

Abstract
Sprague Dawley rats from different vendor colonies display divergent responses in a variety of experimental paradigms. An adjuvant-induced arthritis (AA) model of human rheumatoid arthritis was used to examine immune and endocrine responses to inflammatory challenge in Sprague Dawley rats from Charles River and Harlan colonies. Rats were injected with either complete Freund's adjuvant or physiological saline (control), weights, and paw volumes measured over 15 days, and blood and tissue were collected 16 days post-injection. Overall, Harlan rats developed more severe AA than Charles River rats. In addition, despite comparable corticosterone levels, corticosteroid binding globulin levels were lower in Harlan compared with Charles River rats in the absence of inflammation, suggesting that a lower corticosterone reservoir in Harlan rats may underlie their greater susceptibility to inflammation. With increasing AA severity, there was an increase in plasma corticosterone (total and free) and a decrease in corticosteroid binding globulin in both Charles River and Harlan rats. However, contrasting patterns of cytokine activation were observed in the hind paw, suggesting a reliance on different cytokine networks at different stages of inflammation, with Charles River rats exhibiting increased TNF-α, monocyte chemotactic protein-1 (MCP-1), keratinocyte chemoattractant/growth-regulated oncogene (KC/GRO), and IL-1β in the absence of clinical signs of arthritis, whereas Harlan had increased TNF-α, monocyte chemotactic protein-1, and IL-6 with mild to moderate arthritis. These colony-specific differences in endocrine and immune responses to AA in Sprague Dawley rats must be considered when comparing data from different laboratories and could be exploited to provide insight into physiological changes and therapeutic outcomes in arthritis and other inflammatory disorders.
Rats of the same strain are generally considered comparable, but genetic drift within colonies of a single strain may occur. Significant differences in various measures have been reported among Sprague Dawley (SD) rats from different vendors, including effects of dopamine agonists on sensorimotor gating (1); Pavlovian conditioned behaviors (2); hypoxic responses (3); noradrenergic innervation of the spinal cord (4); neuropathic pain behaviors (5); and metabolic, endocrine, and immune function (6).
Using the adjuvant-induced arthritis (AA) model of rheumatoid arthritis (RA) used extensively in immunological research (7), the current study investigated how the endocrine and immune systems of SD rats from two different vendors (Harlan vs Charles River) respond to an inflammatory challenge. Briefly, AA is induced through an intradermal injection of complete Freund's adjuvant (CFA). Inflammation then develops as a result of an immune response against components of the cartilage matrix, initiated by a T-cell response to an exogenous antigen, likely a component of a mycobacterial 65 kDa heat-shock protein (8), thought to contain a cartilage mimicking epitope (9). This AA model allows examination of a number of parameters, including the percentage of rats that develop inflammation (not all rats will develop AA after adjuvant injection [10]) and the rate of resolution from inflammation, with these factors depending on dose and site of injection (10), sex (11), and strain (12–14) as well as environmental factors (15).
Cytokines and hormones of the hypothalamic-pituitary-adrenal (HPA) axis are among the most consistently studied mediators of inflammation. Furthermore, there is significant interplay between the immune and endocrine systems, with shared receptors, ligands, and regulatory feedback mechanisms (16). Cytokines stimulate the HPA axis after an immune challenge, affecting the release of hormones at the level of the hypothalamus and pituitary gland, which, in turn, has downstream effects on immune function and resolution of inflammation (17). As a result, changes in communication between cytokines and the HPA axis can impair recovery and lead to and/or exacerbate underlying pathological conditions (18). In the AA model, plasma corticosterone levels increase with disease onset, whereas the adrenocorticotropic hormone (ACTH) levels decrease and there is a loss of the corticosterone diurnal rhythm (19). Conversely, in the absence of a functional HPA axis (adrenaletomized animals), AA onset is earlier and more severe, and a fatal course of inflammation often results (20). In addition to HPA axis activation, cytokines increase in the circulation and synovial tissue during active AA, and strong positive correlations have been described between plasma cytokine levels, joint cytokine levels, and joint circumference (21). As a result, agents targeting inflammatory cytokines are used clinically for treatment of RA (22), especially in combination with glucocorticoid-based treatments (23, 24).
Charles River (Hollister, CA) and Harlan (Indianapolis, IN) SD rats exhibit different endocrine and immune responses to inflammatory challenge with lipopolysaccharide (LPS), turpentine, and IL-1β, showing unique ACTH and cytokine (IL-6) responses but no differences in the corticosterone response (6). Moreover, Charles River (Raleigh, NC) SD rats have been shown to have a more pronounced ACTH response to restraint stress when compared to SD rats from Simonsen (Gilroy, CA), with Harlan (Kent, WA) SD rats demonstrating a lack of an ACTH response (25). Differences in corticosteroid binding globulin (CBG), the major transport protein for glucocorticoids, have not been previously reported in specific rat strains, but the effects of differential plasma CBG levels on inflammatory outcomes are suspected based on differences between strains. For example, Fisher rats exhibit higher plasma CBG and corticosterone levels than Lewis rats (26), and generally fail to develop arthritis, a finding linked to appropriate stimulation of the HPA axis by inflammatory mediators and subsequent feedback between HPA and immune systems (27). Based on these findings, the current study tested the hypothesis that differential endocrine and immune responses may underlie the different responses to inflammation of Harlan vs Charles River SD rats.
Materials and Methods
Animals
Adult female SD rats were obtained from Charles River Laboratories International, Inc (St Constant, QC, Canada) and Harlan Laboratories, Inc (Frederick, MD) (n = 29/vendor). Rats were raised in barrier rooms in their respective vendor facilities, under controlled temperatures (21–23°C), with access to autoclaved diets (Charles River Rodent Diet 5075; Harlan Teklad 2018S), and maintained on a 12-hour light, 12-hour dark cycle. Rats were shipped by air under climate-controlled conditions (transport time for Charles River ∼24 h; Harlan, ∼24–48 h) and were pair housed in a colony room on a 12-hour light, 12-hour dark cycle, with controlled temperature (21–22°C) and ad libitum access to standard laboratory chow (Purina Laboratory; Rodent Diet number 5001) and water. All procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of British Columbia Animal Care Committee.
Induction and clinical assessment of AA
Only female rats were used because they exhibit heightened immune responses (28) and increased prevalence of autoimmune disorders (29), including increased susceptibility to experimentally induced arthritis (30, 31). On postnatal day 55–60, rats were anesthetized with isoflurane, and each received two 0.05-mL intradermal injections at the base of the tail. Rats were injected with either CFA (n = 7–8 per vendor per dose) or physiological saline (control; n = 6–7 per vendor). CFA was prepared by grinding Mycobacterium tuberculosis H37 RA (Difco Laboratory) and dissolving the powder in incomplete Freund's adjuvant (15). Two doses of CFA were prepared initially, 12 mg/mL and 3 mg/mL, resulting in a dose of 1.2 or 0.3 mg/rat. These doses were chosen with the aim of identifying a dose that results in mild to moderate arthritis in approximately 50% of rats from each vendor. At 0.3 mg, none of the Charles River rats showed signs of inflammation on day 15 post-injection; however, approximately 40% of the Harlan rats displayed signs of severe inflammation. As a result, two CFA doses were added, 0.6 mg and 0.2 mg/rat, designed to induce mild to moderate inflammation in the Charles River and Harlan rats, respectively. After the injections, the rats were single housed and weighed, and paw volume was measured using a plethysmometer (IITC Life Science Inc).
On days 6, 9, 11, 13, and 15 post-injection, rats from both the CFA- and saline-injected conditions were anesthetized and weighed, paw volume was measured, and clinical signs of arthritis were assessed (Figure 1). To calculate the clinical score, each of the four paws was scored on a 0- to 4-point scale (15). Across doses, rats achieving a clinical score of 8 or greater at any point during the study were classified as developing severe arthritis (Adj/S), whereas rats with a clinical score of 1 or greater but less than 8 were classified as developing mild to moderate arthritis (Adj/M-M).
Figure 1.
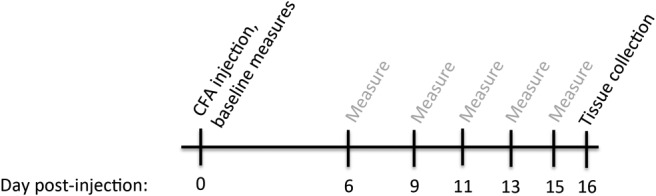
Experimental time line. Rats received intradermal injections of CFA on postnatal days 55–60 (hereafter referred to as d 0). Baseline measurements of body weight, hind paw volume, and clinical score were collected on day 0, and post-injection measurements were collected on days 6, 9, 11, 13, and 15. Tissue and plasma were collected on day 16 post-injection (peak of inflammation).
Termination and tissue collection
On day 16 post-injection, rats were decapitated between 8:00 and 10:30 am, and trunk blood was collected in polystyrene tubes containing EDTA. Blood was centrifuged and plasma was collected and stored at −80°C. Hind paws were removed at the level of the tibiotarsal joint, flash frozen in liquid nitrogen, and stored at −80°C. Vaginal lavage samples were collected and assessed cytologically to determine estrous cycle stage.
Plasma ACTH, corticosterone, and CBG measurements
Plasma ACTH and corticosterone (total) were measured using the ImmuChem double antibody hACTH 125I radioimmunoassay kit and the ImmuChem double antibody corticosterone 125I radioimmunoassay kit (MP Biomedicals, LLC), respectively. The minimum detectable concentrations were 5.7 pg/mL for ACTH, and 7.7 ng/mL for corticosterone, with intra- and interassay coefficients of variation of less than 10% for both assays.
The steroid-binding capacity of CBG was measured using a ligand-saturation assay that uses dextran-coated charcoal (DCC) to separate CBG-bound from free [3H]corticosterone (PerkinElmer Life Sciences) (32, 33). Plasma samples were diluted (1:1500) in phosphate buffered saline and stripped of endogenous steroids by incubation with DCC. Samples were then incubated with 1 nM [3H]corticosterone in the absence or presence of excess corticosterone to monitor nonspecific binding, followed by the measurement of bound [3H]corticosterone after adsorption of free steroid with DCC for 10 minutes at 0°C. All samples were measured in the same assay and the intra-assay variability was less than 10% (33). The dissociation-rate constant (Kd), was determined by Scatchard analysis at 4°C, in which Charles River and Harlan plasma samples were incubated with increasing amounts of [3H]corticosterone, as described previously (32).
Levels of CBG-bound corticosterone were calculated using the mass action equation, according to Boksa (34), using a Kd of 45 nM, as determined for rat CBG at 37°C (35). The CBG-bound corticosterone levels were then used to calculate the free corticosterone levels using the following equation: free corticosterone = (total corticosterone) − (CBG bound corticosterone).
Serpina6 sequencing
Genomic DNA was isolated from blood lymphocytes using the QIAamp DNA minikit (QIAGEN Sciences). PCR amplification was performed on the rat Serpina6 exons and proximal promoter with specific oligonucleotide primer pairs (Table 1) and Platinum Pfx polymerase (Invitrogen). The PCR products were sequenced and results were analyzed using 4Peaks software (version 1.7.2, mekentosj.com).
Table 1.
Oligonucleotide Primers Used to Sequence
| Sequence | PCR and Sequencing Primers (5′–3′) | PCR Product, bp |
|---|---|---|
| Promoter | CCAGCATGCATAAGGTTCAGC | 591 |
| (+550 to −41) | GGTTGTGCTTTGCTGCCAGG | |
| Exon 1 | Forward, CCAGCAAACAAGATTTAGTAGG | 199 |
| Reverse, GCTGTGTTCTGGAGTGCAGC | ||
| Exon 2 | Forward, GCCAATGTGAAGGAAGGATAGG | 862 |
| Reverse, CCAACCTGGTAGAGATTGGC | ||
| Exon 3 | Forward, GCAGGTGGCTGCATAGCTGG | 556 |
| Reverse, GGCTAGAGAACCTCACAGCC | ||
| Exon 4 | Forward, CCTATCCCCAAGTTTAACCAGG | 460 |
| Reverse, GGGTTTGTCATTTGGGACC | ||
| Exon 5 | Forward, GGCTATTTCACCTTCCATGG | 462 |
| Reverse, CCTCTTTCTCAGTGCTCCCTTC |
Hind paw homogenization
Hind paw samples were manually crushed on dry ice, added to tubes containing 1.3 g garnet and 2 mL cold lysis buffer, and homogenized using an Omni Bead Ruptor 24 (Omni International). The homogenates were sonicated three times (5 sec/cycle) on ice and centrifuged, and the supernatants were stored at −20°C for protein and cytokine measurements.
Multiplex cytokine immunoassays and protein quantification
Cytokine assays were performed using a custom Meso Scale Discovery rat cytokine 8-plex panel capable of simultaneously measuring IL-1β, IL-4, IL-6, IL-10, interferon (IFN)-γ, keratinocyte chemoattractant/growth-regulated oncogene (KC/GRO), monocyte chemotactic protein-1 (MCP-1), and TNF-α (catalog number N05IA-1; Meso Scale Discovery). Plates were read using a Sector Imager 2400 and data analyzed using the Meso Scale Discovery Workbench software version 4.0 (Meso Scale Discovery).
Total protein levels were quantified in hind paw tissue homogenates using the Pierce microplate bicinchoninic assay protein assay kit (Pierce Biotechnology). Tissue cytokine levels were adjusted and reported as picograms of cytokine per milligram of protein.
Statistical analyses
Data were analyzed using ANOVAs for the factors of colony and AA severity or colony and dose, as appropriate, with repeated measures as required, followed by Fisher post hoc tests to examine significant main effects and interactions (significant ANOVA and post hoc P values reported in text; F statistic reported in figure legends). The assumption of sphericity was examined using Mauchly's test and when violated, F values were corrected using Greenhouse-Geisser estimates of sphericity. In line with our hypotheses that Charles River and Harlan rats would differ in severity of inflammation and in hormone and cytokine responses, planned pairwise comparisons were carried out as indicated. Differences were considered significant at P ≤ .05, and trends (P > .05 and P < .085) were examined, as appropriate.
Results
Clinical scores reveal a more severe course of AA in Harlan compared with Charles River SD rats at a low (0.3 mg) CFA dose but a similar course at a high (1.2 mg) CFA dose
The mean day of arthritis onset (first day of clinical score >0) did not differ between Harlan and Charles River rats, ranging from a mean of day 11.1 (± 1.20) to 13.0 (± 0.63) post-injection for all CFA doses examined. For comparisons of average clinical scores, only days 11, 13, and 15 post-injection are shown (Figure 2, A and B) because clinical scores were ≤2 prior to day 11.
Figure 2.

Severity of AA after CFA injection in Charles River and Harlan SD rats. A, At 0.3 mg CFA, Harlan rats showed increasing clinical scores over time, with no change in clinical scores over time for Charles River rats (main effect of day, F [2, 26] = 5.62, P < .01; day × colony interaction, F [2, 26] = 6.35, P < .01). On d 15, the average clinical score was greater in the Harlan than Charles River rats (§, P = .016; trend for effect of colony: F (1, 13) = 3.85, P = .072). B, At 1.2 mg CFA, Charles River and Harlan rats both showed increasing clinical scores over time (main effect of day, F [1.16, 16.27] = 10.10, P < .01). No colony-specific differences were detected at this dose. Data are presented as mean ± SEM; n = 7–8 per colony/dose; dotted line = clinical score of 8. Post hoc: *, P < .05, **, P < .01, ***, P < .001 comparison with d 11, unless indicated otherwise; §, P < .05, pairwise comparison between Charles River and Harlan rats. C and D, Arthritis severity profiles (percentage of rats in each category) compared by stacked bar graphs across the three CFA doses for Charles River (C; 0.3, 0.6, 1.2 mg) and Harlan (D; 0.2, 0.3, 1.2 mg) rats. Adj/NA, no clinical signs of AA after CFA injection; Adj/M-M, mild-moderate AA, clinical score <8; Adj/S, severe AA, clinical score ≥8.
At the 0.3-mg CFA dose, Harlan rats had increasing clinical scores over time, which is indicative of increasing arthritis severity, whereas Charles River rats had little to no inflammatory response at this dose (Figure 2A; day × vendor interaction, P = .006). Based on our a priori hypothesis, as well as a trend for a colony difference in the average clinical scores (between-subjects effect, P = .072), planned pairwise comparisons on day 15, at the peak of inflammation, indicated that Harlan rats displayed higher clinical scores than Charles River rats (P = .016), supporting the suggestion of greater arthritis severity in Harlan compared with Charles River rats at the 0.3-mg dose. Comparatively, at the high CFA dose (1.2 mg), colony differences in inflammation were not observed, with both Charles River and Harlan rats showing increasing clinical scores over time (Figure 2B; main effect of day, P = .004). There were no colony differences in the proportion of rats within each stage of the estrous cycle in controls (saline-injected) or CFA-injected rats.
Classification of rats by severity of inflammation supports the finding of more severe arthritis in Harlan compared with Charles River SD rats
To further investigate dose-response differences, an additional low CFA dose (0.2 mg) was tested in Harlan rats, and an additional intermediate CFA dose (0.6 mg) was tested in Charles River rats. As noted in Materials and Methods, separate doses were selected for the Charles River and Harlan rats due to the more severe arthritic profile observed in Harlan rats at the 0.3-mg dose. Stacked bar graphs (Figure 2, C and D) represent the proportion of rats from all CFA doses at each arthritis severity level: no clinical signs of arthritis (Adj/NA), mild to moderate arthritis (Adj/M-M), or severe arthritis (Adj/S). It should be noted that clinical score groupings are based on the highest clinical score reached by a rat during the course of the experiment (d 0–16).
A comparison of severity profiles across the three selected CFA doses demonstrates that at the lowest doses tested in rats from each vendor (Charles River: 0.3 mg; Harlan: 0.2 mg), there were no cases of severe arthritis. However, at the other two doses, fewer Harlan (0.3 and 1.2 mg: 13%) than Charles River (0.6 mg: 33%; 1.2 mg: 38%) rats failed to develop clinical signs of arthritis, and there were more cases of mild to moderate arthritis in Harlan (0.3 mg: 50%; 1.2 mg: 63%) compared with Charles River (0.6 mg: 33%; 1.2 mg: 38%) rats. A direct comparison of Charles River and Harlan rats at the 0.3 mg and 1.2 mg doses (Figure 2, C and D) also indicates increased AA severity in Harlan rats. At the 0.3-mg dose, 38% of Harlan rats developed severe AA compared with 0% of Charles River rats, and 13% of Harlan rats failed to develop AA compared with 43% of Charles River rats. At the 1.2-mg/mL dose, although similar arthritis profiles were observed for Charles River and Harlan rats at the most severe level of inflammation (Adj/S; 25% for both Charles River and Harlan rats), colony differences similar to those at the lower CFA dose were again observed for rats failing to develop arthritis (Charles River: 38%, Harlan: 13%) as well as for those developing mild to moderate inflammation (Charles River: 38%, Harlan: 63%).
Rats with severe inflammation had decreased weight gain and increased paw volume
Weight and paw volume changes over the course of the experiment (measured on d 0–15 post-injection) collapsed across dose are shown in Figure 3. Overall, both body weight and paw volume were significantly higher in Charles River compared with Harlan rats, both on the day of injection and at the peak of inflammation (main effects of colony, P < .001), and as expected, both body weight and paw volume increased as rats grew throughout the experiment (day × colony interaction, P < .001). Not surprisingly, rats developing severe arthritis (Adj/S) exhibited reduced weight gain, and increased paw volume, compared with those with no clinical signs of arthritis (control, Adj/NA) or with mild-moderate arthritis (Adj/M-M) (main effects of AA severity for weight and paw volume, P < .001).
Figure 3.

Body weight and paw volume after CFA injection in Charles River and Harlan SD rats. A, Body weight increased in all rats from d 0 to d 15 (main effect of day, F [1, 49] = 537.49, P < .001), and weights were higher in Charles River vs Harlan rats on both days (day × colony interaction, F [1, 49] = 61.14, P < .001; §§§). Body weight was impacted by severity of arthritis (main effect of AA severity, F [3, 49] = 2.71, P = .05), with lower body weights in rats developing severe arthritis (Adj/S), compared with all other conditions. B, Paw volume increased from d 0 to d 15 (main effect of day, F [1, 49] = 42.24, P < .001) and was also impacted by severity of arthritis (main effect AA severity, F [3, 11.51] = 49, P < .001), with higher paw volumes in Adj/S compared with all other conditions on d 15. Charles River rats also had higher paw volumes than Harlan rats on d 0 and d 15 (main effect of colony, F [1, 49] = 36.04, P < .001; §§§). Data presented as mean ± SEM. Adj/NA: Charles River: n = 14, Harlan: n = 6; Adj/M-M: Charles River: n = 3, Harlan: n = 12; Adj/S: Charles River: n = 4, Harlan: n = 5; control, saline-injected: Charles River: n = 7, Harlan: n = 6. Post hoc: *P < 0.05; **P < 0.01; ***P < 0.001 (comparison to control, unless indicated otherwise). §§§: P < 0.001; post hoc comparisons between colonies.
AA caused an increase in plasma corticosterone and a decrease in CBG in both Charles River and Harlan SD rats but no change in plasma ACTH
On day 16, basal plasma ACTH levels were not different between colonies and did not differ by AA severity (Figure 4A). Plasma corticosterone levels, however, increased with CFA injection (main effect of AA severity, P < .001; Figure 4B), with the highest corticosterone levels in Adj/S rats (Adj/S > control, Adj/NA, P < .05). Free plasma corticosterone also increased with CFA injection (main effect of AA severity, P < .001; Figure 4D) in both Charles River and Harlan SD rats, with patterns of free corticosterone closely paralleling those of total corticosterone.
Figure 4.

Basal ACTH, corticosterone (total and free), and CBG at the peak of inflammation (d 16 post-injection) and characterization of CBG protein by Scatchard Plot. A, Plasma ACTH levels did not change with CFA injection and were not different between colonies. B, Plasma corticosterone levels increased with CFA injection in rats from both colonies (main effect of AA severity, F [3, 49] = 7.00, P < .001). Colony-specific differences were not detected. C, Plasma CBG levels decreased with AA (main effect of AA severity, F [3, 49] = 10.99, P < .001), with differential effects by colony (main effect of colony, F [1, 49] = 9.72, P < .01); Harlan rats show decreased CBG in control and Adj/NA conditions (§§/§) compared with Charles River rats. D, Free corticosterone levels increased with CFA injection in rats from both colonies (main effect of AA severity, F [3, 48] = 8.40, P < .001). Colony-specific differences in free corticosterone levels were not detected. Note: Free corticosterone levels were log transformed for statistical analysis; untransformed corticosterone levels are presented in the figure. Data are presented as mean ± SEM (A–D). Adj/NA: Charles River: n = 14, Harlan: n = 6; Adj/M-M: Charles River: n = 3, Harlan: n = 12; Adj/S: Charles River: n = 4, Harlan: n = 5; control, saline-injected: Charles River: n = 7, Harlan: n = 6. Post hoc: *, P < .05, **, P < .01, ***, P < .001 (comparison with control, unless indicated otherwise); §/§§, P < .05, P < .01, respectively, indicating pairwise comparisons between colonies. E, Scatchard plots indicate no difference in the rate dissociation constant (Kd) between Charles River (Kd = 1.15 nM) and Harlan (Kd = 1.13 nM) control rats, indicating no difference in CBG-corticosterone binding affinities between colonies.
Plasma CBG levels decreased with AA severity (main effect of AA severity, P < .001), with lowest levels detected in Adj/S rats (Adj/S < control, P < .001 for Charles River, P < .05 for Harlan; Figure 4C) and were significantly different between colonies, with lower levels overall in Harlan compared with Charles River (main effect of colony, P < .01). Planned pair-wise comparisons also revealed that even in the absence of AA (control, Adj/NA), CBG levels were significantly lower in Harlan compared with Charles River rats (P < .05; Figure 4B). In addition, the overall plasma CBG profile differed between the colonies, with Charles River showing a more marked drop in CBG levels than Harlan rats. With severe arthritis, however, CBG levels did not differ between Charles River and Harlan rats.
Differences in Serpina6 sequences in Harlan and Charles River SD rats
When the Serpina6 coding sequences for CBG were compared in the two SD rat colonies, we found a synonymous single nucleotide transition (C>T) in exon 2 within the codon for Phe152, and two non-synonymous single nucleotide transitions (A>G) in exon 4 that cause amino acid substitutions (Ile298Met and Met307Val) in Charles River SD rats. However, there were no sequence differences within the 439-bp proximal promoter region we have defined previously (36).
Plasma CBG-corticosterone binding affinities are similar in Charles River and Harlan SD rats
To determine whether the difference in CBG coding sequences between Charles River and Harlan SD rats alters CBG-corticosterone binding affinity, plasma samples from control rats from each colony were subjected to Scatchard analysis, using 3H-corticosterone as the radiolabeled ligand. The results indicated that CBG from Charles River and Harlan rats have similar rate dissociation constants (Kd) for corticosterone, 1.15 nM and 1.13 nM, respectively (Figure 4E), and therefore similar affinities (Ka = 1/Kd).
Hind paw levels of proinflammatory cytokines increased with AA, with Charles River and Harlan SD rats showing different inflammatory profiles
In the hind paw, the levels of five proinflammatory cytokines and chemokines (Figure 5, A–E) increased with the development of AA (main effects of AA severity, P < .002). By contrast, the anti-inflammatory cytokines IL-4 and IL-10 were uniformly undetectable (not shown).
Figure 5.

Levels of cytokines in the hind paw at the peak of inflammation (d 16 post-injection). TNF-α (A) and MCP-1 (B) levels were differentially impacted by arthritis severity in Charles River vs Harlan rats (for TNF-α, main effect of AA severity, F [3, 49] = 20.26, P < .001; colony × AA severity interaction, F [3, 49] = 5.29, P < .01; for MCP-1, main effect of AA severity, F [3, 49] = 20.68, P < .001; trend for colony × AA severity interaction, F [3, 49] = 2.59, P = .064). TNF-α levels were lower in Harlan compared with Charles River control rats (§§), and TNF-α and MCP-1 were lower in Harlan compared with Charles River rats in the absence of AA (§§). C, KC/GRO levels increased in both Charles River and Harlan rats with CFA injection (main effect of AA severity, F [3, 49] = 13.28, P < .001). Overall, KC/GRO levels were lower in Harlan compared with Charles River rats, irrespective of AA severity (main effect of colony, F [1, 49] = 10.23, P < .01; §§). D, IL-1β levels increased with CFA injection, with no differences between colonies (main effect of AA severity, F [3, 49] = 18.54, P < .001). E, IL-6 levels increased with arthritis in all Charles River and Harlan rats that developed clinical signs of arthritis, compared with controls (main effect of AA severity, F [3, 48] = 12.42, P < .001). F, For IFN-γ, data were non-normally distributed after transformation and were not analyzed statistically (percentage of rats with detectable levels within each severity group indicated on graph). Note: Lower limits of detection (LLOD) for cytokine assay are as follows: KC/GRO, 2.62 pg/mL; IFN-γ, 15.8 pg/mL; IL-10, 38.6 pg/mL; IL-1β, 6.7 pg/mL; IL-4, 7.8 pg/mL; IL-6, 35.4 pg/mL; MCP-1, 9.3 pg/mL; TNF-α, 1.7 pg/mL. Cytokine levels were Blom transformed for statistical analysis; untransformed data (pg cytokine/mg total protein) is presented in the figure. Data are presented as mean ± SEM. Adj/NA: Charles River: n = 14, Harlan: n = 6; Adj/M-M: Charles River: n = 3, Harlan: n = 12; Adj/S: Charles River: n = 4, Harlan: n = 5; control, saline-injected: Charles River: n = 7, Harlan: n = 6. Post hoc: *, P < .05, **, P < .01, ***, P < .001 (comparison with control, unless indicated otherwise); §/§§, P < .05, P < .01, respectively; post hoc comparisons between colonies.
Cytokine profiles differed between colonies (Figure 5, A–C). Interestingly, both TNF-α and MCP-1 levels (vendor × AA severity interactions, P < .01 and trend P = .064, respectively) were lower in Harlan than Charles River rats but only in the absence of AA (for both control and Adj/NA, Harlan < Charles River, P < .01, with the exception that for MCP-1, for control, trend P = .08). In addition, both TNF-α and MCP-1 levels were higher in the Adj/NA compared with the control condition in Charles River rats (P < .05) but were higher in the Adj/M-M compared with the control condition in the Harlan rats (P < .001) (Figure 5, A and B), indicating colony differences in the overall cytokine patterns, in a disease state-dependent manner. On the other hand, for KC/GRO, levels were lower overall in Harlan compared with Charles River, independent of AA severity (main effect of colony, P < .01).
For IL-1β and IL-6 (Figure 5, D and E), levels increased in all rats receiving CFA injection (main effects of AA severity, IL-1β: P < .05, IL-6: P < .01), regardless of whether clinical signs of arthritis developed (Adj/NA, Adj/M-M, Adj/S > controls). However, whereas interactions between AA severity and colony were not detected for either IL-1β or IL-6, inspection of Figure 5 suggests that the overall increase of IL-1β in the Adj/NA condition appears to be driven by Charles River rats, whereas the overall increase in IL-6 in the Adj/M-M condition appears to be driven by Harlan rats. This is consistent with the colony specific changes in TNF-α and MCP-1. IFN-γ levels, although appearing to respond to AA, were not detectable in all animals (percentage of rats with detectable levels indicated in the graph) and thus were not normally distributed and were not analyzed statistically.
Plasma cytokine levels were low under control conditions and in the absence of AA but were increased with AA onset
In the plasma, the levels of TNF-α, MCP-1, KC/GRO, IL-6, and IFN-γ all generally increased with the development of AA (Figure 6, A–E). However, the antiinflammatory cytokines measured (IL-4, IL-10), as well as IL-1β, were uniformly undetectable (not shown). Circulating levels of TNF-α (Figure 6A), IL-6 (Figure 6D), and IFN-γ (Figure 6E) increased with severe AA (Adj/S) but in the control, Adj/NA, and/or Adj/M-M groups, cytokine levels were low or undetectable. As a result of the low cytokine levels, the percentage of rats with detectable levels within each severity condition are indicated on the graphs and the data were not analyzed statistically.
Figure 6.

Levels of cytokines in the plasma at the peak of inflammation (d 16 post-injection). TNF-α (A), IL-6 (D), and IFN-γ (E) generally increased in rats with arthritis but were low or undetectable in most disease states other than severe AA (Adj/S), were not normally distributed, and were not analyzed statistically (percentage of rats with detectable levels within each severity group is indicated on the graph). B, MCP-1 increased with CFA injection in both Charles River and Harlan rats (main effect of AA severity, F [3, 48] = 9.68, P < .001). C, KC/GRO levels only increased with severe AA (main effect of AA severity, F [3, 48] = 6.00, P < .01) and overall were lower in Harlan than Charles River rats (main effect of colony, F [1, 48] = 9.21, P < .01; §§). Note: Lower limits of detection (LLOD) for cytokine assays are as follows: KC/GRO, 3.3 pg/mL; IFN-γ, 104.0 pg/mL; IL-10, 32.2 pg/mL; IL-1β, 23.5 pg/mL; IL-4, 8.26 pg/mL; IL-6, 74.4 pg/mL; MCP-1, 5.27 pg/mL; TNF-α, 12.6 pg/mL. Cytokine levels were Blom transformed for statistical analysis; untransformed data (pg cytokine/ml) are presented in the figure. Data are presented as mean ± SEM. Adj/NA: Charles River: n = 14, Harlan: n = 6; Adj/M-M: Charles River: n = 3, Harlan: n = 12; Adj/S: Charles River: n = 4, Harlan: n = 5; control, saline-injected: Charles River: n = 7, Harlan: n = 6. Post hoc: *, P < .05, **, P < .01, ***, P < .001 (comparison with control, unless indicated otherwise); §§, P < .01, post hoc comparisons between colonies.
By contrast, MCP-1 levels increased with CFA injection (Figure 6B; main effect of AA severity, P < .001), and KC/GRO levels increased with severe AA (Figure 6C; main effect of AA severity, P < .01). In addition, KC/GRO levels were lower overall in Harlan compared with Charles River rats (main effect of colony, P < .01).
Discussion
Our data demonstrate differences in both the incidence and severity of inflammation and the endocrine and immune response after the CFA injection in SD rats obtained from Charles River and Harlan Laboratories. Overall, we found that Harlan rats were more susceptible to inflammation, developing a more severe course of arthritis at lower doses of CFA, compared with Charles River rats. In addition, while rats from both vendors showed increasing corticosterone levels with the development of AA, Harlan had lower CBG than Charles River rats in the absence of arthritis (control and Adj/NA groups). As a result, the decrease in plasma CBG that occurs with the development of arthritis was less pronounced in Harlan rats. Moreover, Harlan and Charles River rats exhibited differential cytokine patterns with inflammation in the hind paw. Whereas the SD rats from both colonies showed the highest cytokine/chemokine levels with severe inflammation, Charles River rats responded at lower levels of AA severity. That is, Charles River rats generally showed an increase in cytokine levels to the challenge of CFA injection, even in the absence of clinical signs of arthritis (Adj/NA), whereas Harlan rats generally showed an initial increase only with mild-moderate arthritis. These data suggest that colony-based differences in endocrine and immune measures not only serve as a sensitive index of inflammation but that these differences could be exploited to understand factors responsible for differential responses to inflammatory challenges in general.
Both Charles River and Harlan rats showed increasing basal total and free corticosterone with increasing AA severity, likely due to increased levels of proinflammatory cytokines driving increased HPA axis activity and a loss of the corticosterone diurnal rhythm (19). With the development of severe arthritis, there was also increased variability in corticosterone levels, a finding common to the AA model (37–39), and likely occurs as a result of individual differences in the severity of arthritis. Of note, differential corticosterone profiles were detected between Charles River and Harlan SD rats, with Charles River rats displaying increased mean corticosterone levels (total and free) in the absence of clinical signs of AA (Adj/NA), whereas Harlan rats had a more stepwise increase in corticosterone levels with increasing AA severity, although these differences failed to reach statistical significance. Interestingly, however, the corticosterone profiles mimic the colony-specific cytokine responses discussed below, with Charles River rats showing increased cytokine levels in the Adj/NA group, compared with a stepwise cytokine increase with increasing AA severity for Harlan rats.
Importantly, differences in CBG levels were also detected, with lower CBG in Harlan than Charles River rats under control conditions and in rats with no clinical signs of arthritis (Adj/NA group). This difference may in part explain previous findings of differences in ACTH levels seen after the administration of LPS and turpentine in rats from the same SD colonies studied here (6). Plasma CBG levels were not measured in the latter study, but large increases in plasma corticosterone were observed in all rats treated with LPS or turpentine, irrespective of the SD colony. If CBG levels were different between colonies, as we have now observed, it would be expected that the lower levels of CBG in Harlan SD rats would lead to higher free corticosterone levels, which would be available to feedback onto ACTH, thus explaining the lower ACTH levels in Harlan compared with Charles River SD rats (6). In addition, higher CBG levels in Charles River SD rats could explain why they have increased levels of basal corticosterone (6) because more of the steroid would be bound to CBG. In the present study, although ACTH levels were not statistically different between colonies, mean ACTH levels were lower in Harlan compared with Charles River rats in the Adj/S condition, which may also be suggestive of colony differences in the overall HPA activity.
Differences in the baseline CBG levels between Harlan and Charles River SD rats were not due to differences in CBG steroid-binding affinity despite the presence of two amino acid differences in their CBG sequences. There were also no differences between the two colonies of SD rats in their Serpina6 proximal promoter sequences that contain a CCAAT/enhancer-binding protein-β regulatory site known to be important for the acute phase response (40, 41), Therefore, although we cannot exclude the possibility that the differences in the CBG coding sequences alter the secretion rates of CBG from hepatocytes in vivo, it is possible that differences in transcription factor levels or differences in regulatory sequences outside of the proximal promoter sequence might account for this.
Lower plasma CBG levels in Harlan rats suggest a lower corticosterone reservoir, which could play a key role in the observed increased susceptibility to the immune challenge. It is known that human CBG is targeted by proteases such as neutrophil elastase (42), significantly decreasing its binding capacity for glucocorticoids and allowing for their targeted delivery at sites of inflammation. Immunoreactive CBG degradation products have also been observed in wound fluids of rats subjected to thermal injury (43). Thus, the larger corticosteroid reservoir in the Charles River SD rats may function to more effectively deliver corticosterone to sites of inflammation and result in reduced inflammation in these rats. Of note, with the development of arthritis, CBG levels decreased in SD rats from both colonies, with significant differences in CBG levels detected between controls and rats with severe arthritis. In the severe arthritis state, when CBG levels are significantly lower and total corticosterone levels are significantly higher, there was a substantial increase in free corticosterone, which would also be available at target tissues. The decrease in the plasma CBG levels observed with increasing arthritis severity in both Charles River and Harlan SD rats may reflect either an increase in the plasma clearance of CBG (44) and/or a decrease in hepatic CBG production in response to increases in the plasma levels of corticosterone (45) and proinflammatory cytokines (46). In support of the latter, it has been shown that IL-6 decreases CBG levels, likely through decreased stability of CBG mRNA (47), with TNF-α also causing a decrease in plasma CBG levels (48).
To investigate the possible role of differential activation of cytokine networks in mediating the responses to AA in Charles River and Harlan rats, a subset of key pro- and anti-inflammatory cytokines were quantified in the hind paw and plasma. Activated immune cells produce a wide array of cytokines, which in turn activate other immune cells, resulting in the production of a complex cytokine network, the regulation and dynamics of which are still not yet fully understood (49). With AA and RA, cytokine production leads to matrix metalloproteinase production and osteoclast activation, resulting in the characteristic symptoms of bone destruction and extracellular matrix breakdown (50).
Examination of cytokine levels in the hind paw revealed, as expected, increased levels of proinflammatory cytokines with AA in SD rats from both colonies, with undetectable levels of anti-inflammatory cytokines (IL-4, IL-10) in all rats. However, although Charles River and Harlan SD rats showed similar cytokine/chemokine levels under conditions of severe inflammation, subtle but important differences in hind paw cytokine response profiles were detected for TNF-α, KC/GRO (CXCL1), and MCP-1 (CCL2). In general, TNF-α is involved in systemic inflammation, specifically the acute phase reaction, and is mainly produced by macrophages, whereas KC/GRO and MCP-1 are inflammatory chemokines, KC/GRO being a chemoattractant for neutrophils and MCP-1 being a chemoattractant for monocytes. Increases in TNF-α, KC/GRO, and MCP-1 in the AA paradigm are well established (8, 21, 51), and all three cytokines are critically involved in RA pathogenesis. TNF-α, KC/GRO, and MCP-1 are elevated in synovial fluid of patients with RA (52–55), and anti-TNF agents are currently widely used in RA therapeutics, with agents targeting the KC/GRO pathway also generating interest (49, 54, 56, 57). Interestingly, differences in TNF-α and MCP-1 levels in Charles River vs Harlan SD rats were detected both under control conditions and in adjuvant-injected rats that did not develop arthritis (Adj/NA) but not under conditions of active inflammation (Adj/M-M and Adj/S), whereas KC/GRO levels were lower overall in the Harlan compared with the Charles River rats.
In the plasma, cytokine levels generally increased with AA; however, levels were lower than in the hind paw, with low/undetectable levels for a number of plasma cytokines (TNF-α, IL-6, IFN-γ) in the control, Adj/NA and/or Adj/M-M groups. Similar to what was observed in the hind paw, KC/GRO levels were lower in the plasma of Harlan compared with the Charles River rats. In addition, Charles River rats in the Adj/NA condition had stronger TNF-α and IFN-γ responses than those in the Adj/M-M condition, again paralleling results in the hind paw. As well, the overall plasma cytokine response for KC/GRO and MCP-1 in Harlan animals appears to be lower than that of Charles River animals, with the increase in cytokine levels seen with increasing AA severity likely driven by the response in Charles River rats. Although not analyzed statistically, Harlan rats also appear to produce less IFN-γ and IL-6 than Charles River rats with severe arthritis.
Few studies have probed for colony-based differences in the cytokine response to challenge in a single rat strain (6), but our findings are consistent with more extensive evidence of differential cytokine activation in response to AA in rats from different strains. For example, Banik et al (14) found that whereas the administration of a CFA dose of 0.6 mg/rat to Lewis rats resulted in AA in 100% of rats, there was little to no effect in Wistar and SD rats (14). Even increasing the doses to 1.0–1.2 mg/rat resulted in AA in only 58% and 45% of Wistar and SD rats, respectively (14). Similarly, whereas Lewis rats generally develop more severe AA than SD rats (58), time-course analysis of serum levels of TNF-α has revealed enhanced TNF-α levels in SD (Laboratory Animal Unit, University of Hong Kong, China) over Lewis rats throughout the disease course (12), suggesting that TNF-α is a critical component of the cytokine network in response to AA in SD rats. Moreover, we found increased levels of a number of cytokines in SD rats that did not develop clinical signs of arthritis, including TNF-α, MCP-1, KC/GRO, and IL-1β. Specifically, we found that TNF-α and MCP-1 levels were higher in Charles River than Harlan rats but only in the absence of inflammation (Adj/NA, control). In addition, there were differential TNF-α and MCP-1 profiles between colonies: Charles River rats had increased TNF-α and MCP-1 levels in the Adj/NA compared with control condition, whereas Harlan rats had increased levels TNF-α and MCP-1 levels in the Adj/M-M compared with control condition. Furthermore, although statistical interactions between colony and AA severity were not detected for IL-1β and IL-6, the data suggest that the overall increase in IL-1β in rats with no clinical signs of arthritis (Adj/NA) is driven primarily by the response in Charles River rats, whereas the overall elevation in IL-6 in rats with mild-moderate arthritis (Adj/M-M) is driven primarily by the response in Harlan rats, a finding that parallels the differences detected in TNF-α and MCP-1 profiles. These divergent cytokine profiles between Charles River and Harlan rats suggest that they may rely on different cytokine networks and do so in a disease state-dependent manner.
The finding that even in the absence of clinical signs of arthritis, important physiological differences were observed in a single strain of rat obtained from different vendors is novel and important. Rarely have cytokine levels been assessed in the absence of clinical signs of AA, with measurements more commonly made only in rats developing overt signs of AA and/or by collapsing across groups to simply include a CFA-injected vs a control group. However, our findings of differences in the TNF-α and MCP-1 responses in Charles River vs Harlan SD rats to preclinical AA highlight the importance of separating out these disparate states of preclinical vs clinical inflammation and suggest that the same cytokines may differentially mediate severity of inflammation in rats from the two colonies. Furthermore, preclinical levels of cytokines and other proteins are used as biomarkers and predictors of disease status in RA (59), and our findings on preclinical inflammation, unmasked by using colony specific differences, may provide important insight into the human condition.
Whereas we have shown genetic differences between Charles River and Harlan rats, supporting the hypothesis that genetic drift has occurred between these two SD rat populations, the role of environmental factors, such as diet, early-life rearing conditions, and shipping conditions, cannot be discounted. These environmental factors are all known to impact physiological parameters and naturally differ between vendors. Although early-life environmental conditions may be affecting the epigenetic regulation of endocrine and immune genes (60, 61), the finding of genetic differences between Charles River and Harlan rats supports a genetic drift, with further genetic investigation required to determine the extent of the genetic differences.
Although our studies provide only correlative evidence of colony differences in endocrine and immune function, as well as genetic differences between colonies, the greater severity of inflammation observed in Harlan vs Charles River rats, together with differential plasma CBG levels and patterns of cytokine activation, is novel and provides a foundation for future mechanistic studies. Furthermore, our findings indicate that in addition to strain, the breeding colony of rats must be taken into account in both the experimental design process and the interpretation and generalization of published literature because it may explain conflicting physiological/neurobiological findings reported by different laboratories using SD rats. SD rats are considered a standard and reliable model for RA, being an outbred strain with a heterogenic background, which allows for evaluation of possible genetic factors involved in inflammation and response to treatment (12). Our data demonstrate that the colony of origin should also be considered in the evaluation of anti-arthritic agents. Moreover, rather than viewing colony differences as a limitation, the present data suggest that differences between SD rats from different colonies could be used as a powerful tool for revealing differential endocrine and immune factors underlying inflammation and to provide insight into the pathophysiology of and treatment strategies for RA.
Acknowledgments
We acknowledge Dr Eric Sandberg (Meso Scale Discovery) for his excellent technical assistance in the development of the multiplex cytokine assays. We also thank Dr Edie Dullaghan (Head of Target Validation at the Centre for Drug Research and Development), for generously providing access to their Sector Imager and Leanna Yee (senior technician, Target Validation at the Centre for Drug Research and Development) for her technical assistance.
This work was supported by National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism Grants RO1 AA022460 and R37 AA007789, NeuroDevNet (Canadian Networks of Centres of Excellence), and the Canadian Foundation on Fetal Alcohol Research (to J.W.); Natural Sciences and Engineering Research Council of Canada CGS-D (to T.S.B.), Canadian Institutes for Health Research Operating Grant 133606 (to K.K.S.), a Canadian Institutes for Health Research Doctoral Canada Graduate Scholarship (to M.D.T.), and Operating Grant MOP-111102 from the Canadian Institutes of Health Research (to G.L.H.). G.L.H. holds a Tier 1 Canada Research Chair in Reproductive Health
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AA
- adjuvant-induced arthritis
- CBG
- corticosteroid binding globulin
- CFA
- complete Freund's adjuvant
- DCC
- dextran-coated charcoal
- HPA
- hypothalamic-pituitary-adrenal
- IFN
- interferon
- KC/GRO
- keratinocyte chemoattractant/growth-regulated oncogene
- Kd
- dissociation-rate constant
- LPS
- lipopolysaccharide
- MCP-1
- monocyte chemotactic protein-1
- RA
- rheumatoid arthritis
- SD
- Sprague Dawley.
References
- 1. Swerdlow NR, Martinez ZA, Hanlon FM, et al. Toward understanding the biology of a complex phenotype: rat strain and substrain differences in the sensorimotor gating-disruptive effects of dopamine agonists. J Neurosci. 2000;20(11):4325–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fitzpatrick CJ, Gopalakrishnan S, Cogan ES, et al. Variation in the form of Pavlovian conditioned approach behavior among outbred male Sprague-Dawley rats from different vendors and colonies: sign-tracking vs. goal-tracking. PloS One. 2013;8(10):e75042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fuller DD, Baker TL, Behan M, Mitchell GS. Expression of hypoglossal long-term facilitation differs between substrains of Sprague-Dawley rat. Physiol Genomics. 2001;4(3):175–181. [DOI] [PubMed] [Google Scholar]
- 4. Clark FM, Yeomans DC, Proudfit HK. The noradrenergic innervation of the spinal cord: differences between two substrains of Sprague-Dawley rats determined using retrograde tracers combined with immunocytochemistry. Neurosci Lett. 1991;125(2):155–158. [DOI] [PubMed] [Google Scholar]
- 5. Yoon YW, Lee DH, Lee BH, Chung K, Chung JM. Different strains and substrains of rats show different levels of neuropathic pain behaviors. Exp Brain Res. 1999;129(2):167–171. [DOI] [PubMed] [Google Scholar]
- 6. Turnbull AV, Rivier CL. Sprague-Dawley rats obtained from different vendors exhibit distinct adrenocorticotropin responses to inflammatory stimuli. Neuroendocrinology. 1999;70(3):186–195. [DOI] [PubMed] [Google Scholar]
- 7. Freund J. The mode of action of immunologic adjuvants. Bibl Tuberc. 1956(10):130–148. [PubMed] [Google Scholar]
- 8. Billiau A, Matthys P. Modes of action of Freund's adjuvants in experimental models of autoimmune diseases. J Leukocyte Biol. 2001;70(6):849–860. [PubMed] [Google Scholar]
- 9. Ratkay LG. Investigation of an Adjuvant-Enhanced Model of Murine Arthritis and Its Therapeutic Application. The University of British Columbia, Department of Oral Biology; 1994. (Thesis). [Google Scholar]
- 10. Cai X, Wong YF, Zhou H, et al. Manipulation of the induction of adjuvant arthritis in Sprague-Dawley rats. Inflamm Res. 2006;55(9):368–377. [DOI] [PubMed] [Google Scholar]
- 11. Schuurs AH, Verheul HA. Effects of gender and sex steroids on the immune response. J Steroid Biochem. 1990;35(2):157–172. [DOI] [PubMed] [Google Scholar]
- 12. Cai X, Wong YF, Zhou H, et al. The comparative study of Sprague-Dawley and Lewis rats in adjuvant-induced arthritis. Naunyn Schmied Arch Pharmacol. 2006;373(2):140–147. [DOI] [PubMed] [Google Scholar]
- 13. Swingle KF, Jaques LW, Kvam DC. Differences in the severity of adjuvant arthritis in four strains of rats. Proc Soc Exp Biol Med. 1969;132(2):608–612. [DOI] [PubMed] [Google Scholar]
- 14. Banik RK, Kasai M, Mizumura K. Reexamination of the difference in susceptibility to adjuvant-induced arthritis among LEW/Crj, Slc/Wistar/ST and Slc/SD rats. Exp Anim. 2002;51(2):197–201. [DOI] [PubMed] [Google Scholar]
- 15. Zhang X, Lan N, Bach P, et al. Prenatal alcohol exposure alters the course and severity of adjuvant-induced arthritis in female rats. Brain Behav Immun. 2012;26(3):439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haddad JJ, Saade NE, Safieh-Garabedian B. Cytokines and neuro-immune-endocrine interactions: a role for the hypothalamic-pituitary-adrenal revolving axis. J Neuroimmunol. 2002;133(1–2):1–19. [DOI] [PubMed] [Google Scholar]
- 17. Bumiller A, Gotz F, Rohde W, Dorner G. Effects of repeated injections of interleukin 1β or lipopolysaccharide on the HPA axis in the newborn rat. Cytokine. 1999;11(3):225–230. [DOI] [PubMed] [Google Scholar]
- 18. Rivier C. Stimulatory effect of interleukin-1β on the hypothalamic-pituitary-adrenal axis of the rat: influence of age, gender and circulating sex steroids. J Endocrinol. 1994;140(3):365–372. [DOI] [PubMed] [Google Scholar]
- 19. Sarlis NJ, Chowdrey HS, Stephanou A, Lightman SL. Chronic activation of the hypothalamo-pituitary-adrenal axis and loss of circadian rhythm during adjuvant-induced arthritis in the rat. Endocrinology. 1992;130(4):1775–1779. [DOI] [PubMed] [Google Scholar]
- 20. Harbuz MS, Rees RG, Lightman SL. HPA axis responses to acute stress and adrenalectomy during adjuvant-induced arthritis in the rat. Am J Physiol. 1993;264(1 Pt 2):R179–R185. [DOI] [PubMed] [Google Scholar]
- 21. Szekanecz Z, Halloran MM, Volin MV, et al. Temporal expression of inflammatory cytokines and chemokines in rat adjuvant-induced arthritis. Arthritis Rheum. 2000;43(6):1266–1277. [DOI] [PubMed] [Google Scholar]
- 22. Hueber AJ, Asquith DL, McInnes IB, Miller AM. Embracing novel cytokines in RA—complexity grows as does opportunity! Best Pract Res Clin Rheumatol. 2010;24(4):479–487. [DOI] [PubMed] [Google Scholar]
- 23. Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58(10):2968–2980. [DOI] [PubMed] [Google Scholar]
- 24. Smolen JS, Landewe R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis. 2010;69(6):964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pecoraro N, Ginsberg AB, Warne JP, Gomez F, la Fleur SE, Dallman MF. Diverse basal and stress-related phenotypes of Sprague Dawley rats from three vendors. Physiol Behav. 2006;89(4):598–610. [DOI] [PubMed] [Google Scholar]
- 26. Dhabhar FS, McEwen BS, Spencer RL. Stress response, adrenal steroid receptor levels and corticosteroid-binding globulin levels—a comparison between Sprague-Dawley, Fischer 344 and Lewis rats. Brain Res. 1993;616(1–2):89–98. [DOI] [PubMed] [Google Scholar]
- 27. Joe B, Griffiths MM, Remmers EF, Wilder RL. Animal models of rheumatoid arthritis and related inflammation. Curr Rheumatol Rep. 1999;1(2):139–148. [DOI] [PubMed] [Google Scholar]
- 28. Grossman C. Possible underlying mechanisms of sexual dimorphism in the immune response, fact and hypothesis. J Steroid Biochem. 1989;34(1–6):241–251. [DOI] [PubMed] [Google Scholar]
- 29. Da Silva JA. Sex hormones and glucocorticoids: interactions with the immune system. Ann NY Acad Sci. 1999;876:102–117; discussion 117–118. [DOI] [PubMed] [Google Scholar]
- 30. Holmdahl R. Female preponderance for development of arthritis in rats is influenced by both sex chromosomes and sex steroids. Scand J Immunol. 1995;42(1):104–109. [DOI] [PubMed] [Google Scholar]
- 31. Remmers EF, Joe B, Griffiths MM, et al. Modulation of multiple experimental arthritis models by collagen-induced arthritis quantitative trait loci isolated in congenic rat lines: different effects of non-major histocompatibility complex quantitative trait loci in males and females. Arthritis Rheum. 2002;46(8):2225–2234. [DOI] [PubMed] [Google Scholar]
- 32. Smith CL, Hammond GL. An amino acid substitution in biobreeding rat corticosteroid binding globulin results in reduced steroid binding affinity. J Biol Chem. 1991;266(28):18555–18559. [PubMed] [Google Scholar]
- 33. Hammond GL, Lahteenmaki PL. A versatile method for the determination of serum cortisol binding globulin and sex hormone binding globulin binding capacities. Clin Chim Acta. 1983;132(1):101–110. [DOI] [PubMed] [Google Scholar]
- 34. Boksa P. Early developmental profiles of plasma corticosterone are altered by birth condition in the rat: a comparison of vaginal birth, cesarean section, and cesarean section with added anoxia. Pediatr Res. 1997;41(1):34–43. [DOI] [PubMed] [Google Scholar]
- 35. Perrin FM, Forest MG. Time course of the effect of adrenalectomy on transcortin binding characteristics: appraisal of different methods of calculation. Endocrinology. 1975;96(4):869–878. [DOI] [PubMed] [Google Scholar]
- 36. Underhill DA, Hammond GL. Cis-regulatory elements within the proximal promoter of the rat gene encoding corticosteroid-binding globulin. Gene. 1995;162(2):205–211. [DOI] [PubMed] [Google Scholar]
- 37. Harbuz MS, Conde GL, Marti O, Lightman SL, Jessop DS. The hypothalamic-pituitary-adrenal axis in autoimmunity. Ann NY Acad Sci. 1997;823:214–224. [DOI] [PubMed] [Google Scholar]
- 38. Harbuz MS, Rooney C, Jones M, Ingram CD. Hypothalamo-pituitary-adrenal axis responses to lipopolysaccharide in male and female rats with adjuvant-induced arthritis. Brain Behav Immun. 1999;13(4):335–347. [DOI] [PubMed] [Google Scholar]
- 39. Webster EL, Barrientos RM, Contoreggi C, et al. Corticotropin releasing hormone (CRH) antagonist attenuates adjuvant induced arthritis: role of CRH in peripheral inflammation. J Rheumatol. 2002;29(6):1252–1261. [PubMed] [Google Scholar]
- 40. Verhoog N, Allie-Reid F, Vanden Berghe W, et al. Inhibition of corticosteroid-binding globulin gene expression by glucocorticoids involves C/EBPβ. PloS One. 2014;9(10):e110702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Poli V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem. 1998;273(45):29279–29282. [DOI] [PubMed] [Google Scholar]
- 42. Hammond GL, Smith CL, Paterson NA, Sibbald WJ. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J Clin Endocrinol Metab. 1990;71(1):34–39. [DOI] [PubMed] [Google Scholar]
- 43. Garrel DR, Zhang L, Zhao XF, Hammond GL. Effect of burn injury on corticosteroid-binding globulin levels in plasma and wound fluid. Wound Repair Regen. 1993;1(1):10–14. [DOI] [PubMed] [Google Scholar]
- 44. Mast AE, Enghild JJ, Pizzo SV, Salvesen G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase-complexed, and reactive site cleaved serpins: comparison of α1-proteinase inhibitor, α1-antichymotrypsin, antithrombin III, α2-antiplasmin, angiotensinogen, and ovalbumin. Biochemistry. 1991;30(6):1723–1730. [DOI] [PubMed] [Google Scholar]
- 45. Smith CL, Hammond GL. Hormonal regulation of corticosteroid-binding globulin biosynthesis in the male rat. Endocrinology. 1992;130(4):2245–2251. [DOI] [PubMed] [Google Scholar]
- 46. Bernier J, Jobin N, Emptoz-Bonneton A, Pugeat MM, Garrel DR. Decreased corticosteroid-binding globulin in burn patients: relationship with interleukin-6 and fat in nutritional support. Crit Care Med. 1998;26(3):452–460. [DOI] [PubMed] [Google Scholar]
- 47. Bartalena L, Hammond GL, Farsetti A, Flink IL, Robbins J. Interleukin-6 inhibits corticosteroid-binding globulin synthesis by human hepatoblastoma-derived (Hep G2) cells. Endocrinology. 1993;133(1):291–296. [DOI] [PubMed] [Google Scholar]
- 48. Fleshner M, Silbert L, Deak T, et al. TNF-α-induced corticosterone elevation but not serum protein or corticosteroid binding globulin reduction is vagally mediated. Brain Res Bull. 1997;44(6):701–706. [DOI] [PubMed] [Google Scholar]
- 49. Feldmann M, Maini RN. Anti-TNF α therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–196. [DOI] [PubMed] [Google Scholar]
- 50. Gravallese EM, Goldring SR. Cellular mechanisms and the role of cytokines in bone erosions in rheumatoid arthritis. Arthritis Rheum. 2000;43(10):2143–2151. [DOI] [PubMed] [Google Scholar]
- 51. Stolina M, Bolon B, Middleton S, et al. The evolving systemic and local biomarker milieu at different stages of disease progression in rat adjuvant-induced arthritis. J Clin Immunol. 2009;29(2):158–174. [DOI] [PubMed] [Google Scholar]
- 52. Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. [DOI] [PubMed] [Google Scholar]
- 53. Koch AE, Kunkel SL, Harlow LA, et al. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest. 1992;90(3):772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. FEBS J. 2008;275(18):4448–4455. [DOI] [PubMed] [Google Scholar]
- 55. Harigai M, Hara M, Yoshimura T, Leonard EJ, Inoue K, Kashiwazaki S. Monocyte chemoattractant protein-1 (MCP-1) in inflammatory joint diseases and its involvement in the cytokine network of rheumatoid synovium. Clin Immunol Immunopathol. 1993;69(1):83–91. [DOI] [PubMed] [Google Scholar]
- 56. Maini RN, Taylor PC. Anti-cytokine therapy for rheumatoid arthritis. Annu Rev Med. 2000;51:207–229. [DOI] [PubMed] [Google Scholar]
- 57. Szekanecz Z, Koch AE, Kunkel SL, Strieter RM. Cytokines in rheumatoid arthritis. Potential targets for pharmacological intervention. Drugs Aging. 1998;12(5):377–390. [DOI] [PubMed] [Google Scholar]
- 58. Rosenthale ME. A comparative study of the Lewis and Sprague Dawley rat in adjuvant arthritis. Arch Int Pharmacodyn Ther. 1970;188(1):14–22. [PubMed] [Google Scholar]
- 59. Deane KD, O'Donnell CI, Hueber W, et al. The number of elevated cytokines and chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum. 2010;62(11):3161–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011;31(3):363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Champagne FA, Curley JP. Epigenetic mechanisms mediating the long-term effects of maternal care on development. Neurosci Biobehav Rev. 2009;33(4):593–600. [DOI] [PubMed] [Google Scholar]