

Abstract
Herpes simplex virus (HSV) entry and cell-cell fusion require the envelope proteins gD, gH/gL and gB. We propose that receptor-activated conformational changes to gD activate gH/gL, which then triggers gB (the fusogen) into an active form. To study this dynamic process, we have adapted a dual split protein assay originally developed to study the kinetics of human immunodeficiency virus (HIV) mediated fusion. This assay uses a chimera of split forms of renilla luciferase (RL) and green fluorescent protein (GFP). Effector cells are co-transfected with the glycoproteins and one of the split reporters. Receptor-bearing target cells are transfected with the second reporter. Co-culture results in fusion and restoration of RL, which can convert a membrane permeable substrate into a luminescent product, thereby enabling one to monitor initiation and extent of fusion in live cells in real time. Restoration of GFP can also be studied by fluorescence microscopy. Two sets of split reporters have been developed: the original one allows one to measure fusion kinetics over hours whereas the more recent version was designed to enhance the sensitivity of RL activity allowing one to monitor both initiation and rates of fusion in minutes. Here, we provide a detailed, step-by-step protocol for the optimization of the assay (which we call the SLA for split luciferase assay) using the HSV system. We also show several examples of the power of this assay to examine both the initiation and kinetics of cell-cell fusion by wild type forms of gD, gB, gH/gL of both serotypes of HSV as well as the effect of mutations and antibodies that alter the kinetics of fusion. The SLA can be applied to other viral systems that carry out membrane fusion.
Keywords: split luciferase assay, herpes simplex virus, cell-cell fusion, kinetics of fusion
Introduction
Membrane fusion, the disruption of both the inner and outer layers of plasma or endocytic membranes and viral membranes, is a biological process that enveloped viruses use to enter cells and deliver the capsid containing the viral genome for replication. All viral fusion proteins have distinct conformations before, during and after executing membrane fusion. For many enveloped viruses, receptor binding and/or lowered pH activates the prefusion state of the viral fusion protein to a form that allows the fusion peptide or fusion loop(s) to insert into a target membrane, causing both fusion and conversion of the fusion protein into a postfusion form [1]. In addition, these major conformational changes are assumed to provide the energy needed for membrane fusion [1-3].
The earliest step in herpes simplex virus (HSV) infection, virus entry, is mediated by four glycoproteins contained within the viral envelope: gD, gB and a heterodimer gH/gL. Both serotypes of HSV (HSV-1, the oral form and HSV-2, the genital form) contain similar versions of these four proteins. Their importance is highlighted by the fact that each is required for virus entry and each is a major target of virus neutralizing antibodies [4-8]. The route of HSV entry is cell-type dependent and involves fusion between the virus envelope and either the plasma membrane or an endocytic vesicle [9]. Fusion can be pH dependent or independent [10, 11]. A detailed understanding of the molecular interactions regulating HSV entry can provide important insights into the pathogenesis of HSV and reveal steps that could be targeted by vaccines and therapeutics. Furthermore, because gH/gL and gB are conserved in all herpesviruses, a greater understanding of their function would have broad significance to human health.
Due to a lack of methods to directly study virus-cell fusion, most of what we know about virus entry is based on cell-cell fusion as a surrogate model for virus fusion. We have proposed a model for HSV fusion regulation that involves the sequential activation of gD (via receptor binding), gH/gL and, finally, gB [12-14]. Structural biology of gD [15], gB [16] and gH/gL [17], as well as two protein receptors (HVEM and nectin-1) bound to gD [18, 19] have transformed our thinking about how each protein participates in fusion. Biochemical, visual and in vivo evidence lends support for our hypothesis that fusion is the result of a multistep pathway as diagrammed in Figure 1 [13, 20-24]. First, gD binds one of its receptors, e.g. nectin-1 (step 1). As a result, the C-terminal portion of the gD ectodomain undergoes major conformational changes, uncovering the gD core [2, 7, 18, 25, 26]. This activated form of gD interacts with gH/gL (step 2) and transforms it into a positive regulator that helps gB undergo one or more steps to become an active fusogen (step 3) [12].
Figure 1. Illustration of the HSV cell-cell fusionentry pathway.

HSV cell-cell fusion HSV entry requires the four key glycoproteins (gD, gH, gL and gB) and a cellular receptor. The first step involves gD binding to receptor (nectin-1 is shown), which leads to conformational changes in gD. This activates gD and allows it to interact with the heterodimer, gH/gL (step 2). The gD-gH/gL interaction transforms gH/gL into a positive regulator that helps gB become an active fusogen (step 3). All these steps will ultimately lead to fusion between the viral and host cell membranes.
To study the complex series of events leading to HSV glycoprotein-induced fusion, many labs have employed cell-cell fusion assays, which mimic key aspects of virus-cell fusion. In this case, fusion bypasses the attachment step mediated in the virus by glycoprotein C [27, 28] but still requires gD, gH/gL and gB along with one of the gD receptors. When these four proteins are co-expressed in a cell that bears a gD receptor, fusion results in the formation of multinucleated cells, i.e. syncytia. Syncytium formation can be visualized by Giemsa staining of cell nuclei [29, 30] or by immunofluorescence using antibodies to one or more of the glycoproteins [13, 31]. The microscopic techniques require counting of syncytia and nuclei, which is labor intensive [12, 14, 29, 32].
Alternatively, many laboratories have employed a luciferase gene activation assay (Okuma, Nakamura et al. 1999 ) and this assay was subsequently adapted to the HSV system [30, 33]. In this assay, effector cells are transfected with expression plasmids for the four essential HSV glycoproteins as well as T7 RNA polymerase; target cells expressing receptor are transfected with a vector expressing firefly luciferase under the control of the T7 promoter. When these two populations are co-cultivated, fusion and cell content mixing occur, allowing the T7 polymerase to transcribe the reporter gene which is then translated into active luciferase. After a period of time, cells are lysed, luciferase substrate (membrane impermeable) is added and luminescence is quantified. Because the assay relies on de novo transcription and translation of the reporter gene, there is a long lag (hours) before a measurable signal is achieved. The most common time for measuring luminescence is generally 18h post co-cultivation. Regardless of which of these methods is used, fusion levels can be measured only after cells are fixed or lysed. Jackson et al [34] used this assay to examine fusion kinetics at 5, 8 and 18h post co-cultivation using separate lysates for each time point. The major drawback is that this assay does not allow measurements of the earliest events of fusion, particularly initiation.
To study the dynamic process of HSV glycoprotein induced cell fusion, we have adapted a dual split protein assay originally used to study the kinetics of HIV mediated fusion in live cells [35, 36]. The major similarities of this assay to the original firefly luciferase assay are: 1) luciferase activity is measured as a read-out of fusion and 2) the luminescent signal is generated after co-cultivation of effector and target cells. However, in the split luciferase assay (SLA), the reporter plasmids contain chimeras of the N- or C-terminal portions of both RL and GFP under the control of a CMV promoter (Figure 2A). To measure fusion in the HSV system, effector cells (B78, no gD receptor) are co-transfected with gD, gH, gL, gB and one of the split reporter plasmids (DSP1–7 or RLuc81-7) and the target receptor-bearing target cells are transfected with the reporter plasmid encoding the other split reporter (either DSP8–11 or RLuc88-11) (Figure 2B) [35, 37]. In each case, the split RL and GFP are synthesized prior to co-cultivation. Once the two cell sets are mixed, fusion occurs and this restores both RL activity and GFP fluorescence. The interaction of the two halves of GFP is strong enough to stabilize the weak interaction between the RL fragments. Importantly, there are membrane permeable substrates for RL such as coelenterazine (EnduRen) that can be added to live cells and be converted to a luminescent product. Thus, the kinetics of fusion can be measured in intact cells. In addition, GFP fluorescence can also be used for kinetic measurements, either by direct examination of syncytium formation (fluorescence microscopy) or with a plate reader. However, exposure to light in a plate reader would lead to bleaching of GFP and therefore, a loss of signal. We previously showed that the kinetics of fusion measured by luminescence correlate well with the rates calculated by manually counting GFP fluorescent syncytia [38]. The Rluc8 plasmids differ from the original DSP plasmids in that wild type RL is replaced with a variant that contains eight mutations and has a different split point (Figure 2A) [35, 37]. These changes enhance the sensitivity of the assay by 100 fold, thereby allowing one to evaluate both the initiation and kinetics of fusion within minutes after co-culture [35, 37, 38]. We define initiation of fusion as the time when the luciferase signal becomes significant (3:1 signal to background ratio). The SLA is thus superior to previous methods for measuring cell-cell fusion, combining the advantages of a visual readout of fusion along with a method to measure the kinetics of fusion in real time.
Figure 2. Schematic representation of the split luciferase assay and the constructs used.

(A). Constructs. Both renilla luciferase (RL) and green fluorescent protein (GFP) were split into inactive halves. The N-terminal region of RL was paired with the inactive N-terminal region of GFP. The second half of GFP was cloned to the C-terminal fragment of RL. Two generations of reporter genes were generated (DSP and RLuc8) that are different in the split point in RL and the presence of several mutations in RL ( ) that make luciferase brighter. The weak interaction of the RL halves is augmented by split-GFP and ensures a high efficiency in the recovery of RL activity. (B). Assay. In the HSV system, effector cells are transfected with gB, gD, gH, gL and the plasmid encoding one half of either the DSP or Rluc8 gene. Target cells, bearing receptor are transfected with the other half of the DSP or Rluc8 reporter genes. 24 hours post-transfection, EnduRen substrate is added and target and effector cells are mixed, so that upon fusion, the two inactive halves of DSP or Rluc8 can come together to produce fluorescence and luminescence.
Besides surpassing many of the limitations seen with conventional fusion assays, the SLA is extremely sensitive and has facilitated kinetic studies that examine how mutations in HIV and HSV glycoproteins [38-40] affect the overall rate of fusion and the time of initiation (time when fusion begins post co-cultivation). For example, we used both reporter systems to evaluate the effects of mutations of residues of the gB crown and fusion loops on both initiation and overall rates of fusion [38]. Most fusion loop mutants exhibited kinetics consistent with their fusion phenotype seen in an endpoint luciferase assay [23, 41]. Surprisingly, mutations which appeared to have little or no effect on fusion using that assay exhibited a long lag before fusion began as well as a slow rate of fusion (measured using the Rluc8 plasmids) that evolved into faster ongoing rates as measured with the DSP plasmids [38]. Thus, the SLA has allowed us to identify several previously overlooked residues that make significant contributions to fusion loop function. The crown of gB (FR3) is important in the interaction of gB with a cellular molecule [42]. In addition, several rate of virus entry [43, 44] as well as linker insertion mutations [45] map here. We found that mutations in gB FR3 had two distinct kinetic patterns when compared to wild-type gB: “slow” and “fast”. The rapid kinetics were not due to enhanced cell surface expression [32, 38], thereby emphasizing the complex role FR3 plays in fusion and the fact that hyperfusogenic mutations are not solely associated with the cytoplasmic tail of gB [46]. Moreover, the sensitivity of the SLA has allowed us to add an important parameter when characterizing mutants, i.e. initiation of fusion. We were able to determine that the time of initiation of fusion (content mixing) varies from 7 minutes for wild-type gB, to 3 minutes post co-culture for a hyperfusogenic gB mutant, and to 10 minutes for slow, hypofusogenic mutants [38]. Additionally, we found that HSV mutants that map to the gB ectodomain and exhibit a reduction in the rate of entry [43, 44] also exhibit a slower rate of fusion [38]. The initiation time may help explain why some mutants have a slower or more enhanced rate of fusion but it is also important to note that this does not apply to all mutants. For example, some gB mutants have a faster initiation time, but a wild-type rate of fusion [38]. We posit that mutations can influence the formation of fusion sites but can also affect recruitment of cells into a developing syncytium once fusion has begun, hence explaining the difference between initiation time and rate of fusion.
In a nutshell, the dual split luciferase assay represents a rapid, highly-flexible, sensitive and quantitative platform to continuously monitor initial and ongoing cell-cell fusion driven by such examples as the HSV entry glycoproteins.
2. Materials and Methods
2.1 Selection of reporter gene
The two generations of reporter genes, DSP and RLuc8 [35], are both useful depending on the experiment. For analysis of early events (such as fusion initiation), when it is desirable to measure kinetics in minutes, we recommend the RLuc8 reporter genes, where the signal is measurable within minutes. However, when experiments need to be carried out over several hours to study the later stages of fusion such as changes in the kinetics of cell recruitment into a growing syncytium, we suggest using the DSP constructs. The high sensitivity of the RLuc8 gene could, in extreme instances (such as hyperactive mutants), approach or even exceed the reading capacity of the plate reader. This could lead to an undesired “bleed” of signal into the neighboring wells, especially if multiple samples are to be analyzed. Therefore, we do not recommend using the RLuc8 reporter gene to monitor HSV fusion over an extended period of time.
2.2 Optimization of fusion
To analyze the kinetics of fusion and the events that lead to it, it is important to optimize fusion. The concentration of the constructs that contribute to fusion can alter fusion rates. Therefore, it is necessary to determine the concentration of each construct that yields the highest level of fusion. The purpose is firstly, to determine the correlation between quantity of transfected DNA and amount of protein expressed on the cell surface (as measured by cell-based enzyme-linked immunosorbent assay (CELISA)) required to achieve maximum activity and secondly, to determine the rate limiting protein (as measured by SLA). This becomes particularly important when multiple proteins are involved (such as in the HSV system) and for analyzing mutants that affect the rate of fusion.
Titration of plasmids encoding HSV glycoproteins showed a linear correlation between the amount of DNA transfected and the expression of the respective protein on the cell surface: the more DNA transfected, the more protein detected (Figure 3A). However, the absolute rates are the same. By titrating each DNA construct (while maintaining the rest at a constant concentration) we found that the rate limiting protein was the fusion protein (gB). One possibility is that at lower DNA concentrations, most of the gB expressed on the cell surface is inactive. Transfecting increasing amounts of DNA might increase the proportion of active gB. However, current reagents cannot distinguish between active and inactive gB. gD and gH/gL seemed to work in a catalytic manner (Figure 3B) (Figure 3 is reprinted by permission from the American Society for Microbiology, Journal of Virology, 2013 Nov; 87(21): 11332–11345. DOI: 10.1128/JVI.01700-13) [38]. Maximum fusion rates were achieved with a concentration of 125ng of gD, gH, and gL plasmids and a concentration of 375ng for gB.
Figure 3. Titration of glycoproteins.

(A). Cell sSurface expression (CELISA). B78 cells were transfected with varying concentrations of DNA corresponding to one viral glycoprotein (gB, gD, or gH/gL) while the other three were maintained at 125ng along with one of the DSP plasmids. Data were normalized by setting the absorbance value for cells transfected with 125ng of each glycoprotein (filled bars) to be 100%. (B). Rates of fusion obtained from luciferase activity. B78 cells were transfected the same way as for panel A, and fusion was triggered with C10 cells carrying the second DSP half as shown in Figure 2. Rates were determined by taking the luminescence units (normalized by considering the signal at 7h to be 100%) between 3 and 7h and calculating the slope of that line between these time points. Data from 3 independent experiments were averaged; R2 value was equal to or higher than 0.96. (Copyright © American Society for Microbiology, Journal of Virology, 2013 Nov; 87(21): 11332–11345. DOI: 10.1128/JVI.01700-13)
2.3 Length of time course
The choice of reporter genes depends on how rapidly fusion begins and gives a measurable signal. We now use the original DSP plasmids to examine fusion over long periods of time (exemplified for the wild type glycoproteins in Figure 4A) and we use the Rluc8 plasmids to examine fusion over short periods (Figure 4B, C, D) [38, 47]. To precisely determine the initiation time of fusion caused by the wild-type glycoproteins, we used the RLuc8 reporter genes and recorded luminescence readings every minute for 30min (Figure 4C). A significant signal-to-background ratio (at least 3:1) became apparent as soon as 7min after co-culture, allowing us to define this as the time of fusion initiation for wild-type HSV glycoproteins. As examples of how mutations can affect fusion kinetics, we show data for two gB mutants that are affected in their time of fusion initiation (Figure 4D). The first of these is gBLL871AA, a double mutant in the cytoplasmic tail that was originally identified as a virus that is hyperfusogenic in forming syncytia [46]. Using the Rluc8 plasmids, we showed that gBLL871AA initiates fusion faster than wild-type gB (3min for gBLL871AA vs 7min for wild-type gB) [38], recapitulating the hyperfusogenic phenotype in a cell-cell fusion assay [32, 38]. The second mutant, gBQ584A is located in the crown of gB. We characterized it as having a slow rate of fusion and also a delayed initiation of fusion (10min) [38]. Thus, the SLA is more informative about the phenotype of some mutants than a standard endpoint assay. It allows for the evaluation of the impact of changes in the structure of glycoproteins on both the initiation of fusion and continuing fusion steps.
Figure 4. Length of time course.

(A). Long time course. Curves showing raw luminescence values obtained from a representative experiment with cells transfected with the gB, gD, gH/gL and DSP plasmids versus cells transfected with control plasmid, pCAGGS and DSP for a 6 hour time course. (B). Two hour time course. The Rluc8 reporter genes increase the sensitivity of the assay such that fusion can be monitored within the first 2 hours of co-cultivation when cells were transfected with all four viral glycoproteins. (C). Initiation of fusion. In addition to a 2 hour time course, the Rluc8 plasmids also allow signal to be detected as early as 7 minutes for cells transfected with all four viral glycoproteins. (D). Effect of mutations in gB on fusion initiation. Curves show differences in initiation time between wild-type and mutant gBs.
2.4 Split Luciferase Assay Protocol
2.4.1 Day 1
Seed 5 × 104 effector cells/well (B78 cells) on a white 96-well luciferase plate treated for cell culture. Cells are grown in selective DMEM medium (Gibco) containing 5% fetal calf serum (FCS) and 100μg/ml penicillin/streptomycin. If SLA is paired with CELISA to determine surface expression, cells are plated at the same density on regular, transparent 96-well plate treated for cell culture.
Seed target cells (B78-C10 cells stably expressing nectin-1) on a 6-well plate treated for cell culture at 2 × 105 cells/well; cells are maintained in B78 cells medium supplemented with 500μg/ml G418. Cell density should not exceed 80% confluency the following day.
2.4.2 Day 2
When we first developed the SLA, we paired it, head-to-head with visual assays such as Giemsa and immunofluorescence staining to assess the reliability of the results we saw from the SLA. Since visual assays are typically done in a 24-well plate and the SLA in a 96-well plate, we made sure that the cells on both plates were transfected with similar amounts of DNA, for a correct comparison. As such, the transfection mix was prepared in duplicate: one mixture to be applied to cells aimed for visual examination and the other mixture to be distributed over 3 wells in a 96-well plate for SLA. Therefore, all DNA amounts described for the SLA in this report reflect the concentration of each construct that will be distributed over 3 wells in a 96-well plate and not the concentration per well.
2.4.2.1 Transfection of effector cells
Co-transfect B78 (effector) cells with the following plasmids: 125ng each of the gD, gH, gL, and DSP1–7 (or RLuc81-7) plasmids and 375ng of the gB plasmid. To account for background, we include a negative control: B78 cells transfected with 750ng of empty vector, pCAGGS, so that the amount of DNA used for negative control would be the same as the total amount of DNA used in our samples of interest. For each sample, dilute the plasmids in a 250μl final volume of OPTI-MEM® reduced serum media (Life Technologies) containing 4μl of Lipofectamine 2000 (Life Technologies). Incubate the DNA-lipid complexes for 10 minutes at room temperature. Pipette the 250μl DNA-lipid complex over 3 wells (80μl/well) of effector cells. If SLA is paired with CELISA, the transfection mix is prepared in duplicate so that the same mix can be used for cells on the clear 96-well plate.
2.4.2.2 Transfection of target cells
Transfect B78-C10 cells stably expressing nectin-1 (target) cells with 1μg of DSP8–11 (or RLuc88-11) plasmid. Dilute DNA in 1ml final volume of OPTI-MEM® reduced serum media containing 10μl of Lipofectamine 2000. Incubate the DNA-lipid complexes for 10 minutes at room temperature. Add mix to target cells. Allow transfection of both effector and target cells to proceed for 24 hours.
2.4.3 Day 3
2.4.3.1 Preparing effector cells
Remove transfection mix by suction. Add 40μl fusion medium/well (DMEM without phenol red (Gibco) supplemented with 5% FCS and 50mM HEPES) containing EnduRen substrate (Promega), diluted 1:1000 based on an 80μl final volume in each well after co-cultivation. Incubate for 1 hour at 37ºC.
2.4.3.2 Preparing target cells
Remove transfection mix. Detach cells with 1ml/well pre-warmed versene; wash well with 1ml fusion medium to collect all cells. Transfer cells to a Corning conical tube. Spin cells down for 10 minutes at 1000 rpm/4ºC. Remove supernatant. Resuspend pelleted cells in 500μl of fusion medium. Transfer 40μl target cells over effector cells.
2.4.3.3 Luminescence readings
After the co-cultivation step, place plate immediately (no lid) into a BioTek Synergy 2 plate reader set at 37ºC. This will be “time 0”. If DSP reporter genes are used, readings will be recorded every hour. Therefore, the plate should be returned to a 37ºC incubator until the next read.
When RLuc8 reporters are present, the plate is left in the plate reader set at 37°C and readings are taken continuously either every 1 minute or every 5 minutes. The presence of HEPES helps maintain a constant pH and C02 level. However, the “open plate” configuration can lead to uneven evaporation and therefore uneven signal in a longer time frame. For this reason, we do not recommend continuous readings for more than 2 hours.
2.4.3.4 Measuring cell surface expression (CELISA)
Replace transfection mix with 50μl blocking solution (3% bovine serum albumin diluted in PBS++ (1x Gibco Dulbecco’s PBS containing Ca2+ and Mg2+). Block for 30 minutes. Replace blocking solution with primary antibody (R7 polyclonal antibody IgG for gD; cocktail of monoclonal antibodies (A22, SS10, and SS67) for gB; R137 Pab for gH/gL) diluted 1:1000 in 3% BSA-PBS++. Incubate 1 hour at room temperature. Rinse cells once with PBS++. Fix cells with 3% paraformaldehyde for 30 minutes at room temperature. Remove paraformaldehyde and rinse cells three times with PBS++. Add secondary goat anti-rabbit secondary antibody coupled to horseradish peroxidase for gD and gH/gL (Cell Signaling Technology), or goat anti-mouse antibody coupled to horseradish peroxidase for gB (Cell Signaling Technology) for 1 hour at room temperature; all secondary antibodies are diluted 1:100 in 3% BSA-PBS++. Rinse cells three times with PBS++ and once with 20mM citrate buffer (pH 4.5). Add 100μl ABTS (2,2'-azinobis [3-ethylbenzothiazoline-6-sulfonic acid]) peroxidase substrate (Moss, Inc.). Measure absorbance at 405nm using a plate reader.
2.5 Applications
2.5.1 Synchronizing fusion with soluble protein
Previous studies done in the lab have shown that a soluble, truncated form of gD lacking the transmembrane region and cytoplasmic tail (gD306t) can be used to trigger fusion [12, 13, 24]. This not only allows for synchronization of fusion, but also enables the study of fusion inhibitors as well. By using soluble gD as a trigger, we can analyze the effect of an antibody to a specific glycoprotein for example, by allowing enough time for the antibody to bind before early fusion events are initiated.
A direct comparison of transfected and soluble gD fusion curves shows that in both cases the absolute rates of fusion are identical (Figure 5 A). B78 cells were transfected with gB, gH and gL plasmids as described and 250μg/ml soluble gD [12, 24] was added the next day, at the co-cultivation step. This was run simultaneously with a sample that was transfected with gD. Although the raw values for fusion with soluble gD are somewhat lower than for transfected plasmid (not shown), the assay is sensitive and allows for a titration of gD306t. The rate of fusion is dose-dependent up to a concentration of 30μg/ml, which was as good as 500μg/ml at triggering fusion (Figure 5B). This highlights the catalytic manner in which gD was found to trigger fusion by gB ([38] and Figure 3B). Plotting the data as percent of fusion at a specific time over a range of concentrations (Figure 5C) can be used to determine the concentration that can yield 50% of the maximum fusion level. This way of analyzing data can be particularly useful when the effect of inhibitory compounds on fusion/function of glycoproteins is examined.
Figure 5. Use of soluble gD(gD306t) to trigger fusion between receptor bearing target cells and effector cells bearing gB and gH/gL.

(A) Absolute fusion rates are similar when gD is expressed from a plasmid (A) or delivered as a soluble protein (B). (BC).Dose-dependence of fusion on gD306t concentration. Curves show correlation between fusion levels and different concentrations of gD306t. Concentration ranging between 500 μg/ml and 30μg/ml gave the same signal output as 250μg/ml. (CD).Determining gD306t concentration that yields 50% of maximum fusion level. Data from (C) are plotted as levels of fusion 6 hours post co-cultivation.
2.5.2 Blocking of fusion with inhibitors
An important step in the analysis of function of glycoproteins (either wild-type or mutant) is to determine the effect of various compounds (antibodies, peptides, chemicals) on fusion [12, 14, 48, 49]. Here, we show as a proof of principle the effect of three gD antibodies on fusion: MC1, MC5 and DL11. MC5 and DL11 are neutralizing antibodies [7, 8] whereas MC1 is not [7]. Using the firefly luciferase assay [30, 33], we found that MC1 had no effect while MC5 and DL11 blocked cell-cell fusion (Fig. 6A) confirming their phenotypes in virus-cell fusion. Using the SLA, we found that MC5 and DL11 completely blocked the initiation of fusion whereas MC1 had no effect on either initiation or kinetics of fusion (Fig. 6B) Antibodies, peptides and other inhibitory molecules that target different glycoproteins can be assessed in this manner for their ability to affect initiation and the rate of fusion [48].
Figure 6. Blocking of fusion with antibody.
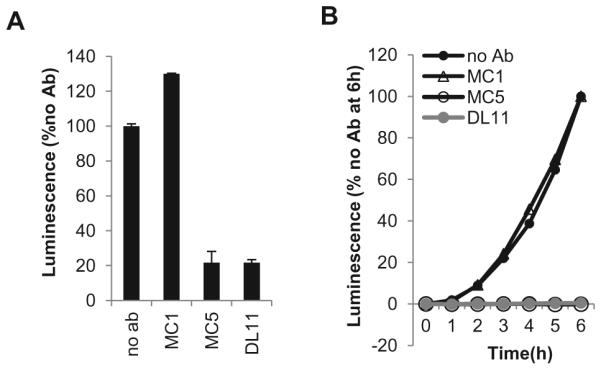
(A). Firefly Luciferase Assay. One population of CHO-K1 cells was transfected with the gD, gH, gL, gB and T7 polymerase plasmids. The other population was transfected with receptor and luciferase plasmids. 6 hours post-transfection the two populations were co-cultivated and 100μg/ml of antibody (MC1, MC5 or DL11) was added. 18 hours post co-cultivation, cells were lysed and luminescence measured. Values were normalized to no antibody control. (B). Split Luciferase Assay. B78 cells were transfected with gB, gH, gL, and one of the DSP plasmids. C10 cells were transfected with the other DSP plasmid. An hour prior to co-cultivation, 30μg/ml soluble gD was pre-incubated with 30μg/ml of either MC1, MC5 or DL11. The gD-Ab mixture was then added at the co-cultivation step. Fusion was monitored over 6 hours. Data were normalized to no antibody control.
2.6 Troubleshooting
Low or no signal during fusion assay is usually linked to suboptimal transfection protocol. Here are suggestions for potential problematic steps.
2.6.1 Adherence of cells to plate
If cells are suspected of detaching, especially during CELISA, culture plates can be treated with gelatin diluted in sterile distilled water. Add 100μl of 0.2% gelatin per well for 1 hour at 37ºC. Aspirate and air dry the plate in a hood (to maintain sterility) until no trace of solution is seen. Then, plate the cells as required.
2.6.2 Cell confluency at the time of transfection (for both target and effector cells)
Cells that have been seeded too heavily or too lightly are transfected with low efficiency. Refer to the transfection reagent’s recommendations to determine cell density. The 96-well white plate used for SLA does not have a clear bottom. We suggest plating effector cells in a clear bottom 96-well replica plate, so that confluency can be checked under a microscope before transfection is performed.
2.6.3 DNA or soluble protein concentration
Varying the concentration of DNA used for transfection can affect transfection efficiency. Perform a titration to determine the concentration of DNA that yields the best signal. If possible, check for the expression of the protein on cell surface as well (by CELISA). Varying the concentration of soluble protein used to trigger fusion can also affect signal. A titration of exogenously added proteins is strongly recommended.
2.6.4 Number of target cells used for co-cultivation
In our hands, 1/12 of a 6 well of confluent target cells (after 24h post transfection) for each well of effector cells worked best. This ratio may be different for other cell lines. Signal production may be improved by increasing the number of target cells used for co-cultivation.
2.6.5 Substrate
EnduRen substrate by Promega comes as a dry pellet that is to be reconstituted in dimethylsulfoxide (DMSO). We found that reconstitution was more efficient when fresh DMSO was used. Once reconstituted, substrate should be aliquoted as single-use aliquots; repeated cycles of freeze-thawing can lead to degradation and therefore a decrease in sensitivity. The substrate is light sensitive and should be added to cells as rapidly as possible and away from bright light. Substrate also precipitates at lower temperatures. Therefore, dilutions should be done in warmed medium and the plate reader should be pre-set to 37ºC before the experiment begins.
2.6.6 Plate reader settings
These will largely vary depending on the machine used. Check the instruction manual for the correct selection of filters and sensitivity. For a BioTek Synergy 2 reader, we used sensitivity set at 150.
2.7 Data analysis
A negative control (where fusion is not expected) is always included to account for background noise. Data can be normalized, after background subtraction, to the last reading of the time course. The linear portion of the curves is used to calculate the rate of fusion. A slope with a R2 value greater than 0.96 is considered a good fit.
Highlights.
The split luciferase assay (SLA) has been adapted from its use to measure kinetics of HIV envelope glycoprotein-mediated fusion to study HSV glycoprotein mediated cell-cell fusion in real time.
The time of fusion initiation and kinetics of fusion can be measured due to the sensitivity of the split reporter system.
The assay offers more information about the phenotypes of mutant forms of HSV glycoproteins than an endpoint luciferase assay.
HSV cell-cell fusion can be triggered with soluble gD and inhibited with neutralizing antibodies.
Acknowledgments
This work was supported by NIH grants R3718289 to GHC, R01AI056045 to RJE and AI076231 to RJE. ZM was supported by a contract research fund from the Ministry of Education, Culture, Sports, Science and Technology for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID).
3. Appendix
3.1 Equipment
A plate reader with temperature control (we used a BioTek reader) is necessary to measure fusion. Temperature control is especially important when using the Rluc8 plasmids.
3.2 Supplies
Standard molecular biology reagents are needed for tissue culture and transfection. A 96-well white plate (Corning catalogue no. 3917) is used for the effector cells in SLA to facilitate measuring luminescence. Clear tissue culture plates are used for checking cell density of effector cells and for CELISA (96-well, Corning catalogue no. 3595) and culturing target cells (6-well, Corning catalogue no. 3516). Plasmids for the DSP and Rluc8 assays are available upon request.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15(7):690–8. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenberg RJ, et al. Herpes virus fusion and entry: a story with many characters. Viruses. 2012;4(5):800–32. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heldwein EE. Entry of herpesviruses into cells: more than one way to pull the trigger. Structure. 2009;17(2):147–9. doi: 10.1016/j.str.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Bender FC, et al. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 2007;81(8):3827–3841. doi: 10.1128/JVI.02710-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gompels UA, et al. Characterization and sequence analyses of antibody-selected antigenic variants of herpes simplex virus show a conformationally complex epitope on glycoprotein H. J Virol. 1991;65(5):2393–401. doi: 10.1128/jvi.65.5.2393-2401.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gompels UA, Minson AC. Antigenic properties and cellular localization of herpes simplex virus glycoprotein H synthesized in a mammalian cell expression system. J. Virol. 1989;63(11):4744–4755. doi: 10.1128/jvi.63.11.4744-4755.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazear E, et al. Antibody-induced conformational changes in herpes simplex virus glycoprotein gD reveal new targets for virus neutralization. J Virol. 2012;86(3):1563–76. doi: 10.1128/JVI.06480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitbeck JC, et al. The major neutralizing antigenic site on herpes simplex virus glycoprotein D overlaps a receptor-binding domain. J. Virol. 1999;73:9879–9890. doi: 10.1128/jvi.73.12.9879-9890.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heldwein EE, Krummenacher C. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci. 2008;65(11):1653–68. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milne RS, et al. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol. 2005;79(11):6655–63. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicola AV, McEvoy AM, Straus SE. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol. 2003;77(9):5324–32. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atanasiu D, et al. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol. 2010;84(23):12292–9. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atanasiu D, et al. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A. 2007;104(47):18718–23. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atanasiu D, et al. Bimolecular complementation defines functional regions of Herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol. 2010;84(8):3825–34. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krummenacher C, et al. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. Embo J. 2005;24(23):4144–53. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heldwein EE, et al. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313(5784):217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 17.Chowdary TK, et al. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol. 2010;17(7):882–8. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carfi A, et al. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Molec. Cell. 2001;8(1):169–179. doi: 10.1016/s1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- 19.Di Giovine P, et al. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 2011;7(9):e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geraghty RJ, Jogger CR, Spear PG. Cellular expression of alphaherpesvirus gD interferes with entry of homologous and heterologous alphaherpesviruses by blocking access to a shared gD receptor. Virology. 2000;268(1):147–158. doi: 10.1006/viro.1999.0157. [DOI] [PubMed] [Google Scholar]
- 21.Avitabile E, Forghieri C, Campadelli-Fiume G. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J Virol. 2007;81(20):11532–7. doi: 10.1128/JVI.01343-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Avitabile E, Forghieri C, Campadelli-Fiume G. Cross talking among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J Virol. 2009 doi: 10.1128/JVI.01287-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hannah BP, et al. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J Virol. 2009;83(13):6825–36. doi: 10.1128/JVI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atanasiu D, et al. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. MBio. 2013;4(2) doi: 10.1128/mBio.00046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krummenacher C, et al. Entry of herpesviruses into cells: the enigma variations. In: Pohlmann S, Simmons G, editors. Viral Entry into Host Cells. Lanes Bioscience; 2007. [Google Scholar]
- 26.Lazear E, et al. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J Virol. 2008;82(2):700–9. doi: 10.1128/JVI.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laquerre S, et al. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J Virol. 1998;72(7):6119–30. doi: 10.1128/jvi.72.7.6119-6130.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tal-Singer R, et al. Interaction of herpes simplex virus glycoprotein gC with mammalian cell surface molecules. J Virol. 1995;69(7):4471–83. doi: 10.1128/jvi.69.7.4471-4483.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Browne H, Bruun B, Minson T. Plasma membrane requirements for cell fusion induced by herpes simplex virus type 1 glycoproteins gB, gD, gH and gL. J Gen Virol. 2001;82:1419–22. doi: 10.1099/0022-1317-82-6-1419. Pt 6. [DOI] [PubMed] [Google Scholar]
- 30.Pertel PE, et al. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology. 2001;279(1):313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 31.Muggeridge MI. Characterization of cell-cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH and gL in transfected cells. J Gen Virol. 2000;81:2017–27. doi: 10.1099/0022-1317-81-8-2017. Pt 8. [DOI] [PubMed] [Google Scholar]
- 32.Silverman JL, Heldwein EE. Mutations in the cytoplasmic tail of herpes simplex virus 1 gH reduce the fusogenicity of gB in transfected cells. J Virol. 2013;87(18):10139–47. doi: 10.1128/JVI.01760-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okuma K, et al. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology. 1999;254(2):235–44. doi: 10.1006/viro.1998.9530. [DOI] [PubMed] [Google Scholar]
- 34.Jackson JO, et al. Insertion mutations in herpes simplex virus 1 glycoprotein H reduce cell surface expression, slow the rate of cell fusion, or abrogate functions in cell fusion and viral entry. J Virol. 2010;84(4):2038–46. doi: 10.1128/JVI.02215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishikawa H, et al. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel. 2012 doi: 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 36.Kondo N, et al. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J Biol Chem. 2010;285(19):14681–8. doi: 10.1074/jbc.M109.067090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loening AM, Fenn TD, Gambhir SS. Crystal structures of the luciferase and green fluorescent protein from Renilla reniformis. J Mol Biol. 2007;374(4):1017–28. doi: 10.1016/j.jmb.2007.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atanasiu D, et al. Dual Split Protein-Based Fusion Assay Reveals that Mutations to Herpes Simplex Virus (HSV) Glycoprotein gB Alter the Kinetics of Cell-Cell Fusion Induced by HSV Entry Glycoproteins. J Virol. 2013;87(21):11332–45. doi: 10.1128/JVI.01700-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long Y, et al. Conserved arginine residue in the membrane-spanning domain of HIV-1 gp41 is required for efficient membrane fusion. Protein Cell. 2011;2(5):369–76. doi: 10.1007/s13238-011-1051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, et al. Co-expression of foreign proteins tethered to HIV-1 envelope glycoprotein on the cell surface by introducing an intervening second membrane-spanning domain. PLoS One. 2014;9(5):e96790. doi: 10.1371/journal.pone.0096790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hannah BP, et al. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J Virol. 2007;81(9):4858–65. doi: 10.1128/JVI.02755-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bender FC, et al. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J Virol. 2005;79(18):11588–97. doi: 10.1128/JVI.79.18.11588-11597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bzik DJ, et al. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: mutations affecting rate of virus entry and cell fusion. Virology. 1984;137(1):185–90. doi: 10.1016/0042-6822(84)90022-9. [DOI] [PubMed] [Google Scholar]
- 44.Highlander SL, et al. Identification of mar mutations in herpes simplex virus type 1 glycoprotein B which alter antigenic structure and function in virus penetration. J Virol. 1989;63(2):730–8. doi: 10.1128/jvi.63.2.730-738.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin E, Spear PG. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc Natl Acad Sci U S A. 2007;104(32):13140–5. doi: 10.1073/pnas.0705926104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beitia Ortiz de Zarate I, et al. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell-cell fusion. J Virol. 2007;81(24):13889–903. doi: 10.1128/JVI.01231-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallagher JR, et al. Functional fluorescent protein insertions in herpes simplex virus gB report on gB conformation before and after execution of membrane fusion. PLoS Pathog. 2014;10(9):e1004373. doi: 10.1371/journal.ppat.1004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toda T, et al. Dynamic appearance of antigenic epitopes effective for viral neutralization during membrane fusion initiated by interactions between HIV-1 envelope proteins and CD4/CXCR4. Immunobiology. 2012;217(9):864–72. doi: 10.1016/j.imbio.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 49.Fuller AO, Spear PG. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc Natl Acad Sci U S A. 1987;84(15):5454–8. doi: 10.1073/pnas.84.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]