

Abstract
We have previously shown that polyclonal natural IgM protects mice from renal IRI by inhibiting the reperfusion inflammatory response. We hypothesized that a potential mechanism involved IgM modulation of dendritic cells as we observed high IgM binding to splenic DC. To test this hypothesis, we pre-treated BMDC with polyclonal murine or human IgM prior to LPS activation and demonstrate that 0.5 × 106 IgM/LPS pretreated BMDC, when injected into WT-B6 mice, 24 hours before renal ischemia, protect mice from developing renal IRI. We show that this switching of LPS activated BMDC to a regulatory phenotype requires modulation of BMDC function that is mediated by IgM binding to non-apoptotic BMDC receptors. Regulatory BMDC require IL-10 and PD1 as well as downregulation of CD40 and p65NF-κB phosphorylation to protect in renal IRI. Blocking the PD1 ligand binding site just before intravenous injection of IgM/LPS pretreated BMDC or using IL-10ko BMDC fails to induce protection. Similarly, IgM/LPS pretreated BMDC are rendered non-protective by increasing CD40 expression and phosphorylation of p65NF-κB. How IgM/LPS regulatory BMDC suppress in-vivo ischemia induced innate inflammation remains to be determined. However, we show that suppression is dependent on other in-vivo regulatory mechanisms in the host i.e. CD25+ T cells, B cells, IL10 and circulating IgM. There was no increase in Foxp3+ Tregs in the spleen either before or after renal IRI. Collectively, these findings show that natural IgM anti-leucocyte antibodies can switch BMDC to a regulatory phenotype despite the presence of LPS that ordinarily induces BMDC maturation.
Introduction
Several observations clearly demonstrate that innate and adaptive immune inflammatory responses are regulated to protect the host from an over exuberant inflammatory response and from initiating autoimmune diseases. One such newly described regulatory mechanism is natural IgM (reviewed in 1). These antibodies are referred to as “natural antibodies” as these antibodies are produced at birth by B1 cells, independently of foreign antigen exposure and without the need for T helper cells. Natural IgM are polyclonal autoantibodies with differing specificities, some of which have been identified, for example, IgM autoantibodies with reactivity to leucocytes, erythrocytes, oxidized lipids, IgG (referred to as IgM rheumatoid factor), apoptotic cell membranes and complement proteins. Autoantibody producing B1 cells, unlike self-reactive T cells, are positively selected for their self-reactivity thus implying that natural autoantibodies are conserved by design (2). Extensive studies on a natural IgM autoantibody reactive to phosphorylcholine (PC), that is present on oxidized lipids, demonstrate that this autoantibody by binding to PC can inhibit atherosclerosis induced by oxidized lipids (reviewed in 3). Additionally, IgM anti-PC, by binding to PC on apoptotic cell membranes, enhances DC phagocytosis of apoptotic cells, thus providing an additional mechanism to inhibit DC maturation. Such a mechanism was exploited by infusing IgM pre-treated apoptotic cells to inhibit inflammation in a murine model of arthritis (3).
Our laboratory has been interested in IgM anti-leucocyte autoantibodies (IgM-ALA) which is a subset of natural IgM that binds to non-apoptotic leucocyte membrane receptors (reviewed in 1). These antibodies are evolutionarily conserved in that they are polyreactive and are encoded from germline genes that are largely non-mutated. Hence these natural IgM antibodies can bind to receptors present on autologous and allogeneic leucocytes as well as receptors present on leucocytes from other species. IgM-ALA consists of several different antibody clones, with most clones being polyreactive. Some of the leucocyte receptors binding to polyclonal natural IgM or to isolated natural IgM clones have been identified e.g. CD4, CD3, CXCR4, CCR5 and TcR (4). The polyreactivity of natural IgM is best exemplified by the monoclonal IgM anti-PC antibody, which not only binds to phosphorylcholine (PC) on oxidized lipids and apoptotic cell membranes but also binds to pneumococcal polysaccharides, ABO blood group antigens and other autoantigens (3, 5). Interestingly, these IgM- ALA are referred to as “cold-reactive” antibodies especially since the cell bound IgM activates complement and causes cell lysis at colder temperatures (<22°C) and not at body temperature (6–8). Prior observations over the last 40 years, particularly in humans, have clearly demonstrated that IgM-ALA levels increase with various infections (viral and parasitic), autoimmune diseases (SLE) and other inflammatory states (sarcoidosis and end stage renal disease) and IgM-ALA levels normalize with control of infection or the inflammatory state (1). We hypothesized that naturally occurring IgM-ALA increase with infective and inflammatory states to regulate the inflammatory response as they possess certain unique characteristics that are suited for such a purpose. Firstly, these 960 KDa pentameric autoantibodies predominantly reside in the vascular compartment and bind to intravascular proteins as well as receptors on endothelial cells and leucocytes. Secondly, they bind to autologous leucocyte receptors involved in initiating the inflammatory response (e.g CD3, CD4, co-stimulatory receptors) and in leucocyte trafficking (e.g chemokine receptors). Thirdly, unlike pathogenic IgG autoantibodies, IgM-ALA autoantibodies have low binding affinity to cells and at body temperature the cell bound IgM does not activate complement. Hence, at body temperature, they are particularly suited to modulate cell receptors without damaging the cell (4, 6–8). Observations showing that patients with high levels of IgM-ALA have (i) significantly less rejections and prolonged kidney and heart survival (9–13) and (ii) have significantly lower titers of anti-HLA antibodies after alloantigen sensitization (8), support such a concept i.e. that IgM-ALA can regulate immune cells that mediate both innate and adaptive immune inflammatory responses. In subsequent studies with murine models of inflammation, we showed that administration of intravenous polyclonal IgM, to increase plasma IgM-ALA levels in wild type C57BL6 ( referred to as WT-B6) mice, attenuated the inflammatory response mediated by innate (renal IRI), adaptive (cardiac transplants) and autoimmune (diabetes mellitus in NOD mice) immune mechanisms (14, 15). Specifically, we showed that protection was mediated by the IgM-ALA subset present in the purified polyclonal IgM preparation as the protective effect on renal IRI was abolished when polyclonal IgM was depleted of IgM-ALA with leucocyte adsorption (14).
The current studies were aimed at determining the mechanism for the observed protective effect of IgM-ALA in the murine model of renal IRI. We hypothesized that IgM-ALA inhibits the innate inflammatory response in part, by modulating dendritic cells (DC) especially since we observed higher binding of IgM to activated splenic DC when compared to activated T cells. To examine this question, we intravenously injected mice with in-vitro cultured bone-marrow dendritic cells (BMDC) that were pretreated with polyclonal IgM and LPS. Our data demonstrate that such in-vitro pretreated BMDC, when injected intravenously, protects WT-B6 mice from renal IRI by inhibiting the inflammatory response to the ischemic kidney. Our data also show that optimal protection requires that BMDC undergo LPS activation to upregulate cytokines and receptors (e.g. IL10, costimulatory molecules) and in addition requires IgM during LPS activation to switch BMDC to function as “regulatory” DC.
Materials and Methods
IgM and IgG purification from plasma
IgM and IgG were purified from heat inactivated (56°C) WT-B6 plasma using size exclusion column chromatography (Sephacryl S-300 HR) as previously described (14) except for modifications detailed below. Care was taken to separate plasma within 4 hrs of obtaining blood and then plasma was sterile filtered to remove any contaminating bacteria. Plasma was stored at 4°C prior to column purification. IgM was not isolated by dialyzing sera in water or by ammonium chloride precipitation as both these techniques yielded IgM with impaired functional activity. Column purified IgM was re-passaged through Sephacryl S-300 to remove other contaminating proteins. Contaminating IgG in the IgM preparation was removed by using agarose beads covalently linked to goat anti-murine IgG. Protein A was not used as contaminating protein A in the IgM preparation also altered lymphocyte function. Purified IgM and IgG was concentrated to 1.3-1.5mg/ml and then dialyzed against RPMI-1640 and sterile filtered prior to use in cultures and for in-vivo use. The effect of IgM on in-vitro cultures was dose dependent and the maximum effect of IgM was observed using IgM at a physiological dose of 10 to 30µg/250 × 103 cells/0.5 ml of culture.
Leucocyte adsorption of polyclonal IgM
1mg of purified polyclonal IgM was diluted to 10ml in media containing 1% FCS and adsorbed with 5 different aliquots of 3 × 108 activated splenic cells suspended in 0.2ml media. Splenic cells were activated for 48 hours with LPS and soluble anti-CD3 to enhance receptor expression and each aliquot of cells was adsorbed with IgM for 45 min at 37°C.. Adsorbed IgM was extensively dialyzed to remove any cytokines, concentrated and sterile filtered. About 60% of the IgM was recovered after the adsorption procedure. This adsorption procedure with activated splenic leucocytes reduced binding of polyclonal IgM to BMDC (data not shown) but completely depleted IgM that bound to soluble recombinant CD40 and PD1 (see Fig 3C).
FIGURE 3.

(A) (Left panel) IgM does not inhibit binding of FITC labelled LPS to BMDC. Here, 20µg of IgM was initially added to 0.2 × 106 BMDC at 4°C for 1 hour and without a wash step, FITC-LPS was added. (Right panel) Polyclonal IgM does not inhibit LPS induced TLR4 downregulation. IgM was added to WT-BMDC for 1 hour at 37°C and without a wash step, LPS was added to activate WT-BMDC for 48 hours. (B) An anti-Fcα/µR blocking antibody (clone TX61) does not prevent IgM mediated downregulation of CD40. Blocking antibody was initially added to WT-BMDC at 4°C for 1hr before adding IgM and LPS. BMDC were then cultured at 37°C for 48hrs. (C) Recombinant soluble PD1-Ig and CD40-Ig (but not PDL1-Ig, CD40L and CD 80-Ig) bind to polyclonal IgM immobilized on sepharose beads. No binding was observed when soluble receptor proteins were added to isotype monoclonal murine IgM or to control polyclonal IgM (i.e., depleted of IgM-ALA with leucocyte adsorption) immobilized on sepharose beads (D) Polyclonal murine IgM does not inhibit binding of soluble CD40L-Ig and soluble PDL1-Ig to their ligands. LPS activated BMDC were pretreated with murine polyclonal IgM and human IgG for 1 hour at 4°C, washed in the cold and then exposed to the ligands at 4°C for 1 hour. Cells were exposed to human IgG to prevent binding of soluble protein via the fused human Fc. PDL1 ko BMDC were used to evaluate binding of soluble PDL-1 as the high expression of PDL-1 receptor on WT-BMDC tended to partially mask the increase in PDL1 expression after soluble PDL1-Ig bound to PD1. Data in Fig 2 are representative examples of at least 3 separate experiments. For flowcytometry panels of BMDC, dead or GR1+ cells were excluded.
Mice
We obtained from NCI labs Balb/c mice and C57BL6 mice (WT-B6) that either expressed CD45.1 or CD45.2 on their leucocytes. B6/S4-IgMko mice, derived from a background of C57BL6 and 129S4 mice (StrainB6; 129S4-Igh-6tm1Che/J) as well as Rag-1 ko derived from a C57BL6 background were obtained from Jackson Labs (14). There was <2 µg/mL IgM in the plasma of B6/S4-IgMko and no detectable IgM in Rag-1ko plasma using an ELISA technique nor could we detect the presence of IgM-ALA when B6/S4-IgMko sera was added to autologous or allogeneic leucocytes (data not shown). In experiments involving B6/S4-IgMko mice, we used litter mates as WT controls i.e. mice with the same C57BL/6 and 129S4 background (referred to as WT-B6/S4). IL-10 ko and IDO ko mice on a C57/BL6 background were from Jackson Laboratories. PDL1 ko mice on C57BL/6 background were a kind gift from Dr Tasuku Honjo (Kyoto University, Japan) and Dr Lieping Chen (Yale University, USA ).
Surgical protocol for kidney ischemia (I) and reperfusion injury (RI)
All experiments were performed in accordance with NIH and Institutional Animal Care and Use Guidelines. The Animal Research Committee of the University of Virginia approved all procedures and protocols. All experiments were performed on 7–10 wk old male mice, weighing approximately 20g. Kidney IRI was performed under anesthesia with bilateral flank incisions as we have previously described (14). Both kidney pedicles, consisting of renal artery and vein, were exposed and cross-clamped for 26 min and then clamps were released and kidneys were reperfused for 24 hrs. Body temperature was maintained at 35 to 36°C (rectal temp) during surgery with heating pads. Kidney pedicles were exposed, but not clamped in sham-operated mice. Post operatively, mice were administered analgesia and maintained at 35 to 37°C.
Assessment of kidney function and histology after renal IRI
Plasma creatinine was determined 24 hours after renal IRI using a colorimetric assay according to the manufacturer’s protocol (Sigma) (14). Kidney histology and quantitation of tubular injury were assessed at 24 hours using previously described techniques (14).
IgM and LPS pretreatment of BMDC
Bone marrow was obtained from murine femurs and cultured in RPMI containing 10% FCS and 2ng/ml of recombinant GM-CSF. Media was changed every 3 days and supplemented with GM-CSF. Non-adherent cells were used and after 8–10 days, >90% of these non-adherent cells expressed moderate to high levels of CD11c/MHC-Class II and <15% were GR1 positive. Pretreatment of BMDC involved culturing 1 × 106 BMDC cells (7–10 days old) in 1ml media in 5ml falcon plastic tubes for 36 to 48 hours in presence of LPS (400ng/ml) with or without IgM (100µg/ml). IgM was added 40 to 60 minutes before LPS. After the culture period, cells were washed x2 to remove unbound LPS and IgM and 0.5 × 106 pretreated BMDC were injected in the tail vein of each mouse. Viability of 48hr pretreated BMDC was >75% as assessed by Live/Dead stain with flowcytometry.
Induction of apoptosis in BMDC
BMDC pretreated with IgM and LPS for 48 hours at 37°C were exposed to 150mJ /cm2 of ultraviolet C irradiation (Stratalinker 2400). Irradiated cells were incubated for 6 hrs at 37°C to allow for IgM binding to apoptotic cells. Cells were washed prior to intravenous injection into mice. More than 75% of cells were apoptotic (Annexin V+) at time of injection.
Pretreatment of mice with BMDC
In these experiments, LPS or IgM pretreated BMDC (0.5 ×106 cells in 0.5ml) were infused via the tail vein. Bilateral renal IRI was performed 24 hours after the infusion. Mice were sacrificed 20 to 24hrs after renal IRI.
Antibodies, soluble recombinant costimulatory receptors and other reagents
The following monoclonal antibodies were used for flowcytometry or immunostaining of tissues and obtained from eBioscience, San Diego, CA: anti-PD1 (clone J43), anti-PDL1 (clone MIH5), anti-CD40 (clone 1C10), anti-CD80 (clone 16-10A1), anti-CD86 (clone GL1), anti-CD11c (clone N418), anti-CD3( clone 145-2C11), anti-CD19 (clone eBio 1D3), anti-GR1 (clone RB6-8C5), anti-IgM (clone II/41). Appropriate fluorochrome conjugated, isotype matched irrelevant mAb were also obtained from eBioscience. Other antibodies obtained from other sources: goat anti-PD1, anti-PDL1, anti-CD40 and anti-CD40L ( R & D systems, Minneapolis, MN), rabbit anti-IDO (Santa Cruz Biotechnology), Isotype IgM control having anti-KLH activity (clone C48-6) was obtained from BD Bioscience, San Jose, CA. anti-Fcα/µR (clone TX61, Biolegend, San Diego,CA), anti-IL10 neutralising Ab (BioXCell, West Lebanon, NH), anti-CD25 depleting Ab (clone PC61, TIB-222- hybridoma), anti-CD20 depleting Ab (clone MB20 (16), Other reagents : LPS- E. Coli 0111:B4 (Sigma-Aldrich, St. Louis, MO), FITC-LPS (Sigma-Aldrich), recombinant fusion proteins PD1-Ig, PDL1-Ig, CD40-Ig, CD40L-Ig, CD80-Ig and CD86-Ig, produced in mammalian cells, were obtained from Sino Biologicals, Beijing, China. All recombinant co-stimulator receptor proteins only contained the extra-cellular domain of the receptors and in addition were fused with human Fc fragment of IgG and histidine tag. CD40L-Ig lacked the histidine tag. ELISA kits for quantitating various cytokines were obtained from eBioscience. Live/Dead fixable Kit was from Life Technology, Grand Island, NY. TUNEL in situ cell death detection kit was obtained from Roche diagnostics, IN.
Immunofluoresence staining of BMDC
9–12 day BMDC were used in these studies. IgM mediated receptor downregulation was evaluated by adding IgM 40 to 60 minutes before LPS at the initiation of the 37°C culture which was terminated after 48 hours. After culture termination, cells were washed and blocked with unlabeled anti-mouse CD16/32 (clone 2.4G2) prior to adding labelled anti-receptor antibody. Binding of IgM to the receptor was evaluated by determining if polyclonal (or isotype control) IgM, when incubated for 1 hour at 4°C to 48 hour LPS activated BMDC, blocked binding of labelled anti-receptor detection antibody or blocked binding of the receptor ligand. In these studies, IgM was added to BMDC that were activated with LPS for 36–48 hrs. Appropriate fluorochrome–conjugated, isotype matched, irrelevant mAbs were used as negative controls. Intracytoplasmic staining was performed using methods as described for splenic leucocytes.
Immunofluorescence staining of splenic leucocytes
Immunofluoresence staining was performed on either unactivated or LPS/anti-CD3 activated spleenocytes. 2 × 105 splenic leucocytes in 0.2ml media were stained with appropriate labelled anti-sera or their isotype control for 40 min at 4°C. Unlabelled anti-CD16/32 was added 30 min prior to adding labelled anti-receptor antibodies to block FcR binding and reduce non-specific binding of labelled anti- receptor antibodies. After a wash step, Live/Dead stain was added and cells were rewashed before fixing with 4% paraformaldehyde. These fixed cells were examined by flowcytometry. In these studies, splenic DC were identified as CD11c high F4/80-GR1− , B cells as CD19+ CD3−, Granulocytes as GR1+F4/80− CD19−. Tregs as CD4+CD25+Foxp3+ and Bregs as CD19+CD5+IL10high.
Immunofluoresence staining of kidney sections
Kidney tissue was fixed and frozen as we have previously described in detail (14). Frozen sections (5µm) of kidney were permeabilized with 0.3% Triton X-100, and non-specific binding was blocked with 10% horse serum and anti-mouse CD16/32. Tissue sections were stained with the following conjugated antibodies: rat anti-neutrophil (clone 7/4, Cederlane Labs, Burlington, NC), rat anti F4/80. Nuclei were visualized using DAPI.
Flowcytometry assay to detect IgM binding to recombinant co-stimulatory receptor protein
Since natural IgM has binding reactivity to human Fc, it became important to remove and block IgM with binding reactivity to human Fc especially since the recombinant receptors were fusion proteins containing human Fc. Hence, in these studies, purified murine polyclonal IgM was initially depleted of IgM anti- human IgG-Fc by interacting the poyclonal IgM with agarose that was precoated with human IgG. IgM (10 µg), depleted of IgM anti-human IgG, was then added to 0.5 million magnetic polystyrene beads precoated with rat IgG anti-murine IgM (Dynal/Life Technologies, Grand Island, NY) to immobilize the polyclonal murine IgM. After a wash step, beads with immobilized IgM were initially incubated with 5µg human IgG-Fc fragment (Jackson Immunoresearch) for 1 hr at 4°C to block any residual IgM with binding reactivity to human Fc. Recombinant fusion protein (4 µg) was then incubated with the immobilized polyclonal IgM for 12 to 14 hours at 4°C. Beads were then washed and stained with a labelled polyclonal anti-receptor antibody to detect the receptor protein that bound to immobilized IgM. Appropriate controls included adding the labelled polyclonal anti-receptor antibody to (i) beads not exposed to IgM but exposed only to recombinant receptor protein or to (ii) beads exposed to isotype monoclonal IgM (anti-KLH) and recombinant receptor protein. In these experiments, each wash step included 3 washes with PBS supplemented with 1% FCS.
Real-time reverse transcriptase-PCR
Kidney specimens were immediately transferred into RNA Later® (Ambion, Austin, TX). RNA and cDNA were prepared from these reagents as we have described previously (14).
Western Blots to detect NF-κB activation in BMDC
Cell lysates isolated from BMDC pretreated with IgM and LPS (400ng/ml) at 37°C for 1 hr were subjected to Western blotting using previously published techniques (17). Tubulin was detected with mouse anti-β-tubulin (clone 3F3-G2, Santa Cruz Biotechnology, Dallas, TX) and a secondary IRDye 800CW labelled goat anti-mouse (LI-COR Biosciences, Lincoln,NE). Phospo-p65NF-κB was detected with a rabbit anti phospho-p65NF-κB and a secondary IRDye 680RD labelled goat anti- rabbit (LI-COR).
LPS activation of splenic cells
LPS (400ng/mL) and/or IgM (30mcg) were added to 400,000 splenic leucocytes in 0.5ml RPMI culture media. IgM was added about 1 hr before LPS. Cells were cultured for 72 hrs at 37°C in 5% CO2. Cytokines in supernatants was quantitated with ELISA kits. Both splenic DC and B cells were activated with LPS.
Statistics
Data were analyzed by 2-tailed t-test unpaired or paired where indicated. P < 0.05 was used to indicate significance.
Results
IgM-ALA exhibits high binding to splenic dendritic cells (DC) and regulates CD40, PD1 and NF-κB expression
In these studies murine splenic leucocytes were pretreated with IgM and binding of IgM to leucocyte subsets was evaluated. Data in Fig 1A, shows that IgM-ALA has higher binding to both non-activated and activated splenic DC and B cells when compared to T cells. The several fold increase in IgM binding to 48 hour activated splenic B cells and BMDC resulted predominantly from IgM binding to cell membrane receptors that were upregulated after cell activation (Fig 1B). However, the increase in IgM binding to both non-apoptotic and apoptotic/dead B cells and BMDC was similar thus suggesting that oxidized neo-determinants exposed on apoptotic cell membranes contributed minimally to the increased binding of IgM. (Fig 1B).
FIGURE 1.

Polyclonal IgM has several fold increased binding to activated splenic B cells and dendritic cells (DC). (A) Splenic leucocytes, either un-activated (left panel) or activated for 48 hours (right panel) were interacted with mouse IgM at 4°C and evaluated for IgM binding using IgG anti-IgM (clone 11/41). Isotype monoclonal IgM with reactivity to KLH did not bind to activated leucocytes (data not shown). IgM binding to B cells was evaluated by blocking intrinsically expressed IgM with unlabeled IgG anti-IgM (clone 11/41). B cells and DC in spleenocytes were activated with LPS while T cells were activated with LPS and soluble anti-CD3. B cells, T cells, and DC were gated by CD19, CD3, and CD11c respectively. (B) Polyclonal IgM has increased binding to non-apoptotic and apoptotic activated splenic B cells and BMDC. Binding of IgM to annexin V positive cells (i.e. which are about 75% propridium (PI) positive) is not increased when compared to non-apoptotic cells. Splenic B cells were activated as in panel A while BMDC were activated for 48 hours with LPS (400ng). Activated cells were interacted with IgM and stained as in Fig 1A. B cells are gated as CD19+.
Since we observed high binding of IgM-ALA to un-activated and LPS activated splenic DC, we next determined if IgM, after binding to DC, modulates antigen presenting receptors and co-stimulatory molecules that are important in initiating an inflammatory response. This question was examined using 6 to 8 day old LPS activated BMDC as splenic leucocytes have very low levels of DC. In these studies, IgM was added to BMDC 30 to 45 min before LPS activation. Firstly, we observed that IgM had minimal or no effect on LPS induced upregulation of antigen presenting receptors (MHC Class-II ) and certain co-stimulatory receptors i.e CD40L, CD86 and PDL1 expressed on BMDC (Fig 2A). Secondly, IgM inhibited the LPS induced upregulation of CD40 (Fig 2B) and this inhibitory effect was observed even when IgM was added 2 hours after LPS activation (Fig 2B). Thirdly, IgM decreased PD1 expression on LPS activated BMDC and this inhibitory effect was not due to IgM blocking the anti-PD1 detection antibody as no decrease in PD1 expression was observed when IgM was added at 4°C to LPS activated cells (Fig 2D). Fourthly, LPS downregulated the expression of CD11c and this inhibitory effect of LPS was not altered by IgM pretreatment (Fig 2A). Finally, LPS and IgM did not alter the low CD45RB expression present on un-activated BMDC (Fig 2A)
FIGURE 2.

(A) Histograms depicting effect of IgM on expression of BMDC receptors. Here, IgM was added 1 hour before BMDC were activated with LPS for 48 hours. (B and D) Polyclonal IgM downmodulates CD40 and PD1 on WT-BMDC. Cells were activated for 48 hrs at 37°C with LPS or LPS and IgM. IgM was added at different times. (− 1hr), (+ 2hr), (+ 48 hr) indicate when IgM was added in reference to LPS i.e. (−) denotes before and (+) denotes after LPS. “(+48 hr, 4°C)” in both panels indicates that IgM was added to 48 hour LPS activated WT-BMDC and incubated at 4°C for 1hour before staining (C) IgM decreases soluble CD40 secretion of LPS activated WT-BMDC. 48 hour supernatants were quantitated for secreted soluble CD40 via ELISA. IgM was added 1hour before LPS activation of cells. (E) Western blot depicting IgM mediated downregulation of LPS induced phosphorylation of p65 NF-κB in BMDC. IgM was added either before or after LPS activation. Data presented in all these figures are representative data of at least 3 to 4 separate experiments. In all experiments, a control monoclonal murine IgM with reactivity to KLH was used for background staining. For panels of BMDC flow cytometry, dead or Gr-1+ cells were excluded. P values were calculated using an unpaired Student’s t test except panel C, which was a paired t Test.
The decreased expression of CD40 during the 48 hour IgM pretreatment, was due to IgM mediated inhibition of LPS induced CD40 upregulation and not due to IgM interfering with the binding of the anti-receptor detection antibody as no decrease in receptor expression was observed when IgM was added at 4°C for 1 hour to BMDC that were previously activated with LPS for 48 hours (Fig 2B). Since BMDC secrete soluble CD40, we also wanted to determine if IgM decreased the secretion of soluble CD40. Fig 2C shows that BMDC secretion of soluble CD40 was also significantly decreased when LPS activated BMDC were cultured for 48 hrs in presence of IgM, thus confirming that the observed decrease in CD40 membrane expression was due to IgM mediated inhibition of CD40 production induced by LPS.
LPS increases phosphorylation of p65NF-κB, which is one of the transcription factors that regulates gene expression of CD40, CD86 and MHC-II (18). Hence, it became important to determine whether IgM mediated inhibition of LPS induced CD40 upregulation, resulted from IgM mediated decrease in p65NF-κB phosphorylation. Data in Fig 2E would support such a mechanism for CD40 downregulation. However IgM did not downregulate CD86 and MHC-II expression (Fig 2A) and it is possible that these receptors are also regulated by other LPS induced transcription factors which are not inhibited by IgM.
Next we examined whether IgM mediated decrease in p65NF-κB phosphorylation resulted from IgM binding to TLR4 or Fcα/µR. Firstly, inhibition of LPS induced p65NF-κB activity, in the presence of IgM, was not due to IgM inhibition of LPS binding to membrane cell receptors as (i) IgM did not inhibit binding of FITC labelled LPS to TLR4 and other membrane cell receptors and did not inhibit LPS induced TLR4 down-modulation (Fig 3A) (ii) IgM did not inhibit several other receptors e.g. MHC II and CD86 upregulated by LPS (Fig 2A) and (iii) IgM inhibited CD40 upregulation even after IgM was added 2 hours after initiation of LPS activation (Fig 2B). Secondly, IgM did not decrease CD40 expression by binding to Fcα/µR as blocking of the Fcα/µR with the receptor blocking antibody (clone TX61), did not prevent CD40 downregulation mediated by IgM (Fig 3B). These data would indicate that IgM inhibits p65NF-κB phosphorylation by binding to other membrane receptors
IgM binds to CD40 and PD1 without inhibiting binding of their natural ligands
We next investigated whether IgM bound to costimulatory molecules, especially CD40 and PD1 that were downregulated by IgM. This question was evaluated by determining whether IgM binds to the recombinant extracellular fragment of these receptors. We therefore used an approach that would preclude binding of polyclonal IgM to human Fc fused to these soluble recombinant receptors and in addition would accommodate the binding characteristics of these polyreactive natural IgM antibodies i.e. binding with relatively low affinity to conformational motifs on antigen and binding with high avidity through polyvalent binding to antigen (4,5). In these studies, polyclonal murine IgM (depleted of IgM with binding to human Fc, see methods) was initially immobilized on polystyrene beads. Soluble recombinant extracellular receptor-Ig was then added to the immobilized IgM and binding of the extracellular receptor to IgM was detected by flowcytometry. With this flowcytometric technique, we could show significant binding of CD40-Ig and PD1-Ig to immobilized polyclonal IgM, but could not demonstrate any binding of PDL1-Ig, CD40L-Ig and CD80-Ig to immobilized polyclonal IgM (Fig 3C). Control polyclonal IgM, depleted of IgM-ALA by leucocyte adsorption, did not bind to soluble immobilized recombinant CD40 and PD1 (Fig 3C) and binding of control polyclonal IgM to recombinant immobilized proteins was similar to that of the monoclonal IgM anti-KLH control (Fig 3C).
We next wanted to determine if IgM binds to a site on CD40 and PD1 that is close to the binding site of their natural ligands. Fig 3D demonstrates no inhibition in binding of CD40L to CD40 or PDL1 to PD1 when LPS activated BMDC pretreated with IgM at 4°C are exposed to these receptor ligands thus indicating that IgM binds to an epitope that is different from the ligand binding site.
Collectively, these studies would indicate that IgM binds to CD40 and PD1 without interfering with the binding of their natural ligands as well as the binding site of the anti-CD40 or anti-PD1 monoclonal antibodies used in flowcytometry. It is therefore possible that IgM modulates BMDC function by binding to BMDC receptors that include CD40 and PD1.
BMDC require in-vitro LPS activation and IgM mediated receptor modulation at 37°C to optimally mediate protection on renal IRI
Reperfusion of blood into an ischemic kidney provokes an innate inflammatory response that enhances kidney tubular injury as detected histologically and with biomarkers for tubular injury i.e. NGAL and Kim-1. Additionally, after renal ischemia reperfusion injury (IRI) there is retention of plasma products (e.g. creatinine) that are normally excreted by the kidney and the severity of renal IRI can be evaluated by quantitating plasma creatinine. In prior studies, we have shown that intravenous infusion of IgM, prior to renal ischemia, increases IgM-ALA plasma levels and protects mice from developing renal IRI by inhibiting the ischemia induced inflammatory response (14). We therefore hypothesized that IgM-ALA inhibits in part, the renal ischemia induced inflammatory response, by switching in-vivo DC to a suppressor phenotype, especially since IgM bound to PD1, CD40 and down-regulated phosphorylation of p65NF-κB and CD40 expression. To test this hypothesis, we infused BMDC pretreated with IgM. Data in Fig 4A indicate that infusion of non-activated WT-B6 BMDC, pretreated with IgM, partially protects mice from renal IRI. However, the protective effect, as determined by plasma creatinine, mRNA for NGAL, Kim-1 and histologically, is more pronounced when IgM pretreated BMDC are also activated with LPS (Fig 4A–C). These data would indicate that BMDC activation is necessary for IgM to optimally induce BMDC to switch to a suppressor phenotype and that cell activation can be induced through TLR4 and other LPS binding receptors. An attractive hypothesis is that LPS activation of DC is needed to upregulate anti-inflammatory cytokines/co-stimulatory receptors and IgM has a role in inhibiting receptors that are important in activating effectors.
FIGURE 4.

(A–C) In-vivo infusion of polyclonal IgM pretreated WT-BMDC protects mice from renal IRI-there is more protection when using WT-BMDC pretreated with both IgM and LPS as evaluated by SCr (A), NGAL and Kim-1 mRNA (B) and H & E histology (C). WT-BMDC for Fig A–C were pretreated with IgM for 1 hour before adding LPS and culturing BMDC at 37°C for 48 hours. (D) LPS activated BMDC pretreated with either mouse or human IgM are protective in renal IRI. (E) IgM pretreated (37°C for 48 hrs) activated splenic B or T lymphocytes are not protective in renal IRI. Magnetic bead isolated pan-B cells were activated with LPS while purified CD3+T cells were activated with insoluble anti-CD3/28. (F) Pretreating 48 hour LPS activated WT-BMDC with IgM for 1hour at 4°C is not protective in IRI. 48hr IgM/LPS pretreated WT-BMDC are non-protective in IRI after being irradiated (3000 Rad) or rendered apoptotic by exposure to UV light. Additionally, polyclonal IgM adsorbed with splenic leucocytes to deplete IgM-ALA clones was not protective in IRI. Data in Fig 3 are means +/− SD for at least 3 mice in each data point. P values were calculated using an unpaired Student’s t Test.
It became important to determine if this protective effect on renal IRI was specific for IgM and/ or BMDC. Data in Fig 4D, shows that protection is specific for IgM as pretreatment of BMDC with either murine or human IgM but not murine IgG protected mice from renal IRI. Secondly, data in Fig 4E shows that IgM mediates its protective effect on renal IRI when LPS activated BMDC, both autologous and allogeneic (Balb/c), are pretreated with IgM and not when LPS activated B cells or anti-CD3/28 activated T cells are pretreated with IgM. Hence these data would indicate that BMDC are rendered protective or switched to a suppressor phenotype when IgM binds to certain membrane receptors on either autologous or allogeneic BMDC.
Since 25 to 30% of infused 48 hour cultured BMDC were apoptotic (Fig 1B), it became important to determine if IgM mediated protection was due to IgM-ALA that bound to apoptotic cells e.g by IgM anti-PC as previously described (3, 19). To test for this possibility, we used 48 hour IgM +LPS pretreated BMDC that were rendered >75% apoptotic with UV light exposure. BMDC were not washed for another 6 hours after UV exposure so as to maximize IgM binding to apoptotic cell receptors. Such IgM pretreated apoptotic cells failed to protect mice from renal IRI (Fig 4E), thus clearly indicating that protection was not mediated by IgM that bound to apoptotic BMDC cells and that IgM pretreated BMDC require to be alive to mediate protection.
Since optimal protection mediated by IgM pretreated BMDC required both LPS activation and binding of IgM to non-apoptotic BMDC receptors, we wanted to investigate if protection, after binding of IgM to BMDC, requires that IgM bind to BMDC receptors and alter or modulate BMDC function and/or phenotype. Three approaches were used to examine this question. Firstly, BMDC were activated with LPS for 48 hours and cells were then incubated with IgM at 4°C for 1 hour to prevent IgM mediated alteration in BMDC function and/or phenotype. Such LPS activated BMDC pretreated with IgM in the cold, failed to protect WT-B6 mice from renal IRI (Fig 4F), clearly demonstrating that IgM, after binding to BMDC receptors, requires to modulate BMDC function and/or phenotype to induce BMDC to acquire a suppressive or regulatory phenotype. In a second approach, 48 hour IgM/LPS pretreated BMDC were irradiated (3,000 rad) and one hour later were intravenously injected into mice. Such irradiated BMDC failed to protect mice from renal IRI (Fig 4F) even though at the time of intravenous injection, these irradiated BMDC exhibited no change in viability or co-stimulatory receptor expression when compared to non- irradiated IgM/LPS pretreated BMDC. These latter findings indicate that BMDC, after IgM pretreatment, also acquire a radiosensitive product that is necessary to mediate suppression. Thirdly, we tested if a control polyclonal IgM, depleted of IgM-ALA including IgM anti-CD40 and anti-PD1 (Fig 3C) was protective in renal IRI. Such leucocyte adsorbed IgM failed to protect mice from renal IRI (Fig 4E), thus indicating that protection mediated by polyclonal IgM requires IgM with high IgM-ALA that includes IgM anti-CD40 and anti-PD1.
Switching LPS activated BMDC to a suppressor phenotype requires IgM induced downregulation of phospho p65NF-κB and CD40
Since maximum protection from renal IRI required BMDC activation and pretreatment with IgM, we hypothesized that BMDC require LPS to switch BMDC from an immature to a mature phenotype and IgM to switch mature BMDC to a regulatory phenotype by inhibiting LPS induced CD40 upregulation. Two approaches were used to determine if IgM mediated CD40 downregulation was necessary. In the first approach, IgM mediated CD40 downregulation was inhibited by simultaneously adding IgM together with the monoclonal anti-CD40 agonistic antibody (clone 1C10) prior to LPS activation (see Fig 5A). In the second approach, CD40 expression was increased on BMDC by adding soluble CD40-Ig together with IgM at the initiation of LPS activation (see Fig 5B). As depicted in Fig 5C and D, the two approaches to increase CD40 expression in the presence of IgM, also activated phosphorylation of p65NF-κB and prevented IgM/LPS pretreated BMDC from protecting kidneys thus indicating that IgM induced downregulation of phosphorylated p65NF-κB and CD40 is required to switch LPS activated BMDC to a regulatory phenotype.
FIGURE 5.

BMDC that protect renal IRI in WT-B6 mice require decreased CD40 membrane expression. Increasing CD40 expression on IgM/LPS BMDC by either preventing CD40 downregulation with the agonistic anti-CD40 antibody (clone 1C10) (A) or by adding soluble CD40-Ig to 48 hour IgM/LPS activated cells (B), inhibits the IgM mediated protection after renal IRI (C) as well as inhibits the IgM mediated downregulation of phospho p65NF-κB (D). Anti-CD40 or soluble CD40-Ig (20µg) was present throughout the 48 hour IgM/LPS pretreatment of BMDC. Addition of isotype control IgG antibody (for clone 1C10) or human IgG (for CD40-Ig) to IgM/LPS pretreated WT-BMDC did not negate the protective effect of these BMDC (data not shown). Dead or Gr-1+ cells were gated out in all panels. P values were calculated using an unpaired Student’s t Test.
Regulatory BMDC require PD1 and IL10 but not IDO to inhibit in-vivo inflammation
We next investigated the inhibitory mechanisms utilized by IgM/LPS pretreated BMDC to exert an anti-inflammatory effect in-vivo. Potential candidates included inhibitory cytokines and costimulatory molecules i.e. IL10, IDO and PD1/PDL1. We initially investigated if the cytokine profile of BMDC that mediated protection in renal IRI favored increased production of IL10 over IL12p40. Hence supernatants from 48 hour cultured BMDC, to be used in in-vivo IRI studies, were examined using ELISA. After LPS activation, BMDC secreted several fold more IL10 as well as IL12p40. However, the protective effect of IgM pretreated BMDC on IRI could not be predicted by IL10/IL12p40 levels or by their ratio in the supernatants (Fig 6A). As depicted in Fig 6A, there was no significant difference in supernatant levels of IL10 or IL12p40 after 48 hours of culture between LPS pretreated versus IgM/LPS pretreated WT-BMDC.
FIGURE 6.
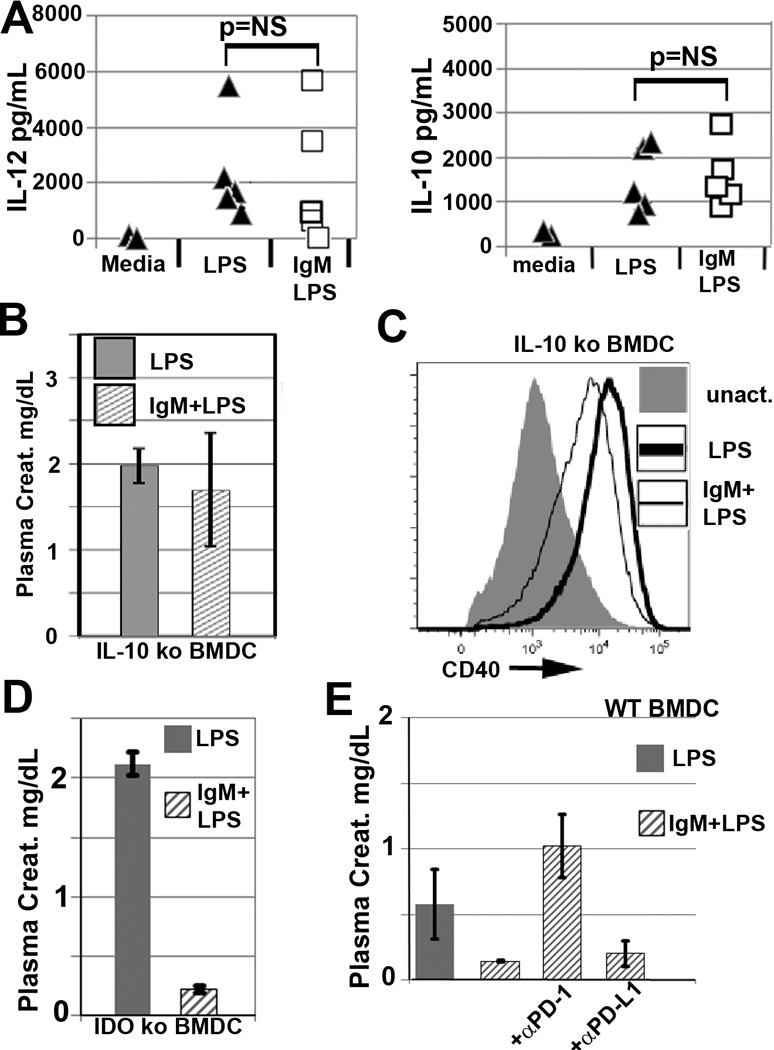
(A) There is no significant difference by a paired t-test when comparing the increase in secreted IL10 and IL12p40 levels present in LPS vs IgM/LPS pretreated BMDC 48 hour culture supernatants. BMDC, in these studies were used in-vivo and some of the data are depicted in Fig 3A. Each supernatant data point is from a different experiment depicting the mean supernatant level of BMDC that were tested with at least 3 mice in each experiment. Cytokine levels were quantitated by commercially available ELISA assays. (B) IL10ko BMDC pretreated with IgM/LPS for 48 hours at 37°C do not acquire a protective phenotype despite CD40 downregulation (C). (D) IDO ko BMDC, pretreated with IgM/LPS for 48 hours at 37°C, retain their ability to acquire a protective phenotype. (E) IgM/LPS pretreated WT-BMDC require the PD1 (but not the PDL1) ligand binding site to mediate their in-vivo anti-inflammatory effect. In these studies, anti-PD-1(clone J43) and anti-PDL1(clone M1H5) antibodies, known to block ligand binding, were added to WT-BMDC that were previously pretreated for 48 hours with IgM/LPS. Blocking antibodies were incubated with BMDC for 1 hour at 4°C, washed and then injected i.v. into mice. Blocking with isotype control antibodies did not alter the protective effect of IgM/LPS pretreated BMDC (data not shown). Each renal IRI data point represents a mean +/− SD from 3–4 mice.
We next investigated whether activated BMDC, with the protective phenotype, required IL10 to mediate protection. This possibility needed to be examined as maximal IgM mediated protection required LPS activation which upregulates both pro and anti-inflammatory cytokines, especially IL10. To test this possibility, BMDC from IL10 ko mice were pretreated with IgM and activated with LPS prior to in-vivo infusion of WT-B6 mice. As depicted in Fig 6B, IgM/LPS pretreated BMDC from IL10 ko mice, failed to protect WT-B6 mice from renal IRI even though IgM downregulated CD40 (Fig 6C). These data would indicate that protection mediated by IgM/LPS pretreated BMDC requires CD40 downregulation and IL10. However it is not necessary for protective BMDC to favor increased production of IL10 over IL12 to mediate this protective effect.
Next, we investigated whether IgM/LPS pretreated BMDC needed the presence of indoleamine 2,3-dioxygenase (IDO) to mediate protection in renal IRI. Both activation of BMDC with LPS and the in-vivo engagement of Treg CTLA-4 with CD80/CD86 on activated BMDC, increase the generation of IDO in the infused BMDC. To examine this question, BMDC from IDOko mice were pretreated with IgM and activated with LPS prior to in-vivo infusion into WT-B6 mice. As depicted in Fig 6D, IgM/LPS pretreated BMDC from IDO ko mice, protected mice from renal IRI thus indicating that IgM/LPS pretreated BMDC did not require IDO or in-vivo interaction with CTLA4 to mediate protection in this innate inflammatory model.
Finally, we investigated whether the IgM/LPS pretreated BMDC required PD1 in addition to CD40 downregulation to mediate an anti-inflammatory effect when injected in-vivo. It became important to study this question especially since IgM bound to PD1 and downregulated this receptor without affecting binding of its ligand, PDL1 (see Fig 1C and 2C,D). This question was examined by using non-agonistic antibodies that are known to block the ligand binding site of either PD1or PDL1 that are co-expressed on these regulatory BMDC. In these studies, receptors on 48 hour IgM/LPS pretreated BMDC were blocked with anti-PD1 or PDL1 antibodies for 1hour at 4°C just before these cells were injected into mice. Fig 6E demonstrates that IgM/LPS pretreated regulatory BMDC require PD1 in-vivo, but not PDL1, to inhibit the in-vivo innate inflammatory response in renal IRI. These studies do not however address the functional role of IgM binding to an epitope on PD1 that is distinct from the binding site of PDL1.
IgM pretreated BMDC require in-vivo regulatory B/T cells and their products to mediate protection against renal IRI
After renal ischemia, DC in the ischemic organ are activated and present endogenously released lipid and glycolipid antigens to NKT cells which in turn activate the innate inflammatory cells, primarily granulocytes and macrophages. The extent of the inflammatory response is regulated by B and T cells with regulatory activity and by their anti-inflammatory products (IL10, natural IgM). We therefore wanted to determine if IgM/LPS pretreated BMDC, when administered in-vivo, mediate protection by directly regulating endogenous DC or other innate inflammatory cells (granulocytes/macrophages/NK cells) or by enhancing regulatory B/T cells and their products. This question was investigated by using RAG-1 ko mice which are not protected from renal IRI (20, 21, 22). As depicted in Fig 7A, IgM/LPS pretreated BMDC were not protective in Rag-1 ko mice, thus indicating that protection requires the presence of B and T cells and/or their products. The latter was examined in WT-B6 mice by depleting IL-10 with an IL10 neutralising antibody, B cells (including Bregs) with anti-CD20 or CD25 positive T cells (including Tregs) with anti-CD25. The IgMko mouse was used to evaluate the role of circulating natural IgM. As depicted in Fig 7B, depletion of any one of these regulators abolished the protective effect of IgM pretreated BMDC on renal IRI. These data would indicate that IgM pretreated BMDC require the presence of circulating IgM, IL-10, B cells and CD25+ T cells to mediate its protective effect. It is possible that IgM pretreated BMDC mediate their protective effect by enhancing the function of these known B/T regulatory cells e.g. by increasing production of IL10 in B/T regulatory cells and inducing B1 cells to increase production of natural IgM.
FIGURE 7.

In-vivo protection mediated by IgM pretreated BMDC require the presence of B and T cells, IL10 and circulating IgM. (A) IgM/LPS pretreated WT-BMDC are not protective in RAG-1ko mice. (B) IgM/LPS pretreated WT-BMDC, when injected into WT-B6 mice, are not protective when these mice are depleted of IL10, B cells and CD25 pos T cells (includingTregs). B cells and CD25pos T cells were depleted 5 days prior to renal IRI by intravenously injecting anti-CD20 (20µg /mouse, clone MB20-11) and anti-CD25 (200µg /mouse, clone PC61) respectively. Anti-IL10 (200µg/mouse) was administered intravenously 24 hours before IRI. Isotype control antibodies did not negate the protective effect of IgM/LPS pretreated WT-BMDC (data not shown). IgMko mice were used to test the protective effect of IgM/LPS pretreated BMDC on renal IRI in IgM deficient mice. Each renal IRI data point represents a mean +/− SD from 3 to 4 mice.
IgM pretreated BMDC protect against Renal IRI by inhibiting leucocyte response to the initial ischemic injury
Renal ischemia leads to release into plasma of DAMPS as well as TLR upregulation in endothelial and renal tubular cells. Activation of TLR by DAMPS leads to increased production of chemokines (e.g. CXCL-1 and MCP-1) by ischemic tubules and up-regulation of adhesion and integrin molecules (e.g ICAM-1) on endothelial cells. DAMPS also mediate activation of DC locally (in the ischemic organ) and systemically. Mature DC present glycolipids to NKT cells which when activated, release pro-inflammatory cytokines to activate and recruit inflammatory cells (23). All these events, especially NKT cell activation, lead to a rapid increase in inflammatory cells, in particular granulocytes, to enter the circulation from bone-marrow and spleen. The activated circulating inflammatory cells express increased levels of integrins to facilitate their adhesion and migration through the ischemic capillaries into the interstitium causing significantly more tubular injury. We wanted to determine if IgM pretreated BMDC protected against renal IRI by inhibiting the inflammatory response during the reperfusion phase.
We show that IgM/LPS pretreated BMDC mediates protection in renal IRI by inhibiting the leukocyte response to the pro-inflammatory mediators produced by the initial ischemic injury (see Fig8C, 3 hrs). This is evidenced by the presence of significantly fewer circulating granulocytes (Fig.8A), as well as minimal CD45+ and GR1+ leucocyte infiltration and decreased ICAM-1 and TLR-4 in the protected kidneys at 20 hours post IRI (fig 8B,C), despite the persistent presence of chemokine mRNA (i.e. CXCL-1 and MCP-1) in the protected ischemic kidney at 20 hours (Fig 8C). However, the current studies do not address how these regulatory IgM/LPS pretreated BMDC inhibit the in-vivo ischemia induced leukocyte response.
FIGURE 8.

There is significantly less inflammatory cell response 20 hrs post ischemia in mice administered IgM/LPS pretreated BMDC as determined by circulating granulocytes (CD45+, GR1+) in blood (see A) and infiltrating CD45+ and GR1+ leucocytes in kidneys (see B). Blood granulocytes in Fig 8A were quantitated from 50 µl of blood with flowcytometry (gated for GR1+ cells) and using Count Bright absolute counting beads (Life Technologies). (C) Kidneys, harvested at 3 hours post ischemia, reveal no significant difference in hypoxia induced mRNA levels for NGAL, TLR4, ICAM-1 and MCP-1 between both groups of mice. However, kidneys harvested at 20 hours post-ischemia reveal no significant decrease in mRNA levels for CXCL1 and MCP-1. Data in this figure are obtained from 3 to 6 mice.
Analysis of splenic cell lymphocyte subpopulations with flowcytometry, either 24 hours after administering BMDC or 20 to 24 hours after the ischemic injury, revealed no differences in absolute numbers of Tregs, Bregs, NKT, B and T cell subsets between the two groups thus indicating that the decreased inflammatory response in mice infused with IgM/LPS pretreated BMDC was not due to an increase in regulatory cells (data not shown). Finally, the decreased inflammatory response in mice infused with IgM/LPS pretreated BMDC cannot be attributed to less tubular damage, induced by the initial hypoxic event, as at 3 hours post ischemia i.e well before infiltration of inflammatory cells, both groups of mice had similar upregulation for hypoxia induced mRNA in their kidneys i.e for NGAL, TLR4, MCP-1 and ICAM (Fig 8C). Taken together, these data would indicate that IgM/LPS pretreated BMDC exert an anti-inflammatory response without necessarily increasing Foxp3+ Tregs or Bregs but through other mechanisms that inhibit the generation of inflammatory cells. Furthermore, infusion of BMDC, pretreated with IgM/LPS, do not protect renal tubular cells from the initial hypoxia induced injury.
Discussion
The current studies were initiated to determine the mechanism by which polyclonal natural IgM inhibits the inflammatory response following renal IRI. We hypothesized that one potential mechanism involved binding of IgM to receptors on dendritic cells (DC) especially since we noted high IgM binding to splenic DC. To test our hypothesis, we pre-treated LPS activated BMDC with polyclonal IgM and demonstrated that 0.5 × 106 of these BMDC, when intravenously injected into WT-B6 mice, 24 hours before renal IRI, protected mice from developing acute kidney injury (AKI). IgM/LPS pretreatment switched the function of BMDC to a “regulatory” DC as evaluated by their in-vivo inhibitory effect on suppressing innate inflammation mediated by renal IRI. Switching of BMDC to a regulatory phenotype requires IgM to actively modulate these cells as the functional change in BMDC occurred only when IgM was added at the initiation of LPS activation and not when IgM was added to BMDC that were previously activated for 48 hours with LPS. This functional change in BMDC is dependent on IgM binding to membrane receptors that inhibit LPS induced phosphorylation of p65NF-κB and CD40 upregulation. The current studies have not identified the IgM binding receptors that are involved in switching non-apoptotic BMDC to a regulatory phenotype. It is possible that IgM induces regulatory function in BMDC by binding to CD40 and PD1. Additionally, we show that the IgM/LPS pretreated regulatory BMDC require PD1, IL10 but not IDO, to mediate their in-vivo inhibitory effect. However, IgM/LPS pretreated BMDC do not require IL10 production to be in excess of IL12 and do not require downregulation of MHC-II or CD80/CD86 to switch to a suppressive phenotype. Importantly, protection from renal IRI is associated with minimal or no increase in circulating granulocytes and with minimal leucocyte infiltration in the ischemic kidney thus indicating that BMDC with the suppressive phenotype mediates protection by inhibiting the ischemia induced innate inflammatory response. Further studies are needed to delineate the in-vivo mechanism by which IgM/LPS induced regulatory BMDC inhibit the ischemia induced innate inflammatory cell response. We however show that suppression of the innate inflammatory cell response by the suppressive BMDC requires the presence of other in-vivo regulatory mechanisms i.e. Tregs, B cells including Bregs, IL10 and circulating IgM. However, protection mediated by IgM/LPS pretreated BMDC was not associated with an increase in Foxp3+ Tregs in the spleen. IgM/LPS pretreated regulatory BMDC were CD11clo, CD45RB+, CD40lo, PD1lo, PDL1hi, CD86hi and MHC-IIhi. Finally we show that both human and murine IgM have the same functional effect on murine BMDC thus indicating that these largely non-mutated and poly-reactive natural autoantibodies are evolutionarily conserved and possibly bind to innate cell receptors that are also evolutionarily conserved.
These studies add to the growing literature where fully mature DC with regulatory function have been shown to exist and studied in-vivo and in-vitro and generated for therapeutic purposes (reviewed in 24, 25). These regulatory DC have been shown to normally exist in the gut and lung mucosa primarily for inducing immune tolerance towards the bowel and lung flora. Mature DC with regulatory function has also been shown to suppress auto and allo-immune inflammatory processes especially in murine models. Additionally, mature DC with regulatory function, induced by several different cancer cells or by certain infectious agents e.g hepatitis C, listeria and leishmaniasis, have also been shown to inhibit immune effector cells involved in the destruction of tumor cells or these infectious agents thus providing a mechanism for tumors to escape immune-surveillance and for infections to chronically persist in-vivo. DC with regulatory function can also be induced in-vitro by exposure of DC to pharmacological agents or cytokines e.g. dexamethasone, rapamycin, vitamin D3, adenosine A2a receptor agonists or IL10 (23, 25). The phenotypic characteristics of DC with regulatory function, whether induced in-vivo or in-vitro, has been found to be heterogeneous with regard to expression of antigen presenting receptors (MHC II, CD1d) , costimulatory molecules and cytokine production. However, in most studies, DC with regulatory function have decreased expression of CD40, CD80, CD86, IL12 and TNFα and an increase in IL10, TGFβ, PGE2,IDO and PD-1 (24, 25, 26). Very little is known about the mechanisms by which infused regulatory DC mediate an anti-inflammatory effect in-vivo. In most studies, mature DC with regulatory function have been shown to induce and increase Tregs (both Foxp3+ and Foxp3-, IL10+) and to enhance deletion/apoptosis of effector T cells (reviewed in 24, 25). Additionally, very little is known regarding the molecular mechanisms that can switch mature DC to DC with regulatory function, especially in-vivo. TGFβ produced by intestinal epithelial cells has been shown to foster the generation of regulatory DC, which induces tolerance towards the normal bowel flora. Similarly, cancer cells can secrete IL10, TGFβ and PGE2, which has been shown to change the functional state of mature DC and T cells.
Observations in the current studies provide another potential physiological mechanism for reprogramming or switching the function of mature DC towards DC with regulatory function. Natural IgM can inhibit upregulation of CD40 expression that occurs with LPS activation of BMDC. Downregulation of CD40, mediated by natural IgM, together with upregulation of IL10 that occurs with LPS activation of BMDC, appears to be necessary to switch mature BMDC towards DC with regulatory function as tested in renal IRI. Increasing membrane CD40 expression on IgM/LPS pretreated BMDC with either soluble recombinant CD40-Ig or with the agonistic anti-CD40 antibody, switched these intrinsically regulatory BMDC to an immune-stimulatory BMDC (fig 5C). Regulatory DC, generated in-vitro with pharmacological agents or cytokines, were found to also express lower levels of CD40 and these findings would add further support to the concept that CD40 downregulation is involved in switching mature BMDC to regulatory BMDC (23, 24). We also show that mere binding of IgM to BMDC at 4°C i.e without downregulating CD40, is not sufficient to switch mature BMDC towards DC with regulatory function (Fig 4F). Secondly, data in Fig 4F would indicate that IgM/LPS activated BMDC acquire other radiosensitive factors besides IgM induced receptor modulation before they can be switched to a regulatory cell. Finally, data in Fig 6 would indicate that IgM/LPS activated BMDC require both IL-10 and PD1 to regulate innate inflammation.
Based on the prevailing paradigm, DC with a regulatory phenotype produce high levels of IL10 and have lower IL12p40 levels (24, 25). However, in our studies, the IgM/LPS pretreated regulatory WT-BMDC protected kidneys from innate inflammation despite no predominance of IL10 over IL12p40 (Fig 6A). Levels of both these cytokines were increased by several fold over unactivated BMDC but there was no difference in the levels of these cytokines between IgM/LPS pretreated BMDC and LPS pretreated BMDC. IL10 was however required for BMDC to switch to a regulatory BMDC as IL10ko BMDC could not be switched to a regulatory phenotype despite pretreatment with IgM, which downregulated CD40 expression (see Fig 6B and C). Our studies would therefore indicate that regulatory bone marrow dendritic cells, involved in controlling innate inflammation, require increased levels of IL10. However, our studies would also indicate that increased levels of IL12p40 do not appear to negate the function of these regulatory BMDC.
The current studies provide some insight into the mechanism for inhibition of the in-vivo innate inflammatory response after administering only 0.5 ×106 regulatory BMDC. IgM/LPS pretreated BMDC failed to protect RAG-1 ko mice against renal IRI (Fig 7A), thus indicating that these regulatory DC require B and/or T cells in-vivo to mediate their protective effect. Additionally, data from the RAG-1ko mice would also indicate that the administered IgM/LPS pretreated BMDC do not directly regulate the resident DC (present in the kidney or spleen or other hematopoietic organs) or effector cells (i.e monocytes, NK cells and granulocytes) mediating inflammation in Rag-1ko mice. In further studies, we show that these administered regulatory BMDC require in-vivo CD25 positive T cells (including Tregs) and B cells and their secretory products (including IgM and IL10) to regulate the innate inflammatory response (Fig 7B). Furthermore, in the current studies, IgM/LPS pretreated BMDC inhibited the ischemia induced innate inflammatory response even though we did not observe an increase in Foxp3+ Tregs in the spleen or show T reg induction in-vitro when IgM/LPS pretreated BMDC were co-cultured with autologous CD4+ T cells in presence of IL2/TGFβ and insoluble anti-CD3/28 (data not shown). Our findings differ from that of other studies, where in autoimmune and allogeneic mediated inflammatory models, regulatory DC, generated by pretreating DC with pharmaceutical agents and/or inhibitory cytokines, induced an increase in Tregs (24, 25). It is possible that there was not enough time in our experimental model for the administered BMDC to increase Tregs, especially since BMDC were administered just 24 hours before performing renal IRI. Another possibility is that regulatory DC could inhibit innate mediated inflammatory responses through other mechanisms. For example, in a similar murine model of renal IRI, 0.5 × 106 regulatory BMDC, generated by pretreatment with an A2aR agonist, suppressed the innate inflammatory response by altering the function of effector cells through mechanism that did not increase Tregs (17). Data in our studies would indicate that the infused IgM/LPS pretreated regulatory BMDC instruct the host regulatory mechanisms via production of anti-inflammatory cytokines (e.g IL10) and the PD1 receptor. A key finding in the current study is the demonstration that PD1 on BMDC is directly involved in suppressing the in-vivo innate inflammatory response. Blocking PD1 on BMDC, previously pretreated with IgM/LPS, negated the protective effect of these regulatory BMDC on renal IRI (see Fig 6E).
Besides providing another technique to develop regulatory DC for therapeutic purposes, these studies could also provide further insight into other mechanisms by which natural IgM-ALA regulates inflammation. We have shown in prior in-vitro studies that IgM-ALA i) inhibits human and murine T cell activation and proliferation in response to anti-CD3 or alloantigens by binding and downregulating CD4 and also by binding to CD3 and inhibiting Zap70 phosphorylation in response to anti-CD3 ii) enhances generation of human and murine Foxp3+ T regs and iii) binds to leucocyte CCR5 and CXCR4 and inhibits leucocyte chemotaxis in response to chemokines (4, 14). The current studies provide another potential mechanism for IgM-ALA to regulate inflammation i.e reprogramming mature activated DC to switch to regulatory DC.
Two observations would lend support for a regulatory mechanism that requires IgM-ALA in addition to other regulatory mechanisms for suppressing inflammation mediated by B cells, NK and T cells as for example in innate and other inflammatory states. Firstly, in the last 40 years, several clinical observations have shown a strong association between increase in IgM-ALA levels and diverse inflammatory disorders e.g infections with different agents and in chronic inflammation such as sarcoidosis, SLE and ESRD (6, 7, 8, 27–30). Levels of IgM-ALA decrease with control of the inflammatory state (27). Secondly, the IgM ko mouse provides another example for the requirement of IgM-ALA in addition to other regulatory mechanisms to control inflammation. This IgM ko mouse has B cells that secrete normal levels of all immunoglobulins except for IgM, has membrane IgM on B cells for antigen recognition and has normal levels of Bregs and Tregs. We have shown that this mouse develops (i) severe renal IRI with minimal ischemia that is insufficient to cause renal IRI in their WT littermates and (ii) develop accelerated and more severe rejection when compared to their WT littermates after transplantation with cardiac allografts mismatched at only the IA MHC locus (14). Replenishing these mice with intravenous IgM that contains IgM-ALA ameliorates the inflammatory process (14). We propose the following hypothesis based on our findings. Ischemic injury or infectious agents activate DC with upregulation of costimulatory molecules (CD80/86, CD40, PD1/PDL1), cytokine production (IL10, IL12) and antigen presenting receptors. B1cells are also activated via TLR especially with infectious agents, and increase production of IgM-ALA, which switches activated mature DC to a regulatory phenotype by undefined mechanisms that include downregulation of p65NF-κB phosphorylation and CD40. Regulatory BMDC require PD1 and IL10 to mediate suppression and together with other in-vivo regulatory mechanisms (e.g.Bregs, Tregs, IL10 and IgM) regulates the inflammatory process.
Footnotes
This work was supported by National Institutes of Health Grant R01DK083406-01A1 (to P.I.L and M.D.O) and K01DK091444 (AB) and by a National Kidney Foundation Fellowship and an American Heart Association career development grant, 11SDG7000007 (AB).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
References
- 1.Lobo PI, Brayman K, Okusa MD. Natural IgM Anti-leucocyte Autoantibodies (IgM-ALA) Regulate Inflammation Induced by Innate and Adaptive Immune Mechanisms. J of Clin. Immunol. 2014;34(Suppl 1):S22–S29. doi: 10.1007/s10875-014-0027-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayakawa KI, Asano M, Shinton SA, Gui M, Allman D, Stewart CL, Silver J, Hardy RR. Positive selection of natural autoreactive B cells. Science. 1999;285(5424):113–116. doi: 10.1126/science.285.5424.113. [DOI] [PubMed] [Google Scholar]
- 3.Grönwall C, Vas J, Silverman G. Protective Roles of Natural IgM Antibodies. Front Immunol. 2012;3:66. doi: 10.3389/fimmu.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lobo PI, Schlegel KH, Spencer CE, Okusa MD, Chisholm C, Mchedlishvili N, Park A, Christ C, Burtner C. Naturally occurring IgM anti-leukocyte autoantibodies (IgM-ALA) inhibit T cell activation and chemotaxis. J of Immunol. 2008;180:1780–1791. doi: 10.4049/jimmunol.180.3.1780. [DOI] [PubMed] [Google Scholar]
- 5.Baxendale HE, Johnson M, Stephens RCM, Yuste J, Klein N, Brown JS, Goldblatt D. Natural human antibodies to pneumococcus have distinctive molecular characteristics and protect against pneumococcal disease. Clin. Exptl. Immunol. 2007;151:51–60. doi: 10.1111/j.1365-2249.2007.03535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terasaki PI, Mottironi VD, Barnett EV. Cytotoxins in disease. N. Eng. J. Med. 1970;283:724–727. doi: 10.1056/NEJM197010012831403. [DOI] [PubMed] [Google Scholar]
- 7.Winfield JB, Winchester RJ, Wernet P, Fu SM, Kunkle HG. Nature of cold-reactive antibodies to lymphocyte surface. Arthritis Rheum. 1975;18:1–8. doi: 10.1002/art.1780180101. [DOI] [PubMed] [Google Scholar]
- 8.Lobo PI. Nature of autolymphocytotoxins present in renal hemodialysis patients. Transplantation. 1981;68:1855–1858. doi: 10.1097/00007890-198109000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Klouda PT, Jeannet M. Cold and warm antibodies and graft survival in kidney allograft recipients. Lancet. 1976:1876–1878. doi: 10.1016/s0140-6736(76)92094-8. [DOI] [PubMed] [Google Scholar]
- 10.Lobo PI, Rudolf L, Westervelt FB. Enhanced kidney allograft survival across a positive crossmatch (Cx) arising from B-cell specific and cold reactive antibodies. Proc. Dial, Transpl. Forum. 1977;7:4–6. [PubMed] [Google Scholar]
- 11.Iwaki Y, Park PI, Terasaki MS, Billing R. Enhancement of human kidney allografts by cold B-lymphocyte cytotoxins. Lancet. 1978;1:1228–1229. doi: 10.1016/s0140-6736(78)92464-9. [DOI] [PubMed] [Google Scholar]
- 12.Kerman RH, Susskind B, Buyse I. Flow cytometry-detected IgG is not a contraindication to renal transplantation: IgM may be beneficial to outcome. Transplantation. 1999;68:1855–1858. doi: 10.1097/00007890-199912270-00007. [DOI] [PubMed] [Google Scholar]
- 13.Przybylowski PM, Balogna M, Radovancevic B, Frazier OH, Susskind B, Van Buren C, Katz S, Kahan BD, Kerman R. The role of flow cytometry-detected IgG and IgM anti-donor antibodies in cardiac allograft recipients. Transplantation. 1999;67:258–262. doi: 10.1097/00007890-199901270-00012. [DOI] [PubMed] [Google Scholar]
- 14.Lobo PI, Bajwa A, Schlegel KH, Vengal J, Lee SJ, Huang L, Ye H, Deshmukh U, Wang T, Pei H, Okusa MD. Natural IgM anti-leukocyte autoantibodies attenuate excess inflammation mediated by innate and adaptive immune mechanisms involving Th-17. J. of Immunol. 2012;188:1675–1685. doi: 10.4049/jimmunol.1101762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chhabra P, Schlegel K, Okusa MD, Lobo PI, Brayman K. Naturally Occurring Immunoglobulin M (nIgM) Autoantibodies Prevent Autoimmune Diabetes and Mitigate Inflammation After Transplantation. Annals of Surgery. 2012;256(4):634–641. doi: 10.1097/SLA.0b013e31826b4ba9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamaguchi Y, Uchida J, Cain DW, Venturi GM, Poe JC, Haas KM, Tedder TF. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J. of Immunol. 2005;7:4389–4399. doi: 10.4049/jimmunol.174.7.4389. [DOI] [PubMed] [Google Scholar]
- 17.Bajwa A, Huang L, Kurmaeva E, Gigliotti J, Ye H, Miller J, Rosin D, Lobo PI, Okusa MD. Sphingosine 1-Phosphate-3 Deficient Dendritic Cells Modulate Splenic Responses to Ischemia-Reperfusion Injury. J. Am Soc Nephrol. 2015;27 doi: 10.1681/ASN.2015010095. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinz M, Löser P, Mathas S, Krappmann D, Dörken B, Scheidereit C. Constitutive Nf-kB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hidgkin/Reed-Sternberg cells. Blood. 2001;97:2798–2807. doi: 10.1182/blood.v97.9.2798. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Khanna S, Goodyear CS, Park YB, Raz E, Thiel S, Gronwall C, Vas J, Boyle DL, Corr M, Kono DH, Silverman GJ. Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis. J. of Immunol. 2009;183:1346–1359. doi: 10.4049/jimmunol.0900948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ. Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am J Physiol Renal Physiol. 2002;282(2):F352–F357. doi: 10.1152/ajprenal.00160.2001. [DOI] [PubMed] [Google Scholar]
- 21.Burne-Taney MJ, Yokota-Ikeda N, Rabb H. Effects of combined T- and B-cell deficiency on murine ischemia reperfusion injury. Am J Transplant. 2005;5(6):1186–1193. doi: 10.1111/j.1600-6143.2005.00815.x. [DOI] [PubMed] [Google Scholar]
- 22.Gigliotti JC, Huang L, Ye H, Bajwa A, Chattrabhuti K, Lee S, Klibanov AL, Kalantari K, Rosin DL, Okusa MD. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J. Am Soc Nephrol. 2013;24(9):1451–1460. doi: 10.1681/ASN.2013010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Huang L, Ye H, Song S, Bajwa A, Lee SJ, Moser E, Jaworska K, Kinsey G, Day Y, Linden J, Lobo PI, Rosin D, Okusa MD. Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. Journ. Clin. Investigation. 2012;122(11):3931–3942. doi: 10.1172/JCI63170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt S, Nino-Castro A, Schlultze J. Regulatory dendritic cells: there is more than just immune activation. Frontiers in Immunol. 2012;3:274. doi: 10.3389/fimmu.2012.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raïch-Reguéa D, Glancya M, Thomson A. Regulatory dendritic cell therapy: from rodents to clinical application. Immunology Letters. 2014;161(2):216–221. doi: 10.1016/j.imlet.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, Goode EL, Kalli KR, Knutson KL. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol. 2011;186(12):6905–6913. doi: 10.4049/jimmunol.1100274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lobo PI, Surratt PM. Studies on the autoantibody to lymphocytes in sarcoidosis. J. Clin. Lab Immunol. 1979;1:283–288. [PubMed] [Google Scholar]
- 28.Gilbreath MJ, Pavanand K, MacDermott RP, Wells RA, Ussery MA. Characterization of cold reactive lymphocytotoxic antibodies in malaria. Clin. Exp. Immunol. 1983;51:232–238. [PMC free article] [PubMed] [Google Scholar]
- 29.Dorsett B, Cronin W, Chuma V, Iochim HI. Anti-lymphocyte antibodies in patients with the acquired immune deficiency syndrome. Am. J. Med. 1985;78:621–626. doi: 10.1016/0002-9343(85)90405-x. [DOI] [PubMed] [Google Scholar]
- 30.Williams RC, Masur H, Spira TJ. Lymphocyte reactive antibodies in acquired immune deficiency syndrome. J. Clin. Immunol. 1984;4:118–123. doi: 10.1007/BF00915045. [DOI] [PubMed] [Google Scholar]