

Abstract
Melanoma is the most aggressive form of skin cancer. Disrupted intracellular signaling pathways are responsible for melanoma's extraordinary resistance to current chemotherapeutic modalities. The pathophysiologic basis for resistance to both chemo- and radiation therapy is rooted in altered genetic and epigenetic mechanisms that, in turn, result in the impairing of cell death machinery and/or excessive activation of cell growth and survival-dependent pathways. Although most current melanoma therapies target mitochondrial dysregulation, there is increasing evidence that endoplasmic reticulum (ER) stress-associated pathways play a role in the potentiation, initiation and maintenance of cell death machinery and autophagy. This review focuses on the reliability of ER-associated pathways as therapeutic targets for melanoma treatment.
Keywords: Melanoma, Endoplasmic reticulum, Apoptosis, Autophagy, Signaling pathways, Chemotherapy
Core tip: This editorial describes the clinical validity of the endoplasmic reticulum (ER) as therapeutic target for melanoma treatment. In addition, we highlight in this review the mechanistic role of ER stress in the modulation of both apoptosis and autophagy- associated pathways. Drugs that perturb ER function may represent an alternative approach for melanoma treatment. This paper reviews the pervious and current published studies on the reliability of ER-associated pathways as therapeutic targets for melanoma.
INTRODUCTION
Although melanoma accounts for less than 5% of all skin cancers, it exhibits the highest mortality rate of all cutaneous tumors and its incidence is rapidly increasing[1]. The high mortality rate is the result of the propensity of metastatic dissemination throughout the body[2], and the development of resistance mechanisms that permit melanoma to evade normal immune surveillance mechanisms and the anti-tumor effects of chemotherapy[3]. Early detection and surgical excision of early stage disease offers the best hope of cure in patients with primary melanoma[4]. Even with new targeted therapies, the prognosis for advanced metastatic malignant melanoma is poor[5]. The available options for patients with advanced malignant melanoma patients provide limited therapeutic benefit with successful treatments often being measured in months of increased survival rather than years[6-8]. The potential to develop resistance mechanisms that counteract drug-induced apoptosis and evade host immunological responses is particularly devastating[9]. Accordingly, the replacement of single agent chemotherapy with targeted therapies is revolutionizing systemic therapy[10]. Besides the mechanistic role of mitochondrial damage-dependent pathways in the modulation of anti-cancer agent-induced apoptosis of tumor cells, anti-cancer agents can also improve killing efficiency via endoplasmic reticulum (ER) stress-dependent pathways[11-13]. While autophagy-mediated tumor death in response to anti-cancers is clinically relevant, these anti-cancer agents can also induce autophagy-mediated cytoprotective mechanisms[12,13], a pattern of tumor resistance to chemotherapy.
Metastatic melanoma demonstrates particularly poor response rates to single chemotherapeutic agents[14,15]. For instance, dacarbazine (DTIC) demonstrates no impact on survival, though it is considered to be one of the most effective agents that is used as standard therapy for the treatment of metastatic melanoma[16,17]. Other anticancer agents such as cisplatin, carmustine and the vinca alkaloids (e.g., vindesine and vinblastine) fail to show any therapeutic advantage over DTIC[18], though several combination chemotherapy regimens demonstrate a modest increased response rate[19].
Melanoma’s resistance to therapy is the results of an upregulation in pro-survival factors, which potentiate tumor maintenance and progression[20]. One of these factors is the inducible transcription factor NF-κB that is responsible for the regulation of the expression of genes related to apoptosis[21]. It is also, central to the development of tumor resistance to alkylating agents such as DTIC[22-24]. Accordingly, the inhibition of NF-κB pathway may improve the cytotoxic efficacy of alkylating agent-based therapy. To that end, preclinical studies in vitro and in vivo using human melanoma tumor models revealed that the therapeutic efficiency of DTIC or temozolomide is enhanced with the addition of the proteasome inhibitor, bortezomib[25,26].
Traditional mono- or multi-chemotherapy regimens are also associated with the development of significant adverse effects[27,28]. The development of new tumor types in these patients is attributed to the molecular action of the anticancer agents leading to the induction and/or destruction of aberrant signaling pathways.
The molecular action of chemotherapy in tumor cells is commonly associated with phenotypic alterations including cell death and survival-dependent mechanisms including apoptosis and autophagy[12,13].
Apoptosis and autophagy occur in normal cells. These are essential physiological mechanisms required for the maintenance of organismal and cellular homeostasis[29]. Current information about autophagy in melanoma focuses on autophagosome formation and/or autolysosome degradation in response to a variety of therapeutic agents using melanoma derived cell lines[13,30,31]. Chemotherapy induction of autophagy serves to protect melanoma cells from intendent chemotherapy-induced apoptosis. In fact, the induction of autophagy following the treatment of melanoma cells with bortezomib reduces bortezomib-induced apoptosis[13]. Similarly, the induction of autophagy by esomeprazole, a proton pump inhibitor, blocks melanoma cell death[32]. Based on this preclinical evidence, the modulation of autophagy-associated pathways offers a promising treatment strategy to increase treatment efficiency by overcoming melanoma resistance to chemotherapy.
The involvement of ER stress in the modulation of apoptotic mechanisms leading to melanoma cell death has been reported in several studies[12,13,33]. This may result from the induction of BH3 proteins such as Noxa and Puma leading to the inhibition of Bcl-2 localization at the ER membrane, alterations in the distribution of the calcium flux which produce ER stress[13,34].
Although ER stress and autophagy are capable of modulating each other in tumor tissues, their specific function is thought to be tumor type and stage-dependent[34-36]. The clinical potential of ER stress and/or autophagy-associated pathways as therapeutic target for melanoma treatment has been reported in several studies[37-39]. For example, BRAF wild type (wt) melanoma is more sensitive to ER stress-based therapies than melanoma with hyperactivating BRAF mutations[40]. The frequency of BRAF mutation seems to be associated with elevated levels of autophagy in melanoma. Accordingly, ER stress-induced apoptosis of melanoma cells harboring oncogenic BRAF is lower than those observed in BRAF wt melanoma cells[40-42]. Inhibition of autophagy is a good strategy to sensitize BRAF wt melanoma cells to ER stress-mediated apoptosis. In addition, the development of anti-cancer agents based on the enhancement or suppression of these processes may be relevant therapeutic strategies[38,43,44].
Tumor resistance or response to available therapeutic modalities depends on the balance between apoptosis and autophagy-associated mechanisms[45,46]. Although the development of the most available therapeutic approaches focuses on the excessive activation of mitochondrial dysregulation-dependent pathways leading to apoptosis, there is increasing evidence that ER stress-associated pathways represent an important therapeutic target for melanoma treatment[13,47]. Thus, the development of anti-cancer agents with ability to trigger the intrinsic activation of ER stress/unfolded protein response (UPR)-associated pathways may offer a novel therapeutic strategy for tumor treatment. UPR is mediated in response to the enhancement of protein synthesis through the activation of mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK/ERK) pathway that, in turn, induces cell proliferation, a mechanism that can block ER stress-induced apoptosis[48]. Thus, ER stress-dependent pathways have been proposed to represent a new therapeutic target for melanoma treatment[10,49]. Accordingly, the inhibition of oncogenic BRAF (V600E) and/or MEK-attenuated activation of inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6) signaling of the UPR in melanoma cells may sensitize melanoma cells to apoptosis. Our work focuses on the reliability of ER stress-dependent pathways as a therapeutic target for melanoma treatment.
FUNCTION OF ER IN NORMAL AND TUMOR CELLS
ER is a network of tubules and flattened sacs comprising rough and smooth regions that differ in their structure and function[50]. The rough ER is characterized by the existence of ribosomes attached to the cytoplasmic side of the membrane, whereas the smooth ER lack these ribosomes[50]. ER plays a crucial role in normal cellular functioning, by processing of post-translational modification and folding of secretory and membrane proteins. These secretory and membrane proteins are synthesized along the membrane of the rough ER and subsequently are passed onto the Golgi apparatus, where they undergo further post-translational modifications by the attachment of lipid and glucose moieties in a lipidation and glycosylation-dependent manner, respectively[51]. The ability of ER to correctly fold nascent proteins depends on chaperone proteins that, under normal physiological condition, are in excess in the ER lumen[52]. The function of most chaperone proteins is known to be Ca2+-dependent[53]. ER contains a high concentration of Ca2+ and is the only cellular organelle that plays an essential role in intracellular Ca2+ homeostasis[54]. Thus, the escalation of intracellular calcium into the cytoplasm is a signal for pathophysiological alteration of the cells. This pathological phenomenon results from ER stress in response to externally physical or chemical stressors, such as radiation and toxins[55].
ER function is critical for the regulation of many aspects of cell physiology, such as vesicle trafficking, lipid and membrane biogenesis as well as protein targeting and secretion. Normal and tumor cells react rapidly to ER stress via mechanisms mediated by a set of ER stress-associated pathways. The regulation of these pathways is thought to be the consequence of the perturbations in ER function, such as the accumulation of unfolded or misfolded proteins, as well as the accumulation of ER lipid, glycolipid imbalances, or alteration in the ionic or redox conditions in the lumen of ER[56,57]. Three distinct signaling pathways have been identified as ER stress-dependent pathways, namely protein kinase RNA-like endoplasmic reticulum kinase (PERK), ATF6, and IRE1 pathways. The primary purpose of these pathways is implicated to promote cell survival by mechanisms mediated through the reduction of the misfolded protein[58]. Figure 1 outlines the ER stress-associated pathways in normal and tumor cells.
Figure 1.
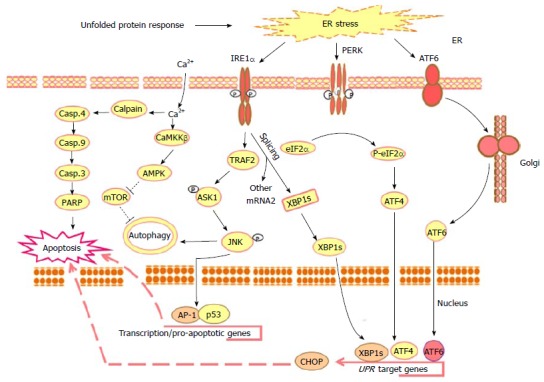
Outline of the main unfolded protein response-mediated mechanisms in response to the molecular action of anti-cancer agents. Upon the accumulation of misfolded proteins in the ER lumen, chaperones, the ER stress sensors PERK, IRE1α and ATF6 become active. The phosphorylation of PERK allows it to assemble in a homo-dimer to form an active form that, in turn, results in the phosphorylation of eukaryotic initiation factor2α (eIF2α) to initiate UPR downstream response leading to reduction of the protein overload to ER by the suppression of the translation and activation of ATF4 together with ER stress associated transcription factors such as, CHOP, PERK, IRE1 and ATF6. Activated PERK phosphorylates the translation initiation factor eIF2α to decrease the protein synthesis and enhance stress-inducible messages, such as ATF4. During ER stress the ATF6 traffics to the Golgi apparatus, where it is cleaved by S1P/S2P proteases. The cleavage of ATF6 from the Golgi membrane facilitates its localization to the nucleus, where it enhances the transcriptional up-regulation of UPR target genes leading to apoptosis. Whereas, activated IRE1α is implicated into several functions: One of these functions is essential to drive the splice mechanism of the XBP-1 mRNA to allow the translation of mature XBP-1 protein that, in turn, functions as a transcription factor to promote the transcription of UPR target genes such as CHOP leading to apoptosis; the other function of the activated IRE1α is to recruit TRAF2 that subsequently mediates the phosphorylation of ASK1 and the activation of its downstream JNK leading to the activation of the transcription factors AP-1 and p53 that are essential for the transcription of the pro-apoptotic genes and genes implicated in the processes of autophagsome formation and autophagy. UPR results also in intracellular calcium release leading to cell death via Calpain/Caspase-4, Caspase-9, caspase-3 and PARP axis or autophagy via CaMKKβ/AMPK axis leading to the inhibition of mTOR and subsequently autophagy. ER: Endoplasmic reticulum; PERK: Protein kinase RNA-like endoplasmic reticulum kinase; IRE1α: Inositol-requiring protein 1; ATF6: Activating transcription factor 6; UPR: Unfolded protein response; XBP-1: X-box binding protein 1; CHOP: CCAAT/enhancer-binding protein (C/EBP) homologous protein; TRAF2: TNF receptor-associated factor 2; ASK1: Apoptosis signal regulating kinase 1; JNK: C-jun-N-terminal kinase; PARP: Poly (ADP-ribose) polymerase; CaMKKβ: Calmodulin-dependent protein kinase kinase-β; AMPK: AMP-activated protein kinase; mTOR: Mammalian target of rapamycin.
INDUCTION OF ER STRESS-ASSOCIATED PATHWAYS BY ANTI-CANCER AGENTS
Dysregulation of ER homeostasis is a primary pathophysiological mechanism responsible for the initiation of an ER stress response that leads to the development of a number of human diseases including cancer[59]. The induction of ER stress by anti-cancer agents and other stimuli has been reported in several studies. The anti-cancer agent’s bortezomib, vinblastine and taxol trigger ER stress in melanoma cells[13,60,61]. Similarly, caffeic acid phenethyl ester, the BH3 mimetic obatoclax and the Abbott Compound ABT-737 have been reported to induce ER stress in melanoma[33,62]. Interestingly, the induction of ER stress in melanoma cells by these agents is correlated with the deregulation of ER stress associated pathways including eukaryotic translation initiation factor 2α (elF2α) and PERK.
ER stress induced activation of PERK leads to the phosphorylation of the elF2α that inhibits the translation and subsequently triggers cell cycle arrest[63]. CHOP (C/EBP homology protein) is downstream of PERK-elF2α-ATF4 and involved in the regulation of the apoptotic proteins of the Bcl-2 family members[64]. ER stress-induced activation of IRE1α is responsible for the regulation of the transcription factor XBP1[65]. Once ER stress is initiated, the conversion of unspliced XBP1 mRNA to mature mRNA is mediated permitting the translation and further modification of this protein to operate as an active transcription factor[66]. The activation of the transcription factor XBP1 is essential for the induction of the transcription of ER-related genes that, in turn, mediate the disposal of unfolded proteins[67]. Although this panel of responses is mainly implicated in restoring ER homeostasis, sustained ER stress is essential for the promotion of apoptosis[68,69]. More importantly, it has been demonstrated that ER stress-associated pathways are involved in the modulation of the anti-cancer agent-induced apoptosis of tumor cells, particularly, in melanoma[13]. In recent years, we and others uncovered the mechanistic role of ER stress-associated pathways such as PERK-ATF4-CHOP/Bim and IRE1α-ASK1-JNK-AP-1/HSF1-HSP70, in the modulation of anti-cancer agent-induced apoptosis of melanoma cells[12,13]. More importantly, we demonstrated that Noxa-induced ER stress triggers apoptosis of melanoma cells via mechanism mediated by ASK1-JNK/p38 axis[58,70]. Also, apoptosis related protein-2 (APR-2)-induced ER-stress drives apoptosis of melanoma cells via mechanism mediated by three parallel pathways, namely IRE1α/tumor necrosis factor receptor-associated factor 2 (TRAF2)-ASK1-JNK/Cytochrome c/caspase-9/caspase-3/PARP, Calpain-caspase-4-/caspase-9/caspase-3/PARP, and PERK-ATF4-CHOP/Bim[58]. Furthermore, bortezomib/vinblastine-induced ER stress in melanoma cells is essential for the induction of cell survival via autophagy-dependent pathways including, IRE1α-ASK1-JNK-AP-1/HSF1-HSP70 axis. More importantly, in our laboratory, we demonstrated that the inhibition of IRE1α-ASK1-JNK-AP-1/HSF1-HSP70 pathways synergistically enhance bortezomib or vinblastine-induced apoptosis of melanoma cells[12,13].
ER stress-mediated pathways to apoptosis in melanoma
It is established that the primary function of ER stress is to restore normal ER homeostasis and to engage cytoprotective mechanisms to counteract or mediate both intra- and extracellular-induced alterations[71]. Therefore, if the induced ER stress is strong or persistent, the ER enhanced dysfunction becomes irreversible and consequently triggers cell death machinery to initiate apoptosis. Thus, the destruction of the ER stress-dependent pathways that are essential for the modulation of the cytoprotective machinery by small molecule-inhibitors would be expected to trigger apoptosis of tumor cells. In addition, the enhancement of key components leading to excessive activation of apoptotic pathways, such as the mammalian IRE1α, could impact the regulation of kinases such as ASK1[62]. The activation of the pro-apoptotic kinase ASK1, the upstream kinase of the JNK pathway, is essential for the regulation of ER stress-induced apoptosis of melanoma in response to chemotherapeutic agents such as vinblastine[12], as well as in response to pro-apoptotic proteins such as the BH- only proteins such as Noxa[70] and APR-2[58]. Unlike various tumor types, particularly, those undergoing prolonged ER stress, the ER stress-dependent pathways such as IRE1α and ATF6 are persistent in melanoma cells[72]. Thus, it is expected that constitutive activation of both IRE1α and ATF6 would be associated with the development of melanoma resistance to anti-cancer agents[72]. Accordingly, the destruction of IRE1α and/or ATF6 signaling pathways has been reported to trigger apoptosis via mechanism mediated by PERK pathway[73]. The role of the PERK pathway in the modulation of ER stress-induced apoptosis has been demonstrated in various tumor types including melanoma via mechanism mediated by the BH3-only protein Bim[58,73].
Although the UPR is established as a cyto- protective response, excessive and/or persistent activation of ER stress-associated pathways can also trigger apoptosis[74]. However, the mechanism whereby UPR switches from the cyto- protection to apoptosis is thought to be the consequence of the attenuation of IRE1α and/or ATF6 activities[72,75]. The resistance of melanoma cells to most anti-cancer therapies during the course of anti-cancer-induced ER stress is attributed to the fact that the melanoma cells have adapted to ER stress. Although the molecular mechanisms that describe the contribution of ER stress in melanoma survival has been established, several studies revealed that the resistance of melanoma cells to ER stress-induced apoptosis results from the prolonged activation of the IRE1α and ATF6 pathways that, in turn, lead to the attenuation of the PERK signaling pathway[72]. Accordingly, the knockdown of IRE1α or ATF6 sensitizes melanoma cells to ER stress-induced apoptosis[33]. To that end, the destruction of the IRE1α/XBP-1 pathway along ER stress is expected to overcome melanoma resistance to ER stress inducers. The involvement of IRE1α in the activation of PI3K/Akt pathway together with the induction of Mcl-1 expression has been suggested to play an essential role in the modulation ER stress-induced survival of melanoma cells[76]. ATF6 is involved in the transcriptional regulation of both GRP78 and XBP-1 and thereby plays an important role in melanoma resistance to ER stress-induced apoptosis[77]. In conclusion, the differential response of various tumor types to PERK activation seems to rest on cellular factors and/or cell growth and survival pathways-dependent activation. Although the importance of IRE1/XBP-1 axis in tumor growth and survival has been established[78,79], its mechanistic role in the promotion of the XBP1 splicing processes and the subsequent effect on the components of the downstream signaling pathway have not been well characterized. More importantly, the activation of IRE1 kinase has been reported to be essential for the activation of c-Jun-N-terminal kinase, JNK and NF-κB pathways besides its role in the modulation of the induced unfolded protein response[79,80]. Upon the induction of ER stress, IRE1 kinase becomes capable of recruiting TRAF2. This results in the activation of both JNK and NF-κB pathways[81]. The mechanisms, involved in the modulation of ER stress are outlined in Figure 1.
ER stress-mediated pathways to autophagy in melanoma
Autophagy is a highly conserved degradation pathway that is responsible for the elimination of damaged cellular components. This process is implicated in several physiological and pathological processes leading to cell survival or cell death, and is characterized by the formation of double membrane autophagosomes[82]. The functional role of autophagy in melanoma has been reported in the context of in vitro analysis of chemotherapy-mediated effects in established melanoma cells[12,13]. We recently demonstrated that the induction of autophagic machinery in response to bortezomib protects melanoma cells from bortezomib-induced apoptosis[13]. Also, the induction of autophagy in response to the treatment with esomeprazole, a proton pump inhibitor, plays an essential role in the delay of melanoma cell death[32]. More importantly, the inhibition of ER stress-induced autophagy by the knockdown of Mcl-1, heat shock protein 70 (HSP70) or the inhibition of Bcl-2 potentiates bortezomib-induced apoptosis of melanoma cells[13,83]. Also, the combination of autophagy inhibitors 3-MA, bafilomycin A1 (BafA1) or LY294002 with the antiproliferative agents such as sanguilutine has the potential to reduce melanoma cell viability[84]. Orlotti et al[85] addressed the essential role of autophagy in the protection of melanoma cells from G-quadruplex ligand-induced-ER stress associated with DNA damage. Also, G-quadruplex ligand-induced autophagy has been suggested to be the consequence of Ataxia Telangiectasia mutated-dependent DNA damage response as well as the transactivation of the cyclin-dependent kinase inhibitor 1A[84]. Although its mechanistic role in tumor survival and resistance to treatment with chemo-and radiotherapy has been established, autophagy can also enhance the killing efficiency of chemotherapy-based treatments in various tumor types including melanoma[86]. In recent years, autophagic cell death, also known as type II apoptosis, gained more attention, as a potential therapeutic target for tumor treatment. Soares et al[87], demonstrated that the combination of Cl-IB-MECA inhibitor and paclitaxel can induce mTOR-dependent autophagic cell death, as well as caspase-dependent and/or independent apoptosis in melanoma cells. In addition, the potential of the micro-tubule poison, JG-03-14, to cause cytotoxic effects in melanoma cells both in vitro and in vivo via autophagy-dependent mechanism has been approved[88]. Thus, chemotherapeutic agents, whose cytotoxicity is mediated by autophagy-dependent mechanisms are considered to be suitable therapeutic approaches, particularly for tumors conferring resistance to anti-cancer agents-induced apoptosis. In addition, the identification of ER stress-associated pathways as a link between BRAF signaling and cytoprotective autophagy provides a potential therapeutic target for melanoma treatment[46]. Anti-cancer agents-induced autophagy is mostly resistant to several kinase inhibitors, particularly, those targeting the link between autophagic machinery and PI3K/AKT/mTOR pathway[89,90].
The common genetic alterations leading to the development of malignant melanoma are widely established to be the consequence of the activating mutations in NRAS and BRAF proto-oncogenes[91,92]. Also, genome-wide mutation detection in melanoma derived cell lines and primary tumors revealed significant alterations in the BRAF gene[93]. The most identified mutations were found to affect a single residue (V600E) that is located in the kinase activation domain of BRAF[94,95]. The importance of BRAF mutation is attributed to the potential role of RAF serine/threonine kinases, the most important key signaling components in the RAS pathways[96]. The clinical relevance of BRAF in melanoma is based on its mechanistic role in the activation of melanocytes in cAMP-dependent pathway in response to α-melanocyte-stimulating hormone-mediated activation of melanocortin receptor 1[97]. Accordingly, the mutation in the BRAF gene with its consequent impact on melanoma development and progression has gained increasing attention as a therapeutic target in melanoma. The development of a broad-spectrum of kinase inhibitors confirmed the clinical relevance of the inhibition of BRAF as an efficient therapeutic strategy for melanoma treatment. These kinase inhibitors have demonstrated the ability to inhibit BRAF, mutant BRAFV600E, and CRAF[98]. The most potent BRAF inhibitors, vemurafenib and dabrafenib, have demonstrated antitumor activity for advanced melanoma in phase III trials, particularly in patients with BRAF mutations[99]. Also, MEK inhibitors, such as trametinib, showed significant antitumor activity in melanoma patients with a V600 BRAF mutation[100]. Other MEK inhibitors, such as Binimetinib, exhibit antitumor activity in patients with advanced melanoma, who demonstrate NRAS mutation[101]. Most importantly, the combination of BRAF inhibitors such as dabrafenib with MEK inhibitors such as trametinib have enhanced therapeutic benefits when compared with the response rate to dabrafenib alone[102]. Despite the demonstration of therapeutic progress by both BRAF and MEK inhibitors, most patients with metastatic melanoma fail to achieve a clinical cure[103]. The development of more effective therapeutics for advanced metastatic melanoma requires a direct evaluation of novel and innovative therapies. The roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and the implications for sensitivity to treatment of melanoma are outlined in Figure 2.
Figure 2.
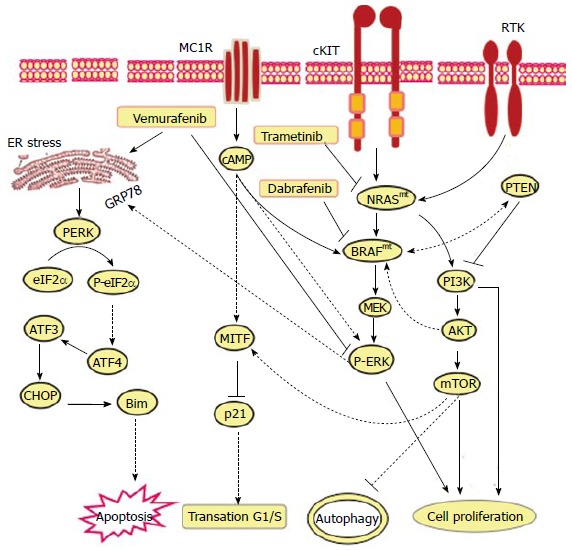
Proposed models for the mechanistic role of endoplasmic reticulum stress in the modulation of the anti-tumor efficiency of vemurafenib, dabrafenib and trametenib in melanoma patients harboring activating neuroblastoma RAS viral (v-ras) oncogene homolog or rapidly accelerated fibrosarcoma murine sarcoma viral (v-raf) oncogene homolog B mutations. The main function of MC1R, TRK on melanoma cell is to transmit extracellular signaling that is essential for the activation of RAS-RAF-MEK-ERK and PI3K-AKT-mTOR pathways to mediate various cellular functions including melanoma initiation, progression and resistance. Thus, the exposure of melanoma (BRAFmt and NRASmt) cells to either Vemurafenib, Dabrafenib or Trametenib, respectively, results in inhibition of MC1R, cKIT and TRK-mediated activation of RAS-RAF-MEK-ERK and PI3K-AKT-mTOR pathways. As consequence the inhibition of RAS-RAF-MEK-ERK and PI3K-AKT-mTOR pathways results in the execution melanoma cell death via mechanism-mediated by ER stress-dependent pathways including PERK-eIF2α-ATF4-ATF3-CHOP-Bim pathway. NRAS: Neuroblastoma Rat Sarcoma viral (v-ras) oncogene homolog; BRAF: Rapidly Accelerated Fibrosarcoma murine sarcoma viral (v-raf) oncogene homolog B; MC1R: Melanocortin 1 receptor; TRK: CKIT and receptor tyrosine kinase; PI3K: Phosphatidylinositol-4,5-bisphosphate 3-kinase; AKT: Protein kinase B.
While MAP kinase pathways modulate autophagy-associated cell death[104], accumulated evidence demonstrates that autophagy also plays a role in the promotion of tumor resistance and survival via MAP kinase pathway-dependent mechanisms[13,105-107]. Specifically, the induction of cytoprotective autophagy counteracts MAP kinase-mediated pathways to apoptosis in response to chemotherapy-based treatments[13,108]. The presence of autophagosomes in tumor cells undergoing apoptosis in response to the treatment with chemotherapy is evidence for the ability of tumor cells to evade the cytotoxicity via autophagy-dependent pathways[109]. Thus, the inhibition of autophagic machinery induced by chemotherapeutics, such as bortezomib, may prove to be an effective therapeutic strategy[13]. In addition, these pathways may play a role in ER stress suppression of the anti-tumor efficiency of vemurafenib, dabrafenib and trametenib in melanoma patients harboring activating NRAS or BRAF mutations (Figure 2).
Anti-cancer agents affecting ER stress-associated pathways to apoptosis of melanoma
There are a number of United States Food and Drug Administration-approved anti-cancer agents that influence key components of ER stress-dependent pathways. For example, the ruthenium-derived compounds trigger the expression of ER stress proteins such as, Bip, XBP1, PDI, and CHOP leading to tumor growth inhibition or cell death[110,111]. Also, the anti-cancer agent 2-Hydroxyoleic acid triggers ER stress and autophagy in various human glioma cell lines[112]. Furthermore, the inhibition of the proteasome system with bortezomib overcomes resistance in a variety of tumors via mechanisms mediated by the accumulation of misfolded proteins that overwhelm the ER-associated degradation pathway that produce ER stress[113]. This mechanism is well described in multiple myeloma (MM) cells that constitutively express ER stress-associated survival factors that are essential for propagation and maintenance of MM cells[114,115]. Thus, proteasome inhibitors induce apoptosis in MM because the UPR is unable to mediate the degradation of the misfolded proteins[116]. In fact, compared to other cell lines, MM cells are the most sensitive to proteasome inhibitors-induced apoptosis via mechanism mediated by the activation of UPR-associated pathways including PERK and ATF4, and the pro-apoptotic target, CHOP[117].
The involvement of ER stress in the modulation of melanoma cell death in response to the treatment with anti-cancer agents has been studied extensively. For example, Syed et al[118], demonstrated that fisetin-induced apoptosis of melanoma cells is mediated by ER stress-associated pathways such as IRE1α, XBP1s, ATF4 and GRP78[119]. Also, the small molecule inhibitor honokiol, a potent anti-tumorigenic compound, has been shown to trigger apoptosis of melanoma cells via a mechanism mediated by the binding of honokiol to the unfolded ATPase domain of GRP78 leading to the induction of ER stress and pro-apoptotic associated pathways. Beck et al[120], addressed an important role for ER stress-associated pathways in the modulation of the anti-cancer agents. For example, in patients with BRAFV600E-mutated melanoma vemurafenib- induced apoptosis is associated with increased levels of the spliced isoform of the transcription factor, XBP1, a marker for the induction of ER stress, and with increased phosphorylation of the translation initiation factor eIF2α. Also, ER stressors such as diallyl trisulfide play a role in the sensitization of melanoma cells to death receptor- induced apoptosis[121]. Moreover, the role of ER stress-associated pathways in the modulation of the anti-tumor activity of the natural marine compound, 11-dehydrosinulariolide has been demonstrated[122]. Interestingly, the 11-dehydrosinulariolide compound was found to trigger apoptosis of melanoma cells via mechanism-mediated by both PERK/eIF2α/ATF4/CHOP and ATF6/CHOP pathways[122]. In another study, Hiscutt et al[123], demonstrated that knockdown of the X-linked inhibitor of apoptosis protein (XIAP) enhances both fenretinide and bortezomib-induced apoptosis of metastatic melanoma cells via ER stress-mediated pathways. Also, melanoma under ER stress shows more susceptibility to obatoclax-induced apoptosis[124]. Moreover, the role of ER stress-associated signaling pathways, GRP78, ATF6, IRE1α, and PERK/eIF2α has been reported to be essential for docatxel-induced apoptosis of melanoma[125]. More importantly, it has been suggested that the constitutively activated MEK/ERK pathway results in resistance of melanoma cells to ER stress-induced apoptosis. Accordingly, Jiang et al[48], demonstrated that the inhibition of MEK by U0126 inhibitor or by the knockdown of MEK1 by its specific siRNA sensitizes melanoma cells to tunicamycin- or thapsigargin-induced apoptosis. Also, the induction of ER stress by Tunicamycin can sensitize human melanoma cells to tumor necrosis factor-related apoptosis in response to ligand-induced apoptosis[126].
CONCLUSION
Although it has been demonstrated that ER stress-dependent pathways play a significant role in the regulation of tumor initiation and resistance, it is more difficult to confirm the hypothesis that ER is a valid therapeutic target for tumor treatment. The induction of UPR is a cellular mechanism that reduces or prevents the cytotoxic effect of anti-cancer treatment. Accordingly, the destruction of key UPR components should provide an effective therapeutic strategy for melanoma treatment. Moreover, a functional analysis of UPR-mediated pathways, particularly those which are essential for cell survival or cell death, may help to identify key molecules of the aberrant pathways whose excessive activation and/or inhibition may overcome melanoma resistance to standard treatments. In addition, gaining an understanding of the molecular mechanisms of UPR may provide insight into the development of therapeutic strategies such as the development of small molecule inhibitors to control melanoma through the modulation of UPR signaling. Just as most current melanoma therapies were developed following a functional analysis of their ability to trigger mitochondrial dysregulation, ER stress-dependent pathways could provide new therapeutic targets designed to effect key components of aberrant signaling pathways.
Footnotes
Conflict-of-interest statement: Authors have no conflict-of-interest to declare.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 28, 2015
First decision: May 13, 2015
Article in press: September 8, 2015
P- Reviewer: Mendes RE, Siu PM, Sugawara I S- Editor: Tian YL L- Editor: A E- Editor: Wu HL
References
- 1.Chinembiri TN, du Plessis LH, Gerber M, Hamman JH, du Plessis J. Review of natural compounds for potential skin cancer treatment. Molecules. 2014;19:11679–11721. doi: 10.3390/molecules190811679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray C, D’Intino Y, MacCormick R, Nassar B, Walsh N. Melanosis in association with metastatic malignant melanoma: report of a case and a unifying concept of pathogenesis. Am J Dermatopathol. 1999;21:28–30. doi: 10.1097/00000372-199902000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Čeović R, Smolković N, Pašić A, Kostović K, Hrsan D. Multiple basal cell carcinomas of lower legs with stasis dermatitis: a therapeutic challenge. Acta Dermatovenerol Croat. 2012;20:191–196. [PubMed] [Google Scholar]
- 4.Essner R. Surgical treatment of malignant melanoma. Surg Clin North Am. 2003;83:109–156. doi: 10.1016/S0039-6109(02)00205-0. [DOI] [PubMed] [Google Scholar]
- 5.Gipponi M, Solari N, Giovinazzo D, Queirolo P, Bertoglio S, Villa G, Gualco M, Bleidl D, Cafiero F. The role of sentinel lymph node biopsy in patients with local recurrence or in-transit metastasis of melanoma. Anticancer Res. 2014;34:3197–3203. [PubMed] [Google Scholar]
- 6.Larkin J, Del Vecchio M, Ascierto PA, Krajsova I, Schachter J, Neyns B, Espinosa E, Garbe C, Sileni VC, Gogas H, et al. Vemurafenib in patients with BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety study. Lancet Oncol. 2014;15:436–444. doi: 10.1016/S1470-2045(14)70051-8. [DOI] [PubMed] [Google Scholar]
- 7.Berrocal A, Arance A, Lopez Martin JA, Soriano V, Muñoz E, Alonso L, Espinosa E, Lopez Criado P, Valdivia J, Martin Algarra S. Ipilimumab for advanced melanoma: experience from the Spanish Expanded Access Program. Melanoma Res. 2014;24:577–583. doi: 10.1097/CMR.0000000000000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Culos KA, Cuellar S. Novel targets in the treatment of advanced melanoma: new first-line treatment options. Ann Pharmacother. 2013;47:519–526. doi: 10.1345/aph.1R614. [DOI] [PubMed] [Google Scholar]
- 9.Güven K, Kittler H, Wolff K, Pehamberger H. Cisplatin and carboplatin combination as second-line chemotherapy in dacarbazine-resistant melanoma patients. Melanoma Res. 2001;11:411–415. doi: 10.1097/00008390-200108000-00012. [DOI] [PubMed] [Google Scholar]
- 10.Martin S, Hill DS, Paton JC, Paton AW, Birch-Machin MA, Lovat PE, Redfern CP. Targeting GRP78 to enhance melanoma cell death. Pigment Cell Melanoma Res. 2010;23:675–682. doi: 10.1111/j.1755-148X.2010.00731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Khattouti A, Selimovic D, Haïkel Y, Megahed M, Gomez CR, Hassan M. Identification and analysis of CD133(+) melanoma stem-like cells conferring resistance to taxol: An insight into the mechanisms of their resistance and response. Cancer Lett. 2014;343:123–133. doi: 10.1016/j.canlet.2013.09.024. [DOI] [PubMed] [Google Scholar]
- 12.Selimovic D, Badura HE, El-Khattouti A, Soell M, Porzig BB, Spernger A, Ghanjati F, Santourlidis S, Haikel Y, Hassan M. Vinblastine-induced apoptosis of melanoma cells is mediated by Ras homologous A protein (Rho A) via mitochondrial and non-mitochondrial-dependent mechanisms. Apoptosis. 2013;18:980–997. doi: 10.1007/s10495-013-0844-4. [DOI] [PubMed] [Google Scholar]
- 13.Selimovic D, Porzig BB, El-Khattouti A, Badura HE, Ahmad M, Ghanjati F, Santourlidis S, Haikel Y, Hassan M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell Signal. 2013;25:308–318. doi: 10.1016/j.cellsig.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 14.Bajetta E, Del Vecchio M, Vitali M, Martinetti A, Ferrari L, Queirolo P, Sertoli MR, Cainelli T, Cellerino R, Cascinelli N. A feasibility study using polychemotherapy (cisplatin + vindesine + dacarbazine) plus interferon-alpha or monochemotherapy with dacarbazine plus interferon-alpha in metastatic melanoma. Tumori. 2001;87:219–222. doi: 10.1177/030089160108700402. [DOI] [PubMed] [Google Scholar]
- 15.Meckbach D, Keim U, Richter S, Leiter U, Eigentler TK, Bauer J, Pflugfelder A, Büttner P, Garbe C, Weide B. BRAF-V600 mutations have no prognostic impact in stage IV melanoma patients treated with monochemotherapy. PLoS One. 2014;9:e89218. doi: 10.1371/journal.pone.0089218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CK, Jung M, Choi HJ, Kim HR, Kim HS, Roh MR, Ahn JB, Chung HC, Heo SJ, Rha SY, et al. Results of a Phase II Study to Evaluate the Efficacy of Docetaxel and Carboplatin in Metastatic Malignant Melanoma Patients Who Failed First-Line Therapy Containing Dacarbazine. Cancer Res Treat. 2015;47:781–789. doi: 10.4143/crt.2014.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linardou H, Pentheroudakis G, Varthalitis I, Gogas H, Pectasides D, Makatsoris T, Fountzilas G, Bafaloukos D. Predictive biomarkers to chemotherapy in patients with advanced melanoma receiving the combination of cisplatin--vinblastine--temozolomide (PVT) as first-line treatment: a study of the Hellenic Cooperative Oncology Group (HECOG) Anticancer Res. 2015;35:1105–1113. [PubMed] [Google Scholar]
- 18.Lens MB, Eisen TG. Systemic chemotherapy in the treatment of malignant melanoma. Expert Opin Pharmacother. 2003;4:2205–2211. doi: 10.1517/14656566.4.12.2205. [DOI] [PubMed] [Google Scholar]
- 19.Queirolo P, Picasso V, Spagnolo F. Combined BRAF and MEK inhibition for the treatment of BRAF-mutated metastatic melanoma. Cancer Treat Rev. 2015;41:519–526. doi: 10.1016/j.ctrv.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Thu YM, Su Y, Yang J, Splittgerber R, Na S, Boyd A, Mosse C, Simons C, Richmond A. NF-κB inducing kinase (NIK) modulates melanoma tumorigenesis by regulating expression of pro-survival factors through the β-catenin pathway. Oncogene. 2012;31:2580–2592. doi: 10.1038/onc.2011.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim A, Son M, Kim KI, Yang Y, Song EY, Lee HG, Lim JS. Elevation of intracellular cyclic AMP inhibits NF-kappaB-mediated thymosin beta4 expression in melanoma cells. Exp Cell Res. 2009;315:3325–3335. doi: 10.1016/j.yexcr.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 22.Sims JT, Ganguly SS, Bennett H, Friend JW, Tepe J, Plattner R. Imatinib reverses doxorubicin resistance by affecting activation of STAT3-dependent NF-κB and HSP27/p38/AKT pathways and by inhibiting ABCB1. PLoS One. 2013;8:e55509. doi: 10.1371/journal.pone.0055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levati L, Ruffini F, Muzi A, Umezawa K, Graziani G, D’Atri S, Lacal PM. Placenta growth factor induces melanoma resistance to temozolomide through a mechanism that involves the activation of the transcription factor NF-κB. Int J Oncol. 2011;38:241–247. [PubMed] [Google Scholar]
- 24.Lev DC, Ruiz M, Mills L, McGary EC, Price JE, Bar-Eli M. Dacarbazine causes transcriptional up-regulation of interleukin 8 and vascular endothelial growth factor in melanoma cells: a possible escape mechanism from chemotherapy. Mol Cancer Ther. 2003;2:753–763. [PubMed] [Google Scholar]
- 25.Poklepovic A, Youssefian LE, Winning M, Birdsell CA, Crosby NA, Ramakrishnan V, Ernstoff MS, Roberts JD. Phase I trial of bortezomib and dacarbazine in melanoma and soft tissue sarcoma. Invest New Drugs. 2013;31:937–942. doi: 10.1007/s10637-012-9913-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su Y, Amiri KI, Horton LW, Yu Y, Ayers GD, Koehler E, Kelley MC, Puzanov I, Richmond A, Sosman JA. A phase I trial of bortezomib with temozolomide in patients with advanced melanoma: toxicities, antitumor effects, and modulation of therapeutic targets. Clin Cancer Res. 2010;16:348–357. doi: 10.1158/1078-0432.CCR-09-2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macdonald JB, Macdonald B, Golitz LE, LoRusso P, Sekulic A. Cutaneous adverse effects of targeted therapies: Part II: Inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221–236; quiz 237-238. doi: 10.1016/j.jaad.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 28.Kreuter A, van Eijk T, Lehmann P, Fischer M, Horn T, Assaf C, Schley G, Herbst R, Kellner I, Weisbrich C, et al. Electrochemotherapy in advanced skin tumors and cutaneous metastases - a retrospective multicenter analysis. J Dtsch Dermatol Ges. 2015;13:308–315. doi: 10.1111/ddg.12583. [DOI] [PubMed] [Google Scholar]
- 29.Chu SC, Hsieh YS, Yu CC, Lai YY, Chen PN. Thymoquinone induces cell death in human squamous carcinoma cells via caspase activation-dependent apoptosis and LC3-II activation-dependent autophagy. PLoS One. 2014;9:e101579. doi: 10.1371/journal.pone.0101579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wengrod J, Wang D, Weiss S, Zhong H, Osman I, Gardner LB. Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci Signal. 2015;8:ra27. doi: 10.1126/scisignal.aaa0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong H, Tian L, Li R, Pei C, Fu Y, Dong X, Xia F, Wang C, Li W, Guo X, et al. IFNg-induced Irgm1 promotes tumorigenesis of melanoma via dual regulation of apoptosis and Bif-1-dependent autophagy. Oncogene. 2015;34:5363–5371. doi: 10.1038/onc.2014.459. [DOI] [PubMed] [Google Scholar]
- 32.Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F, Venturi G, Della Mina P, Pattingre S, Rivoltini L, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 2010;1:e87. doi: 10.1038/cddis.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wroblewski D, Jiang CC, Croft A, Farrelly ML, Zhang XD, Hersey P. OBATOCLAX and ABT-737 induce ER stress responses in human melanoma cells that limit induction of apoptosis. PLoS One. 2013;8:e84073. doi: 10.1371/journal.pone.0084073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang GJ, Deng JS, Huang SS, Wang SY, Chang YS, Kuo YH. Bioassay guided isolation and identification of anti-inflammatory active compounds from the root of Ficus formosana. J Agric Food Chem. 2013;61:11008–11015. doi: 10.1021/jf4033766. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Ma M, Teng J, Zhang J, Zhou S, Zhang Y, Wu E, Ding X. Chronic exposure to cerebrospinal fluid of multiple system atrophy in neuroblastoma and glioblastoma cells induces cytotoxicity via ER stress and autophagy activation. Oncotarget. 2015;6:13278–13294. doi: 10.18632/oncotarget.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Y, Yang J, Zhao J, Xiao C, Xu C, Xiang Y. The switch from ER stress-induced apoptosis to autophagy via ROS-mediated JNK/p62 signals: A survival mechanism in methotrexate-resistant choriocarcinoma cells. Exp Cell Res. 2015;334:207–218. doi: 10.1016/j.yexcr.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Bertrand L, Toborek M. Dysregulation of Endoplasmic Reticulum Stress and Autophagic Responses by the Antiretroviral Drug Efavirenz. Mol Pharmacol. 2015;88:304–315. doi: 10.1124/mol.115.098590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu H, He Z, Simon HU. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin Cancer Biol. 2013;23:352–360. doi: 10.1016/j.semcancer.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Fecher LA, Amaravadi RK, Schuchter LM, Flaherty KT. Drug targeting of oncogenic pathways in melanoma. Hematol Oncol Clin North Am. 2009;23:599–618, x. doi: 10.1016/j.hoc.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 40.Roulstone V, Pedersen M, Kyula J, Mansfield D, Khan AA, McEntee G, Wilkinson M, Karapanagiotou E, Coffey M, Marais R, et al. BRAF- and MEK-Targeted Small Molecule Inhibitors Exert Enhanced Antimelanoma Effects in Combination With Oncolytic Reovirus Through ER Stress. Mol Ther. 2015;23:931–942. doi: 10.1038/mt.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodall ML, Wang T, Martin KR, Kortus MG, Kauffman AL, Trent JM, Gately S, MacKeigan JP. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy. 2014;10:1120–1136. doi: 10.4161/auto.28594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ho AL, Musi E, Ambrosini G, Nair JS, Deraje Vasudeva S, de Stanchina E, Schwartz GK. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PLoS One. 2012;7:e40439. doi: 10.1371/journal.pone.0040439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chatterjee SJ, Pandey S. Chemo-resistant melanoma sensitized by tamoxifen to low dose curcumin treatment through induction of apoptosis and autophagy. Cancer Biol Ther. 2011;11:216–228. doi: 10.4161/cbt.11.2.13798. [DOI] [PubMed] [Google Scholar]
- 44.Chatterjee S, Willis N, Locks SM, Mott JH, Kelly CG. Dosimetric and radiobiological comparison of helical tomotherapy, forward-planned intensity-modulated radiotherapy and two-phase conformal plans for radical radiotherapy treatment of head and neck squamous cell carcinomas. Br J Radiol. 2011;84:1083–1090. doi: 10.1259/bjr/53812025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohana-Kumaran N, Hill DS, Allen JD, Haass NK. Targeting the intrinsic apoptosis pathway as a strategy for melanoma therapy. Pigment Cell Melanoma Res. 2014;27:525–539. doi: 10.1111/pcmr.12242. [DOI] [PubMed] [Google Scholar]
- 46.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124:1406–1417. doi: 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K, Huang P, Abbruzzese JL, McConkey DJ. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005;65:11658–11666. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]
- 48.Jiang CC, Chen LH, Gillespie S, Wang YF, Kiejda KA, Zhang XD, Hersey P. Inhibition of MEK sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res. 2007;67:9750–9761. doi: 10.1158/0008-5472.CAN-07-2047. [DOI] [PubMed] [Google Scholar]
- 49.Bakhshi J, Weinstein L, Poksay KS, Nishinaga B, Bredesen DE, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in mouse melanoma cells: effect of curcumin. Apoptosis. 2008;13:904–914. doi: 10.1007/s10495-008-0221-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park SH, Blackstone C. Further assembly required: construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 2010;11:515–521. doi: 10.1038/embor.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen X, Karnovsky A, Sans MD, Andrews PC, Williams JA. Molecular characterization of the endoplasmic reticulum: insights from proteomic studies. Proteomics. 2010;10:4040–4052. doi: 10.1002/pmic.201000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Voisine C, Pedersen JS, Morimoto RI. Chaperone networks: tipping the balance in protein folding diseases. Neurobiol Dis. 2010;40:12–20. doi: 10.1016/j.nbd.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stutzmann GE, Mattson MP. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol Rev. 2011;63:700–727. doi: 10.1124/pr.110.003814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–290. doi: 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kadowaki H, Nishitoh H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes (Basel) 2013;4:306–333. doi: 10.3390/genes4030306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selimovic D, Ahmad M, El-Khattouti A, Hannig M, Haïkel Y, Hassan M. Apoptosis-related protein-2 triggers melanoma cell death by a mechanism including both endoplasmic reticulum stress and mitochondrial dysregulation. Carcinogenesis. 2011;32:1268–1278. doi: 10.1093/carcin/bgr112. [DOI] [PubMed] [Google Scholar]
- 59.Bi K, Nishihara K, Machleidt T, Hermanson S, Wang J, Sakamuru S, Huang R, Xia M. Identification of known drugs targeting the endoplasmic reticulum stress response. Anal Bioanal Chem. 2015;407:5343–5351. doi: 10.1007/s00216-015-8694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi Y, Yang Y, Hoang B, Bardeleben C, Holmes B, Gera J, Lichtenstein A. Therapeutic potential of targeting IRES-dependent c-myc translation in multiple myeloma cells during ER stress. Oncogene. 2015:May 11; Epub ahead of print. doi: 10.1038/onc.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Selimovic D, Hassan M, Haikel Y, Hengge UR. Taxol-induced mitochondrial stress in melanoma cells is mediated by activation of c-Jun N-terminal kinase (JNK) and p38 pathways via uncoupling protein 2. Cell Signal. 2008;20:311–322. doi: 10.1016/j.cellsig.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 62.El-Khattouti A, Sheehan NT, Monico J, Drummond HA, Haikel Y, Brodell RT, Megahed M, Hassan M. CD133⁺ melanoma subpopulation acquired resistance to caffeic acid phenethyl ester-induced apoptosis is attributed to the elevated expression of ABCB5: significance for melanoma treatment. Cancer Lett. 2015;357:83–104. doi: 10.1016/j.canlet.2014.10.043. [DOI] [PubMed] [Google Scholar]
- 63.Suga N, Gao E, Zhang Y, Ma X, Olsen JF. The corticofugal system for hearing: recent progress. Proc Natl Acad Sci USA. 2000;97:11807–11814. doi: 10.1073/pnas.97.22.11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30:16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tohmonda T, Yoda M, Mizuochi H, Morioka H, Matsumoto M, Urano F, Toyama Y, Horiuchi K. The IRE1α-XBP1 pathway positively regulates parathyroid hormone (PTH)/PTH-related peptide receptor expression and is involved in pth-induced osteoclastogenesis. J Biol Chem. 2013;288:1691–1695. doi: 10.1074/jbc.C112.424606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Back SH, Lee K, Vink E, Kaufman RJ. Cytoplasmic IRE1alpha-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J Biol Chem. 2006;281:18691–18706. doi: 10.1074/jbc.M602030200. [DOI] [PubMed] [Google Scholar]
- 67.Vaeteewoottacharn K, Kariya R, Matsuda K, Taura M, Wongkham C, Wongkham S, Okada S. Perturbation of proteasome function by bortezomib leading to ER stress-induced apoptotic cell death in cholangiocarcinoma. J Cancer Res Clin Oncol. 2013;139:1551–1562. doi: 10.1007/s00432-013-1473-6. [DOI] [PubMed] [Google Scholar]
- 68.Allagnat F, Christulia F, Ortis F, Pirot P, Lortz S, Lenzen S, Eizirik DL, Cardozo AK. Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia. 2010;53:1120–1130. doi: 10.1007/s00125-010-1699-7. [DOI] [PubMed] [Google Scholar]
- 69.Prasanthi JR, Larson T, Schommer J, Ghribi O. Silencing GADD153/CHOP gene expression protects against Alzheimer’s disease-like pathology induced by 27-hydroxycholesterol in rabbit hippocampus. PLoS One. 2011;6:e26420. doi: 10.1371/journal.pone.0026420. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Hassan M, Alaoui A, Feyen O, Mirmohammadsadegh A, Essmann F, Tannapfel A, Gulbins E, Schulze-Osthoff K, Hengge UR. The BH3-only member Noxa causes apoptosis in melanoma cells by multiple pathways. Oncogene. 2008;27:4557–4568. doi: 10.1038/onc.2008.90. [DOI] [PubMed] [Google Scholar]
- 71.Xi H, Barredo JC, Merchan JR, Lampidis TJ. Endoplasmic reticulum stress induced by 2-deoxyglucose but not glucose starvation activates AMPK through CaMKKβ leading to autophagy. Biochem Pharmacol. 2013;85:1463–1477. doi: 10.1016/j.bcp.2013.02.037. [DOI] [PubMed] [Google Scholar]
- 72.Tay KH, Luan Q, Croft A, Jiang CC, Jin L, Zhang XD, Tseng HY. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell Signal. 2014;26:287–294. doi: 10.1016/j.cellsig.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 73.Croft A, Tay KH, Boyd SC, Guo ST, Jiang CC, Lai F, Tseng HY, Jin L, Rizos H, Hersey P, et al. Oncogenic activation of MEK/ERK primes melanoma cells for adaptation to endoplasmic reticulum stress. J Invest Dermatol. 2014;134:488–497. doi: 10.1038/jid.2013.325. [DOI] [PubMed] [Google Scholar]
- 74.Huang TT, Lin HC, Chen CC, Lu CC, Wei CF, Wu TS, Liu FG, Lai HC. Resveratrol induces apoptosis of human nasopharyngeal carcinoma cells via activation of multiple apoptotic pathways. J Cell Physiol. 2011;226:720–728. doi: 10.1002/jcp.22391. [DOI] [PubMed] [Google Scholar]
- 75.Han X, Zhou J, Zhang P, Song F, Jiang R, Li M, Xia F, Guo FJ. IRE1α dissociates with BiP and inhibits ER stress-mediated apoptosis in cartilage development. Cell Signal. 2013;25:2136–2146. doi: 10.1016/j.cellsig.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 76.Kim HS, Kim TJ, Yoo YM. Melatonin combined with endoplasmic reticulum stress induces cell death via the PI3K/Akt/mTOR pathway in B16F10 melanoma cells. PLoS One. 2014;9:e92627. doi: 10.1371/journal.pone.0092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu MC, Gong HY, Lin GH, Hu SY, Chen MH, Huang SJ, Liao CF, Wu JL. XBP-1, a key regulator of unfolded protein response, activates transcription of IGF1 and Akt phosphorylation in zebrafish embryonic cell line. Biochem Biophys Res Commun. 2007;359:778–783. doi: 10.1016/j.bbrc.2007.05.183. [DOI] [PubMed] [Google Scholar]
- 78.Thorpe JA, Schwarze SR. IRE1alpha controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress Chaperones. 2010;15:497–508. doi: 10.1007/s12192-009-0163-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guichard C, Pedruzzi E, Fay M, Marie JC, Braut-Boucher F, Daniel F, Grodet A, Gougerot-Pocidalo MA, Chastre E, Kotelevets L, et al. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis. 2006;27:1812–1827. doi: 10.1093/carcin/bgl009. [DOI] [PubMed] [Google Scholar]
- 80.Zhang H, Nakajima S, Kato H, Gu L, Yoshitomi T, Nagai K, Shinmori H, Kokubo S, Kitamura M. Selective, potent blockade of the IRE1 and ATF6 pathways by 4-phenylbutyric acid analogues. Br J Pharmacol. 2013;170:822–834. doi: 10.1111/bph.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang C, Kawauchi J, Adachi MT, Hashimoto Y, Oshiro S, Aso T, Kitajima S. Activation of JNK and transcriptional repressor ATF3/LRF1 through the IRE1/TRAF2 pathway is implicated in human vascular endothelial cell death by homocysteine. Biochem Biophys Res Commun. 2001;289:718–724. doi: 10.1006/bbrc.2001.6044. [DOI] [PubMed] [Google Scholar]
- 82.Law BY, Chan WK, Xu SW, Wang JR, Bai LP, Liu L, Wong VK. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci Rep. 2014;4:5510. doi: 10.1038/srep05510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Armstrong JL, Corazzari M, Martin S, Pagliarini V, Falasca L, Hill DS, Ellis N, Al Sabah S, Redfern CP, Fimia GM, et al. Oncogenic B-RAF signaling in melanoma impairs the therapeutic advantage of autophagy inhibition. Clin Cancer Res. 2011;17:2216–2226. doi: 10.1158/1078-0432.CCR-10-3003. [DOI] [PubMed] [Google Scholar]
- 84.Xie Z, Xie Y, Xu Y, Zhou H, Xu W, Dong Q. Bafilomycin A1 inhibits autophagy and induces apoptosis in MG63 osteosarcoma cells. Mol Med Rep. 2014;10:1103–1107. doi: 10.3892/mmr.2014.2281. [DOI] [PubMed] [Google Scholar]
- 85.Orlotti NI, Cimino-Reale G, Borghini E, Pennati M, Sissi C, Perrone F, Palumbo M, Daidone MG, Folini M, Zaffaroni N. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy. 2012;8:1185–1196. doi: 10.4161/auto.20519. [DOI] [PubMed] [Google Scholar]
- 86.Rebecca VW, Massaro RR, Fedorenko IV, Sondak VK, Anderson AR, Kim E, Amaravadi RK, Maria-Engler SS, Messina JL, Gibney GT, et al. Inhibition of autophagy enhances the effects of the AKT inhibitor MK-2206 when combined with paclitaxel and carboplatin in BRAF wild-type melanoma. Pigment Cell Melanoma Res. 2014;27:465–478. doi: 10.1111/pcmr.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Soares AS, Costa VM, Diniz C, Fresco P. Combination of Cl-IB-MECA with paclitaxel is a highly effective cytotoxic therapy causing mTOR-dependent autophagy and mitotic catastrophe on human melanoma cells. J Cancer Res Clin Oncol. 2014;140:921–935. doi: 10.1007/s00432-014-1645-z. [DOI] [PubMed] [Google Scholar]
- 88.Biggers JW, Nguyen T, Di X, Gupton JT, Henderson SC, Emery SM, Alotaibi M, White KL, Brown R, Almenara J, et al. Autophagy, cell death and sustained senescence arrest in B16/F10 melanoma cells and HCT-116 colon carcinoma cells in response to the novel microtubule poison, JG-03-14. Cancer Chemother Pharmacol. 2013;71:441–455. doi: 10.1007/s00280-012-2024-6. [DOI] [PubMed] [Google Scholar]
- 89.Zhang L, Wang H, Xu J, Zhu J, Ding K. Inhibition of cathepsin S induces autophagy and apoptosis in human glioblastoma cell lines through ROS-mediated PI3K/AKT/mTOR/p70S6K and JNK signaling pathways. Toxicol Lett. 2014;228:248–259. doi: 10.1016/j.toxlet.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 90.Xie X, White EP, Mehnert JM. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One. 2013;8:e55096. doi: 10.1371/journal.pone.0055096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Platz A, Egyhazi S, Ringborg U, Hansson J. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol Oncol. 2008;1:395–405. doi: 10.1016/j.molonc.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–341. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, Molla A, Vegetti C, Nonaka D, Mortarini R, Parmiani G, et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene. 2006;25:3357–3364. doi: 10.1038/sj.onc.1209379. [DOI] [PubMed] [Google Scholar]
- 94.Eisenhardt AE, Olbrich H, Röring M, Janzarik W, Anh TN, Cin H, Remke M, Witt H, Korshunov A, Pfister SM, et al. Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int J Cancer. 2011;129:2297–2303. doi: 10.1002/ijc.25893. [DOI] [PubMed] [Google Scholar]
- 95.Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, Ono T, Albertson DG, Pinkel D, Bastian BC. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95:1878–1890. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- 96.Deichmann M, Thome M, Benner A, Kirschner M, Hassanzadeh J, Kurzen H. Preponderance of the oncogenic V599E and V599K mutations in B-raf kinase domain is enhanced in melanoma cutaneous/subcutaneous metastases. BMC Cancer. 2005;5:58. doi: 10.1186/1471-2407-5-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, Bastian BC, Springer C, Marais R. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006;66:9483–9491. doi: 10.1158/0008-5472.CAN-05-4227. [DOI] [PubMed] [Google Scholar]
- 98.Qin J, Xin H, Nickoloff BJ. Specifically targeting ERK1 or ERK2 kills melanoma cells. J Transl Med. 2012;10:15. doi: 10.1186/1479-5876-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peters S, Bouchaab H, Zimmerman S, Bucher M, Gaide O, Letovanec I, Homicsko K, Michielin O. Dramatic response of vemurafenib-induced cutaneous lesions upon switch to dual BRAF/MEK inhibition in a metastatic melanoma patient. Melanoma Res. 2014;24:496–500. doi: 10.1097/CMR.0000000000000055. [DOI] [PubMed] [Google Scholar]
- 100.Anforth R, Carlos G, Clements A, Kefford R, Fernandez-Peñas P. Cutaneous adverse events in patients treated with BRAF inhibitor-based therapies for metastatic melanoma for longer than 52 weeks. Br J Dermatol. 2015;172:239–243. doi: 10.1111/bjd.13200. [DOI] [PubMed] [Google Scholar]
- 101.Urner-Bloch U, Urner M, Stieger P, Galliker N, Winterton N, Zubel A, Moutouh-de Parseval L, Dummer R, Goldinger SM. Transient MEK inhibitor-associated retinopathy in metastatic melanoma. Ann Oncol. 2014;25:1437–1441. doi: 10.1093/annonc/mdu169. [DOI] [PubMed] [Google Scholar]
- 102.Uribe P, Anforth RM, Kefford RF, Fernandez-Peñas P. Acneiform eruption in a patient with metastatic melanoma after ceasing combination dabrafenib/trametinib therapy. Melanoma Res. 2014;24:501–503. doi: 10.1097/CMR.0000000000000096. [DOI] [PubMed] [Google Scholar]
- 103.Okwan-Duodu D, Pollack BP, Lawson D, Khan MK. Role of radiation therapy as immune activator in the era of modern immunotherapy for metastatic malignant melanoma. Am J Clin Oncol. 2015;38:119–125. doi: 10.1097/COC.0b013e3182940dc3. [DOI] [PubMed] [Google Scholar]
- 104.Maddodi N, Huang W, Havighurst T, Kim K, Longley BJ, Setaluri V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J Invest Dermatol. 2010;130:1657–1667. doi: 10.1038/jid.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lv S, Wang X, Zhang N, Sun M, Qi W, Li Y, Yang Q. Autophagy facilitates the development of resistance to the tumor necrosis factor superfamily member TRAIL in breast cancer. Int J Oncol. 2015;46:1286–1294. doi: 10.3892/ijo.2014.2812. [DOI] [PubMed] [Google Scholar]
- 107.He H, Zang LH, Feng YS, Chen LX, Kang N, Tashiro S, Onodera S, Qiu F, Ikejima T. Physalin A induces apoptosis via p53-Noxa-mediated ROS generation, and autophagy plays a protective role against apoptosis through p38-NF-κB survival pathway in A375-S2 cells. J Ethnopharmacol. 2013;148:544–555. doi: 10.1016/j.jep.2013.04.051. [DOI] [PubMed] [Google Scholar]
- 108.Escalante AM, McGrath RT, Karolak MR, Dorr RT, Lynch RM, Landowski TH. Preventing the autophagic survival response by inhibition of calpain enhances the cytotoxic activity of bortezomib in vitro and in vivo. Cancer Chemother Pharmacol. 2013;71:1567–1576. doi: 10.1007/s00280-013-2156-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meng X, Leyva ML, Jenny M, Gross I, Benosman S, Fricker B, Harlepp S, Hébraud P, Boos A, Wlosik P, et al. A ruthenium-containing organometallic compound reduces tumor growth through induction of the endoplasmic reticulum stress gene CHOP. Cancer Res. 2009;69:5458–5466. doi: 10.1158/0008-5472.CAN-08-4408. [DOI] [PubMed] [Google Scholar]
- 111.Gaiddon C, Jeannequin P, Bischoff P, Pfeffer M, Sirlin C, Loeffler JP. Ruthenium (II)-derived organometallic compounds induce cytostatic and cytotoxic effects on mammalian cancer cell lines through p53-dependent and p53-independent mechanisms. J Pharmacol Exp Ther. 2005;315:1403–1411. doi: 10.1124/jpet.105.089342. [DOI] [PubMed] [Google Scholar]
- 112.Marcilla-Etxenike A, Martín ML, Noguera-Salvà MA, García-Verdugo JM, Soriano-Navarro M, Dey I, Escribá PV, Busquets X. 2-Hydroxyoleic acid induces ER stress and autophagy in various human glioma cell lines. PLoS One. 2012;7:e48235. doi: 10.1371/journal.pone.0048235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brem GJ, Mylonas I, Brüning A. Eeyarestatin causes cervical cancer cell sensitization to bortezomib treatment by augmenting ER stress and CHOP expression. Gynecol Oncol. 2013;128:383–390. doi: 10.1016/j.ygyno.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 114.Cea M, Cagnetta A, Fulciniti M, Tai YT, Hideshima T, Chauhan D, Roccaro A, Sacco A, Calimeri T, Cottini F, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. 2012;120:3519–3529. doi: 10.1182/blood-2012-03-416776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Manni S, Brancalion A, Mandato E, Tubi LQ, Colpo A, Pizzi M, Cappellesso R, Zaffino F, Di Maggio SA, Cabrelle A, et al. Protein kinase CK2 inhibition down modulates the NF-κB and STAT3 survival pathways, enhances the cellular proteotoxic stress and synergistically boosts the cytotoxic effect of bortezomib on multiple myeloma and mantle cell lymphoma cells. PLoS One. 2013;8:e75280. doi: 10.1371/journal.pone.0075280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jiang Q, Li F, Shi K, Wu P, An J, Yang Y, Xu C. Involvement of p38 in signal switching from autophagy to apoptosis via the PERK/eIF2α/ATF4 axis in selenite-treated NB4 cells. Cell Death Dis. 2014;5:e1270. doi: 10.1038/cddis.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Syed DN, Lall RK, Chamcheu JC, Haidar O, Mukhtar H. Involvement of ER stress and activation of apoptotic pathways in fisetin induced cytotoxicity in human melanoma. Arch Biochem Biophys. 2014;563:108–117. doi: 10.1016/j.abb.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martin S, Lamb HK, Brady C, Lefkove B, Bonner MY, Thompson P, Lovat PE, Arbiser JL, Hawkins AR, Redfern CP. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP78. Br J Cancer. 2013;109:433–443. doi: 10.1038/bjc.2013.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Beck D, Niessner H, Smalley KS, Flaherty K, Paraiso KH, Busch C, Sinnberg T, Vasseur S, Iovanna JL, Drießen S, et al. Vemurafenib potently induces endoplasmic reticulum stress-mediated apoptosis in BRAFV600E melanoma cells. Sci Signal. 2013;6:ra7. doi: 10.1126/scisignal.2003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Murai M, Inoue T, Suzuki-Karasaki M, Ochiai T, Ra C, Nishida S, Suzuki-Karasaki Y. Diallyl trisulfide sensitizes human melanoma cells to TRAIL-induced cell death by promoting endoplasmic reticulum-mediated apoptosis. Int J Oncol. 2012;41:2029–2037. doi: 10.3892/ijo.2012.1656. [DOI] [PubMed] [Google Scholar]
- 122.Su TR, Tsai FJ, Lin JJ, Huang HH, Chiu CC, Su JH, Yang YT, Chen JY, Wong BS, Wu YJ. Induction of apoptosis by 11-dehydrosinulariolide via mitochondrial dysregulation and ER stress pathways in human melanoma cells. Mar Drugs. 2012;10:1883–1898. doi: 10.3390/md10081883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hiscutt EL, Hill DS, Martin S, Kerr R, Harbottle A, Birch-Machin M, Redfern CP, Fulda S, Armstrong JL, Lovat PE. Targeting X-linked inhibitor of apoptosis protein to increase the efficacy of endoplasmic reticulum stress-induced apoptosis for melanoma therapy. J Invest Dermatol. 2010;130:2250–2258. doi: 10.1038/jid.2010.146. [DOI] [PubMed] [Google Scholar]
- 124.Jiang CC, Wroblewski D, Yang F, Hersey P, Zhang XD. Human melanoma cells under endoplasmic reticulum stress are more susceptible to apoptosis induced by the BH3 mimetic obatoclax. Neoplasia. 2009;11:945–955. doi: 10.1593/neo.09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mhaidat NM, Thorne R, Zhang XD, Hersey P. Involvement of endoplasmic reticulum stress in Docetaxel-induced JNK-dependent apoptosis of human melanoma. Apoptosis. 2008;13:1505–1512. doi: 10.1007/s10495-008-0276-8. [DOI] [PubMed] [Google Scholar]
- 126.Jiang CC, Chen LH, Gillespie S, Kiejda KA, Mhaidat N, Wang YF, Thorne R, Zhang XD, Hersey P. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res. 2007;67:5880–5888. doi: 10.1158/0008-5472.CAN-07-0213. [DOI] [PubMed] [Google Scholar]