

Abstract
RF9, a reported antagonist of the mammalian gonadotropin-inhibitory hormone receptor, stimulates gonadotropin secretion in mammals. Recent studies have suggested that the stimulatory effect of RF9 on gonadotropin secretion relies on intact kisspeptin receptor (KISS1R) signaling, but the underlying mechanisms remain to be elucidated. Using Chinese Hamster Ovary cells stably transfected with KISS1R, we show that RF9 binds specifically to KISS1R, with a Kd of 1.6 × 10−5M, and stimulates an increase in intracellular calcium and inositol phosphate accumulation in a KISS1R-dependent manner, with EC50 values of 3.0 × 10−6M and 1.6 × 10−7M, respectively. RF9 also stimulated ERK phosphorylation, with a time course similar to that of kisspeptin-10. RFRP-3, the putative endogenous ligand for NPFFR1, did not stimulate inositol phosphate accumulation or pERK, nor did it alter responses to of kisspeptin-10 or RF9. In agreement with these in vitro data, we found that RF9 stimulated a robust LH increase in Npffr1−/− mice, similar to that in wild-type littermates, whereas the stimulatory effect of RF9 was markedly reduced in Kiss1r−/− and double Kiss1r−/−/Npfrr1−/− mice. The stimulatory effect of RF9 on LH secretion was restored by the selective rescue of Kiss1r expression in GnRH neurons, in Kiss1r−/−T mice. Taken together, our study demonstrates that RF9 acts primarily as a KISS1R agonist, but not as an allosteric modulator, to stimulate LH secretion. Our findings raise questions regarding the utility of RF9 for assessing NPFF1R function and de-emphasize a predominant role of this signaling system in central regulation of reproduction.
RFamide-related peptide-3 (RFRP-3) and its receptor, neuropeptide FF receptor 1 (NPFFR1; also termed GPR147) are the mammalian orthologs of avian GnIH and its receptor (1–4), which negatively regulate reproduction. RFRP-3 was isolated and identified from extracts of the human hypothalamus by immunoaffinity purification (4). Neuronal mapping indicated that RFRP-3 neurons and fibers were observable in brain areas known to control the hypothalamic-pituitary-gonadal axis, as well as feeding and reproductive behaviors (2, 3, 5). Dual-label immunohistochemistry revealed that RFRP-3 fibers projected to approximately 26% of GnRH neurons in male and diestrous female mice, and to 19% of kisspeptin neurons in proestrous female mice (6). Intracerebroventricular (ICV) injection of high concentrations of RFRP-3 (500 ng) significantly suppressed male rat sexual behavior (5). In addition, ICV injection of RFRP-3 suppressed LH secretion in rats and ovariectomized Syrian hamsters (3, 5). The inhibitory effect of RFRP-3 on gonadotropin secretion was observed following either central or peripheral administration of RFRP-3 (7). However, the purely inhibitory effect of RFRP-3 on reproduction was questioned following findings that RFRP-3 displayed mixed actions on the firing rate of GnRH-green fluorescent protein-tagged neurons, 41% of them being inhibited and 12% activated by RFRP-3 (8).
RF9 (1-adamantane carbonyl-Arg-Phe-NH2), a derivative of the RFamide dipeptide shared by this family of neuropeptides, was identified as an antagonist of NPFFR1 (9). It specifically inhibited binding of 125I-neuropeptide FF (NPFF; the first mammalian RFamide peptide purified and also a ligand of NPFFR1 (10) to NPFFR1 and to the family member NPFFR2 (GPR74), and ICV injection of RF9 prevented the increase in blood pressure and heart rate elicited by NPFF. In animal studies of reproduction, RF9 demonstrated stimulatory effects on gonadotropin release (11–14). In the ewe, the increase in plasma LH concentrations induced by RF9 was blocked by pretreatment with a GnRH antagonist (11). Similar findings that a GnRH antagonist inhibited the stimulatory effect of RF9 on LH secretion were documented in castrated male rats (6), suggesting that the site of action of RF9 is upstream of the pituitary gland. Consensus eventually emerged that RF9 acts predominantly at the level of or upstream of GnRH neurons (13–15). RF9 blocked the inhibitory effects of NPFF on pacemaker activity of GnRH neurons (15) and reversed the inhibitory effects of T on GnRH release frequency from brain slices, as measured by fast-scan cyclic voltammetry to detect directly the oxidation of secreted GnRH in mice (13). These effects were attributed primarily to its antagonistic action on NPFFR1, suggesting that RF9 blocked an endogenous inhibitory RFRP-3 tone (6, 11, 13).
Our recent study showed that Npffr1−/− mice displayed increased litter sizes and resistance to metabolically induced gonadotropin suppression, consistent with an inhibitory role of Npffr1 signaling in the central regulation of reproduction in rodents (16). However, in our earlier study, the gonadotropin response to RF9 was significantly blunted in Kiss1r−/− male mice (14). Additional evidence is emerging to suggest that the action of RF9 on GnRH and gonadotropin secretion does not rely solely on its antagonistic effects on NPFFR1 signaling, and recent studies provide support for the hypothesis that the stimulatory effects of RF9 on GnRH neurons rely on intact KISS1R signaling (17, 18). To date, no studies have addressed whether RF9 has a direct effect on KISS1R signaling. To determine whether RF9 acts directly on KISS1R, we performed binding assays in vitro to evaluate specific binding of RF9 to KISS1R. We also performed signal transduction assays to evaluate the possible direct action of RF9 on KISS1R signaling. Furthermore, to better delineate the pathways of RF9's effects on the gonadotropic axis in vivo, we performed studies in Npffr1−/−, Kiss1r−/−, and double Npffr1−/−/Kiss1r−/− mice, as well as in Kiss1r−/−-null mice with selective rescue of Kiss1r expression in GnRH cells (Kiss1r−/−T). These studies provide convincing evidence that RF9 is an agonist of KISS1R and that its stimulatory effects on gonadotropin secretion occur primarily through its activation of KISS1R signaling.
Materials and Methods
Reagents
Radioisotopes, 125I-kisspeptin-10, and myo-[2-3H]-inositol were purchased from PerkinElmer; cell culture medium was from Mediatech, Inc. Kisspeptin-10 (KP10) was synthesized by the Tufts University Core Facility. RF9 and RFRP-3 were purchased from Tocris Bioscience. The Fluo-4 Direct Kit was from Invitrogen.
Cell culture
The generation of a stable cell line, Chinese Hamster Ovary (CHO) cells expressing human KISS1R (CHO-KISS1R) were previously described (19–21). CHO-KISS1R cells were maintained in Dulbecco's Modified Eagle's Medium and Ham's F-12 Nutrient Mixture (DME/F12) containing 10% fetal bovine serum and 800 mg/L geneticin. The parental CHO cell line, CHO K1 (purchased from ATCC), was maintained in DME/F12 containing 10% fetal bovine serum but without geneticin. Cells were cultured in geneticin-free media for 24 hours prior to experiments.
Binding assays
Competition binding assays were conducted in CHO-KISS1R and parental CHO K1 cells maintained in DME/F12 with 10% fetal bovine serum and 800 mg/L geneticin. The cells (5 × 106/well) were plated in 12-well plates overnight. The next day, the CHO-KISS1R cells were washed with DME/F12 containing 0.5% BSA (binding medium) and incubated with 0.05nM 125I-kisspeptin-10 (125I-KP10) in the presence of increasing concentrations (10−11 to 10−4M) of unlabeled KP10 or RF9 for 20 minutes at room temperature. For CHO K1 cells, the binding assays were performed in the presence of fixed doses of KP10 (10−6M), RF9 (10−5M), and RFRP-3 (10−5M) 125I-KP10 and the unlabeled KP10 or RF9 were premixed and added to the cells simultaneously. After 20 minutes, the cells were placed on ice, washed three times with ice-cold PBS, lysed in 0.5 mL 1M NaOH, and counted using a γ-counter (PerkinElmer 1470 Automatic Gamma Counter). Data were analyzed and plotted using GraphPad Prism (http://www.graphpad.com/scientific-software/prism/) one site homologous competition with depletion. Specific binding at each point is represented as the percentage of binding relative to binding in the absence of unlabeled reagent.
Calcium assays
CHO-KISS1R and the parental CHO K1 cells (1 × 104/well) were plated into black-walled, clear-bottomed 96-well plates. The next day, the cells were washed with DME/F12 and incubated in serum-free DME/F12 for 2 hours at 37°C. The Fluo-4 Direct Kit (Invitrogen) was used to measure intracellular calcium concentrations ([Ca2+]i) as previously described (21). To test for agonist activity, CHO-KISS1R cells were stimulated with increasing concentrations of KP10 (10−11 to 10−7M) or RF9 (10−9 to 10−4 M). CHO K1 cells were also treated with KP10 (10nM) or RF9 (10−4M) as negative controls. All peptides were dissolved in dimethylsulfoxide with a final concentration of 0.1% dimethylsulfoxide. The change in fluorescence intensity following addition of ligand was measured using a POLARstar OPTIMA multifunction plate reader (BMG Labtech). Basal activities were measured twice prior to treatment. After treatment, the first measurement was made 5 seconds after addition of the ligand and then repeated every 10 seconds for 30 cycles. The intracellular calcium response was determined by calculating the ratio of the difference between the maximum and minimum fluorescence units over the minimum fluorescence units, as recommended by the manufacturer's protocol. The ratios were graphed as the percentage of the ratio for the maximal dose of KP10.
Inositol phosphate assay
Inositol phosphates (IPs) were measured as previously described (22, 23). Briefly, CHO-KISS1R or CHO K1 cells (5 × 104/well) were plated in 24-well plates. The next day, the cells were cultured in serum- and inositol-free DMEM (1 mL/well) containing 0.5 μCi of myo-[2-3H]-inositol (PerkinElmer) overnight. The following day, medium was replaced with fresh inositol-free DMEM (1 mL/well) containing 10mM LiCl. Fifteen minutes later, the cells were stimulated with increasing concentrations of KP10 (10−10 to 3 × 10−6M) or RF9 (10− to 10−5M), in the presence or absence of 10−5M RFRP-3 or 10−6M RF9 as indicated, for 1 hour. After stimulation, the cells were lysed in 1 mL ice-cold 20mM formic acid. The lysates were collected and neutralized to pH 7.5 with 300 μL solution containing 7.5mM HEPES and 150mM KOH. 3H-IP products were isolated through prepared AG 1-X8 resin (Bio-Rad) anion exchange columns by washing with 5 mL of H2O, and then 5 mL solution containing 5mM borax and 60mM sodium formate, then eluting in 3 mL elution solution containing 0.9M ammonium formate and 0.1M formic acid. The radioactivity in the eluates was measured in a scintillation counter (Beckman).
ERK phosphorylation assay
CHO-KISS1R and CHO K1 cells (1 × 106/well) were plated into six-well plates. The next day, the cells were incubated in serum-free DME overnight at 37°C. The cells were then incubated with different agents for varying times, as indicated in the Results section. After treatment, the cells were lysed in 100 μL ice-cold radioimmunoprecipitation assay lysis buffer containing protease and phosphatase inhibitors. The lysates were centrifuged for 10 minutes. The lysates were resolved on SDS-PAGE gels, and the resolved proteins were electrophoretically transferred to nitrocellulose membranes. After blocking with 5% fat-free milk in Tris-buffered saline with Tween 20, the blots were incubated overnight at 4°C with anti-pERK (Cell Signaling) or anti-ERK (Santa Cruz) antibody, the membranes were then washed and incubated with a secondary antirabbit IgG antibody (Cell Signaling). The proteins were visualized using the chemiluminescent HRP antibody detection reagent (Denville Scientific, Inc.) and the Kodak Image Station 4000 mm Pro system. The intensity of the bands was quantified using the image software provided by the Kodak Image Station 4000 mm Pro system.
Animal studies
All experimental protocols were approved by the Córdoba University Ethical Committee of animal experimentation and conducted in accordance with the European Union guidelines for use of experimental animals. Generation and genotyping of Npffr1−/−, Kiss1r−/−, Kiss1r−/−T, and double Kiss1r−/−/Npffr1−/− mice were described previously (14, 16, 24). Npffr1−/− and Kiss1r−/− mouse lines are devoid of Rfrp-3 and kisspeptin signaling, respectively (14, 16), whereas the Kiss1r−/−T mouse is characterized by the selective rescue of expression of Kiss1r in GnRH cells (24). For hormonal analyses, blood samples (200 μL) were obtained using standard procedures in our laboratory, by jugular venipuncture before (basal) and 15 minutes after RF9 administration. RF9 (5 nmol/5 μL) was administered intracerebro-ventricularly as described previously (14, 16). Adult (3–4-month-old) male and female mice of the indicated genotypes were used. To exclude the possibility that defective gonadotropin responses to the various stimuli might result from insufficient pituitary responsiveness to GnRH due to low endogenous tone, which is characteristic of Kiss1r−/− animals, Kiss1r−/− and double Kiss1r−/−/Npffr1−/− mice were subjected to a protocol of GnRH priming for 2 days before RF9 administration, to heighten pituitary responsiveness, as previously described (14). Because of the technical challenges of generating sufficient numbers of homozygous double-knockout Kiss1r−/−/Npffr1−/− mice, we were only able to generate sufficient numbers of females, but not males, for this genotype, so that our double knockout data are derived only from female mice. To allow delivery of drugs into the lateral cerebral ventricle, the cannulae were lowered to a depth of 2 mm beneath the surface of the skull, with an insertion point at 1 mm posterior and 1.2 mm lateral to bregma (16). LH levels in blood samples were assayed using a double-antibody method and RIA kits supplied by the National Institutes of Health (Dr A. F. Parlow, National Hormone and Peptide Program). Rat LH-I-10 was labeled with 125I using Iodo-gen tubes, as per manufacturer instructions (Pierce Chemical Co.), following previously validated protocols at our laboratory (7, 12). The validity of this assay for accurate detection of circulating LH levels in males has been previously documented (14, 16). Hormone concentrations were expressed using reference preparation LH-RP-3, the luteinizing hormone standard preparation provided by National Institute of Diabetes and Digestive and Kidney Diseases. Intra- and interassay coefficients of variation were <8% and 10%, respectively. The sensitivity of the assay was 5 pg/tube for LH. Accuracy of hormone determinations was confirmed by assessment of mouse serum samples of known concentrations, used as external controls.
Statistics
GraphPad Prism software was used to analyze binding data by nonlinear regression using a sigmoidal curve fit with a variable slope, and to compare curves for statistical significant differences. The results of binding, [Ca2+]i, IP, and ERK assays shown are pooled data from three independent experiments. Statistical analysis of the quantitative pERK results was performed by one-way ANOVA followed by multiple comparisons to compare the mean of each group with the mean of the control group (Fisher's least significant difference test). Hormonal data are expressed as the mean ± SEM for each group. Group sizes for hormonal determinations were as follows: male Npffr1−/− (Vehicle, N = 10; RF9, N = 7); male Npffr1+/+ (wild type [WT]; Vehicle, N = 10; RF9, N = 9); female Npffr1−/− (Vehicle, N = 10; RF9, N = 9); female Npffr1+/+ (WT; Vehicle, N = 10; RF9, N = 9); male Kiss1r−/− (Vehicle, N = 9; RF9, N = 10); male Kiss1r−/−T (Vehicle, N = 4; RF9, N = 4); female Kiss1r−/− (Vehicle, N = 6; RF9, N = 7); female Kiss1r−/−T (Vehicle, N = 5; RF9, N = 5); and female Kiss1r−/−/Npffr1−/− (Vehicle, N = 5; RF9, N = 5). For tests using Kiss1r-null mice, littermate WT male (Vehicle, N = 8; RF9, N = 9) and female (Vehicle, N = 9; RF9, N = 10) mice were used as controls. ANOVA followed by post-hoc Student-Newman-Keuls and Bonferroni tests were used to assess variation between experimental groups. Similar results were obtained with both post-hoc tests. The significance level was set at P ≤ .05.
Results
RF9 displaces specific 125I-KP10 binding to KISS1R in a dose-dependent manner
To determine whether RF9 could bind specifically to KISS1R, radiolabeled competition binding assays were performed using 125I-KP10 together with increasing concentrations of unlabeled KP10 or RF9. KP10 displaced 125I-KP10 in a dose-dependent manner as expected, with a binding affinity (Kd) of 1.1 × 10−8M (Figure 1), consistent with previous studies (22, 25, 26). RF9 was also able to displace 125I-KP10 binding in a dose-dependent manner, indicating competitive binding of RF9 to KISS1R. The Kd of RF9 to KISS1R was 1.6 × 10−5M. Of note, no specific binding of 125I-KP10 to parental CHO K1 cells was detected, with no displacement of 125I-KP10 by RF9 (10−5M), RFRP-3 (10−5M), or KP10 itself (10−6M) (Supplemental Figure 1).
Figure 1.
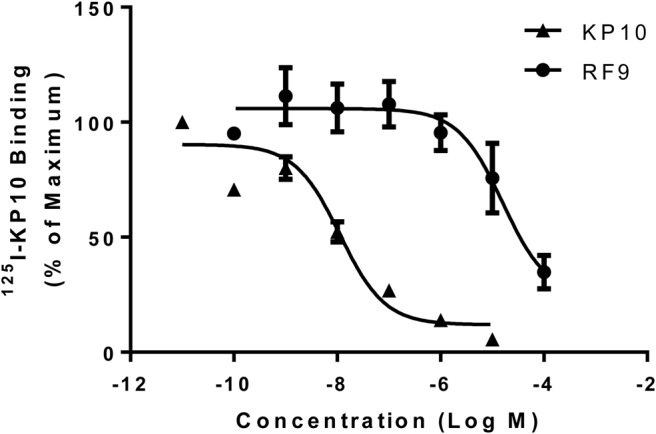
RF9 binds specifically to KISS1R. Competitive displacement binding assays were performed in CHO-KISS1R cells incubated with 0.05nM 125I-KP10 and increasing concentrations of either unlabeled KP10 or RF9. Data represent mean ± SEM from three independent experiments.
RF9 acts as a KISS1R agonist to stimulate increases in intracellular calcium levels in CHO-KISS1R cells
The ability of RF9 to compete for and displace binding of kisspeptin to KISS1R suggested that RF9 may act as a KISS1R agonist or antagonist. To test for this possibility, CHO-KISS1R cells were treated with increasing concentrations of KP10 (10−11M to 10−7M) or RF9 (10−9M to 10−4M). KP10 stimulation resulted in a dose-dependent increase in [Ca2+]i levels (EC50 8.8 × 10−9M) as expected (Figure 2). RF9 also had a stimulatory effect on intracellular calcium concentrations, albeit requiring higher concentrations than for KP10 (EC50 3.0 × 10−6M). KP10 and RF9 had no effects on [Ca2+]i in CHO K1 cells, confirming that their effects are mediated specifically through KISS1R activation (Supplemental Figure 2). These results suggest that RF9 acts as a KISS1R agonist to stimulate KISS1R-mediated increases in [Ca2+]i.
Figure 2.
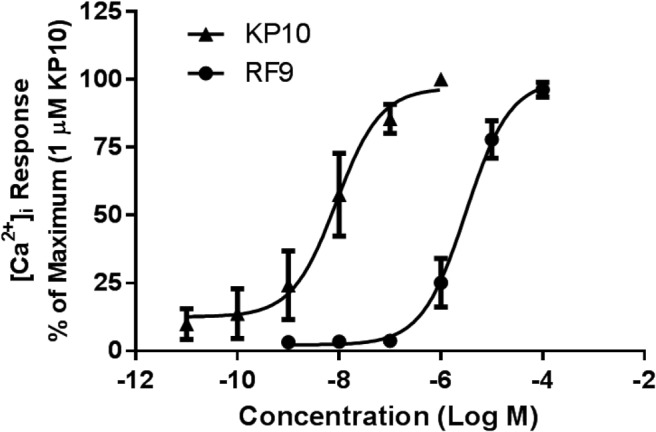
RF9 stimulates KISS1R-mediated increases in [Ca2+]i.[Ca2+]i responses were measured in CHO-KISS1R cells using a fluo-4 [Ca2+]i assay. CHO-KISS1R cells were treated with increasing concentrations of RF9 or KP10 as indicated. Data are shown as the percent of the maximum response and represent mean ± SEM from three independent experiments.
RF9 induces intracellular IP accumulation and ERK phosphorylation in CHO-KISS1R cells
One of the key signal transduction pathways for KISS1R is the phospholipase C-mediated IP3 pathway. To evaluate the role of RF9 in activating the KISS1R-mediated IP3 signaling pathway, we studied the effects of RF9 on IP accumulation in CHO-KISS1R cells and compared the effects with those of KP10. CHO-KISS1R cells were treated with increasing concentrations of KP10 (10−10 to 10−7M), or RF9 (10−8 to 10−5M). Consistent with previous studies (20, 23), KP10 stimulation resulted in a dose-dependent increase in IP accumulation (EC50 4.0 × 10−10M) (Figure 3). Similarly, RF9 treatment resulted in a dose-dependent increase in IP accumulation (EC50 1.6 × 10−7M) (Figure 3). Notably, coadministration of 10−6M RF9 with increasing doses of KP10 did not produce a synergistic effect on IP accumulation, indicating that RF9 acts as an agonist rather than as an allosteric modulator of KISS1R signaling, because an allosteric modulator would be expected to shift the dose response curve (27). Given that RF9 was identified as an NPFF1R antagonist, we next tested the effect of RFRP-3, the endogenous ligand of NPFFR1, on KISS1R signaling. RFRP-3 (10−7 to 10−5M) did not alter IP accumulation in either CHO-KISS1R (Figure 3) or in CHO K1 cells (data not shown). Cotreatment with 10−5M RFRP-3 did not affect the dose-response curves for either KP10 or RF9 (Figure 3), suggesting that the stimulatory effects of KP10 and RF9 on IP accumulation in CHO-KISS1R are due to a direct action on KISS1R rather than through actions on NPFFR1 and are independent of actions of RFRP-3 on NPFFR1. Our finding that neither KP10 (10−7M) nor RF9 (10−5M) stimulated IP accumulation in CHO K1 cells, which do not express KISS1R, further supports this statement (Supplemental Figure 3).
Figure 3.

RF9 stimulates KISS1R-mediated increases in IP accumulation. Intracellular IP accumulation was measured as described in the Methods. CHO-KISS1R cells were treated with increasing concentrations of KP10 or RF9 in the presence or absence of 10−5M RFRP-3, or with increasing concentrations of RFRP-3 alone. In addition, cells were treated with increasing concentrations of KP10 in the presence of 10−6M RF9. Intracellular IP levels are expressed as counts per minute. Data are shown as the percent of the KP10-induced maximal response and represent the mean ± SEM from at least three independent experiments.
We also evaluated the effect of RF9 on another known KISS1R-mediated signaling pathway, ERK phosphorylation, in CHO-KISS1R cells. Compared with control levels, ERK phosphorylation was significantly increased following stimulation for 2 minutes by RF9 and KP10 alone, as well as by combinations of KP10 + RFRP-3, RF9 + RFRP-3, and KP10 + RF9, in CHO-KISS1R cells (Figure 4A, left panel; and 4B). The time course of RF9-induced ERK phosphorylation was similar to that induced by KP10, with higher ERK phosphorylation 2 minutes after ligand stimulation than 30 minutes after stimulation (Figure 4A, left panel; and 4B). Cotreatment of CHO-KISS1R cells with KP10 and RF9 did not significantly increase pERK comparing to that induced by KP10 alone, demonstrating no synergistic effects. RFRP-3 alone did not induce ERK phosphorylation, nor did it significantly blunt KP10- or RF9-stimulated ERK phosphorylation. In CHO K1 cells, neither KP10, RF9, nor RFRP-3 stimulated ERK phosphorylation (Figure 4A, right panel).
Figure 4.

RF9 stimulates ERK phosphorylation. A, CHO-KISS1R (left panel) and CHO K1 (right panel) cells were treated with 10−7M KP10 or 10−6M RF9 in the presence or absence of 10−5M RFRP-3, or with 10−5 M RFRP-3 alone, as indicated. Cell lysates were collected for Western blot analysis after 2 or 30 minutes of treatment. The image shown is a representative blot from at least three independent experiments performed with similar results. B, The Western blot data were quantified and the results of three independent experiments were pooled and are depicted as a bar graph, as pERK1/2 normalized for total extracellular-signal-regulated kinase 1/2 levels. Statistical analysis of the quantitative pERK results was performed by one-way ANOVA followed by multiple comparisons to compare the mean of each group with the mean of the control group (Fisher's LSD test). *, P < .05; **, P < .01; ***, P < .001.
RF9 stimulates robust LH secretory responses in Npffr1−/−-null mice
We performed comparative in vivo studies in Npffr1−/− mice and WT littermate controls to evaluate the physiological role of the Npffr1 in mediating the effects of RF9 on LH secretion. Surprisingly, RF9 administration resulted in robust stimulation of LH secretion in both male and female Npffr1−/− mice. In fact, compared with WT mice, RF9 stimulated an even greater level of LH secretion in Npffr1−/− mice, in both males and females (Figure 5). The findings clearly show that the stimulatory effect of RF9 on LH secretion in the mouse can occur in the absence of Npffr1 and, hence, does not require the presence of intact Npffr1 signaling.
Figure 5.

RF9 stimulates robust increases in LH in both WT and Npffr1−/− male (A) and female (B) mice. LH levels were measured in WT and Npffr1−/− mice 15 min after ICV injection of RF9 (5 nmol/5 μL) or vehicle. Each experimental group was composed of 7–10 mice. Statistical significance was analyzed by ANOVA followed by post-hoc Student-Newman-Keuls tests. **, P < .01 vs corresponding unstimulated groups of the same genotype; a, P < .05 vs corresponding WT groups.
The stimulatory effects of RF9 on LH secretion are reduced in Kiss1r−/− and Kiss1r−/−/Npffr1−/− mice but restored by rescuing Kiss1r expression selectively in GnRH neurons
In Kiss1r−/− male and female mice, baseline LH levels were significantly decreased compared with WT controls and RF9 stimulated only modest increases in LH secretion (Figure 6). These findings are consistent with our previous studies in male mice (14). Rescue of Kiss1r expression selectively in GnRH neurons restored the LH response to RF9 to levels seen in WT mice of both sexes (Figure 6). Similarly to Kiss1r−/− mice, double Kiss1r−/−/Npffr1−/− female mice, lacking both the stimulatory and inhibitory receptors, had low baseline LH levels and displayed only modest, albeit significant responses to RF9 stimulation, which were similar in magnitude to those detected in Kiss1r−/− mice. These findings provide further support that Npffr1 has a minimal role in RF9-mediated gonadotropin secretion in rodents. Taken together, our findings support an action of RF9 to stimulate gonadotropin secretion through activation of KISS1R.
Figure 6.

Stimulation of LH secretion by RF9 is blunted in Kiss1r−/− and Kiss1r−/−/Npffr1−/− mice but restored in Kiss1r−/−T mice with selective reactivation of Kiss1r expression in GnRH neurons. LH levels were measured in WT, Kiss1r−/−, Kiss1r−/−T, and Kiss1r−/−/Npffr1−/− mice. Blood samples were collected 15 min after ICV injection of RF9 (5 nmol/5 μL). Each experimental group was composed of 4–10 mice. All tests were performed in both male (A) and female (B) mice, except for Kiss1r−/−/Npffr1−/− mice, which were only studied in females. Statistical significance was analyzed by ANOVA followed by post-hoc Student-Newman-Keuls tests. *, P < .05; **, P < .01 vs corresponding unstimulated groups of the same genotype; a, P < .01 vs corresponding WT values.
Discussion
The discovery of GnIH in the year 2000 established the dual control system of the hypothalamic-pituitary-gonadal axis in birds (1). GnIH was found to act directly on the pituitary to inhibit LH and FSH secretion in avian species (1). Subsequent studies established that GnIH negatively regulates avian reproduction by inhibiting gonadotropin synthesis and secretion from gonadotrophs and/or acting on GnRH neurons via the GnIH receptor (GPR147) (28). The role of GnIH in the control of seasonal changes in avian reproduction was documented as well (29, 30).
The discovery of GnIH in birds subsequently drove the search for its mammalian ortholog. Based on the structure of GnIH, belonging to the family of RFamide peptides, a search of mammalian gene data banks identified three RFamide-related peptides, RFRP-1, RFRP-2, and RFRP-3 in mammals (28, 31). Further study suggested that RFRP-3 is the putative mammalian ortholog of GnIH (32). RFRP-3 decreased gonadotropin secretion in ovine (33, 34), bovine (35) rat (7), and hamster (36) species. Unlike birds, in which GnIH acts primarily on pituitary gonadotrophs, increasing evidence suggests that RFRP-3 acts on and/or upstream of GnRH neurons via its receptor, NPFFR1 (6, 15, 37, 38). Considerable debate, however, has arisen regarding its physiological relevance to the central regulation of reproduction in mammals. On the one hand, administration of RFRP-3 reduced gonadotropin release in rats (5, 7, 39) and cows (35). However, RFRP-3 was found to inhibit 41% of GnRH neurons, but also to activate 12% of them (8). In addition, functional studies have shown that acute central administration of RFRP-3 in the male Syrian hamster induces a marked increase in gonadotropin secretion and T production (36, 40).
The identification of RF9 as an antagonist of RFRP-3 (9) inspired investigators to use it as a tool to interrogate the roles of RFRP-3 and its receptor, NPFFR1, in the regulation of mammalian reproduction. Consequently, RF9 was found to stimulate gonadotropin release in sheep (11) and rats, and it was proposed that RF9 was antagonizing a tonic inhibitory effect of RFRP-3 (6, 12). In addition, RF9 blocked the inhibitory effect of RFRP-3 on the pacemaker activity of GnRH neurons (15). Nonetheless, a central effect of RF9 that relied purely on its antagonistic effect on NPFFR1 was questioned (41).
In our previous report, central injection of RF9 stimulated gonadotropin secretion in both male and female rats (12). Interestingly, whereas the stimulatory effect of RF9 on gonadotropin release was observed in WT mice, the effect was significantly blunted in Kiss1r−/− mice (14), suggesting a role for Kiss1r in the actions of RF9 on gonadotropins. Similarly, a more recent study noted that the excitatory effects of RF9 on GnRH neuronal firing were not observed in Kiss1r−/− mice (17). Furthermore, it has been shown recently that a kisspeptin antagonist, kp234, blocked the RF9-induced LH stimulation in female rats (18). In our current study, we have shown that RF9, like KP10, caused a dose-dependent displacement of 125I-KP10 binding in CHO-KISS1R cells. This finding provides direct evidence that RF9 binds specifically to KISS1R. An earlier study also documented specific binding of RF9 to KISS1R but failed to observe an RF9-induced [Ca2+]i response, even at a high concentration of 10−5M RF9, in KISS1R-expressing CHO cells (42). In this study, we have found that both RF9 and KP10 stimulated a robust [Ca2+]i response in a dose-dependent manner in CHO-KISS1R cells, but did not increase [Ca2+]i in control CHO cells lacking KISS1R. In the previous study (42), a different (FLIPR) assay was used to measure the [Ca2+]i response in the KISS1R-expressing CHO cells, and it is unclear whether transient or stable transfection was used. The sensitivity to detect the [Ca2+]i response may be affected by different assays and receptor expression techniques. The specific binding to KISS1R and the activation of KISS1R-dependent intracellular signaling pathways, as indicated by the calcium response, support a role for RF9 as a KISS1R agonist, binding directly to and activating the kisspeptin receptor.
Our data demonstrating RF9-stimulated IP accumulation further support this agonistic activity. An interaction between RFRP-3 and KISS1R was excluded because RFRP-3 alone did not increase IP accumulation and had no effect on KP10-stimulated IP accumulation in CHO-KISS1R cells. Importantly, our IP accumulation studies showed that RFRP-3 did not alter RF9-stimulated IP accumulation in CHO-KISS1R cells. This finding suggests that in CHO-KISS1R cells, RF9-induced IP production does not occur through an antagonistic effect on NPFFR1. In contrast, the finding that KP10 and RF9 did not produce a synergistic effect on IP induction or ERK phosphorylation suggests that the effect of RF9 is through a direct action on KISS1R rather than via allosteric actions on KISS1R or through crosstalk between the receptors, given that in both of these scenarios a synergistic effect would typically be seen. In the current study, we did not perform an assay to confirm the positive bioactivity of RFRP-3, but a recent study clearly showed that RFRP-3 inhibited forskolin-induced cAMP production in NPFF1R-transfected CHO cells. Moreover, in that study, RF9 failed to reverse the inhibitory effect of RFRP-3 on forskolin-induced cAMP production in the NPFF1R-transfected CHO cells (43).
We have also shown that both KP10 and RF9 activate ERK phosphorylation, another signal transduction pathway known to be activated by kisspeptin via KISS1R signaling (23). Again, RFRP-3 itself did not induce ERK phosphorylation, nor did it alter the activation of ERK phosphorylation by RF9 or KP10. Furthermore, the similar kinetics of ERK phosphorylation in response to RF9 or KP10 further supports a similar signaling pathway for both of these ligands. Taken together, our data demonstrate that the pattern of activation of signal transduction cascades by RF9 closely matches the pattern activated by kisspeptin, thus providing convincing evidence that RF9 acts as a KISS1R agonist, albeit not as potent as kisspeptin.
In animal studies, robust stimulation of gonadotropin release by RF9 has been well documented (11, 12). Given our findings that RF9 acts as an agonist of KISS1R in vitro, combined with previous data that RF9 is an antagonist of NPFFR1, one may question whether the stimulation of gonadotropin release in response to RF9 in vivo is the result of an agonistic effect on KISS1R, an antagonistic effect on NPFFR1, or a combination of both effects. In this study, we have taken advantage of different genetically modified mouse models to address the above possibilities. Our studies demonstrate that the stimulation of LH release by RF9 is maintained in Npffr1−/− mice (hence in the absence of Npffr1 signaling), at levels comparable, if not somewhat higher, to those in WT mice. These data suggest that the actions of RF9 on LH release are independent of its antagonistic effects on Npffr1. Moreover, our recent study showed that Npffr1−/− mice displayed only a modest reproductive phenotype, suggesting that Npffr1 normally plays only a small role in the regulation of reproduction in mice (16). In contrast, the stimulatory effect of RF9 on LH release was significantly blunted in Kiss1r−/− mice, indicating that the RF9-stimulated gonadotropin release relies to a large extent on its agonistic effect on Kiss1r.
This conclusion is further supported by the finding that the selective rescue of Kiss1r expression in GnRH neurons on a Kiss1r-null background (in Kiss1r−/−T mice) restored the stimulatory effect of RF9 on LH release. These in vivo observations are in good agreement with a previous study showing that RF9 acts directly on GnRH neurons to activate GnRH neuronal firing (17). Nonetheless, actions of RF9 upstream (or independent) of GnRH neurons cannot be entirely excluded. It is interesting to note that the stimulation of LH secretion by RF9 was not completely abolished in Kiss1r−/− mice, suggesting that RF9 may act through additional alternative mechanisms to produce these residual stimulatory effects on gonadotropin secretion. In fact, a recent study suggested that kisspeptin can act through a Kiss1r-independent pathway to inhibit firing of non-GnRH neurons from the arcuate nucleus, and this effect was blocked by high concentrations of RF9 (44). Whether this mechanism applies for the gonadotropin-releasing action of RF9 reported here warrants further investigation. In any event, given that the modest stimulatory effect of RF9 on LH secretion in Kiss1r−/− mice was similar in magnitude to that observed in Kiss1r−/−/Npffr1−/− double-knockout mice, it is unlikely that Npffr1 plays a major role in this modest residual LH response. In our in vivo studies, we did not see a significant response of FSH secretion to either kisspeptin or RF9 treatment (data not shown). This may be attributed to the time point of sample collection we used, of 15 minutes after ICV injection of the reagents, which may not be optimal for stimulation of FSH in mice (45).
In conclusion, our data, derived from in vitro studies in a heterologous cell model, demonstrate that RF9 binds specifically to KISS1R and activates KISS1R-mediated intracellular signal transduction cascades. Our findings from these in vitro studies are supported by another recent report (43). Our in vivo data using genetically modified mouse models are consistent with our in vitro findings, and show that the stimulatory effects of RF9 on LH secretion do not rely on its antagonistic effects on Npffr1, but rather that Kiss1r plays a key role in mediating RF9-stimulated LH secretion. Our study suggests that using RF9 as a tool to study antagonistic effects on Npffr1 may lead to misleading findings. Given that RF9 is a chemically modified dipeptide, we are studying its agonistic effect on Kiss1r pharmacologically rather than physiologically. Nonetheless, the results help us to understand the underlying mechanisms of action. As RF9 is a much smaller molecule than kisspeptin, the cost of synthesis and the stability of RF9 and/or its derivatives may be more favorable than those of kisspeptin, which may provide a clinical or therapeutic advantage.
Acknowledgments
We acknowledge that the Kiss1r−/−T mouse line was transferred to us by Dr Milen Kirilov and Professor Gunther Schütz (Heidelberg, Germany).
This research was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, through cooperative agreement U54 HD28138 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research and by R01 HD19938 (to U.B.K.), by NICHD/NIH K08 HD070957 (to L.M.), and by Grants BFU2011–025021 and BFU2014–57581-P (Ministerio de Economía y Competitividad, Spain; cofunded with EU funds from FEDER Program) and Project P12-FQM-01943 (Junta de Andalucía, Spain) (to M.T.-S.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CHO
- Chinese Hamster Ovary
- CHO-KISS1R
- Chinese Hamster Ovary cells expressing human KISS1R
- DME/F12
- Dulbecco's Modified Eagle's Medium and Ham's F-12 Nutrient Mixture
- ICV
- intracerebroventricular
- IP
- Inositol phosphate
- KISS1R
- kisspeptin receptor
- KP10
- kisspeptin-10
- NPFF
- neuropeptide FF
- RF9
- 1-adamantane carbonyl-Arg-Phe-NH2
- RFRP-3
- famide-related peptide-3
- WT
- wild type.
References
- 1. Tsutsui K, Saigoh E, Ukena K, et al. A novel avian hypothalamic peptide inhibiting gonadotropin release. Biochem Biophys Res Commun. 2000;275:661–667. [DOI] [PubMed] [Google Scholar]
- 2. Ukena K, Tsutsui K. Distribution of novel RFamide-related peptide-like immunoreactivity in the mouse central nervous system. Neurosci Lett. 2001;300:153–156. [DOI] [PubMed] [Google Scholar]
- 3. Kriegsfeld LJ, Mei DF, Bentley GE, et al. Identification and characterization of a gonadotropin-inhibitory system in the brains of mammals. Proc Natl Acad Sci U S A. 2006;103:2410–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ubuka T, Morgan K, Pawson AJ, et al. Identification of human GnIH homologs, RFRP-1 and RFRP-3, and the cognate receptor, GPR147 in the human hypothalamic pituitary axis. PLoS One. 2009;4:e8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson MA, Tsutsui K, Fraley GS. Rat RFamide-related peptide-3 stimulates GH secretion, inhibits LH secretion, and has variable effects on sex behavior in the adult male rat. Horm Behav. 2007;51:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rizwan MZ, Poling MC, Corr M, et al. RFamide-related peptide-3 receptor gene expression in GnRH and kisspeptin neurons and GnRH-dependent mechanism of action. Endocrinology. 2012;153:3770–3779. [DOI] [PubMed] [Google Scholar]
- 7. Pineda R, Garcia-Galiano D, Sanchez-Garrido MA, et al. Characterization of the inhibitory roles of RFRP3, the mammalian ortholog of GnIH, in the control of gonadotropin secretion in the rat: In vivo and in vitro studies. Am J Physiol Endocrinol Metab. 2010;299:E39–E46. [DOI] [PubMed] [Google Scholar]
- 8. Ducret E, Anderson GM, Herbison AE. RFamide-related peptide-3, a mammalian gonadotropin-inhibitory hormone ortholog, regulates gonadotropin-releasing hormone neuron firing in the mouse. Endocrinology. 2009;150:2799–2804. [DOI] [PubMed] [Google Scholar]
- 9. Simonin F, Schmitt M, Laulin JP, et al. RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc Natl Acad Sci U S A. 2006;103:466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang HY, Fratta W, Majane EA, Costa E. Isolation, sequencing, synthesis, and pharmacological characterization of two brain neuropeptides that modulate the action of morphine. Proc Natl Acad Sci U S A. 1985;82:7757–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caraty A, Blomenröhr M, Vogel GM, Lomet D, Briant C, Beltramo M. RF9 powerfully stimulates gonadotrophin secretion in the ewe: Evidence for a seasonal threshold of sensitivity. J Neuroendocrinol. 2012;24:725–736. [DOI] [PubMed] [Google Scholar]
- 12. Pineda R, Garcia-Galiano D, Sanchez-Garrido MA, et al. Characterization of the potent gonadotropin-releasing activity of RF9, a selective antagonist of RF-amide-related peptides and neuropeptide FF receptors: Physiological and pharmacological implications. Endocrinology. 2010;151:1902–1913. [DOI] [PubMed] [Google Scholar]
- 13. Glanowska KM, Burger LL, Moenter SM. Development of gonadotropin-releasing hormone secretion and pituitary response. J Neurosci. 2014;34:15060–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. García -Galiano D, van Ingen Schenau D, Leon S, Krajnc-Franken MA, et al. Kisspeptin signaling is indispensable for neurokinin B, but not glutamate, stimulation of gonadotropin secretion in mice. Endocrinology. 2012;153:316–328. [DOI] [PubMed] [Google Scholar]
- 15. Saito TH, Nakane R, Akazome Y, Abe H, Oka Y. Electrophysiological analysis of the inhibitory effects of FMRFamide-like peptides on the pacemaker activity of gonadotropin-releasing hormone neurons. J Neurophysiol. 2010;104:3518–3529. [DOI] [PubMed] [Google Scholar]
- 16. León S, García-Galiano D, Ruiz-Pino F, et al. Physiological roles of gonadotropin-inhibitory hormone signaling in the control of mammalian reproductive axis: Studies in the NPFF1 receptor null mouse. Endocrinology. 2014;155:2953–2965. [DOI] [PubMed] [Google Scholar]
- 17. Liu X, Herbison AE. RF9 excitation of GnRH neurons is dependent upon Kiss1r in the adult male and female mouse. Endocrinology. 2014;155:4915–4924. [DOI] [PubMed] [Google Scholar]
- 18. Sahin Z, Canpolat S, Ozcan M, Ozgocer T, Kelestimur H. Kisspeptin antagonist prevents RF9-induced reproductive changes in female rats. Reproduction. 2015;149(5):465–473. [DOI] [PubMed] [Google Scholar]
- 19. Kuohung W, Burnett M, Mukhtyar D, et al. A high-throughput small-molecule ligand screen targeted to agonists and antagonists of the G-protein-coupled receptor GPR54. J Biomol Screen. 2010;15:508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. [DOI] [PubMed] [Google Scholar]
- 21. Min L, Soltis K, Reis AC, et al. Dynamic kisspeptin receptor trafficking modulates kisspeptin-mediated calcium signaling. Mol Endocrinol. 2014;28:16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bianco SD, Vandepas L, Correa-Medina M, et al. KISS1R intracellular trafficking and degradation: Effect of the Arg386Pro disease-associated mutation. Endocrinology. 2011;152:1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahow M, Min L, Pampillo M, et al. KISS1R signals independently of Gαq/11 and triggers LH secretion via the β-arrestin pathway in the male mouse. Endocrinology. 2014;155:4433–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirilov M, Clarkson J, Liu X, et al. Dependence of fertility on kisspeptin-Gpr54 signaling at the GnRH neuron. Nat Commun. 2013;4:2492. [DOI] [PubMed] [Google Scholar]
- 25. Ohtaki T, Shintani Y, Honda S, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–617. [DOI] [PubMed] [Google Scholar]
- 26. Kotani M, Detheux M, Vandenbogaerde A, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276:34631–34636. [DOI] [PubMed] [Google Scholar]
- 27. May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. [DOI] [PubMed] [Google Scholar]
- 28. Kriegsfeld LJ, Ubuka T, Bentley GE, Tsutsui K. Seasonal control of gonadotropin-inhibitory hormone (GnIH) in birds and mammals. Front Neuroendocrinol. 2015;37:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bentley GE, Perfito N, Ukena K, Tsutsui K, Wingfield JC. Gonadotropin-inhibitory peptide in song sparrows (Melospiza melodia) in different reproductive conditions, and in house sparrows (Passer domesticus) relative to chicken-gonadotropin-releasing hormone. J Neuroendocrinol. 2003;15:794–802. [DOI] [PubMed] [Google Scholar]
- 30. Chowdhury VS, Yamamoto K, Ubuka T, Bentley GE, Hattori A, Tsutsui K. Melatonin stimulates the release of gonadotropin-inhibitory hormone by the avian hypothalamus. Endocrinology. 2010;151:271–280. [DOI] [PubMed] [Google Scholar]
- 31. Tsutsui K, Osugi T. Evolutionary origin and divergence of GnIH and its homologous peptides. Gen Comp Endocrinol. 2009;161:30–33. [DOI] [PubMed] [Google Scholar]
- 32. Kriegsfeld LJ, Gibson EM, Williams WP, 3rd, et al. The roles of RFamide-related peptide-3 in mammalian reproductive function and behaviour. J Neuroendocrinol. 2010;22:692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clarke IJ, Sari IP, Qi Y, et al. Potent action of RFamide-related peptide-3 on pituitary gonadotropes indicative of a hypophysiotropic role in the negative regulation of gonadotropin secretion. Endocrinology. 2008;149:5811–5821. [DOI] [PubMed] [Google Scholar]
- 34. Sari IP, Rao A, Smith JT, Tilbrook AJ, Clarke IJ. Effect of RF-amide-related peptide-3 on luteinizing hormone and follicle-stimulating hormone synthesis and secretion in ovine pituitary gonadotropes. Endocrinology. 2009;150:5549–5556. [DOI] [PubMed] [Google Scholar]
- 35. Kadokawa H, Shibata M, Tanaka Y, et al. Bovine C-terminal octapeptide of RFamide-related peptide-3 suppresses luteinizing hormone (LH) secretion from the pituitary as well as pulsatile LH secretion in bovines. Domest Anim Endocrinol. 2009;36:219–224. [DOI] [PubMed] [Google Scholar]
- 36. Ubuka T, Inoue K, Fukuda Y, et al. Identification, expression, and physiological functions of Siberian hamster gonadotropin-inhibitory hormone. Endocrinology. 2012;153:373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gojska NM, Friedman Z, Belsham DD. Direct regulation of gonadotrophin-releasing hormone (GnRH) transcription by RF-amide-related peptide-3 and kisspeptin in a novel GnRH-secreting cell line, mHypoA-GnRH/GFP. J Neuroendocrinol. 2014;26:888–897. [DOI] [PubMed] [Google Scholar]
- 38. Poling MC, Kim J, Dhamija S, Kauffman AS. Development, sex steroid regulation, and phenotypic characterization of RFamide-related peptide (Rfrp) gene expression and RFamide receptors in the mouse hypothalamus. Endocrinology. 2012;153:1827–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murakami M, Matsuzaki T, Iwasa T, et al. Hypophysiotropic role of RFamide-related peptide-3 in the inhibition of LH secretion in female rats. J Endocrinol. 2008;199:105–112. [DOI] [PubMed] [Google Scholar]
- 40. Ancel C, Bentsen AH, Sébert ME, Tena-Sempere M, Mikkelsen JD, Simonneaux V. Stimulatory effect of RFRP-3 on the gonadotrophic axis in the male Syrian hamster: The exception proves the rule. Endocrinology. 2012;153:1352–1363. [DOI] [PubMed] [Google Scholar]
- 41. Maletínská L, Tichá A, Nagelová V, et al. Neuropeptide FF analog RF9 is not an antagonist of NPFF receptor and decreases food intake in mice after its central and peripheral administration. Brain Res. 2013;1498:33–40. [DOI] [PubMed] [Google Scholar]
- 42. Oishi S, Misu R, Tomita K, et al. Activation of Neuropeptide FF Receptors by Kisspeptin Receptor Ligands. ACS Med Chem Lett. 2011;2:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim JS, Brownjohn PW, Dyer BS, et al. Anxiogenic and Stressor Effects of the Hypothalamic Neuropeptide RFRP-3 are Overcome by a Novel Antagonist. Endocrinology. 2015:en20151532. [DOI] [PubMed] [Google Scholar]
- 44. Liu X, Herbison A. Kisspeptin regulation of arcuate neuron excitability in kisspeptin receptor knockout mice. Endocrinology. 2015;156:1815–1827. [DOI] [PubMed] [Google Scholar]
- 45. Thompson EL, Patterson M, Murphy KG, et al. Central and peripheral administration of kisspeptin-10 stimulates the hypothalamic-pituitary-gonadal axis. J Neuroendocrinol. 2004;16:850–858. [DOI] [PubMed] [Google Scholar]