

Abstract
Vitamin D receptor (VDR)-mediated 1,25-dihydroxyvitamin D3 (1,25(OH)2D3)-dependent gene expression is compromised in the VDR null mouse. The biological consequences include: hypocalcemia, hypophosphatemia, elevated parathyroid hormone (PTH) and 1,25(OH)2D3, and consequential skeletal abnormalities. CYP24A1 is a cytochrome P450 enzyme that is involved in the side chain oxidation and destruction of both 1,25(OH)2D3 and 25-hydroxyvitamin D3 (25-OH-D3). In the current studies, we used liquid chromatography-tandem mass spectrometry technology to compare the metabolic profiles of VDR null mice fed either a normal or a calcium and phosphate-enriched rescue diet and to assess the consequence of transgenic expression of either mouse or human VDR genes in the same background. Serum 1,25(OH)2D3 levels in VDR null mice on normal chow were highly elevated (>3000 pg/mL) coincident with undetectable levels of catabolites such as 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone normally observed in wild-type mice. The rescue diet corrected serum Ca++, PTH, and 1,25(OH)2D3 values and restored basal expression of Cyp24a1 as evidenced by both renal expression of Cyp24a1 and detection of 24,25-(OH)2D3 and the 25-OH-D3-26,23-lactone. Unexpectedly, this diet also resulted in supranormal levels of 3-epi-24,25-(OH)2D3 and 3-epi-25-OH-D3-26,23-lactone. The reappearance of serum 24,25-(OH)2D3 and renal Cyp24a1 expression after rescue suggests that basal levels of Cyp24a1 may be repressed by high PTH. Introduction of transgenes for either mouse or human VDR also normalized vitamin D metabolism in VDR null mice, whereas this metabolic pattern was unaffected by a transgene encoding a ligand binding-deficient mutant (L233S) human VDR. We conclude that liquid chromatography-tandem mass spectrometry-based metabolic profiling is an ideal analytical method to study mouse models with alterations in calcium/phosphate homeostasis.
The biological actions of vitamin D are mediated by the hormone 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), which acts through a transcriptional mechanism that involves the vitamin D receptor (VDR) (1, 2). The production of 1,25(OH)2D3 is accomplished through the actions of CYP27B1, a specific mitochondrial cytochrome P450 enzyme whose renal expression is coordinately regulated by parathyroid hormone (PTH), fibroblast growth factor 23, 1,25(OH)2D3, and likely other factors as well. The VDR null mouse is devoid of all vitamin D-dependent gene expression and exhibits a number of calcium and phosphate related defects that include rickets (3, 4). One of the principal 1,25(OH)2D3-induced, VDR-mediated effects of vitamin D is to stimulate an autoregulatory feedback loop that involves the superinduction of CYP24A1, a gene that also encodes a cytochrome P450 enzyme (5). Although not restricted to expression exclusively in the kidney, this enzyme catalyzes the 24- and 23-hydroxylation of both 25-hydroxyvitamin D3 (25-OH-D3) and 1,25(OH)2D3, which initiates further degradation of vitamin D metabolites into side chain-truncated catabolic by-products destined for excretion (6). Early studies of mouse and human CYP24A1 suggest that although basal expression of these genes is generally low to undetectable in most target cells in vitro and in tissues in vivo, treatment with 1,25(OH)2D3 causes a rapid and dramatic up-regulation of both Cyp24a1 mRNA and protein beginning several hours after exposure (7, 8). In contrast, however, basal levels of CYP24A1 expression in the kidney are much higher. These levels appear responsible for the circulating levels of 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone that are routinely detected in the blood (9). The factors and the mechanisms responsible for the higher basal level of CYP24A1 expression in the kidney are unknown but are thought to involve residual activity of the VDR (10–12). These actions at the CYP24A1 gene are in addition to those that are exerted on renal CYP27B1 to suppress the expression of this gene and reduce the synthesis of 1,25(OH)2D3 under conditions in which the circulating levels of the hormone are elevated (13, 14).
The recent development of liquid chromatography-tandem mass spectrometry (LC-MS/MS) for simultaneous assay of several vitamin D metabolites, including 25-OH-D3, 24,25-(OH)2D3, and 25-OH-D3-26,23-lactone (15), is now facilitating the study of Cyp24a1 expression in vivo through measurement of serum levels of metabolic products that appear in individual mice after physiological or pathological manipulation. Previous studies of the VDR null mouse suggest that abrogation of VDR-mediated gene expression leads to a complete loss of not only basal and 1,25(OH)2D3-inducible Cyp24a1 expression, but the loss of an important negative feedback loop at the Cyp27b1 gene that by RIA results in a dramatic increase in circulating 1,25(OH)2D3 levels (3, 4). Further work using this VDR null mouse model has also revealed that high 1,25(OH)2D3 and low Ca++ levels can be corrected by feeding a high calcium, phosphate, and lactose (rescue) diet beginning at 16–21 days of age (12, 16). It is presumed from an observed increase in kidney Cyp24a1 mRNA measurements that serum 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone levels return to normal in animals maintained on this rescue diet as well, although this has not been shown (12). Interestingly, the rescue diet does not appear to induce the same network of genes that is regulated by vitamin D and responsible for the transepithelial uptake of calcium in the intestine (17).
An alternative approach to normalizing vitamin D metabolic profiles in VDR null mice would likely be to genetically reintroduce VDR transgenes into this particular mouse model. We have recently reported that both mouse and human BAC transgenes that span the regulatory and transcriptional loci comprising the 2 VDR genes are capable of recapitulating the tissue-specific expression of these genetic regions and fully rescuing the skeletal and numerous other phenotypic defects associated with the VDR null mouse (3, 18). The effectiveness of this rescue suggests but does not prove that vitamin D metabolic profiles are also normalized in these genetically modified mice. In contrast, however, a mouse strain genetically modified to express a human form of the VDR that is unable to bind 1,25(OH)2D3 (L233S) was incapable of restoring systemic mineral homeostasis, suppressing PTH or rescuing the skeletal phenotype of the VDR null mouse (19), although the noted ligand-independent effect of the VDR to prevent alopecia and perhaps other biology remains (19–21). Under this latter transgenic condition, we predict that vitamin D metabolic profiles are likely to remain aberrant.
In the current studies, we utilize LC-MS/MS technology to quantify vitamin D metabolite concentrations and to establish vitamin D profiles in individual mice under several sets of conditions. We show that elevated 1,25(OH)2D3 levels are indeed characteristic of VDR null mice, thereby confirming earlier studies. We then examined the time-dependent consequence of rescue diet administration on blood levels of Ca++, PTH, and vitamin D metabolites in the VDR null mice, and on the expression of both Cyp27b1 and Cyp24a1 mRNAs. The rescue diet temporally restored all appropriate parameters of mineral homeostasis in the background of the VDR null mouse, including blood levels of Ca++, phosphate and PTH, the renal expression of Cyp27b1, and most interestingly, the induction of renal Cyp24a1. Importantly, this diet also normalized vitamin D metabolite profiles in a fashion consistent with the down-regulation of Cyp27b1 and the up-regulation of Cyp24a1. Finally, we show that VDR null mice transgenic for either the wild-type mouse VDR (WT-mVDR) or wild-type human VDR (WT-hVDR) but not the ligand binding-defective form of the protein exhibited normal vitamin D metabolic profiles as well.
Materials and Methods
Animals
C57BL/6 mice (Harlan) and transgenic mice were fed a standard rodent chow (5008; Harlan Teklad). The VDR null strain was produced previously through targeted ablation of the second zinc finger of VDR DNA-binding domain (3). VDR null mice were fed a high-calcium and high-phosphate rescue diet containing 20% lactose and vitamin D (TD.96348; Harlan Teklad) beginning at weaning to maintain normal calcium levels. Animals were sacrificed at various times between 2 and 6 months, and blood and tissues were collected for biochemical analyses and RNA measurements, respectively. We have previously described the generation of bacterial artificial chromosome-based transgenic mice containing either WT-mVDR or WT-hVDR (18) as well as a transgenic mouse contain a BAC transgene expressing a human ligand binding-defective VDR (L233S-hVDR) (19). The transgenic strains were maintained as heterozygotes through outbreeding with C57BL/6 mice. These mice were crossed into the VDR null background as described previously (18). For gene expression analyses, 1,25(OH)2D3 (10 ng/g body weight) or PTH (1–84) (230 ng/g body weight) together with their vehicle controls were injected into mice ip, and tissues were collected 6 or 1 hour later, respectively, as previously described (22). Both genders were used equally for these studies. No correlation between the sexes was observed for any of the experimental parameters measured. Mice were exposed to a 12-hour light, 12-hour dark cycle. All animal studies were reviewed and approved by the Research Animal Care and Use Committee of the University of Wisconsin-Madison.
Reverse transcription-polymerase chain reaction
Tissues were collected in TRI reagent (Molecular Research Center) and homogenized using a PowerGen Model 125 Homogenizer (Fischer Scientific) to prepare total RNA following the manufacturer's protocol. Total RNAs were subjected to reverse transcription using the high capacity cDNA Reverse Transcription kit (Applied Biosystems). Gene expression was assessed by quantitative PCR using TaqMan primers (Applied Biosystems).
Serum analysis
Blood was collected from euthanized mice via cardiac puncture. Calcium and phosphate concentrations were determined in isolated serum using QuantiChrom Calcium and QuantiChrom Phosphate Assay kits (Bioassay Systems) as indicated by the manufacturer. Serum PTH concentrations were determined in EDTA-plasma using Mouse Intact PTH ELISA kits (Immutopics) according to the manufacturer. Vitamin D metabolite profiles were generated using LC-MS/MS (Model Acquity LC/Xevo TQ-S LC-MS/MS system; Waters Corp) after derivatization with the dienophile, 4-[2-(3,4-dihydro-6,7-dimethoxy-4-methyl-3-oxo-2-quinoxalinyl)ethyl]-3H-1,2,4-triazole-3,5(4H)-dione (DMEQ-TAD), as described recently (15). Briefly, this technique involves the simultaneous detection and quantification of a number of vitamin D metabolites from the analysis of 100 μL samples from individual mice. Multiple reaction monitoring (MRM) enables quantitative detection of several specific daughter fragments representing various monohydroxy (MRM = m/z 746→468) and dihydroxyvitamin D3 (MRM = m/z 762→468) metabolites and 25-OH-D3-26,23-lactone (MRM = m/z 774→468) during triplicate LC-MS/MS chromatographic runs of the same sample extract. Vitamin D metabolites were identified by their specific MRM and their chromatographic retention time on LC and quantified using added deuterated internal standards. Specific details of LC-MS/MS conditions and deuterated standards are provided in an earlier publication (15).
Generation of 3-epi-24,25-(OH)2D3 and 3-epi-25-OH-D3-26,23-lactone
3-epi-25-Hydroxyvitamin D3 (3-epi-25-OH-D3) (Isosciences) (5μM) was incubated for 12–24 hours with Chinese hamster lung fibroblasts (V79) stably transfected with hCYP24A1 as described previously (23). The resulting lipid extract was subjected to HPLC on Zorbax-SIL to resolve the substrate from the products, and the main metabolite, presumed to be 3-epi-24,25-(OH)2D3,was purified on further rounds of HPLC and then analyzed by LC-MS/MS to confirm its molecular mass and purity. Similarly, authentic 3-epi-25-OH-D3-26,23-lactone was generated from 3-epi-25-OH-D3 using the V79 cell line overexpressing the V391L/A326G mutant form of human CYP24A1 (23).
Statistical analyses
Data of serum analyses and gene expression are presented as the mean ± SEM. Student's unpaired t test was used to identify significant differences (P < .05).
Results
The diets used in these studies contained no significant quantities of vitamin D2 resulting in minimally detectable serum metabolites of vitamin D2. Accordingly, LC-MS/MS analysis focused on vitamin D3 metabolites (Tables 1 and 2). As reported in several previous studies, the VDR null mice reared on normal chow show a dramatic increase in serum 1,25(OH)2D3 concentrations to ng/mL levels close to 100 times the values found in wild-type mice; a corresponding fall in serum 25-OH-D3 levels (4, 24, 25) and dramatically reduced 24,25-(OH)2D3 concentrations. The chromatograms from VDR null and wild-type mice, shown in Figure 1A, illustrate the change in the dihydroxyvitamin D metabolite MRM profile with 2 adduct peaks representing DMEQ-TAD derivatives of 24,25-(OH)2D3 at 1.52 and 2.29 minutes and 2 adduct peaks representing DMEQ-TAD derivatives of 1,25(OH)2D3 at 2.41 and 2.80 minutes. Shown in Figure 1B is the MRM representing the molecular mass of the 25-OH-D3-26,23-lactone (m/z 774→468) metabolite, which shows 2 adduct peaks representing DMEQ-TAD derivatives of 25-OH-D3-26,23-lactone at 1.44 and 2.24 minutes. 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone are virtually undetectable in the VDR null mouse on normal chow. In an attempt to better resolve the residual 24,25-(OH)2D3 peak from the 1,25(OH)2D3 peak in the VDR null mouse, we used a variety of chromatography conditions, but under all conditions, a portion of the putative 24,25-(OH)2D3 peak still comigrated with authentic 24,25-(OH)2D3, whereas the 2 adduct peaks of 1,25(OH)2D3 were better resolved (data not shown). When the lipid extract was treated with sodium m-periodate, which is known to cleave vitamin D metabolites with vicinal hydroxyls (26), the metabolic profile was simplified such that the residual putative 24,25-(OH)2D3 peak was eliminated and only the adduct peaks for 1,25(OH)2D3 remained (data not shown).
Table 1.
Major Serum Vitamin D Metabolites in VDR-KO and Transgenic Mouse Models
| Genotype/Diet | n | Age (mo) | 25-OH-D3 (ng/mL) | 24,25-(OH)2D3 (ng/mL) | 25-OH-D3: 24,25-(OH)2D3 (Ratio) | 25-OH-D3-26,23-Lactone (ng/mL) | 25-OH-D3: 25-OH-D3-26,23-Lactone (Ratio) | 1,25(OH)2D3 (pg/mL) |
|---|---|---|---|---|---|---|---|---|
| Wild type + ND | 9 | 2 | 20.2 ± 3.2 | 9.6 ± 2.1 | 2.2 ± 0.2 | 3.0 ± 1.1 | 3.6 ± 1.7 | <300 |
| VDR-KO + ND | 8 | 2 | 8.2 ± 0.9 | 0.3 ± <0.1 | 47.8 ± 8.1 | 0.1 ± <0.1 | 98.0 ± 37 | 3140 ± 470 |
| VDR-KO + RD | 16 | 2 | 10.1 ± 1.8 | 0.1 ± 0.1 | 110.4 ± 30.0 | 0.1 ± <0.1 | 170.1 ± 55.1 | 3420 ± 630 |
| VDR-KO + RDa | 1 | 4 | 10.4 | 0.9 | 11.6 | 2.8 | 3.7 | <300 |
| VDR-KO + RD | 1 | 4 | 9.9 | 0.8 | 12.7 | 2.7 | 3.7 | <300 |
| VDR-KO + RDa | 1 | 4 | 18.1 | 0.2 | 89.4 | <0.1 | 304.3 | 3650 |
| VDR-KO + RD | 1 | 4 | 11.1 | 0.1 | 77.8 | <0.1 | 228.9 | 5160 |
| VDR-KO + RD | 1 | 4 | 15.2 | 0.2 | 87.5 | <0.1 | 314.7 | 2630 |
| VDR-KO + RD | 1 | 6 | 9.9 | 0.7 | 15.2 | 2.2 | 4.5 | <300 |
| VDR-KO + RD | 1 | 6 | 11.6 | 1.1 | 9.6 | 3.2 | 3.6 | <300 |
| VDR-KO + RD | 1 | 6 | 16.0 | 0.2 | 93.9 | <0.1 | 330.1 | 2850 |
| VDR-KO/Tgb mVDR + ND | 7 | 2 | 18.4 ± 4.7 | 8.5 ± 1.8 | 2.1 ± 0.2 | 3.4 ± 0.5 | 5.4 ± 0.8 | <300 |
| VDR-KO/Tg hVDR + ND | 3 | 2 | 22.2 ± 1.2 | 8.9 ± 1.6 | 1.9 ± 0.2 | 2.9 ± 0.3 | 5.9 ± 1.1 | <300 |
| VDR-KO/Tg L233S-hVDR | 5 | 2 | 6.7 ± 0.8 | 0.1 ± <0.1 | 62.3 ± 0.3 | <0.1 | 139.5 ± 16.4 | 3303 ± 656 |
Abbreviations: KO, knockout; ND, normal diet; RD, rescue diet containing high calcium/high lactose.
Chromatographic traces for these mice are presented in Figure 3.
Transgenic animals (Tg) with mouse(m), human(h), or L233S-modified hVDR (ND).
Table 2.
Serum 3-epi Metabolites of Vitamin D in VDR-KO and Transgenic Mouse Models
| Genotype/Diet | n | Age (mo) | 3-epi-25-OH-D3 (ng/mL) | 3-epi-25-OH-D3 (%a) | 3-epi-24,25-(OH)2D3 (ng/mL) | 3-epi-25-OH-D3-26,23-Lactone (ng/mL) |
|---|---|---|---|---|---|---|
| Wild type + ND | 9 | 2 | 1.4 ± 0.5 | 6.7 ± 2.8 | 0.7 ± 0.2 | 0.2 ± <0.1 |
| VDR-KO + ND | 8 | 2 | <0.1 | 1.0 ± 1.0 | <0.1 | 0.1 ± <0.1 |
| VDR-KO + RD | 16 | 2 | 0.7 ± 0.2 | 6.0 ± 1.1 | <0.1 | <0.1 |
| VDR-KO + RDb | 1 | 4 | 1.7 | 13.7 | 2.1 | 0.5 |
| VDR-KO + RD | 1 | 4 | 1.2 | 11.1 | 1.9 | 0.5 |
| VDR-KO + RDb | 1 | 4 | 1.4 | 7.0 | <0.1 | <0.1 |
| VDR-KO + RD | 1 | 4 | 1.2 | 9.5 | <0.1 | <0.1 |
| VDR-KO + RD | 1 | 4 | 1.9 | 11.1 | <0.1 | <0.1 |
| VDR-KO + RD | 1 | 6 | 1.3 | 11.6 | 1.3 | 0.3 |
| VDR-KO + RD | 1 | 6 | 1.7 | 12.6 | 1.9 | 0.4 |
| VDR-KO + RD | 1 | 6 | 1.7 | 9.5 | <0.1 | <0.1 |
| VDR-KO/Tgc mVDR + ND | 7 | 2 | 1.6 ± 0.4 | 8.4 ± 2.6 | 0.8 ± 0.1 | 0.2 ± <0.1 |
| VDR-KO/Tg hVDR + ND | 3 | 2 | 2.2 ± 1.3 | 9.9 ± 4.1 | 0.9 ± 0.7 | 0.2 ± <0.1 |
| VDR-KO/Tg L233S-hVDR | 5 | 2 | 0.2 ± 0.1 | 2.6 ± 0.6 | <0.1 | <0.1 |
Abbreviations: KO, knockout; ND, normal diet; RD, rescue diet containing high calcium/high lactose.
[3-epi-25-OH-D3] expressed as a percentage of 3-epi-25-OH-D3+25-OH-D3.
Chromatographic traces for these mice are presented in Figure 3.
Transgenic animals (Tg) with mouse(m), human(h), or L233S-modified hVDR (ND).
Figure 1.

LC-MS/MS-based vitamin D metabolic profiles of serum extracts of normal and VDR null mice fed normal chow. Vitamin D metabolites are extracted, derivatized with DMEQ-TAD, and then subjected to LC-MS/MS as described in Materials and Methods. Depicted are selected MRMs representing (A) the dihydroxyvitamin D3 daughter fragments at m/z 762→468 and (B) the 25-OH-D3-26,23-lactone daughter fragment at m/z 762→468. Note that each vitamin D metabolite generates 2 adduct peaks when derivatized with DMEQ-TAD; the peaks can be resolved on the LC column. Retention times of metabolites, based upon authentic standards, are provided in the text.
Administration of the synthetic rescue diet to VDR null mice led to rapid normalization of both Ca++ and phosphate levels within 2 months that was sustained as the mice aged (Figure 2A and data not shown). This diet-induced normalization of serum Ca++ and phosphate was likely responsible for the time-dependent reduction in the level of PTH that was seen as a function of time on the diet and that was eventually normalized to 40–50 ng/mL (Figure 2B). These changes were also accompanied by a progressive restoration of normal serum vitamin D metabolite profiles (Figure 3A and Table 1). This included the reappearance of both 24,25-(OH)2D3 (Figure 3A) and the 25-OH-D3-26,23-lactone (Figure 3B) as well as a reduction in the levels of 1,25(OH)2D3 (<300 pg/mL) in most the mice evaluated individually after 4 or 6 months on the rescue diet (Table 1). Interestingly, several of the mice at each of these latter time points retained both elevated levels of PTH and aberrant levels of vitamin D metabolites that included low levels of 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone and high levels of 1,25(OH)2D3 (3000 pg/mL or greater) (Table 1). These data suggest that despite normalization of blood Ca++ and phosphate levels, complete rescue, as determined by circulating levels of 1,25(OH)2D3, was not fully achieved for all of these mice, highlighting the importance of measuring this parameter together with other vitamin D metabolites in individual mice. Nevertheless, these changes in the levels of key vitamin D metabolites correlated generally with those that were observed at the level of Cyp27b1 and Cyp24a1 expression (Figure 2C). Of particular note was the recovery in fully rescued mice of renal Cyp24a1 expression when PTH levels were reduced in spite of the fact that the VDR remained absent (Figure 2C). This finding is consistent with the hypothesis that in addition to the accepted ability of PTH to induce the expression of Cyp27b1, it may also suppress the expression of Cyp24a1 (Figure 4). We also note that the time course of normalization of the systemic, enzymatic, and vitamin D metabolite profiles was extended relative to many of the studies described in the literature (4, 16). Somewhat surprisingly, the elevation in Cyp24a1 expression in the rescued mice also corresponded to an increase in the production of 3-epi-24,25-(OH)2D3 (1.69 and 1.78 min) and 3-epi-25-OH-D3-26,23-lactone (1.68 min) relative to wild-type animals, as evidenced by the higher quantities of these metabolites in the chromatograms shown in Figure 3, A and B, respectively, and documented in Table 2. The putative serum 3-epi-24,25-(OH)2D3 and 3-epi-25-OH-D3-26,23-lactone peaks comigrated identically with authentic 3-epi-24,25-(OH)2D3 and 3-epi-25-OH-D3-26,23-lactone produced in Cyp24a1 expression systems in vitro (data not shown).
Figure 2.

Serum analyses and renal gene expression in wild-type and VDR null mice on either normal chow diet or a high Ca++, phosphate, and 20% lactose diet. Serum Ca++ and phosphate (A), PTH (B), and Cyp27b1 and Cyp24a1 mRNA levels relative to Gapdh mRNA (C) levels in wild-type mice on normal chow diet, VDR null mice on normal chow diet, and VDR null mice on the high Ca++, phosphate, and 20% lactose diet were measured at the indicated ages. Wild-type animals were also treated with either vehicle or 1,25(OH)2D3 (10-ng/g bw) and Cyp27b1 and Cyp24a1 mRNA levels relative to Gapdh mRNA were measured 6 hours later (C). Ca++, phosphate, PTH, and gene expression levels are presented as the mean ± SEM (n = 5–7). WT, wild-type mice; VDR KO or KO, VDR null mice; ND, normal chow diet; RD, high Ca++, phosphate, and 20% lactose diet; veh, vehicle-treated sample; 1,25, 1,25(OH)2D3-treated samples. *, P < .05, vs wild-type controls.
Figure 3.
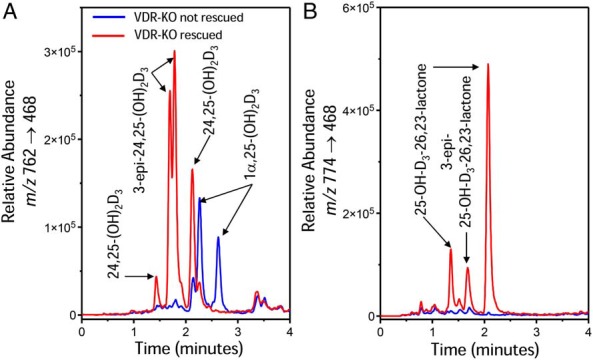
LC-MS/MS-based vitamin D metabolic profiles of serum extracts of VDR null mice (4-mo-old mice as indicated in Tables 1 and 2) fed high-calcium/high-lactose rescue diet. Vitamin D metabolites are extracted, derivatized with DMEQ-TAD, and then subjected to LC-MS/MS as described in Materials and Methods. Depicted are selected MRMs representing (A) the dihydroxyvitamin D3 daughter fragments at m/z 762→468 and (B) the 25-OH-D3-26,23-lactone daughter fragment at m/z 762→468. Note that each vitamin D metabolite generates 2 adduct peaks when derivatized with DMEQ-TAD; the peaks can be resolved on the LC column. Retention times of metabolites, based upon authentic standards, are provided in the text.
Figure 4.
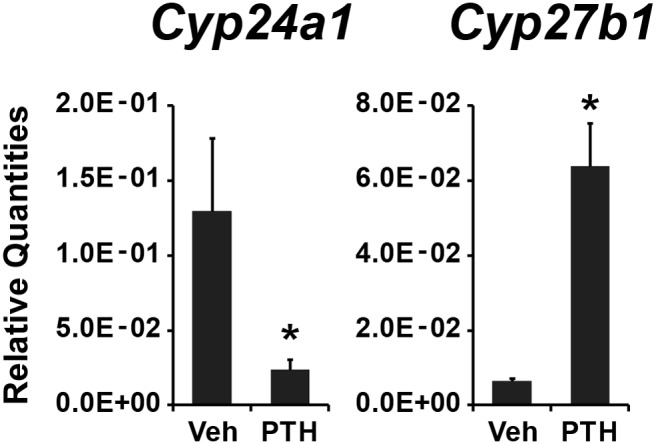
PTH induces the expression of Cyp27b1 and suppresses the expression of Cyp24a1. Cyp24a1 and Cyp27b1 expression levels were measured in kidney from wild-type mice treated with either vehicle (Veh) or PTH. Cyp24a1 or Cyp27b1 mRNA levels were normalized to Gapdh, and the relative quantities were presented as the mean ± SEM (n = 6). *, P < .05, vs vehicle-treated samples.
Figure 5 depicts the vitamin D metabolic profiles observed in VDR null mice maintained on normal chow that genetically express either WT-mVDR, WT-hVDR, or L233S-hVDR transgenes. The WT-mVDR and WT-hVDR transgenes reestablish normal levels of inducible Cyp24a1 expression as indicated by the significant production of both 24,25-(OH)2D3 (Figure 5A) and 25-OH-D3-26,23-lactone (Figure 5B) and suppressed Cyp27b1 expression as indicated by the normalization of circulating levels of 1,25(OH)2D3 (Figure 5A). In contrast, expression of the ligand binding-defective L233S-hVDR transgene failed to restore appropriate levels of these metabolites and the metabolic profile remained dominated by high levels of 1,25(OH)2D3 (Figure 5A). Quantitative analysis of the vitamin D metabolites in these genetically rescued mice is documented in Table 1. In addition, serum 25-OH-D3 was also significantly lower in VDR null mice on normal chow, in mice maintained on the rescue diet and in mice expressing the L233S transgene (25-OH-D3 = 8.2, 10.1, and 6.7 ng/mL). These levels of 25-OH-D3 contrast with the normal levels of 25-OH-D3 that are seen in wild-type mice on normal chow and in VDR null mice expressing either the WT-mVDR or WT-hVDR transgenes (25-OH-D3 = 20.2, 18.4, and 22.2 ng/mL, respectively). Accordingly, VDR null mice on normal chow and those expressing the L233S-hVDR transgene exhibited similar serum 1,25(OH)2D3 levels of 3000–3300 pg/mL. Because the differences in the levels of 24,25-(OH)2D3 were small and both the ratio of 25-OH-D3 to 24,25-(OH)2D3 and the ratio of 25-OH-D3 to 25-OH-D3-26,23-lactone between wild-type mice and those expressing either the mouse or hVDR transgenes were not statistically significant, the data suggest that both mouse and hVDRs are capable of restoring vitamin D metabolism equally well.
Figure 5.

LC-MS/MS-based vitamin D metabolic profiles of serum extracts of normal and VDR null mice on chow diet expressing either mVDR, hVDR, or the L233S-hVDR for their corresponding BAC transgenes mutant hVDRs. Vitamin D metabolites are extracted, derivatized with DMEQ-TAD, and then subjected to LC-MS/MS as described in Materials and Methods. Depicted are selected MRMs representing (A) the dihydroxyvitamin D3 daughter fragments at m/z 762→468 and (B) the 25-OH-D3-26,23-lactone daughter fragment at m/z 762→468. Note that each vitamin D metabolite generates 2 adduct peaks when derivatized with DMEQ-TAD; the peaks can be resolved on the LC column. Retention times of metabolites are provided in the text based upon authentic standards.
Discussion
This study used LC-MS/MS to confirm that the VDR null mouse exhibits elevated 1,25(OH)2D3 levels that can be corrected by the administration of a high Ca++ and high-phosphate rescue diet supplemented with lactose. As expected, a parallel down-regulation of renal Cyp27b1 expression also occurred in response to this diet. Interestingly, normalization of 1,25(OH)2D3 levels was accompanied by the reappearance of both 24,25-(OH)2D3 and the 25-OH-D3-26,23-lactone. The increase in these metabolites had been inferred from previous measurements of renal Cyp24a1 mRNA expression (12) but has not been previously reported in this model. Importantly, our data demonstrate not only an absolute increase in renal Cyp24a1 expression in response to the rescue diet, as previously shown by Panda et al (12), that is consistent with a rise in the level of its metabolic product as seen in our LC-MS/MS-derived serum data, but also a progressive increase in basal Cyp24a1 expression as a consequence of time on the rescue diet. This observation is somewhat surprising, given the finding that the loss of VDR-dependent gene expression in VDR null mice leads to both a loss of basal expression of Cyp24a1 in the kidney and a loss of Cyp24a1 expression in all tissues in response to 1,25(OH)2D3 as well (10–12). In the absence of the VDR, therefore, neither 24,25-(OH)2D3 nor the 25-OH-D3-26,23-lactone should be detectable regardless of dietary administration. We show here, however, that although renal Cyp24a1 is not detected in VDR null mice fed a normal diet and that the loss of its expression is reflected in the absence of circulating 24,25-(OH)2D3 and the 25-OH-D3-26,23-lactone, administration of the rescue diet results in a partial recovery of these metabolites in the blood which is likely mediated by the progressive time-dependent up-regulation of renal Cyp24a1 expression. Based upon this finding, the return of basal Cyp24a1 expression may be due to changes related to the normalization of Ca++ and/or phosphate homeostasis or the down-regulation of PTH. Although unknown factors could also be involved, it is clear that basal expression of renal Cyp24a1 is not fundamentally dependent upon the VDR itself.
Over the past few decades, researchers have postulated effects of both Ca++ and PTH on the renal vitamin D axis, but few of these have been suggested to represent direct effects of these factors on CYP24A1 gene expression. Reinhardt and Horst (27) demonstrated an antagonistic effect of PTH on 1,25(OH)2D3-mediated induction of Vdr and Cyp24a1 expression in cultured bone cells in vitro. Shinki et al (28) used thyroparathyroidectomized rats to demonstrate that renal but not intestinal Cyp24a1 mRNA expression occurred more rapidly in the absence of PTH and slower when PTH or cAMP were administered. Zierold et al (29) proposed that PTH directly suppressed Cyp24a1 expression in vitro by altering its mRNA turnover, although these studies were conducted in a highly engineered cultured cell line. The observations provided in Figure 4 therefore support the idea that high PTH levels may be directly involved in the suppression of Cyp24a1 mRNA levels, although how this occurs mechanistically is unclear. Such an action of PTH appears logical given the fact that this action would reinforce the known physiological role of the hormone in the induction of Cyp27b1 expression seen in the figure by suppressing the levels of the enzyme known to catabolize 1,25(OH)2D3 during times of Ca++ and phosphate shortage.
It is uncertain why dietary rescue of the VDR null mouse appears in this study to result in the normalization of Ca++ and phosphate at 2 months but requires additional time to suppress the circulating levels of both PTH and 1,25(OH)2D3 and to down-regulate renal Cyp27b1 expression. Indeed, in earlier reports, the results suggested that a period of 10 weeks was sufficient to correct most systemic parameters of Ca++, phosphate, and PTH when the diet was introduced to pups at 16 days of age (16, 25) and at weaning to rescue both the expression of Cyp27b1 and circulating levels of 1,25(OH)2D3 as well (24). On the other hand, although Panda et al (12) reported normalization of all of these parameters using the same rescue diet, exposure to this diet was conducted over a slightly longer period of time and the animals were analyzed at 4 months. Nevertheless, more recent studies by other investigators have shown some degree of temporal variability in circulating PTH and/or 1,25(OH)2D3 normalization similar to that we describe in the current study (30–32). For example, although the rescue diet generally restored Ca++ and phosphate levels at both 3 and 9 months in the study by Andrukhova et al (32), neither PTH nor 1,25(OH)2D3 levels were fully normalized even at 9 months. In a more extreme case, the studies of Song et al failed to demonstrate any significant efficacy of the rescue diet to restore appropriate PTH and 1,25(OH)2D3 levels in VDR null mice at 60 days, although Ca++ and phosphate levels were appropriately normalized within this time frame (30). The rescue diet in our case was introduced at 21 days and was identical to that reported in all of these earlier studies, although a follow-up trial that was initiated at 16 days of age and analyzed at 2 months resulted in a similar incomplete pattern. Surprisingly, PTH levels in VDR null pups at 16 days of age were approximately 10-fold higher than in normal mice (data not shown). These data are interesting in view of an earlier finding that VDR null fetuses are normocalcemic and phosphatemic, yet exhibit both higher 1,25(OH)2D3 levels and correspondingly elevated renal Cyp27b1 mRNA levels (33). It is worth noting that the ability of the LC-MS/MS method to detect vitamin D metabolites in small amounts of serum facilitated direct examination of the blood levels in each mouse. As a result, we observed that some of the mice that were held on the rescue diet for 4 months were fully normalized with regard to the levels of circulating vitamin D metabolites and Cyp27b1 expression, whereas these parameters remained high in others. An additional question is why a delay exists in restoring appropriate levels of PTH and 1,25(OH)2D3 in spite of the rapid normalization of blood Ca++ and phosphate. We hypothesize that although mineral levels are rapidly replenished under rescue diet conditions, the absence of the VDR creates a scenario under which neither blood PTH levels nor renal Cyp27b1 expression can be rapidly down-regulated by normal negative feedback loops that are normally mediated by the receptor's actions in both the parathyroid gland and the kidney, respectively. Irrespective of the reasons for this apparent delay in the rescue process, however, we have been able to use this to our advantage to understand features of the time-dependent rescue afforded by this high-Ca++, phosphate, and high-lactose diet.
Another unexpected finding was the appearance of 2 previously unidentified vitamin D metabolites, 3-epi-24,25-(OH)2D3 and 3-epi-25-OH-D3-26,23-lactone, that emerged along with the reappearance of 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone in diet-rescued mice. These 3-epi metabolites could arise through direct 3-epimerization of 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone or via hydroxylation of 3-epi-25-OH-D3, which was also detected in the VDR null mice. Whatever the source of 3-epi-vitamin D metabolites, our results suggest that the putative 3-epimerase enzyme is not regulated through VDR-mediated gene expression. The 2 adduct peaks for 3-epi-24,25-(OH)2D3 are particularly pronounced (see Figure 3A) and easily exceeded the sizes of the 2 24,25-(OH)2D3 adduct peaks. It should be stressed here that accurate quantitative LC-MS/MS requires the availability of deuterated internal standards. Because none of these were available for measuring the 3-epi-metabolites here, the values measured for the 3-epi forms represent only estimates. Based upon the fact that 3-epi-25-OH-D3 provides a less sensitive signal than 25-OH-D3 during LC-MS/MS, however, these values for 3-epi-24,25-(OH)2D3 and 3-epi-25-OH-D3-26,23-lactone may be underestimates of their true levels. Nevertheless, we still deemed the appearance of these 3-epi-metabolites important enough to confirm their identity by comparison with authentic standards generated in a CYP24A1-overexpressing mammalian cell system. We have identified the 2 serum metabolites by comparing LC retention times with authentic standards.
The above findings leave open the obvious question as to why dietary rescue of VDR null mice leads to the synthesis of these vitamin D metabolites with and without 3-epimerization. Interestingly, it has been postulated over the years that in addition to their catabolic status, both 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone may exert anabolic effects in tissues. For example, it has been suggested that 24,25-(OH)2D3 may play a role in bone fracture repair (34). This is an unlikely potential function for 24,25-(OH)2D3 here, however, because although both growth and mineralization of rachitic bone is certain to occur during the diet-induced recovery period, it would involve activity at the growth plate that is unrelated to bone fracture repair. Some 26,23-lactone metabolites circulate due to a strong binding affinity for DBP (35, 36), and some metabolites even possess VDR-binding properties suggesting that they could function as VDR antagonists to block 1,25(OH)2D3 mediated gene expression (37). Our data suggest that further examination of the production and function of the 3-epi vitamin D metabolites in rodent physiology may be warranted.
The vitamin D metabolic profiles of VDR null mice are normalized as a result of the transgenic expression of mVDR or hVDR, thereby confirming that normal vitamin D metabolism has been restored. These results together with our recent work that indicate from a broad phenotypic perspective that these mice are fully rescued is consistent with the disappearance of the rachitic phenotype (18). In contrast, the transgene expressing a mutant VDR incapable of binding ligand (L233S-hVDR) failed to restore normal vitamin D metabolism as indicated by retention of grossly elevated serum 1,25(OH)2D3 and the rachitic phenotype found in the VDR null mouse (19). As a result, we conclude that the use of large BAC transgenes that retain both the regulatory features and VDR transcription unit within the clones recapitulate tissue-specific expression of the endogenous VDR gene and fully rescue the normal mouse phenotype both in qualitative terms relative to the nature of the CYP24A1-mediated hydroxylation reactions (eg, the degree of 24,25-(OH)2D3 and 25-OH-D3-26,23-lactone formation) and in terms of phenotypic rescue of the VDR null mouse. Thus, these transgenic rescue mice will be useful for future studies of the activities of both the mouse and human VDR genes.
The work described here reinforces our view that LC-MS/MS using small 100-μL aliquots of serum (or less) from individual mice represents a novel method to study vitamin D metabolism in animal models. The assay focus is no longer on a single metabolite at a time but rather on the simultaneous measurement of multiple metabolites, thereby allowing for the determination of ratios of related metabolites (eg, 25-OH-D3:24,25-(OH)2D3). This approach has now been used for examining the Cyp2r1 null mouse, the Cyp27a1 null mouse, the Cyp2r1/ Cyp27a1 null mouse (38), the Cyp27b1 null mouse (39), the Cyp24a1 null mouse (15), and now the VDR null mouse and its rescued counterparts, in all cases with novel findings. In each example, the consistency of metabolite levels between individual mouse strains is impressive, suggesting that biological variation between mice with a similar genetic background at the same age and on the same diet is around 10% or less. The problem of data being skewed by pooling of “outliers” with greatly altered metabolism or erroneous genotyping is clearly avoided when employing this approach. Although the accurate assessment of 1,25(OH)2D3 levels in wild-type mice currently requires serum/plasma volumes of around 200 μL, improvements in assay technology, LC-MS/MS instrumentation, and dienophile design are anticipated to make this approach to metabolite analysis feasible for virtually all samples in the near future.
Acknowledgments
Through a Queen's University/Waters Corp agreement, Waters Corp generously provided the LC-MS/MS instrumentation used in these studies.
Animal husbandry aspects of this work were supported by the National Institutes of Arthritis, Musculoskeletal and Skin Diseases Grant AR-045173 and the National Institute of Diabetes Digestive and Kidney Diseases Grant DK-072281 (to J.W.P.). Analytical aspects of this work were supported by the National Institute of Standards and Technology and NIH-ODS as a part of the Vitamin D Standardization Program (VDSP) Grant 60NANB13D203, and the European Rare Diseases Consortium (E-Rare-2)/Canadian Institutes for Health Research Grant ERA-132931 (to G.J.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- DMEQ-TAD
- 4-[2-(3,4-dihydro-6,7-dimethoxy-4-methyl-3-oxo-2-quinoxalinyl)ethyl]-3H-1,2,4-triazole-3,5(4H)-dione
- 3-epi-25-OH-D3
- 3-epi-25-hydroxyvitamin D3
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MRM
- multiple reaction monitoring
- 1,25(OH)2D3
- 1,25-dihydroxyvitamin D3
- 25-OH-D3
- 25-hydroxyvitamin D3
- PTH
- parathyroid hormone
- VDR
- vitamin D receptor
- WT-hVDR
- wild-type human VDR
- WT-mVDR
- wild-type mouse VDR.
References
- 1. Jurutka PW, Bartik L, Whitfield GK, et al. Vitamin D receptor: key roles in bone mineral pathophysiology, molecular mechanism of action, and novel nutritional ligands. J Bone Miner Res. 2007;22(suppl 2):V2–V10. [DOI] [PubMed] [Google Scholar]
- 2. Pike JW. Genome-wide principles of gene regulation by the vitamin D receptor and its activating ligand. Mol Cell Endocrinol. 2011;347:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li YC, Pirro AE, Amling M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci USA. 1997;94:9831–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoshizawa T, Handa Y, Uematsu Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16:391–396. [DOI] [PubMed] [Google Scholar]
- 5. Ohyama Y, Noshiro M, Okuda K. Cloning and expression of cDNA encoding 25-hydroxyvitamin D3 24-hydroxylase. FEBS Lett. 1991;278:195–198. [DOI] [PubMed] [Google Scholar]
- 6. Jones G, Prosser DE, Kaufmann M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch Biochem Biophys. 2012;523:9–18. [DOI] [PubMed] [Google Scholar]
- 7. Nishimura A, Shinki T, Jin CH, et al. Regulation of messenger ribonucleic acid expression of 1 α,25-dihydroxyvitamin D3-24-hydroxylase in rat osteoblasts. Endocrinology. 1994;134:1794–1799. [DOI] [PubMed] [Google Scholar]
- 8. Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev. 1998;78:1193–1231. [DOI] [PubMed] [Google Scholar]
- 9. Sakaki T, Sawada N, Komai K, et al. Dual metabolic pathway of 25-hydroxyvitamin D3 catalyzed by human CYP24. Eur J Biochem. 2000;267:6158–6165. [DOI] [PubMed] [Google Scholar]
- 10. Li X, Zheng W, Li YC. Altered gene expression profile in the kidney of vitamin D receptor knockout mice. J Cell Biochem. 2003;89:709–719. [DOI] [PubMed] [Google Scholar]
- 11. Cui M, Li Q, Johnson R, Fleet JC. Villin promoter-mediated transgenic expression of transient receptor potential cation channel, subfamily V, member 6 (TRPV6) increases intestinal calcium absorption in wild-type and vitamin D receptor knockout mice. J Bone Miner Res. 2012;27:2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Panda DK, Miao D, Bolivar I, et al. Inactivation of the 25-hydroxyvitamin D 1α-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279:16754–16766. [DOI] [PubMed] [Google Scholar]
- 13. Tanaka Y, DeLuca H. Rat renal 25-hydroxyvitamin D3 1- and 24-hydroxylases: their in vivo regulation. Am J Physiol. 1984;246:E168–E173. [DOI] [PubMed] [Google Scholar]
- 14. Murayama A, Takeyama K, Kitanaka S, et al. Positive and negative regulations of the renal 25-hydroxyvitamin D3 1α-hydroxylase gene by parathyroid hormone, calcitonin, and 1α,25(OH)2D3 in intact animals. Endocrinology. 1999;140:2224–2231. [DOI] [PubMed] [Google Scholar]
- 15. Kaufmann M, Gallagher JC, Peacock M, et al. Clinical utility of simultaneous quantitation of 25-hydroxyvitamin D and 24,25-dihydroxyvitamin D by LC-MS/MS involving derivatization with DMEQ-TAD. J Clin Endocrinol Metab. 2014;99:2567–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li YC, Amling M, Pirro AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139:4391–4396. [DOI] [PubMed] [Google Scholar]
- 17. Lee SM, Riley EM, Meyer MB, et al. 1,25-Dihydroxyvitamin D3 controls a cohort of vitamin D receptor target genes in the proximal intestine that is enriched for calcium regulating components. J Biol Chem. 2015;290:18199–18215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee SM, Bishop KA, Goellner JJ, O'Brien CA, Pike JW. Mouse and human BAC transgenes recapitulate tissue-specific expression of the vitamin D receptor in mice and rescue the VDR-null phenotype. Endocrinology. 2014;155:2064–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee SM, Goellner JJ, O'Brien CA, Pike JW. A humanized mouse model of hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Endocrinology. 2014;155:4137–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee SM, Pike JW. The vitamin D receptor functions as a transcription regulator in the absence of 1,25-dihydroxyvitamin D3 [published online August 29, 2015]. J Steroid Biochem Mol Biol. doi: 10.1016/j.jsbmb.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huet T, Laverny G, Ciesielski F, et al. A vitamin D receptor selectively activated by gemini analogs reveals ligand dependent and independent effects. Cell Rep. 2015;10:516–526. [DOI] [PubMed] [Google Scholar]
- 22. Onal M, Bishop KA, St John HC, et al. A DNA segment spanning the mouse Tnfsf11 transcription unit and its upstream regulatory domain rescues the pleiotropic biologic phenotype of the RANKL null mouse. J Bone Miner Res. 2015;30:855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaufmann M, Prosser DE, Jones G. Bioengineering anabolic vitamin D-25-hydroxylase activity into the human vitamin D catabolic enzyme, cytochrome P450 CYP24A1, by a V391L mutation. J Biol Chem. 2011;286:28729–28737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Cromphaut SJ, Dewerchin M, Hoenderop JG, et al. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci USA. 2001;98:13324–13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Erben RG, Soegiarto DW, Weber K, et al. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol Endocrinol. 2002;16:1524–1537. [DOI] [PubMed] [Google Scholar]
- 26. Miller BE, Chin DP, Jones G. 1,25-Dihydroxyvitamin D3 metabolism in a human osteosarcoma cell line and human bone cells. J Bone Miner Res. 1990;5:597–608. [DOI] [PubMed] [Google Scholar]
- 27. Reinhardt TA, Horst RL. Parathyroid hormone down-regulates 1,25-dihydroxyvitamin D receptors (VDR) and VDR messenger ribonucleic acid in vitro and blocks homologous up-regulation of VDR in vivo. Endocrinology. 1990;127:942–948. [DOI] [PubMed] [Google Scholar]
- 28. Shinki T, Jin CH, Nishimura A, et al. Parathyroid hormone inhibits 25-hydroxyvitamin D3-24-hydroxylase mRNA expression stimulated by 1 α,25-dihydroxyvitamin D3 in rat kidney but not in intestine. J Biol Chem. 1992;267:13757–13762. [PubMed] [Google Scholar]
- 29. Zierold C, Mings JA, DeLuca HF. Parathyroid hormone regulates 25-hydroxyvitamin D(3)-24-hydroxylase mRNA by altering its stability. Proc Natl Acad Sci USA. 2001;98:13572–13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song Y, Kato S, Fleet JC. Vitamin D receptor (VDR) knockout mice reveal VDR-independent regulation of intestinal calcium absorption and ECaC2 and calbindin D9k mRNA. J Nutr. 2003;133:374–380. [DOI] [PubMed] [Google Scholar]
- 31. Nakagawa K, Kawaura A, Kato S, Takeda E, Okano T. Metastatic growth of lung cancer cells is extremely reduced in vitamin D receptor knockout mice. J Steroid Biochem Mol Biol. 2004;89–90:545–547. [DOI] [PubMed] [Google Scholar]
- 32. Andrukhova O, Slavic S, Zeitz U, et al. Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol Endocrinol. 2014;28:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kovacs CS, Woodland ML, Fudge NJ, Friel JK. The vitamin D receptor is not required for fetal mineral homeostasis or for the regulation of placental calcium transfer in mice. Am J Physiol Endocrinol Metab. 2005;289:E133–E144. [DOI] [PubMed] [Google Scholar]
- 34. St-Arnaud R, Naja RP. Vitamin D metabolism, cartilage and bone fracture repair. Mol Cell Endocrinol. 2011;347:48–54. [DOI] [PubMed] [Google Scholar]
- 35. Wichmann JK, DeLuca HF, Schnoes HK, Horst RL, Shepard RM, Jorgensen NA. 25-Hydroxyvitamin D3 26,23-lactone: a new in vivo metabolite of vitamin D. Biochemistry. 1979;18:4775–4780. [DOI] [PubMed] [Google Scholar]
- 36. Horst RL. 25-OHD3–26,23-lactone: a metabolite of vitamin D3 that is 5 times more potent than 25-OHD3 in the rat plasma competitive protein binding radioassay. Biochem Biophys Res Commun. 1979;89:286–293. [DOI] [PubMed] [Google Scholar]
- 37. Peräkylä M, Molnár F, Carlberg C. A structural basis for the species-specific antagonism of 26,23-lactones on vitamin D signaling. Chem Biol. 2004;11:1147–1156. [DOI] [PubMed] [Google Scholar]
- 38. Zhu JG, Ochalek JT, Kaufmann M, Jones G, Deluca HF. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc Natl Acad Sci USA. 2013;110:15650–15655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaufmann M, St-Arnaud R, Molin A, Kottler M-L, Jones G. Of knockout mice and men: use of LC-MS/MS to compare vitamin D metabolic profiles of Cyp27b1-null mice and patients with VDDR-type 1. 17th Vitamin D Workshop, 2014; Chicago, IL. [Google Scholar]