Keith Bailey for their assistance with optimizing immunoprecipitation and western blots. We would also like to thank Mark Ginsberg and Marina Slepak for providing the α4-Y991A mice. We appreciate the generous provision of monoclonal antibodies by Don Staunton of CysThera and Eli Lilly Corp.
Footnotes
References
- 1.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. The Journal of clinical investigation. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghattas A, Griffiths HR, Devitt A, Lip GY, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: where are we now? Journal of the American College of Cardiology. 2013;62:1541–1551. doi: 10.1016/j.jacc.2013.07.043. [DOI] [PubMed] [Google Scholar]
- 3.Wu H, Gower RM, Wang H, Perrard XY, Ma R, Bullard DC, Burns AR, Paul A, Smith CW, Simon SI, Ballantyne CM. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–2717. doi: 10.1161/CIRCULATIONAHA.108.823740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. The Journal of clinical investigation. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nature medicine. 2013;19:1166–1172. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veniant MM, Withycombe S, Young SG. Lipoprotein size and atherosclerosis susceptibility in Apoe(-/-) and Ldlr(-/-) mice. Arteriosclerosis, thrombosis, and vascular biology. 2001;21:1567–1570. doi: 10.1161/hq1001.097780. [DOI] [PubMed] [Google Scholar]
- 7.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. Journal of Clinical Investigation. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saja MF, Baudino L, Jackson WD, Cook HT, Malik TH, Fossati-Jimack L, Ruseva M, Pickering MC, Woollard KJ, Botto M. Triglyceride-Rich Lipoproteins Modulate the Distribution and Extravasation of Ly6C/Gr1 Monocytes. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gower RM, Wu H, Foster GA, Devaraj S, Jialal I, Ballantyne CM, Knowlton AA, Simon SI. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:160–166. doi: 10.1161/ATVBAHA.110.215434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster GA, Gower RM, Stanhope KL, Havel PJ, Simon SI, Armstrong EJ. On-chip phenotypic analysis of inflammatory monocytes in atherogenesis and myocardial infarction. PNAS. 2013;110:13944–13949. doi: 10.1073/pnas.1300651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rullo J, Becker H, Hyduk SJ, Wong JC, Digby G, Arora PD, Cano AP, Hartwig J, McCulloch CA, Cybulsky MI. Actin polymerization stabilizes alpha4beta1 integrin anchors that mediate monocyte adhesion. The Journal of cell biology. 2012;197:115–129. doi: 10.1083/jcb.201107140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu C, Celik E, Rico F, Moy VT. Elongated membrane tethers, individually anchored by high affinity alpha4beta1/VCAM-1 complexes, are the quantal units of monocyte arrests. PloS one. 2013;8:e64187. doi: 10.1371/journal.pone.0064187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigal A, Bleijs DA, Grabovsky V, van Vliet SJ, Dwir O, Figdor CG, van Kooyk Y, Alon R. The LFA-1 integrin supports rolling adhesions on ICAM-1 under physiological shear flow in a permissive cellular environment. Journal of immunology. 2000;165:442–452. doi: 10.4049/jimmunol.165.1.442. [DOI] [PubMed] [Google Scholar]
- 14.Sadhu C, Ting HJ, Lipsky B, Hensley K, Garcia-Martinez LF, Simon SI, Staunton DE. CD11c/CD18: novel ligands and a role in delayed-type hypersensitivity. Journal of leukocyte biology. 2007;81:1395–1403. doi: 10.1189/jlb.1106680. [DOI] [PubMed] [Google Scholar]
- 15.Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH, Smith CW, Ballantyne CM. CD11c expression in adipose tissue and blood and its role in diet-induced obesity. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:186–192. doi: 10.1161/ATVBAHA.109.198044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadhu C, Hendrickson L, Dick KO, Potter TG, Staunton DE. Novel Tools for Functional Analysis of CD11c: Activation-Specific, Activation - Independent, and Activating Antibodies. Journal of Immunoassay and Immunochemistry. 2007;29:42–57. doi: 10.1080/15321810701735062. [DOI] [PubMed] [Google Scholar]
- 18.Gower RM, Wu H, Foster GA, Devaraj S, Jialal I, Ballantyne CM, Knowlton AA, Simon SI. CD11c/CD18 Expression Is Upregulated on Blood Monocytes During Hypertriglyceridemia and Enhances Adhesion to Vascular Cell Adhesion Molecule-1. Arteriosclerosis, thrombosis, and vascular biology. 2010;31:160–166. doi: 10.1161/ATVBAHA.110.215434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin F, Saadi W, Rhee SW, Wang SJ, Mittal S, Jeon NL. Generation of dynamic temporal and spatial concentration gradients using microfluidic devices. Lab on a chip. 2004;4:164–167. doi: 10.1039/b313600k. [DOI] [PubMed] [Google Scholar]
- 20.Samady H, Eshtehardi P, McDaniel MC, Suo J, Dhawan SS, Maynard C, Timmins LH, Quyyumi AA, Giddens DP. Coronary Artery Wall Shear Stress Is Associated With Progression and Transformation of Atherosclerotic Plaque and Arterial Remodeling in Patients With Coronary Artery Disease. Circulation. 2011;124:779–788. doi: 10.1161/CIRCULATIONAHA.111.021824. [DOI] [PubMed] [Google Scholar]
- 21.Tsang YT, Neelamegham S, Hu Y, Berg EL, Burns AR, Smith CW, Simon SI. Synergy between L-selectin signaling and chemotactic activation during neutrophil adhesion and transmigration. Journal of immunology. 1997;159:4566–4577. [PubMed] [Google Scholar]
- 22.Simon SI, Chambers JD, Sklar LA. Flow cytometric analysis and modeling of cell-cell adhesive interactions: the neutrophil as a model. The Journal of cell biology. 1990;111:2747–2756. doi: 10.1083/jcb.111.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moghadasian MH, McManus BM, Nguyen LB, Shefer S, Nadji M, Godin DV, Green TJ, Hill J, Yang Y, Scudamore CH, Frohlich JJ. Pathophysiology of apolipoprotein E deficiency in mice: relevance to apo E-related disorders in humans. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:2623–2630. doi: 10.1096/fj.01-0463com. [DOI] [PubMed] [Google Scholar]
- 24.Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 25.Porter JC, Hogg N. Integrin cross talk: activation of lymphocyte function-associated antigen-1 on human T cells alters alpha4beta1- and alpha5beta1-mediated function. The Journal of cell biology. 1997;138:1437–1447. doi: 10.1083/jcb.138.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leitinger B, Hogg N. The involvement of lipid rafts in the regulation of integrin function. J Cell Sci. 2002;115:963–972. doi: 10.1242/jcs.115.5.963. [DOI] [PubMed] [Google Scholar]
- 27.Xu T, Liu W, Luo J, Li C, Ba X, Ampah KK, Wang X, Jiang Y, Zeng X. Lipid Raft is required for PSGL-1 ligation induced HL-60 cell adhesion on ICAM-1. PloS one. 2013;8:e81807. doi: 10.1371/journal.pone.0081807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiely JM, Hu Y, Garcia-Cardena G, Gimbrone MA., Jr Lipid raft localization of cell surface E-selectin is required for ligation-induced activation of phospholipase C gamma. Journal of immunology. 2003;171:3216–3224. doi: 10.4049/jimmunol.171.6.3216. [DOI] [PubMed] [Google Scholar]
- 29.Ting HJ, Stice JP, Schaff UY, Hui DY, Rutledge JC, Knowlton AA, Passerini AG, Simon SI. Triglyceride-rich lipoproteins prime aortic endothelium for an enhanced inflammatory response to tumor necrosis factor-alpha. Circulation research. 2007;100:381–390. doi: 10.1161/01.RES.0000258023.76515.a3. [DOI] [PubMed] [Google Scholar]
- 30.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circulation research. 2000;87:153–159. doi: 10.1161/01.res.87.2.153. [DOI] [PubMed] [Google Scholar]
- 31.Wang YI, Schulze J, Raymond N, Tomita T, Tam K, Simon SI, Passerini AG. Endothelial inflammation correlates with subject triglycerides and waist size after a high-fat meal. Am J Physiol Heart Circ Physiol. 2011;300:H784–791. doi: 10.1152/ajpheart.01036.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyduk SJ, Oh J, Xiao H, Chen M, Cybulsky MI. Paxillin selectively associates with constitutive and chemoattractant-induced high-affinity alpha4beta1 integrins: implications for integrin signaling. Blood. 2004;104:2818–2824. doi: 10.1182/blood-2003-12-4402. [DOI] [PubMed] [Google Scholar]
- 33.Kummer C, Petrich BG, Rose DM, Ginsberg MH. A small molecule that inhibits the interaction of paxillin and alpha 4 integrin inhibits accumulation of mononuclear leukocytes at a site of inflammation. The Journal of biological chemistry. 2010;285:9462–9469. doi: 10.1074/jbc.M109.066993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyun YM, Chung HL, McGrath JL, Waugh RE, Kim M. Activated integrin VLA-4 localizes to the lamellipodia and mediates T cell migration on VCAM-1. Journal of immunology. 2009;183:359–369. doi: 10.4049/jimmunol.0803388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose DM, Liu S, Woodside DG, Han J, Schlaepfer DD, Ginsberg MH. Paxillin binding to the alpha 4 integrin subunit stimulates LFA-1 (integrin alpha L beta 2)-dependent T cell migration by augmenting the activation of focal adhesion kinase/proline-rich tyrosine kinase-2. Journal of immunology. 2003;170:5912–5918. doi: 10.4049/jimmunol.170.12.5912. [DOI] [PubMed] [Google Scholar]
- 36.Sadhu C, Hendrickson L, Dick KO, Potter TG, Staunton DE. Novel tools for functional analysis of CD11c: activation-specific, activation-independent, and activating antibodies. Journal of immunoassay & immunochemistry. 2008;29:42–57. doi: 10.1080/15321810701735062. [DOI] [PubMed] [Google Scholar]
- 37.Willeke T, Schymeinsky J, Prange P, Zahler S, Walzog B. A role for Syk-kinase in the control of the binding cycle of the beta2 integrins (CD11/CD18) in human polymorphonuclear neutrophils. Journal of leukocyte biology. 2003;74:260–269. doi: 10.1189/jlb.0102016. [DOI] [PubMed] [Google Scholar]
- 38.Frommhold D, Mannigel I, Schymeinsky J, Mocsai A, Poeschl J, Walzog B, Sperandio M. Spleen tyrosine kinase Syk is critical for sustained leukocyte adhesion during inflammation in vivo. BMC immunology. 2007;8:31. doi: 10.1186/1471-2172-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldfinger LE, Han J, Kiosses WB, Howe AK, Ginsberg MH. Spatial restriction of alpha4 integrin phosphorylation regulates lamellipodial stability and alpha4beta1-dependent cell migration. The Journal of cell biology. 2003;162:731–741. doi: 10.1083/jcb.200304031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wantha S, Alard JE, Megens RT, van der Does AM, Doring Y, Drechsler M, Pham CT, Wang MW, Wang JM, Gallo RL, von Hundelshausen P, Lindbom L, Hackeng T, Weber C, Soehnlein O. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circulation research. 2013;112:792–801. doi: 10.1161/CIRCRESAHA.112.300666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. Journal of Experimental Medicine. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L, Dai Perrard X, Perrard JL, Yang D, Xiao X, Teng BB, Simon SI, Ballantyne CM, Wu H. Foamy Monocytes Form Early and Contribute to Nascent Atherosclerosis in Mice With Hypercholesterolemia. Arteriosclerosis, thrombosis, and vascular biology. 2015 doi: 10.1161/ATVBAHA.115.305609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller LJ, Bainton DF, Borregaard N, Springer TA. Stimulated mobilization of monocyte Mac-1 and p150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. The Journal of clinical investigation. 1987;80:535–544. doi: 10.1172/JCI113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zwartz G, Chigaev A, Foutz T, Larson RS, Posner R, Sklar LA. Relationship between molecular and cellular dissociation rates for VLA-4/VCAM-1 interaction in the absence of shear stress. Biophysical journal. 2004;86:1243–1252. doi: 10.1016/S0006-3495(04)74198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.March ME, Long EO. beta2 integrin induces TCRzeta-Syk-phospholipase C-gamma phosphorylation and paxillin-dependent granule polarization in human NK cells. Journal of immunology. 2011;186:2998–3005. doi: 10.4049/jimmunol.1002438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andrews RP, Kepley CL, Youssef L, Wilson BS, Oliver JM. Regulation of the very late antigen-4-mediated adhesive activity of normal and nonreleaser basophils: roles for Src, Syk, and phosphatidylinositol 3-kinase. Journal of leukocyte biology. 2001;70:776–782. [PubMed] [Google Scholar]
- 48.Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene. 2001;20:6459–6472. doi: 10.1038/sj.onc.1204786. [DOI] [PubMed] [Google Scholar]
- 49.Baker RG, Hsu CJ, Lee D, Jordan MS, Maltzman JS, Hammer DA, Baumgart T, Koretzky GA. The adapter protein SLP-76 mediates “outside-in” integrin signaling and function in T cells. Molecular and cellular biology. 2009;29:5578–5589. doi: 10.1128/MCB.00283-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arai A, Nosaka Y, Kohsaka H, Miyasaka N, Miura O. CrkL activates integrin-mediated hematopoietic cell adhesion through the guanine nucleotide exchange factor C3G. Blood. 1999;93:3713–3722. [PubMed] [Google Scholar]
- 51.Fernandez RR, Suchard SJS. Syk activation is required for spreading and H2O2 release in adherent human neutrophils. Journal of immunology. 1998;160:5154–5162. [PubMed] [Google Scholar]
- 52.Caron E, Self AJ, Hall A. The GTPase Rap1 controls functional activation of macrophage integrin alphaMbeta2 by LPS and other inflammatory mediators. Current biology : CB. 2000;10:974–978. doi: 10.1016/s0960-9822(00)00641-2. [DOI] [PubMed] [Google Scholar]
- 53.Rullo J, Becker H, Hyduk SJ, Wong JC, Digby G, Arora PD, Cano AP, Hartwig J, McCulloch CA, Cybulsky MI. Actin polymerization stabilizes 4 1 integrin anchors that mediate monocyte adhesion. The Journal of cell biology. 2012;197:115–129. doi: 10.1083/jcb.201107140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller YI, Choi SH, Wiesner P, Bae YS. The SYK side of TLR4: signalling mechanisms in response to LPS and minimally oxidized LDL. British journal of pharmacology. 2012;167:990–999. doi: 10.1111/j.1476-5381.2012.02097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stadtmann A, Germena G, Block H, Boras M, Rossaint J, Sundd P, Lefort C, Fisher CI, Buscher K, Gelschefarth B, Urzainqui A, Gerke V, Ley K, Zarbock A. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. The Journal of experimental medicine. 2013;210:2171–2180. doi: 10.1084/jem.20130664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kahn ML, Koretzky GA. Integrins and ITAMs: more than just good neighbors. Nature immunology. 2006;7:1286–1288. doi: 10.1038/ni1206-1286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.