

Abstract
Many inhibitory interneurones in the hippocampus release the neuropeptide somatostatin (SST) which inhibits neuronal excitability through Gi/Go-coupled receptors. To investigate the signaling pathways underlying the SST inhibition of neuronal excitability in the hippocampus, we performed perforated patch-clamp recordings from CA1 pyramidal neurones in acute brain slices from P14-P18 mice. Bath application of 1 μM SST reversibly reduces the frequency of action potential firing in response to depolarising current steps, and is associated with neuronal hyperpolarisation and a reduction in membrane resistance. This effect is mediated by potassium channels with KCNK-like pharmacology. In addition, in slices that have been cultured in vitro for seven days or more, SST also produces a hyperpolarisation independent reduction in action potential firing, which can be also observed in acute slices when the Ser/Thr protein phosphatases PP2A and PP4 are inhibited selectively with fostriecin. This hyperpolarisation independent effect of SST appears to be mediated by G-protein activated inwardly rectifying K+ (GIRK) channels. Knockdown of protein phosphatase 5, by Cre recombinase mediated deletion of the floxed Ppp5c gene, blocks the hyperpolarisation independent effect of SST, and reduces the hyperpolarisation dependent effect in a manner consistent with increased SST receptor desensitisation. Thus, reversible protein phosphorylation provides a mechanism to enhance or diminish the inhibitory effect of SST, which could allow system level regulation of circuit excitability in the hippocampus.
Keywords: Somatostatin, neuronal excitability, protein phosphatases, hippocampus, K+ channels
1. Introduction
Somatostatin (SST) is an endogenous peptide that is produced in many tissues in the body. In the central nervous system SST is released from a subpopulation of GABAergic interneurones that play an important role in the inhibition of neuronal excitability and regulation of network activity (Royer et al., 2012; Lovett-Barron et al., 2014; Katona et al., 2014). SST also plays a role in modulating synaptic transmission through a reduction in presynaptic glutamate release (Tallent & Siggins, 1997), as well as reducing epileptiform bursting and seizure activity (Tallent & Siggins, 1999; Qiu et al., 2008). Reductions in SST interneurones are associated with aging and neurodegenerative diseases (Koh et al., 2014; Martel et al., 2014), so understanding SST signaling could help in ameliorating their effects.
All five SST receptor genes encode proteins that signal through pertussis toxin sensitive Gi/Go proteins (Patel & Srikant, 1997), which typically inhibit adenylyl cyclase activity. While SST receptor activation inhibits adenylyl cyclase activity in neurones (Schettini et al., 1989, Raynor & Reisine, 1992), many of the effects of other Gi/Go coupled neurotransmitters on excitability persist in the presence of excess exogenous cyclic AMP (Nicoll et al. 1990), implicating other signaling pathways in their actions. Since Gi/Go signaling often antagonizes Gs and Gq signaling through protein kinases, Armstrong & White (1992) postulated that SST might signal through protein phosphatases, which are known to regulate the trafficking and activity of many ion channels (Herzig & Neumann, 2000). Therefore it is of interest to investigate whether protein phosphatases may be involved in mediating or modulating the effect of SST in neurones.
In the hippocampus, which shows a high level of SST binding, the oriens-lacunosum moleculare and oriens-bistratified interneurones release SST which acts on SST receptors found on the soma and dendrites of CA1 pyramidal neurones (Schulz et al., 2000; Selmer et al., 2000, Weckbecker et al., 2003, Viollet et al., 2008). In the rodent hippocampus SST hyperpolarises pyramidal neurones and reduces action potential firing (Pittman & Siggins, 1981, Moore et al., 1988). Although SST has been reported to inhibit N-type voltage gated Ca2+ channels (Ishibashi & Akaike, 1995; Baratta et al., 2002), previous studies of SST action on neurones have focused on K+ channel regulation. In CA1 pyramidal neurones, application of SST has been shown to increase the amplitude of the M-current (Moore et al., 1988; Schweitzer et al., 1998; Qiu et al., 2008), but it is thought that the SST induced hyperpolarisation is, at least in part, mediated by voltage-independent K+ channels (Schweitzer et al., 1998). In other brain regions SST receptors have also been shown to activate inward rectifying K+ channels (Inuoe et al., 1988; Sun et al., 2002; Meis et al., 2005); however, it is still unclear whether these channels are activated by SST in the hippocampus (Moore et al., 1988; Schweitzer et al., 1998).
To study the SST signaling pathways we have used perforated patch-clamp recordings which limit the disruption to the intracellular composition that occurs with whole-cell recording. Here we show that SST induces a hyperpolarisation dependent reduction in action potential firing through KCNK-like channel activation, as well as a hyperpolarisation independent effect, which appears to be mediated by G-protein activated inwardly rectifying K+ (GIRK) channels. The expression of both mechanisms of SST action are regulated by Ser/Thr protein phosphatase activity.
2. Methods
2.1 Preparation of acute slices
Experiments were performed after approval by the NIEHS Animal Care and Use Committee. Animals were sacrificed by an overdose of isoflurane anaesthesia (500ml/m3) followed by decapitation. Acute coronal hippocampal slices (250 μm) were prepared from postnatal day 14–18 C57Bl/6 mice obtained from Charles River. The brain was removed and placed in an ice-cold high sucrose solution composed of (in mM); sucrose (240), KCl (2), NaHCO3 (26), NaH2PO4 (1.25), glucose (10), MgCl2 (3) and CaCl2 (1) saturated with 95% O2 and 5% CO2. Hippocampal slices were cut using a vibroslicer and placed into a holding chamber of oxygenated artificial cerebrospinal fluid (aCSF), and allowed to recover for at least 1 hour at room temperature. Artificial cerebrospinal fluid was composed of (in mM); NaCl (124), NaHCO3 (26), KCl (2.5), NaH2PO4 (1.25), MgCl2 (2), D-glucose (17) and CaCl2 (2).
2.2 Preparation of cultured slices
Coronal hippocampal slices (250 μm) were prepared from postnatal day 7–9 C57Bl/6 mice with a floxed Ppp5c gene (see supplementary fig. 2). The animals were anesthetized, decapitated and the brain was removed and placed in an ice-cold MEM-based cutting solution (Invitrogen) with the addition of (in mM); NaHCO3 (26.2), MgCl2 (3), HEPES (25), glucose (10), Tris (10). Coronal slices were cut using a vibroslicer and the hippocampus dissected out. The hippocampal slices were placed on Corning transwell permeable supports and bathed from below with a 2:1 mixture of basal medium Eagle (Sigma) and Earle’s balanced salts solution (Sigma) supplemented with (in mM) NaCl (20), NaHCO3 (5), CaCl2 (0.2), MgSO4 (1.7), glucose (48), HEPES (26.7), ascorbic acid (0.5), 5% horse serum (Invitrogen), 10ml/l penicillin-streptomycin (Invitrogen), and 1.32 mg/ml insulin (Sigma). The cells were cultured at 34 °C and the culturing solution changed every 2–3 days. On the day following dissection the slices were infected with AAV-hsyn-iCRE-tdTomato to induce Cre mediated deletion of the PP5 gene in neurones from the floxed mice. The virus was mixed 1:1 with 10% sucrose solution, and 0.5 μl of solution was injected onto each slice. Slices were maintained in vitro for 7–12 days before recordings were made.
2.3 Perforated patch recordings
For perforated patch-clamp recordings slices were held in a recording chamber at room temperature (~21 °C) and perfused at 2 ml/min with oxygenated aCS F. Glass recording pipettes with a resistance of 4–7 MΩ had the tip filled with regular whole-cell solution composed of (in mM) K-gluconate (120), KCl (10), HEPES (40), EGTA (0.2), Na2-ATP (2), Na-GTP (0.3), and MgCl2 (3), and were then backfilled with whole-cell solution containing 0.2 mM nystatin. The preparation was visualized using a Zeiss Examiner A1 microscope and a 40x/1.0 differential interference contrast objective. Pyramidal cells in the CA1 subfield of the hippocampus were identified visually. Recordings were made using an Axopatch 200B amplifier (Axon instruments) and Clampex 10.2 software for data acquisition and Clampfit 10.2 for re-analysis. Responses were digitized at 20 kHz and filtered at 2 kHz. Cells were used if their resting membrane potential was more negative than −50 mV, average RMP was −59.4 ± 0.4 mV (125 cells). The series resistance was compensated by 95%. For current-clamp experiments current steps were applied both with the cells at their resting membrane potentials and also with a slow constant current injection that was adjusted throughout the recording to effectively clamp the cells membrane potential at −70 mV. Current steps of 500 ms duration where given in 20 pA increments from −100 pA to +240 pA at a frequency of 1 Hz. The hyperpolarising current steps were used to measure the input resistance throughout the experiment. To study the SST induced current, cells were voltage-clamped and 500 ms duration ramps from −20 mV to −140 mV given in the presence of 1 μM tetrodotoxin.
2.4 Analysis
Where time course data and example traces are shown we have used the lowest current step that produced at least 5 action potentials during baseline. Values plotted are the mean number of action potentials fired with the error bars giving the standard error of the mean. Values in the text are the mean percentage change in action potential firing at the end of SST application relative to the last point of baseline ± standard error of the mean. For statistical analysis we compared the difference in firing between the end of the SST application to the last point of baseline and where appropriate the percentage change was compared to interleaved control groups. Prism software was used to perform paired/unpaired Students t-tests and ANOVAs, followed by Bonferroni post hoc tests, as appropriate. Sample size (n) is the number of animals used.
2.5 Compounds
Somatostatin, barium chloride, bupivacaine hydrochloride, quinidine and linopirdine were obtained from SigmaAldrich. Fostriecin and UCL 1684 ditrifluoroacetate hydrate were obtained from Santa Cruz Biotechnology. Tertiapin Q, paxilline, forskolin and IMBX were from R&D systems. Primaquine diphosphate and E4031 were from Abcam. All compounds were stored as frozen stock solutions and added to the aCSF for bath perfusion, except for bupivacaine and quinidine which were dissolved directly into the aCSF.
3. Results
3.1 SST produces a hyperpolarisation dependent reduction in action potential firing
When CA1 pyramidal cells are current-clamped at their resting membrane potential through nystatin-perforated patches, short, 500 ms, depolarising steps of current injection elicit spiking (Fig. 1A). Larger currents elicit more spikes until the response plateaus at 30–40 spikes/s (Fig. 1B). Bath application of 1 μM SST for 10 min rapidly inhibits spiking in response to current injection (Fig. 1A). When the number of action potentials fired by each current step is plotted against the current amplitude, SST application shifted the curve to the right (n=6, Fig. 1B), indicating a reduction in action potential firing at the lower current steps. The higher frequency firing elicited by larger current steps was unaffected by SST application (Fig. 1B). The smallest current step that fired at least five action potentials during the baseline period for each experiment was analysed, and the SST group was compared to a control group treated with vehicle, over time. SST application resulted in a reversible 85 ± 9 % reduction in action potential firing over time (2-way repeated measures ANOVA with Bonferroni post hoc p<0.01, n=6, Fig. 1C), while over the same time period there was no change in control recordings (4 ± 7 %, Bonferroni post hoc p>0.05, n=6).
Figure 1. Somatostatin induces a hyperpolarisation dependent reduction in action potential firing in CA1 pyramidal cells.

Action potential firing induced by current steps, applied to cells at their RMP, is reversibly reduced by a 10 min application of 1 μM SST. A) Example traces showing the effect of SST on the lowest current step that fires at least 5 action potentials during the baseline period and on the −100 pA hyperpolarising step. B) The mean number of action potentials fired at each current step before and after SST application (n=6). C) Pooled time course data giving the mean number of action potentials fired over time (using the lowest current step that gives at least 5 action potentials during baseline), with SST application (n=6), compared to control recordings with no drug (n=6). SST induced a hyperpolarisation of the membrane potential (D) and a reduction in membrane resistance (E), compared to control recordings. The SST induced effect on action potential firing is blocked when current steps are applied to cells where the resting membrane potential is maintained at −70 mV (F, G & H; n=6). F) Example traces G) mean number of action potentials fired at each current step before and after SST application and H) pooled time course data of the SST effect compared to control recordings.
The reduction in action potential firing induced by SST is associated with a significant hyperpolarisation of ~5 mV of the RMP (2-way repeated measures ANOVA with Bonferroni post hoc p<0.01, Fig. 1D), while there was no change in the RMP in control recordings (Bonferroni post hoc, p>0.05). SST also induced a significant reduction of 21 ± 2 % in the membrane resistance (2-way repeated measures ANOVA with Bonferroni post hoc p<0.01, Fig 1A, E), while control recordings showed a non-significant increase (7 ± 3 %) in membrane resistance over the same period (Bonferroni post hoc p>0.05). When current steps were applied to cells where a slow constant current was injected to maintain the resting membrane potential at −70 mV, SST failed to reduce action potential firing (Fig. 1F, G), confirming that the reduction in firing in response to SST is dependent solely on neuronal hyperpolarisation. Indeed, following a 10 min application of 1 μM SST there was no significant change over time in action potential firing from cells with their RMP maintained at −70 mV (6 ± 6 %, n=6) when compared to control recordings where no SST was applied (6 ± 8 %, n=6; 2-way repeated ANOVA p>0.05, Fig. 2H).
Figure 2. The SST effect appears to be mediated by a KCNK-like (leak) K+ channel.

A) The SST induced reduction in action potential firing (n=15) is blocked by 0.5 mM BaCl2 (n=5), but not by inhibitors of the Kv7/M-current channels (10 μM linopirdine, n=8), ether-a-go-go-related K+ channels (30 μM E4031, n=7), G-protein activated inwardly rectifying K+ channels (200 nM tertiapin Q, n=8), small conductance Ca2+-activated K+ channels (100 nM UCL-1684, n=6), or the large conductance Ca2+-activated K+ channels (10 μM paxilline, n=8). B) Example recording showing the effect of SST on the current induced by a voltage ramp from −140 mV to −20 mV (see inset) with the SST induced current (SST minus Baseline) shown in black. C) Pooled data showing that the SST induced current (n=6) is blocked in the presence of either 200 μM bupivacaine (n=5) or 100 μM quinidine (n=6), blockers of the KCNK-like K+ channels. This result is summarised in A) showing the SST induced current is reduced at typical resting membrane potentials (−60 to −65 mV) by both bupivacaine and quinidine.
3.2 The hyperpolarisation dependent effect of SST is mediated by KCNK-like channels
The neuronal hyperpolarisation and reduction in input resistance induced by SST suggests that SST may be activating a K+ channel; to identify the ion channels involved we compared the effect of SST in the presence of various K+ channels blockers. The reduction of 84 ± 8% in action potential firing by SST (n=15) was prevented by extracellular application of 0.5 mM barium chloride (3 ± 8%, n=5, 1-way ANOVA with Bonferroni post hoc p<0.01, Fig. 2A), which blocks many potassium channels. In contrast, more selective inhibitors of the Kv7/M-current channels (10 μM linopirdine, n=8), ether-a-go-go-related K+ channels (30 μM E4031, n=7), G-protein activated inwardly rectifying K+ channels (200 nM tertiapin Q, n=8), small conductance Ca2+-activated K+ channels (100 nM UCL-1684, n=6), or the large conductance Ca2+-activated K+ channels (10 μM paxilline, n=8) failed to significantly reduce the hyperpolarisation dependent effect of SST application (Bonferroni post hocs p>0.05) (Fig. 2A).
Since the effect of SST is dependent on neuronal hyperpolarisation we then went on to investigate whether activation of the leak potassium (KCNK) channels, which regulate membrane potential, was mediating the SST effect. KCNK channel blockers alone significantly disrupt the resting membrane potential and action potential firing, implying a the high level of tonic activity of these channels, so we employed an alternative approach, using voltage ramps, to assess the effect of KCNK channel blockers on the SST induced current. Application of SST increased the current seen under voltage-clamp in response to voltage ramps from −140 to −20 mV. Subtracting the baseline current from the current in the presence of SST revealed a SST-induced current with a reversal potential of −95 mV (Fig. 2B, C). The SST induced outward current of 11 ± 2 pA at −60 to −65 mV (n=6), was significantly reduced in the presence of 100 μM quinidine to 4 ± 2 pA (n=6) and in the presence of 200 μM bupivacaine to −0.2 ± 1 pA (n=5) (1-way ANOVA with Bonferroni post hocs p<0.01, Fig. 2A, C). This suggests that the hyperpolarisation mediated reduction in action potential firing is due to increased activation of voltage-independent KCNK-like channels (Enyedi & Czirjak, 2010).
3.3 The hyperpolarisation dependent effect of SST is not mediated by adenylyl cyclase signaling
SST receptors activate pertussis toxin-sensitive G proteins, Gi and Go, which inhibit adenylyl cyclase activity. To determine if the adenylyl cyclase pathway was involved in the SST inhibition of pyramidal neurone firing, 20 μM forskolin was applied to stimulate adenylyl cyclase activity and 100 μM IBMX was applied to inhibit the breakdown of cAMP, for 15 min before and during the SST application. When applied in the presence of forskolin and IBMX, SST induced a 93 ± 7 % reduction in action potential firing (n=5) which was not significantly different from interleaved control experiments where SST was applied in the presence of DMSO (n=5, Students unpaired t-test, p>0.05, Fig. 3A). Thus, the effect of SST on firing is not mediated by reducing cAMP levels.
Figure 3. The involvement of adenylyl cyclase and protein phosphatases in SST signaling.
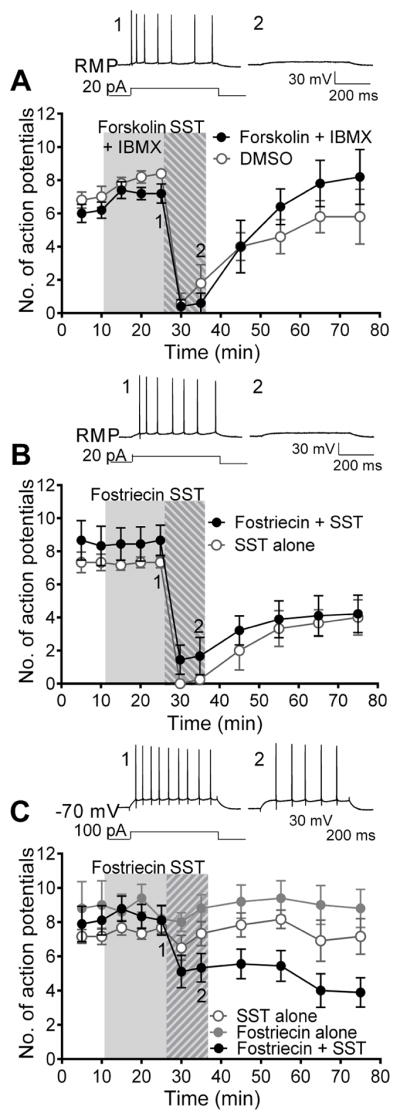
A) Elevation cAMP levels with 20 μM forskolin and 100 μM IBMX does not prevent the SST induced reduction in action potential firing (n=5) seen in DMSO control experiments (n=5). B) Blockade of protein phosphatase 2A and 4 (PP2A/PP4) with 100 nM fostriecin does not prevent the SST induced inhibition of action potential firing (n=9) seen in control experiments (n=6). C) When cells RMP is maintained at −70 mV SST application alone has no effect on action potential firing (n=6), however in the presence of the PP2A/PP4 inhibitor, 100 nM fostriecin, SST induces a reduction in action potential firing (n=9). 100 nM fostriecin alone has no effect on action potential firing (n=5).
3.4 Inhibition of protein phosphatases reveals a hyperpolarisation independent effect of SST
It has been previously suggested that protein phosphatases may play a role in SST signaling (Armstrong & White) so we decided to investigate this hypothesis. In preliminary pharmacological tests of protein phosphatase involvement in SST action on acute slices, we tested 1 μM okadaic acid (data not shown) and 100 nM fostriecin. Okadaic acid, a general inhibitor of Ser/Thr protein phosphatases, gave similar effects to the more selective inhibitor fostriecin, which blocks PP2A along with PP4. Therefore we used the more selective inhibitor, fostriecin. The hyperpolarisation dependent reduction in action potential firing induced by SST was unchanged in the presence of fostriecin (81 ± 13 %, n=9, Fig. 3B) when compared to the effect of SST in interleaved control experiments (n=6, Students unpaired t-test, p>0.05). However, fostriecin application to acute slices for 15 min before and during SST application revealed a second effect of SST, on action potential firing, which was independent of neuronal hyperpolarisation. In cells where a slow constant current was used to maintain the resting membrane potential at −70 mV in the presence of 100 nM fostriecin SST reduced action potential firing by 36 ± 9 % (n=9, Students paired t-test, p<0.01, Fig. 3C). In interleaved experiments, SST application to control cells with their RMP maintained at −70 mV, to exclude the hyperpolarising effect of SST, induced no significant reduction in action potential firing in the absence of fostriecin (5 ± 7 %, n=6, Students paired t-test, p>0.05, Fig. 3C). Fostriecin application alone also had no effect on action potential firing (109 ± 9%, n=5, Fig. 3C). The voltage responses to the negative current steps revealed the hyperpolarisation independent effect at −70 mV is mediated by an increase in membrane conductance (supplementary fig. 1).
Investigating the mechanism underlying this second inhibitory action of SST on firing revealed that the effect appeared to be mediated by G-protein activated inwardly rectifying K+ (GIRK) channels. In the presence of 200 nM tertiapin Q, which inhibits GIRK activation (Jin & Lu, 1999), SST did not significantly reduce action potential firing (5 ± 4 %, n=8, Students paired t-test, p>0.05, Fig 4A). In order to investigate whether the reduction in action potential firing was due to insertion of GIRK channels into the plasma membrane, as opposed to increased activation of existing GIRK channels, we applied fostriecin and SST in the presence of 100 μM primaquine, which blocks endosome recycling and inhibits channel insertion (Chung et al. 2009). Slices were pre-incubated in primaquine for at least one hour and then continually perfused with primaquine throughout the recordings. In the presence of primaquine when the cells RMP was held at −70 mV the application of SST with fostriecin did not induce a significant reduction in action potential firing (5 ± 5 %, n=8, Students paired t-test, p>0.05, Fig. 4B). This block of the hyperpolarisation independent effect of SST cannot be explained by an effect of primaquine on SST receptor levels at the plasma membrane because in the same cells, in the presence of primaquine, SST still induces a hyperpolarisation dependent reduction in action potential firing of 85 ± 8 % when the cells are at resting membrane potential (n=8, Students paired t-test, p<0.01, Fig. 4C). Thus the hyperpolarisation independent effect of SST in the presence of fostriecin appears to be mediated by GIRK channels, likely through the insertion of these channels into the plasma membrane, which is known to be regulated by reversible protein phosphorylation (Chung et al., 2009).
Figure 4. The hyperpolarisation independent effect of SST appears to be mediated by the insertion of GIRK channels into the plasma membrane.

A) The G-protein activated inwardly rectifying K+ (GIRK) channel blocker, 200 nM tertiapin, blocks the hyperpolarisation independent effect of SST (n=8), seen in interleaved control experiments (n=6). B) This hyperpolarisation independent effect of SST in the presence of fostriecin, seen in cells with RMP maintained at −70 mV, is also blocked by 100 μM primaquine, an inhibitor of endosome recycling (n=8), when compared to interleaved control experiments (n=6). C) In the same cells 100 μM primaquine has no effect on the hyperpolarisation dependent reduction in spiking induced by SST at RMP (n=8), which is no different to that seen in interleaved control experiments (n=6).
3.5 The role of PP5 in somatostatin action on cultured slices
Protein phosphase 5 is also a potential candidate for playing a role in SST signaling. SST signaling in pituitary tumour cells involves arachidonic acid metabolites and a protein phosphatase (Duerson et al., 1996) and arachidonic acid has been shown to stimulate PP5 activity (Skinner et al., 1997). To determine if PP5 was involved in mediating the SST effect, we cultured brain slices from mice with a floxed PP5 gene, and infected the slices in vitro with an AAV virus expressing td-tomato and Cre recombinase to delete the ppp5c gene and block synthesis of the PP5 protein (see supplementary figs. 2 & 3). Seven to twelve days after infection, recordings were made from uninfected control cells in infected slices and from virally infected cells that had cre-mediated deletion of the PP5 gene and were labeled with td-tomato (Fig 5A). In the cultured slices, the initial hyperpolarisation dependent reduction in firing, in cells at RMP, produced by SST was just as large as in acute slices, regardless of whether PP5 had been knocked down (Fig. 5B). Thus, after 5 min in SST, spiking was reduced 93 ± 7 % (n=6) in uninfected neurones and 80 ± 12 % (n=5) in infected neurones, which was not significantly different (Students unpaired t-test, p>0.05). However, in the infected neurones the firing began to recover back to baseline levels during the SST application. At the end of the 10 min SST application, there was only a 27 ± 12 % reduction in action potential firing remaining in the infected neurones, which was significantly less than the 90 ± 10 % reduction remaining in uninfected cells (Students unpaired t-test, p<0.01, Fig 5B). Thus, knockdown of PP5 does not reduce the initial response to SST but hastens its recovery in the continued presence of SST, as if the response were desensitising more rapidly in the absence of PP5 activity.
Figure 5. The effect of PP5 knockdown on SST signaling in cultured slices.

Slices were cultured from mice with a floxed PP5 gene and were virally infected with AAV-iCRE-tdtomato to induce Cre deletion of the PP5 gene in some cells. A) Fluorescence image superimposed on a differential interference contrast image to show td-tomato fluorescence in CA1 cells expressing the virus and non-fluorescent cells used for control recordings. Examples of infected and uninfected cells are indicted with white and black arrows, respectively. B) SST application gave a hyperpolarisation dependent reduction in action potential firing in uninfected cells (n=6), similar to that seen in acute slices. In cells with PP5 gene deletion (n=6) SST application resulted in only a transient reduction in action potential firing, with action potential firing returning to baseline levels by the end of the 10 min SST application (n=5). C) When cells RMP was maintained at −70 mV SST application alone produces a hyperpolarisation independent reduction in action potential firing in uninfected cells (n=6). This effect is abolished in cells with PP5 deletion (n=5). D) Schematic summarising both the hyperpolarisation dependent and hyperpolarisation independent effects of SST, and the influence of protein phosphatases.
Interestingly, in cultured slices when recording from uninfected cells with their RMP maintained at −70 mV, SST application alone produced a significant hyperpolarisation independent reduction in action potential firing of 43 ± 8 % (n=6, Students paired t-test, p<0.01, Fig. 5C). This effect did not require pretreatment with fostriecin but was eliminated completely in cells that had been infected virally with Cre recombinase to delete the Ppp5c gene. In the absence of PP5 there was no change in action potential firing following SST application in cells that were maintained at −70 mV (0 ± 0 %, n=5, Fig. 5C).
4. Discussion
The simplest hypothesis to explain our data is illustrated in Fig. 5D. We have shown that Ser/Thr protein phosphatase activity plays an important role in determining how SST affects action potential firing. In acute slices, SST stimulates KCNK-like channels, which hyperpolarise the neurones and reduce action potential firing. In cultured slices or following the inhibition of protein phosphatase 2A/4 activity in acute slices, SST application also appears to stimulate the insertion of GIRK channels into the membrane, which reduces action potential firing in a hyperpolarisation independent manner. Both these effects of SST are dependent on the activity of PP5.
4.1 Hyperpolarisation dependent effect of SST
We show that 1 μM SST reduces the excitability of CA1 pyramidal neurones in acute hippocampal slices through neuronal hyperpolarisation, which confirms previous studies on CA1 pyramidal cells (Pittman & Siggins, 1981; Moore et al., 1988). The reduction in action potential firing in acute slices depends completely on neuronal hyperpolarisation; when changes in the membrane potential are prevented by giving a slow constant current to maintain the RMP at −70 mV, SST does not inhibit firing. This hyperpolarisation dependent effect of SST provides a potential mechanism to dampen excitability and maintain neuronal firing within a functional range. The reduction in firing is blocked completely by 0.5 mM barium, implicating potassium channel activation in the response. In support of this hypothesis, SST application reduced the cells’ input resistance by an amount comparable to that observed in previous studies (Pittman & Siggins, 1981; Moore et al., 1988). This confirms that the inhibitory effect of SST is due to the opening of ion channels, rather than the inhibition of a depolarising current, such as Ih. Using voltage ramps, we show that SST does indeed induce a small current, which reversed at −95 mV. Thus, like a previous study on CA1 neurones in acute slices (Schweitzer et al., 1998), we conclude that at resting membrane potential SST predominantly activates a hyperpolarising leak current mediated by voltage-independent potassium channels.
The “two-pore” family of KCNK channels (Enyedi & Czirjak, 2010), are now recognised as the molecular basis for such hyperpolarising leak currents, and indeed in our cells the SST induced current was blocked by KCNK channel inhibitors, bupivacaine and quinidine. While it is not clear which subclass of KCNK channels are activated by SST, several subclasses are expressed in the hippocampal CA1 region (Talley et al., 2001) and have properties consistent with our results including activation by GPCR signaling through Gi/Go, insensitivity to cAMP modulation and sensitivity to block by submillimolar barium (Patel & Honoré, 2001; Fearon et al., 2003; Deng et al., 2007; Xiao et al., 2009).
Other studies have reported that SST increases the activity of other potassium channels, such as the M channels (Kv7) to reduce epileptiform activity (Moore et al., 1988; Schweitzer et al., 1993; Qiu et al., 2008). However, in our study using perforated patches to record from metabolically intact neurones, we found that the inhibition of firing by SST was not significantly reduced by linopirdine, an inhibitor of Kv7 channels. This was not surprising as we recorded from cells with a typical RMP of −60 mV, while M-current activation by SST is usually only seen at more depolarised potentials (Schweitzer et al., 1998). SST has also been reported to activate large conductance Ca2+-activated K+ channels and reduce action potential firing in neostriatal neurones (Galarraga et al., 2007). We found, however, neither small nor large conductance Ca2+-activated K+ channels were required for the SST induced hyperpolarisation. Similarly Wang et al., 1989 found no SST activation of Ca2+-activated K+ channels in neocortical neurones. In addition we found that ERG channel inhibitors did not reduce the effect of SST on firing. In some brain regions SST receptors activate inward rectifying K+ channels (Inoue et al., 1988; Sodickson & Bean, 1998; Meis et al., 2005; Sun et al., 2002). While Moore et al. 1988, suggested SST may activate an inwardly rectifying K+ channel in CA1 pyramidal neurones, we found no significant involvement of GIRK channels in the SST induced hyperpolarisation dependent inhibition of firing in acute slices. Instead our data, from perforated patch recordings, are consistent with a previous study by Schweitzer et al., 1998, who used sharp microelectrodes and found no evidence for SST activation of either GIRK channels or Ca2+-activated K+ channels in CA1 pyramidal neurones.
SST receptors signal through Gi/Go proteins, which inhibit adenylyl cyclase activity, and previous studies have found that SST inhibits adenylyl cyclase activity in hippocampal neurones (Schettini et al., 1989, Raynor & Reisine, 1992). We used forskolin, which activates adenylyl cyclase, in combination with a phosphodiesterase inhibitor, IBMX, which prevents cAMP degradation, to elevate cAMP levels, and therefore occlude any effect of SST mediated by inhibition of adenylyl cyclase. However, we found no reduction in the inhibitory effect of SST suggesting that the adenylyl cyclase pathway is not involved in mediating the effects of SST on excitability. Similarly, previous studies found that neither the SST induced hyperpolarisation (Schweitzer et al., 1993) nor the SST modulation of ion channels (Ishibashi & Akaike, 1995; Wang et al., 1989) were mediated by a reduction in cAMP/PKA signaling. In various different tissues SST has been shown to signal through many different pathways involving PLA2/arachidonic acid metabolites, protein tyrosine kinases, Na+/H+ exchanger, PLC/IP3 and MAPK (Weckbecker et al., 2003). Of these signaling pathways there are two that are promising potential candidates for the modulation of KCNK channels. Firstly the arachidonic acid metabolites, which have been previously implicated in both SST signaling (Schweitzer et al., 1993; Duerson et al., 1996), can also regulate KCNK channels (Honoré & Patel, 2001). Secondly, somatostatin has been shown to inhibit Na+/H+ exchangers leading to intracellular acidification (Barber et al., 1989), which is known to activate the pH sensitive KCNK channels (Honoré & Patel, 2001).
It has been reported that Ser/Thr protein phosphatases play a role in SST signaling and the regulation of ion channels (Armstrong & White 1992; Herzig & Neumann, 2000). Therefore, we investigated whether Ser/Thr protein phosphatases are involved in the effect of SST reported here. Fostriecin, and also okadaic acid (data not shown), have no effect on the hyperpolarisation dependent inhibition of firing induced by SST, suggesting that PP1, PP2A and PP4 are not involved in mediating this SST effect. However, the pretreatment with fostriecin did reveal a secondary effect of SST on excitability in the acute slices, which was hyperpolarisation independent.
4.2 Hyperpolarisation independent effect of SST
In acute slices SST application to cells where the RMP was maintained at −70 mV did not inhibit the firing induced by subsequent current steps. However, when PP2A and PP4 were inhibited with fostriecin (Honkanen & Golden, 2002), we saw a second inhibitory effect of SST in cells with their RMP maintained at −70 mV. This hyperpolarisation independent effect was blocked by 200 nM tertiapin Q indicating that the effect is mediated by GIRK channels (Jin & Lu, 1999). As mentioned previously, SST receptors in some brain regions have been reported to increase inwardly rectifying channel activity (Inoue et al., 1988; Sodickson & Bean, 1998; Meis et al., 2005; Sun et al., 2002); although the involvement of GIRK channel activation following SST receptor activation in the hippocampal CA1 region has been unclear (Moore et al., 1988; Schweitzer et al., 1998).
The hyperpolarisation independent effect on CA1 neurones that we observed was also blocked by the endosome recycling blocker, primaquine. Therefore, this effect appears to be due to the insertion of GIRK channels into the plasma membrane, which leads to an increase in membrane conductance, and a reduction in action potential firing. A similar phosphorylation-dependent insertion of GIRK channels, in dissociated hippocampal neurones has been reported previously (Chung et al., 2009). In that study the phosphatase involved in stimulating GIRK insertion had the pharmacology of PP1, but the concentration of fostriecin that we used in our study would be expected to block PP2A or PP4, but not PP1 (Honkanen & Golden, 2002).
While the hyperpolarisation dependent effect of SST is likely to predominate, this hyperpolarisation independent effect may also act to dampen neuronal excitability in situations where PP2A or PP4 activity is reduced, independently of the RMP of the neurones.
4.3 The regulation of somatostatin signaling by PP5
Finally we investigated the role of the Ser/Thr protein phosphatase, PP5, in SST signaling. PP5 was discovered to be an arachidonic acid stimulated protein phosphatase in the brain (Skinner et al., 1997) after work showing that SST signaling involved arachidonic acid metabolites and a protein phosphatase (Armstrong & White, 1992; Duerson et al., 1996). We took advantage of a novel mouse strain in which the gene encoding PP5, Ppp5c, had been flanked with loxP sites. By infecting neurones in cultured slices with an AAV virus expressing the Cre recombinase and a fluorescent protein, we could selectively delete expression of PP5 in individual neurones, using adjacent non-infected neurones as controls. However, to our surprise, not only was the hyperpolarisation dependent effect of SST present, but also the hyperpolarisation independent effect was already present in the uninfected cells in cultured slices that retained PP5 activity, without inhibition of PP2A/4. This was not due to a genetic difference because SST had the same effect on acute slices from the floxed mice as it did on the wild types (data not shown). Furthermore, although the cultured slices were prepared from mice that were one week younger (P7-9) than the acute slices, when we recorded SST responses in the acute slices from the younger animals, we did not observe a hyperpolarisation independent inhibition of firing (data not shown). Thus, the difference must result from some effect of the culture conditions, perhaps on PP2A activity, but we did not test that possibility.
In any case, we discovered that PP5 was required for both forms of SST inhibition. The hyperpolarisation independent effect of SST, in cells with RMP maintained at −70 mV, was completely absent in the infected neurones which had no PP5 activity. In contrast, in infected neurones which lack PP5, the hyperpolarisation dependent effect of SST on firing was just as large as it was in uninfected neurones. However, the response was much briefer, with firing returning to baseline levels by the end of the 10 min SST application. Given the recovery of action potential firing during the continual application of SST it appears that the removal of PP5 activity is accelerating SST receptor desensitisation. There are five subtypes of SST receptors (SSTR1-5), all of these subtypes apart from SSTR4 are found to undergo desensitisation, and internalisation in some cases, in response to receptor phosphorylation at serine and threonine residues by G-protein coupled receptor kinases and protein kinase C (Hipkin et al., 1997; Roth et al., 1997; Young Shim et al., 2006; Liu et al., 2009; Ghosh & Schonbrunn, 2011; Petrich et al., 2013). Thus, we cannot be sure whether there is a more direct role for PP5 in the hyperpolarisation independent effect of SST or whether the hyperpolarisation independent effect is mediated by a different SST receptor subtype which desensitises so rapidly that no response ever develops.
4.4 Conclusions and implications
We have described two mechanisms involving two different classes of potassium channels through which SST regulates neuronal excitability. The expression of both of these mechanisms appears to be controlled by protein phosphatase activity. Thus, the ability of reversible protein phosphorylation to enhance or diminish the inhibitory effect of SST could have interesting implications for systems level regulation of activity in the hippocampus (Royer et al., 2012; Lovett-Barron et al., 2014; Katona et al., 2014). In addition, the changes in protein phosphatases and SST signaling that are associated with aging and neurodegeneration (Veeranna et al., 2011; Braithwaite et al., 2012; Koh et al., 2014; Martel et al., 2014) might be more closely interrelated than previously understood.
Supplementary Material
Supplementary Figure 1. Somatostatin induced an increase in membrane conductance. The voltage responses to current steps are shown with the cell at A) RMP and B) with the RMP adjusted to −70 mV. SST application alone reduces the voltage responses; a greater reduction is seen at RMP than at −70 mV (n=6). SST application in the presence of fostriecin reduces the voltage response further at −70 mV than SST alone, while having no additional effect at RMP (n=9). C) Summary of the change in membrane resistance.
Supplementary Figure 2. Creating a C57Bl/6 mouse strain with a floxed PPP5C gene.
Two homology arms (shown in blue) and the conditional KO region, which contained exons 7-13 (shown in light green) were generated by PCR from a BAC (RP23-351I20) and, after a RED cloning/gap repair manipulation, cloned into the 3loxP3NwCD vector. The final vector was obtained by standard molecular cloning. Aside from the homology arms and the KO region, the final vector also contains loxP sequences flanking the conditional KO region (~3.5 kb) and the Neo expression cassette for positive selection of recombinant ES cells. The final ES cell targeting vector was linearized with Not I before electroporation and contained a diptheria toxin gene (DTA) for negative selection of random integration. Screening of the offspring from chimera breedings and later matings was done by PCR. The primers were designed so that one primer pair will detect heterozygous, homozygous and wild type alleles. The nucleotide sequences of this primer pair are: N30 ck3F: 5′-AAGCAATAGGACCCTGCCATAGGAAG-3′
N30 35R: 5′-AACAGTAGCAACGGAGGACTCCCAG-3′
The wild type PCR product has a length of 0.61kb, whereas the recombinant allele will give a PCR product of 0.75kb in length.
Supplementary Figure 3. Validation of PPP5c excision by Cre recombinase.
Dissociated cultures of hippocampal neurons from PP5 conditional floxed homozygote mice were infected in vitro with a lentivirus encoding green fluorescent protein (GFP) alone or fused to the Cre recombinase, which was inserted 5′ to GFP. Total protein from cellular homogenates were run on SDS gels and blotted with a rabbit antibody (1:1000) against the linker region of the PP5 protein (Bahl et al., 2001 Molec. Brain Res. 90:101-9) and then with a secondary anti-rabbit antibody (1:3000). The PP5 protein was detected at ~63kD. From left to right, the four lanes show increasing amounts of viral vector: Control (GFP alone), 50, 200, 500 μl of Cre-GFP virus. The highest concentration of virus eliminates PP5 expression.
Highlights.
Somatostatin (SST) reduces action potential firing in hippocampal CA1 neurones.
SST activates KCNK-like channels which hyperpolarise the neurones reducing firing.
PP2A/PP4 inhibition unmasks a second effect of SST mediated by GIRK channels.
Deletion of PP5 reduces both of the inhibitory effects of SST.
The inhibitory effects of SST can be bidirectionally modulated by phosphorylation.
Acknowledgments
This work was supported by the NIH Intramural program award Z01-ES080043 to D.L.A.
The AAV-iCRE-td-tomato virus, which was used for deletion of PP5, was produced by Bernd Gloss. The td-tomato fluorescence/D.I.C. image was taken by Marie Posch.
Abbreviations
- GIRK
G-protein activated inwardly rectifying K+ channels
- PP
Protein phosphatase
- RMP
Resting membrane potential
- SST
Somatostatin
- Ser/Thr
Serine/Threonine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Armstrong DL, White RE. An enzymatic mechanism for potassium channel stimulation through pertussis-toxin-sensitive G proteins. Trends Neurosci. 1992;15(10):403–408. doi: 10.1016/0166-2236(92)90192-b. [DOI] [PubMed] [Google Scholar]
- Baratta MV, Lamp T, Tallent MK. Somatostatin depresses long-term potentiation and Ca2+ signaling in mouse dentate gyrus. J Neurophysiol. 2002;88:3078–3086. doi: 10.1152/jn.00398.2002. [DOI] [PubMed] [Google Scholar]
- Barber DL, McGuire ME, Ganz MB. β-adrenergic and somatostatin receptors regulate Na-H exchange independent of cAMP. J Biol Chem. 1989;264:21038–21042. [PubMed] [Google Scholar]
- Baxter AW, Wyllie DJ. Phosphatidylinositol 3 kinase activation and AMPA receptor subunit trafficking underlie the potentiation of miniature EPSC amplitudes triggered by the activation of L-type calcium channels. J Neurosci. 2006;26(20):5456–5469. doi: 10.1523/JNEUROSCI.4101-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC. Protein phosphatases and Alzheimer’s disease. Prog Mol Biol Transl Sci. 2012;106:343–379. doi: 10.1016/B978-0-12-396456-4.00012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Qian X, Ehlers M, Jan YN, Jan LY. Neuronal activity regulates phosphorylation-dependent surface delivery of G protein-activated inwardly rectifying potassium channels. Proc Natl Acad Sci USA. 2009;106(2):629–634. doi: 10.1073/pnas.0811615106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Poudel SKS, Rojanathammanee L, Porter JE, Lei S. Serotonin inhibits neuronal excitability by activating two-pore domain K+ channels in the entorhinal cortex. Mol Pharmacol. 2007;72(1):208–218. doi: 10.1124/mol.107.034389. [DOI] [PubMed] [Google Scholar]
- Duerson K, White RE, Jiang F, Schonbrunn A, Armstrong DL. Somatostatin stimulates BKCa channels in rat pituitary tumor cells through lipoxygenase metabolites of arachidonic acid. Neuropharmacology. 1996;35(7):949–961. doi: 10.1016/0028-3908(96)00131-1. [DOI] [PubMed] [Google Scholar]
- Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90(2):559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- Fearon IM, Zhang M, Vollmer C, Nurse CA. GABA mediates auroreceptor feedback inhibition in the rat carotid body via presynaptic GABAB receptors and TASK-1. J Physiol. 2003;553:83–94. doi: 10.1113/jphysiol.2003.048298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarraga E, Vilchis C, Tkatch T, Salgado H, Tecuapetla F, Perez-Rosello T, Perez-Garci E, Hernandez-Echeagaray E, Surmeier DJ, Bargas J. Somatostatinergic modulation of firing pattern and calcium activated potassium currents in medium spiny neostriatal neurons. Neuroscience. 2007;146(2):537–554. doi: 10.1016/j.neuroscience.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Schonbrunn A. Differential temporal and spatial regulation of somatostatin receptor phosphorylation and dephosphorylation. J Biol Chem. 2011;286(15):13561–13573. doi: 10.1074/jbc.M110.215723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80(1):173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- Hipkin RW, Friedman J, Clark RB, Eppler CM, Schonbrunn A. Agonist-induced desensitization, internalization, and phosphorylation of the sst2A somatostatin receptor. J Biol Chem. 1997;272(21):13869–13876. doi: 10.1074/jbc.272.21.13869. [DOI] [PubMed] [Google Scholar]
- Honkanen RE, Golden T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem. 2002;9(22):2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- Inoue M, Nakajima S, Nakajima Y. Somatostatin induces an inward rectification in rat locus coeruleus neurons through a pertussis toxin-sensitive mechanism. J Physiol. 1988;407:177–198. doi: 10.1113/jphysiol.1988.sp017409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishbashi H, Akaike N. Somatostatin modulates high-voltage-activated Ca2+ channels in freshly dissociated rat hippocampal neurons. J Neurophysiol. 1995;74(3):1028–1036. doi: 10.1152/jn.1995.74.3.1028. [DOI] [PubMed] [Google Scholar]
- Jin W, Lu Z. Synthesis of a stable form of tertiapin: a high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1999;38(43):14286–14293. doi: 10.1021/bi991205r. [DOI] [PubMed] [Google Scholar]
- Katona L, Lapray D, Viney TJ, Oulhaj A, Borhegyi Z, Micklem BR, Klausberger T, Somogyi P. Sleep and movement differentiates actions of two types of somatostatin-expressing GABAergic interneuron in rat hippocampus. Neuron. 2014;82(4):872–886. doi: 10.1016/j.neuron.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MT, Spiegel AM, Gallagher M. Age-associated changes in hippocampal- dependent cognition in diversity outbred mice. Hippocampus. 2014;24(11):1300–1307. doi: 10.1002/hipo.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Bee MS, Schonbrunn A. Site specificity of agonist and second messenger-activated kinases for somatostatin receptor subtype 2A (Sst2A) phosphorylation. Mol Pharmacol. 2009;76(1):68–80. doi: 10.1124/mol.108.054262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR, Turi GF, Hen R, Zemelman BV, Losonczy A. Dendritic inhibition in the hippocampus supports fear learning. Science. 2014;343(6173):857–863. doi: 10.1126/science.1247485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel G, Dutar P, Epelbaum J, Viollet C. Somatostatinergic systems: an update on brain functions in normal and pathological aging. Frontiers Endocrinol. 2012;3:154. doi: 10.3389/fendo.2012.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meis S, Sosulina L, Schulz S, Hollt V, Pape HC. Mechanisms of somatostatin-evoked responses in neurons of the rat lateral amygdala. Eur J Neurosci. 2005;21:755–762. doi: 10.1111/j.1460-9568.2005.03922.x. [DOI] [PubMed] [Google Scholar]
- Molley SS, Thomas L, Kamibayashi C, Mumby MC, Thomas G. Regulation of endosome sorting by a specific PP2A isoform. J Cell Biol. 1998;142:1399–1411. doi: 10.1083/jcb.142.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SD, Madamba SG, Joels M, Siggins GR. Somatostatin augments the M-current in hippocampal neurons. Science. 1988;239:278–280. doi: 10.1126/science.2892268. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiol Rev. 1990;70(2):513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001;24(6):339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- Patel YC, Srikant CB. Somatostatin receptors. Trends Endocrin Metab. 1997;8(10):398–405. doi: 10.1016/s1043-2760(97)00168-9. [DOI] [PubMed] [Google Scholar]
- Petrich A, Mann A, Kliewer A, Nagal F, Strigli A, Märtins JC, Pöll F, Schulz S. Phosphorylation of threonine 333 regulates trafficking of the human sst5 somatostatin receptor. Mol Endocrinol. 2013;27(4):671–681. doi: 10.1210/me.2012-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman QJ, Siggins GR. Somatostatin hyperpolarizes hippocampal pyramidal cells in vitro. Brain research. 1981;221:402–408. doi: 10.1016/0006-8993(81)90791-5. [DOI] [PubMed] [Google Scholar]
- Qiu C, Zeyda T, Johnson B, Hochgeschwender U, de Lecea L, Tallent MK. Somatostatin receptor subtype 4 couples to the M-current to regulate seizures. J Neurosci. 2008;28(14):3567–3576. doi: 10.1523/JNEUROSCI.4679-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor K, Reisine T. Differential coupling of somatostatin1 receptors to adenylyl cyclase in the rat striatum vs. the pituitary and other regions of the rat brain. J Pharmacol Exp Ther. 1992;260(2):841–848. [PubMed] [Google Scholar]
- Royer S, Zemelman BV, Losonczy A, Kim J, Chance F, Magee JC, Buzsáki G. Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci. 2012;15(5):769–775. doi: 10.1038/nn.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Kreienkamp HJ, Meyerhof W, Richter D. Phosphorylation of four amino acid residues in the carboxyl terminus of the rat somatostatin receptor subtype 3 is crucial for its desensitization and internalization. J Biol Chem. 1997;272(38):23769–23774. doi: 10.1074/jbc.272.38.23769. [DOI] [PubMed] [Google Scholar]
- Schettini G, Florio T, Meucci O, Landolfi E, Grimaldi M, Ventra C, Marino A. Somatostatin inhibition of adenylate cyclase activity in different brain areas. Brain Res. 1989;492(1–2):65–71. doi: 10.1016/0006-8993(89)90889-5. [DOI] [PubMed] [Google Scholar]
- Schulz S, Handel M, Schreff M, Schmidt H, Hollt V. Localization of five somatostatin receptors in the rat central nervous system using subtype-specific antibodies. J Physiol. 2000;94:259–264. doi: 10.1016/s0928-4257(00)00212-6. [DOI] [PubMed] [Google Scholar]
- Schweitzer P, Madamba S, Champagnat J, Siggins GR. Somatostatin inhibition of hippocampal CA1 pyramidal neurons: mediation by arachidonic acid and its metabolites. J Neurosci. 1993;3(5):2033–2049. doi: 10.1523/JNEUROSCI.13-05-02033.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer P, Madamba SG, Siggins GR. Somatostatin increases a voltage-insensitive K+ conductance in rat CA1 hippocampal neurons. J Neurophysiol. 1998;79(3):1230–1238. doi: 10.1152/jn.1998.79.3.1230. [DOI] [PubMed] [Google Scholar]
- Selmer I-S, Schindler M, Allen JP, Humphrey PPA, Emson PC. Advances in understanding neuronal somatostatin receptors. Regul Peptides. 2000;90:1–18. doi: 10.1016/s0167-0115(00)00108-7. [DOI] [PubMed] [Google Scholar]
- Skinner J, Sinclair C, Romeo C, Armstrong D, Charbonneau H, Rossie S. Purification of a fatty acid-stimulated protein-serine/threonine phosphatase from bovine brain and its identification as a homolog of protein phosphatase 5. J Biol Chem. 1997;272(36):22464–22471. doi: 10.1074/jbc.272.36.22464. [DOI] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. Neurotransmitter activation of inwardly rectifying potassium current in dissociated hippocampal CA3 neurons: Interactions among multiple receptors. J Neurosci. 1998;18(20):8153–8162. doi: 10.1523/JNEUROSCI.18-20-08153.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR, Prince DA. Somatostatin inhibits thalamic network oscillations in vitro: actions on the GABAergic neurons of the reticular nucleus. J Neurosci. 2002;22(13):5374–5386. doi: 10.1523/JNEUROSCI.22-13-05374.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallent MK, Siggins GR. Somatostatin depresses excitatory but not inhibitory neurotransmission in rat CA1 hippocampus. J Neurophysiol. 1997;78:3008–3018. doi: 10.1152/jn.1997.78.6.3008. [DOI] [PubMed] [Google Scholar]
- Tallent MK, Siggins GR. Somatostatin acts in CA1 and CA3 to reduce hippocampal epileptiform activity. J Neurophysiol. 1999;81(4):1626–1635. doi: 10.1152/jn.1999.81.4.1626. [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA. CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci. 2001;21(19):7491–7505. doi: 10.1523/JNEUROSCI.21-19-07491.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranna Yang DS, Lee JH, Vinod KY, Stavrides P, Amin ND, Pant HC, Nixon RA. Declining phosphatases underlie aging-related hyperphosphorylation of neurofilaments. Neurobiol Aging. 2011;32(11):2016–2029. doi: 10.1016/j.neurobiolaging.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet C, Lepousez G, Loudes C, Videau C, Simon A, Epelbaum J. Somatostatinergic systems in brain: networks and functions. Mol cell Endocrinol. 2008;286:75–87. doi: 10.1016/j.mce.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Wang HL, Bogen C, Reisine T, Dichter M. Somatostatin-14 and somatostatin-28 induce opposite effects on potassium currents in rat neocortical neurons. Proc Natl Acad Sci USA. 1989;86:9616–9620. doi: 10.1073/pnas.86.23.9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckbecker G, Lewis I, Albert R, Schmid HA, Hoyer D, Bruns C. Opportunities in somatostatin research: biological, chemical and therapeutic aspects. Nature reviews drug discovery. 2003;2:999–1017. doi: 10.1038/nrd1255. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Deng PY, Rojanathammanee L, Yang C, Grisanti L, Permpoonputtana K, Weinshenker D, Doze VA, Porter JE, Lei S. Noradrenergic depression of neuronal excitability in the entorhinal cortex via activation of TREK-2 K+ channels. J Biol Chem. 2009;284(16):10980–10991. doi: 10.1074/jbc.M806760200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young Shim E, Jung Kim H, Kim MJ, Rhie DJ, Jo YH, Kim MS, June Hahn S, Lee MY, Yoon SH. Desensitization of somatostatin-induced inhibition of low extracellular magnesium concentration-induced calcium spikes in cultured rat hippocampal neurons. Brain Research. 2006;1111(1):61–71. doi: 10.1016/j.brainres.2006.06.081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Somatostatin induced an increase in membrane conductance. The voltage responses to current steps are shown with the cell at A) RMP and B) with the RMP adjusted to −70 mV. SST application alone reduces the voltage responses; a greater reduction is seen at RMP than at −70 mV (n=6). SST application in the presence of fostriecin reduces the voltage response further at −70 mV than SST alone, while having no additional effect at RMP (n=9). C) Summary of the change in membrane resistance.
Supplementary Figure 2. Creating a C57Bl/6 mouse strain with a floxed PPP5C gene.
Two homology arms (shown in blue) and the conditional KO region, which contained exons 7-13 (shown in light green) were generated by PCR from a BAC (RP23-351I20) and, after a RED cloning/gap repair manipulation, cloned into the 3loxP3NwCD vector. The final vector was obtained by standard molecular cloning. Aside from the homology arms and the KO region, the final vector also contains loxP sequences flanking the conditional KO region (~3.5 kb) and the Neo expression cassette for positive selection of recombinant ES cells. The final ES cell targeting vector was linearized with Not I before electroporation and contained a diptheria toxin gene (DTA) for negative selection of random integration. Screening of the offspring from chimera breedings and later matings was done by PCR. The primers were designed so that one primer pair will detect heterozygous, homozygous and wild type alleles. The nucleotide sequences of this primer pair are: N30 ck3F: 5′-AAGCAATAGGACCCTGCCATAGGAAG-3′
N30 35R: 5′-AACAGTAGCAACGGAGGACTCCCAG-3′
The wild type PCR product has a length of 0.61kb, whereas the recombinant allele will give a PCR product of 0.75kb in length.
Supplementary Figure 3. Validation of PPP5c excision by Cre recombinase.
Dissociated cultures of hippocampal neurons from PP5 conditional floxed homozygote mice were infected in vitro with a lentivirus encoding green fluorescent protein (GFP) alone or fused to the Cre recombinase, which was inserted 5′ to GFP. Total protein from cellular homogenates were run on SDS gels and blotted with a rabbit antibody (1:1000) against the linker region of the PP5 protein (Bahl et al., 2001 Molec. Brain Res. 90:101-9) and then with a secondary anti-rabbit antibody (1:3000). The PP5 protein was detected at ~63kD. From left to right, the four lanes show increasing amounts of viral vector: Control (GFP alone), 50, 200, 500 μl of Cre-GFP virus. The highest concentration of virus eliminates PP5 expression.