

Abstract
Microorganism identification by MALDI TOF mass-spectrometry is based on the comparison of the mass spectrum of the studied organism with those of reference strains. It is a rapid and reliable method. However, commercial databases and programs are mostly designed for identification of clinically important strains and can be used only for particular mass spectrometer models. The need for open platforms and reference databases is obvious. In this study we describe a geometric approach for microorganism identification by mass spectra and demonstrate its capabilities by analyzing 24 strains belonging to the Bacillus pumilus group. This method is based on representing mass spectra as points on a multidimensional space, which allows us to use geometric distances to compare the spectra. Delimitation of microorganisms performed by geometric approach correlates well with the results of molecular phylogenetic analysis and clustering using Biotyper 3.1. All three methods used allowed us to reliably divide the strains into two groups corresponding to closely related species, Bacillus pumilus and Bacillus altitudinis. The method developed by us will be implemented in a Web interface designed for using open reference databases for microorganism identification. The data is available at http://www.bionet.nsc.ru/mbl/database/database.html.
When studying bacterial strains isolated from extreme environments, we require rapid and reliable identification of bacterial strains, including those of the genus Bacillus. The genus Bacillus contains Gram-positive aerobic or facultative anaerobic rod-shaped bacteria that form intracellular spores. It includes over 80 valid species1. Representatives of this genus are abundant in soil, air, and water, and are widely used as sources of industrial enzymes for food, textile and chemical industries2. They are also used as expression hosts for recombinant genes3, as well as a source of recombinant genes4. Bacillus strains are promising for agriculture as plant growth promoting rhizobacteria5 and for usage in decontamination systems6,7.
In the last 30 years systematics of the genus was substantially revised. Some species were isolated into new genera: Alicyclobacillus, Paenibacillus, Aneurinibacillus, Brevibacillus, Halobacillus, Virgibacillus, Filobacillus, and Jeotgalibacillus. Also, the genus Bacillus contains several closely related species group, whose delimitation is difficult. For example, the Bacillus cereus group includes Bacillus cereus, Bacillus anthracis, and Bacillus thuringiensis, which are genetically very similar, but nevertheless are considered separate species due to different pathegenicity8. Another example is the Bacillus subtilis group, which contains the species Bacillus subtilis subsp. subtilis, Bacillus amyloliquefaciens, Bacillus licheniformis, Bacillus atrophaeus, Bacillus mojavensis, Bacillus vallismortis, Bacillus subtilis subsp. spizizenii, and Bacillus sonorensis. The 16S rRNA gene sequences of these species have over 99% sequence similarity, so they cannot be distinguished based on it alone9. A new species, Bacillus safensis, was isolated from B. pumilus based on the sequence of the gyrB gene10, and three more species were described using the polyphasic taxonomy approach: Bacillus altitudinis, Bacillus stratosphaericus, and Bacillus aerophilus11. These species have closely related 16S rRNA gene sequences and form the B. pumilus group12.
Classic methods of microorganism identification, such as biochemical tests and DNA sequencing, are time-consuming and labor-intensive. The more recently developed approach that uses MALDI TOF mass-spectrometry is simple and rapid. It is based on the comparison of protein spectrum of the studied specimen to a database of reference spectra13. Several studies demonstrated high reproducibility of this method provided standard protocols are used14,15,16,17. Identification accuracy is known to tolerate varying growth conditions17,18,19.
Effective application of the mass-spectrometry approach requires a comprehensive reference database, as well as specific software for comparison of spectra. Several commercial platforms are currently available: Biotyper (Bruker Daltonicks), Saramis (Shimadzu), Microbelynx (Waters Corporation), and Andromas. These platforms are designed for particular models of mass spectrometers and are rather expensive. They are also oriented towards clinical diagnostics and contain predominantly pathogenic species and strains. Due to these limitation, research groups have to design their own “in house” databases, mathematical algorithms and software. Some commercial products, such as Statgraphics Plus 5.1 (Statpoint Technologies) and BioNumerics 6.0 (Applied-Maths) allow one to create “in house” databases, but open platforms including user-filled databases and software for mass spectra analysis are required for effective development of the field20.
One of the first attempts to create such a database was the BGP-database21 (http://bgp.sourceforge.net). Another example is the Spectra bank (http://www.spectrabank.org), which is a database of mass lists characteristic for species or strains. It currently contains about 200 specimens. Characteristic mass lists may be compared using SPECLUST22, which performs cluster analysis by building a dendrogram. However, it cannot do database searches and does not adjust for relative peak intensity.
Geometrical methods that represent source data as points in a multidimensional space are widely used for mass-spectrometry data. They are mainly applied in cluster analysis and graphical representation of breaking spectra into groups using such methods as PCA and MDS23,24. Metrical distances, such as Euclidean distances, can be used as criteria for comparing unknown spectra with databases. For example, the Hamming23 and Mahalanobis25 distances were proposed.
Since our main goal is to develop a platform for an open database for microorganism identification, the mathematical algorithms have to be simple and not calculation-intensive, but at the same time significantly precise. We used the following algorithm implemented using the JACOBI-4 program developed in ICiG SB RAS26: (1) A set of data represented as (m/z; intensity) is transferred on an (x; y) coordinate plane, where xi are discrete with a specified interval, yi are calculated for each xi node according the peak curve formula, which allows one to represent the spectrum as a vector in multidimensional space and to calculate a centroid for set of vector for tested microorganisms. This transformation allows one to apply all geometric methods; (2) A matrix of Euclidean distances and Jaccard coefficients (JC) representing measures of spectra dissimilarity/similarity is calculated for the obtained vectors. The value of 1 − JC is a metric27, and is therefore suitable for our goals; (3) The Principal Coordinates method (PCo) and dendrogram construction was used to perform cluster analysis and data visualization.
The aim of this study was to test if the approach presented in this study could be used to identify closely related species. As an example we took a set of B. pumilus group strains that had over 98% sequence similarity for the 16S rRNA gene. For the 24 studied strains, we obtained mass spectra series and calculated centroids for cluster analysis. These data were compared with the results of 16S rRNA gene sequencing and MALDI Biotyper 3.1 program (Bruker Daltonics) analysis. The obtained centroids were used as a reference database for identification of two replicates of the studied strains. Identification was based on Euclidean distances and JC used as similarity measures.
Results
16S rRNA analysis
Since we could not reliably identify the strains using the GenBank database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) based on 16S rRNA gene sequences, we compared them to type strain sequences of the Bacillus pumilus group: B. pumilus (AY456263), B. altitudinis (AJ831842), and B. safensis (AB681259) (Fig. 1a). B. stratosphaericus and B. aerophilus were excluded from the analysis, because their 16S rRNA sequences were identical to B. altitudinis and these strains were absent from microbial strain catalogues. Two groups referred to as the A and P groups were detected in our dataset with the bootstrap support of 96. The A group included strains isolated from Kamchatka thermal springs (KT), rhizosphere of higher plants from the Novosibirsk oblast (RG), as well as the B. altitudinis type strain (AJ831842). The P group contained strains from saline lakes of the Novosibirsk oblast (NR), complex ore deposits of the Kemerovo oblast, and type strains of B. pumilus (AY456263) and B. safensis (AB681259). Within the groups, strains were closely related, except for the Cd1 and KH2 strains, which formed a single subcluster within the A group with the bootstrap support of 73. Visualization of the sequences in BioEdit allowed us to find six marker substitutions (Fig. 1b) that distinguished these clusters. Other substitutions were uninformative. Sequences of the 16S rRNA genes of the strains isolated from Kamchatka thermal springs (KT) and from the rhizosphere of higher plants from the Novosibirsk oblast (RG) were identical to B. altitudinis AJ831842, while those of the strains from saline lakes of the Novosibirsk oblast (NR) and complex ore deposits of the Kemerovo oblast (KR) did not differ from B. pumilus (AY456263). B. safensis (AB681259) differed by a single nucleotide substitution from B. pumilus (AY456263), which allowed us to identify NR and KR strains as B. pumilus (Fig. 1b). The KH2 and Cd1 strains had identical patterns of these six marker substitutions to the B. altitudinis (AJ831842) type strain and the rest of the A group. The sequence of B. safensis type strain (AB681259) differed from that of B. pumilus (AY456263) by one marker substitution. Based on this, we identified the representatives of the A group as B. altitudinis, and of the P group, to B. pumilus.
Figure 1. Phylogenetic analysis.

(a) Phylogenetic tree constructed using the Maximum Likelihood algorithm for the 16S rRNA gene sequences. Numbers above branches indicate bootstrap support. Sequence distances represent the number of substitutions per 1000 nucleotides. The following type strains sequences were used for comparison: B. pumilus (AY456263), B. safensis (AB681259), and B. altitudinis (AJ831842); B. licheniformis (EF433410) was used as an outgroup. (b) Fragments of the alignment containing marker substitutions. Numbers in the bottom line indicate their positions in the B. altitudinis sequence (AJ831842).
Analysis of mass spectra
Mass spectra were obtained and processed as described in the Materials and Methods section. For statistical significance 12 samples were taken for each strain, and three independent spectra were taken for each sample. Most of the obtained spectra contained 50 to 60 mass peaks in the 2–10 kDa range. The matrix of Euclidean distances among centroids, transformed using the principal coordinates analysis, was visualized as two 2-dimensional plots in the PCo1, PCo2 and PCo1, PCo3 coordinates (Fig. 2a,b). In both projections, the two groups differ by the position on the PCo1 axis that provides the largest amount of information. B. altitudinis strains fell into the A group, and B. pumulus strains, into the P group. Welsh’s t- test was used to verify the separation of strain centroids on the groups A or P (t = 11.16; p < 10−6; df = 22). The distance between the A and P sample centers along the PCo1 axis was 2.4, and standard deviations for these samples were 0.57 and 0.39, respectively. We should also note that the P group had higher variance along the PCo2 axes PCo3 in comparison to the A group, which indicates that it has higher heterogeneity. Cluster analysis was performed by constructing a dendrogram by the Ward’s method using all 23 coordinates obtained by transforming the matrix of Euclidean distances (Fig. 2c). A phyloproteomic dendrogram built using Biotyper 3.1 is shown for comparison (Fig. 2d). Both dendrograms demonstrate two clusters that correspond to the A and P groups on the plot (Fig. 2a,b) and the 16S rRNA gene tree (Fig. 1a).
Figure 2. Phyloproteomic analysis.

Position of spectra centroids on the principal coordinates plane: (a) PCo1, PCo2; (b) PCo1, PCo3. The proportions of the total variance for those axes were 29.2% for PCo1, 13.2% for PCo2, and 10.4% for PCo3, which sums up to 52.9%. The groups A and P are framed. Dendrograms constructed based on the distances among spectra centroids by the Ward’s method (c) and by MSPs clustering in Biotyper 3.1 (d). Strains were divided into two clusters that correspond to B. altitudinis (A) and B. pumilus (P); (e) Wet-lab experiment: identification of the studied centroids for two biological replicates using JC, Euclidean distances, and Biotyper 3.1 (cutoff criteria - 2.0, wich is defined Bruker as “secure genus identification, probable species identification”). (e) Wet-lab experiment: identification of the studied centroids for two biological replicates using JC, Euclidean distances, and Biotyper 3.1 (cutoff criteria - 2.0, wich is defined Bruker as “secure genus identification, probable species identification”). Strain-level match - case when centroid of tested specimen and closest centroid in data base belong to the one and the same strain.
We performed a SPECLUST analysis which yielded a list of common and group-specific peaks. Eight peaks were found in all studied strains : 3048, 3621, 4914, 5208, 6622, 7242, 7729, and 9830 Da. The A group was distinguished by the presence of the 6671 Da peak, while the P group had three characteristic peaks: 3765, 4589, and 6870 Da. Averaged mass spectra visualized in gel view demonstrate that the A and P groups differ by the presence of group-specific peaks, as well as by their relative intensities (Fig. 3). For example, the 4914, 6622, 7729, and 9830 Da peaks have higher intensities in the A group. Several high-intensity peaks at 6032, 6048, 6063, 6099, and 6117 Da demonstrated no group specificity. B. altitudinis strains had only the 6099 Da peak, while a B. pumilus strain may have any of these peaks. This is likely the cause of high dispersion along the PCo2 and PCo3 axes. We suggest that these peaks represent a single protein that is highly polymorphic in B. pumilus group.
Figure 3. Gel view of averaged mass spectra.

Common peaks are indicated by asterisks; peaks characteristic for the A group, by (a); peaks characteristic for the P group, by (p). A group of high-intensity peaks in the 6032–6117 Da range is framed.
Cross-validation was performed by two methods: by excluding one of each ten random sets of spectra samples and by excluding all spectra for a random strain. For the first method, spectra were correctly assigned to centroids in 96.7% of cases. Accuracy of group determination (A or P) was 99.9%.
The obtained centroids were used as a reference database and a training data set for calculating a cutoff criterion for identification of the studied strains. JC cutoff criterion value was 0.278 for the A group and 0.158 for the P group, and for the Euclidean distances, 3.35 and 3.01, respectively. We performed two independent wet-lab experiments for method verification. Figure 2e demonstrates results of strain verification using our reference database and Biotyper 3.1. Identification accuracy was 100% for Euclidean distances and 98% for JC. We should note that the single unidentified specimen was on the border of identification correctness; its score was 0.277, while cutoff value for that group was 0.278. Matches at the strain level also have been taken into account, when the centroid of tested specimen and the closest centroid in database were obtained for one and the same strain. The rate of these matches was 33.3% for JC and Biotyper. For the Euclidean distance it was 20.8%.
Discussion
Protein profiling using MALDI TOF mass spectrometry proved to be reliable for identification of closely related species, including Bacillus spp.28,29,30,31. However, it is currently restricted by the absence of free databases and software, and by the fact that commercial databases are mostly oriented towards strains encountered in clinical practice. New data can be added to these databases only by the manufacturer. Therefore, creation of open platforms and databases is an urgent task.
This goal requires a simple and reliable method with high fidelity that allows one to use a broad spectrum of analysis techniques. In this study, we propose an algorithm for representing mass spectra as vectors in a multidimensional Euclidean space. Many geometric methods can be used with this algorithm. We chose Euclidean distance as the simplest and most straightforward approach for the Euclidean space. As an alternative, we also implemented Jaccard coefficients, which allow one to calculate spectra similarity. In addition, 1 − JC is a geometric distance, which makes it suitable for such geometric methods as PCo.
As a model for testing this approach, we used 24 closely related strains of the B. pumilus group from the collection of the Institute of Cytology and Genetics SB RAS, which had over 98% sequence similarity for the 16S rRNA gene. The studied strains were isolated from various extreme habitats in various regions of Russia, including thermal springs, saline lakes, complex ore deposits, etc. For these species we found characteristic peaks on mass spectra and characteristic nucleotide substitutions in the 16S rRNA gene. 16S rRNA sequences were used to validate the results of mass spectrometry analysis. The mass spectra centroids of the studied strains were separated into two groups, which was confirmed by the Welsh’s t-test, even if only the first coordinate of the PCo matrix (PCo1) are used. These groups correspond to the two clusters detected on the phylogenetic tree. The dendrogram constructed by the Ward method using all 23 coordinates also yields similar results. We performed an additional analysis of mass-spectrometry data using Biotyper 3.0 as an extra check. All three methods used allowed us to reliably distinguish between two groups that correspond to two species, B. pumilus (P) and B. altitudinus (A).
The obtained reference database was used for identification of the studied microorganisms by wet-lab experiments. Identification accuracy was 98% for JC and 100% for Euclidean distances. Biotyper 3.1 analysis had identification accuracy of 100% when using 2.0 as cutoff score, which is defined by Bruker as “secure genus identification, probable species identification”. The score of the unidentified KG16(3) strain for the JC analysis was located at the border of cutoff value for the A group. In this study we had separate cutoff values for each species, because the A group centroids were significantly more compact in the 1 − JC geometric space than the P group centroids (data not shown). So cutoff score for the A group was more stringent (0.278) than for the P group (0.158). If cutoff values were averaged for the two groups, JC algorithm had 100% identification accuracy. Also a higher rate of matches at strain level for JC than for the Euclidean distance may offer a better performance of this method for strain-level identification.
Therefore, we demonstrated that the approach proposed in this study is suitable for identification of closely related species based on their mass spectra using B. pumilus and B. altitudinis as a model. Theoretically representing mass spectra as vectors in the Euclidean space allows one to use virtually unlimited number of coordinates for each centroid, which enables us to use both peak lists and raw mass spectra describing the spectrum as a curve as source data. It will allow us to take peak form into consideration and abandon peak peaking algorithm. The centroids, Euclidean distances, and strain descriptions are available in the Internet: http://www.bionet.nsc.ru/mbl/database/database.html.
Methods
Strain description
From the collection of extremophilic microorganisms of ICiG SB RAS we selected 24 strains that were identified as belonging to the B. pumilus group based on morphological and biochemical characteristics. This group contained strains isolated from extreme environments form various regions of Russia, including thermal springs, saline lakes, complex ore deposits, etc (Table 1).
Table 1. List of the studied strains.
| Strain | Cultivation Medium | GenBank | Strain source | Geochemical characteristics | Coordinates |
|---|---|---|---|---|---|
| O48 | LB | KP699776 | NR, Solenoye l. (48). Water sample. | 190; 15–20; 8, 0 | 54°14′N78°13′E |
| O32 | LB | KP699772 | NR, Gorkoye l. (42). Bottom sediments. | 280; 15–20; 7, 7 | 54°17′N77°27′E |
| O19 | LB | KP699775 | NR, Dolgoye l. (44). Water sample. | 43; 15–20; 8, 3 | 54°10′N77°56′E |
| O6 | LB | KP699774 | NR, Solenoye l. (48). Water sample. | 190; 15–20; 8, 0 | 54°14′N78°13′E |
| 47(6)il | S4 | KP699765 | NR, Khorosheye l. (47). Bottom sediments. | 99; 15–20; 9, 2 | 54°05′N77°51′E |
| 51(3)w | LB | KP699766 | NR, Gorkoye l. (51). Water sample. | 49; 15–20; 8, 9 | 54°12′N77°02′E |
| O41 | LB | KP699767 | NR, Krugloe l. (45). Bottom sediments. | 290; 15–20; 7, 7 | 54°08′N77°56′E |
| O33 | LB | KP699768 | NR, Gorkoye l. (42). Bottom sediments. | 280; 15–20; 7, 7 | 54°17′N77°27′E |
| 48(1)w | LB | KP699778 | NR, Solenoye l. (48). Water sample. | 114; 15–20; 8, 0 | 54°14′N78°13′E |
| 46(5)il | LB | KP699764 | NR, Razboynoye l. (46). Bottom sediments. | 14, 7; 15–20; 8, 7 | 54°07′N77°55′E |
| 51(1)il | LB | KP699773 | NR, Gorkoye l. (51). Bottom sediments. | 133; 15–20; 8, 0 | 54°12′N77°02′E |
| 51(5)il | LB | KP699777 | NR, Gorkoye l. (51). Bottom sediments. | 49; 15–20; 8, 9 | 54°12′N77°02′E |
| 42(6)w | LB | KP699769 | NR, Gorkoye l. (42). Water sample. | 160; 15–20; 7, 6 | 54°17′N77°27′E |
| 3U | MPA | KP699770 | KR, A swamp near t. Ursk. Bottom sediments. | N.D.; 25; 2, 6 | 54°27′N85°24′E |
| 10U | MPA | KP699771 | KR, River Ur near t. Ursk. Water sample. | N.D.; 18; 7, 7 | 54°27′N85°24′E |
| KH6 | MPA | KP699782 | KT, Geyser valley, G-16 (thermal cauldron). | N.D., 62; 3, 6 | 54°25′N160°7′E |
| KU3-5(2) | MPA | KP699787 | KT, Uzon caldera, U3-5 (Oil field). | 0, 4; 79; 4, 7 | 54°30′N160°0′E |
| KH2 | MPA | KP699786 | KT, Uzon caldera, U3-4-8 (Oil field). | 1, 1; 63; 2, 9 | 54°30′N160°0′E |
| KG16(2) | MPA | KP699780 | KT, Geyser valley, G-16 (thermal cauldron). | N.D., 62; 3, 6 | 54°25′N160°7′E |
| K6dt | MPA | KP699783 | KT, Uzon caldera, U-4 (Bannoye lake). | 0, 2; 36; 4, 6 | 54°30′N160°0′E |
| KH3 | MPA | KP699785 | KT, Uzon caldera, Uskv2. | 0, 3; 95; 8, 4 | 54°30′N160°0′E |
| KG16(3) | MPA | KP699781 | KT, Geyser valley, G-16 (thermal cauldron). | N.D., 62; 3, 6 | 54°25′N160°7′E |
| Cd1 | MPA | KP699779 | RG, Novosibirsk water storage basin. | <1, 0; 21; N.D. | 55°04′N82°92′E |
| Cu1 | MPA | KP699784 | RG, Novosibirsk water storage basin. | <1, 0; 21; N.D. | 55°04′N82°92′E |
NR, Novosibirsk region, saline lakes (l.); KR, Kemerovo region, complex ore deposits; KT, Kamchatka thermal springs; RG, rhizosphere of the water hyacinth, Novosibirsk water storage basin. For mass spectrometry, bacterial strains were grown at 37 °C on the following agar media: Luria-Bertani broth (LB); meat-peptone agar (MPA), and S4 medium containing 1 g/l NaCl, 5 g/l MgCl2, 1 g/l KCl, 1 g/l CaCl2, 4 g/l tripthone, 2 g/l yeast extract. Geochemical characteristics of the source environments: salinity (g/l); temperature (C); pH.
16S rRNA analysis
A fragment of the ribosomal 16S rRNA gene was amplified using universal bacterial primers 16S-8-f-B (5′-AGRGTTTGATCCT GGCTCA-3′) and 16S-1350-r-B (5′-GACGGGCGGTGTGTACAAG-3′) in a 30 microlitre volume on a MyCycler thermal cycler (BioRad) using TaqSE DNA polymerase (SibEnzyme, Novosibirsk) according to manufacturers’ instructions. Amplified products were purified using the PCR purification KIT (Fermentas). DNA sequencing was performed using the BigDye teminator 3.1 kit (Applied Biosystems) according to manufaturers’ instructions in the SB RAS Genomics Core Facility. Sequences were analyzed and edited using the BioEdit program (http://www.mbio.ncsu.edu/BioEdit/bioedit). The following type strain sequences were used for phylogenetic analysis: B. pumilus (AY456263), B. altitudinis (AJ831842), and B. safensis (AB681259); B. licheniformis (EF433410) was used as an outgroup. Sequences were aligned by ClustalW232 and phylogenetic trees were constructed using the Maximum Likelihood algorithm33 implemented in MEGA 6.034. All 16S rRNA sequences obtained by us were deposited in GenBank (Table 1).
Mass spectrometry analysis
Twelve separate colonies were taken for each strain. Colonies were transferred to 1.5 ml Eppendorf tube by a microbial transfer loop and resuspended in 300 microliters of deionized water. For inactivation of bacterial cells, 900 microliters of ethanol was added; cells were resuspended and sedimented by centrifugation for 2 min at 15600 g. Supernatant was removed and the sediment was dried for 5 min in an Eppendorf vacuum concentrator. Bacterial cell walls were destroyed by the addition of 50 microliters of 70% formic acid. Proteins were extracted by the addition of 50 microliters of acetonitrile followed by vigorous vortexing. The mixture was centrifuged for 2 min at 15600 g, and the supernatant was transferred into a clean tube for mass spectrometry analysis.
For mass spectrometry analysis, 1 microliter of protein extract was transferred to a stainless steel plate and allowed to dry at room temperature. Afterwards, 1 microliter of matrix (6 mg/ml of α-cyano-4-hydroxy-cinnamic acid in acetonitrile/water/trifluoroacetic acid solution (50:47.5:2.5, v/v)) was added. Spectra were obtained using an Ultraflex III MALDI TOF/TOF mass spectrometer (Bruker Daltonics) in the linear positive mode with laser frequency of 100 Hz in the 2000–20000 Da mass range. The voltage at the accelerating electrode was 25 kV; IS2 voltage, 23.45 kV; lens voltage, 6 kV; no extraction delay was made.
For each colony we obtained three spectra by summing 500 laser pulses (5 × 100 pulses from various positions of the target cell). Calibration was performed using Escherichia coli proteins: RL36 - 4365.3 Da, RS22 - 5096.8 Da, RL34 - 5381.4 Da, RL32 - 6315.0 Da, RL29 - 7274.5 Da, RS19 - 10300.1 Da.
A total of 36 spectra were obtained for each strain. Visual inspection was performed for all spectra in addition to computer analysis.
Phyloproteomic analysis of mass spectrometry data
Flattening, baseline extraction, and peak picking for the obtained mass spectra were performed using mMass35 (www.mmass.org).
Mass lists obtained in mMass were processed using the following algorithm:
A set of spectra is represented as {(xj, yj), j = 1…Lm; m = 1…M}, where xj is the peak number, yj is the function of signal intensity, Lm is the number of peaks for each spectrum, M is the number of spectra. Spectra are divided into Q classes.
Each spectrum is projected on a grid uniform in the x-coordinate. For each spectrum, the grid has the same boundaries (Xbeg, Xend), numbers of points N, and window half width K measured in points. The parameters Xbeg, Xend, N, and K are set by the user. Grid spacing h and half-width size w are calculated based on input parameters:
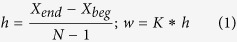 |
The following condition must be met for each spectrum: Xbeg < Xmin − w; Xend > Xmax + w, where Xmin and Xmax are the minimum and maximum values for the x-coordinate for each spectrum.
For each mesh point i we find all x-coordinates xj in the [ih − w, ih + w] range with nonzero yj signal intensities. For each framed signal its impact on the point i is calculated by the formula:
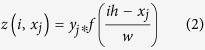 |
where f(x) = 1 − 3x2 + 2|x|3 when |x| ≤ 1 and f(x) = 0 otherwise.
The summarized impact zi on point i is calculated by the formula:
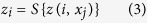 |
for each xj within the window [ih − w, ih + w], where S is the combination method (sum, averaging, maximum). As the result of this algorithm, a vectors sized N was obtained for each spectrum.
In this work we used the following parameters: Xbeg = 1000; Xend = 15000; N = 14000; K = 5; combination method was averaging.
When spectra were projected on the grid, centroids for every of Q classes were calculated. A matrix of Euclidean distances between all centroids was computed and processed by the Principal Coordinates analysis (PCo), which allowed us to represent all class centroids as points in a multidimensional Euclidean space with dimension no more than Q − 1. Welsh’s t-criterion was used to validate the studied groups on the PCo plot. Dendrogram were built using the Ward’s method by the PAST program36.
Jaccard coefficients are calculated according to the formula37:
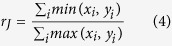 |
where x = {xi : xi ≥ 0}, y = {yi : yi ≥ 0} are vector representations of spectra.
Peak frequencies were analyzed using SPECLUST with the ≪peak in common≫ procedure. Search was performed in the 2 to 10 kDa range, which provides the optimum peak reproducibility with the “Width in peak match score” parameter value of 5 Da. In addition, the spectra were analyzed visually.
In addition, “main spectra” (MSPs) were generated in Biotyper 3.1 for the obtained mass spectra, and a phyloproteomic diagram was built using standard parameters18.
Wet-lab experiment
For wet-lab experiments all studied strains were grown under the same conditions as for the training sample (Table 1). For mass spectrometry we took three replicates for each specimen, one spectrum per each replicate. Specimen centroids were calculated as described in the Phyloproteomic analysis of mass-spectrometry data section.
The aim of the experiments was to assign each tested specimen either to A or P groups, or to a separate species. Cutoff radius of the group i (A or P) were calculated using the following formula38:
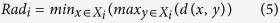 |
where Xi is the set of i centroids; d(x, y) is the distance between spectra x and y.
A centroid of tested specimen belongs to the “attraction zone” of group i if its distance from at least one centroid of this group does not exceed Radi. If specimen belong to one or more “attraction zones” of different groups i its assign to the group of the closest centroid. If a specimen does not belong to “attraction zone” of any group in the database, it is treated as an unknown species.
Additional Information
How to cite this article: Starostin, K. V. et al. Identification of Bacillus strains by MALDI TOF MS using geometric approach. Sci. Rep. 5, 16989; doi: 10.1038/srep16989 (2015).
Acknowledgments
Work of K.V.S., E.A.D., A.V.B., A.S.R. and S.E.P. was supported by Budget Project VI.58.1.3. Work of V.M.E. was supported by Russian Scientific Foundation (Project No 14-24-00123). The authors would like to express the gratitude to the staff of the Kronotsky State Biosphere Reserve for assistance in the work in Uzon caldera.
Footnotes
Author Contributions K.V.S. and E.A.D. performed MS experiments and data analysis. A.V.B. performed microbial culture isolation and cultivation. Mathematical algorithm was developed by V.M.E. A.S.R. performed 16s rRNA sequencing. S.E.P. supervised this project. All authors were involved in writing the manuscript.
References
- Blackwood K. S., Turenne C. Y., Harmsen D. & Kabani A. M. Reassessment of sequence-based targets for identification of bacillus species. Journal of Clinical Microbiology 42, 1626–1630 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallmey M., Singh A. & Ward O. P. Developments in the use of bacillus species for industrial production. Canadian journal of microbiology 50, 1–17 (2004). [DOI] [PubMed] [Google Scholar]
- Vary P. S. et al. Bacillus megaterium-from simple soil bacterium to industrial protein production host. Applied Microbiology and Biotechnology 76, 957–967 (2007). [DOI] [PubMed] [Google Scholar]
- Pan J., Huang Q. & Zhang Y. Gene cloning and expression of an alkaline serine protease with dehairing function from bacillus pumilus. Current microbiology 49, 165–169 (2004). [DOI] [PubMed] [Google Scholar]
- Sunar K., Dey P., Chakraborty U. & Chakraborty B. Biocontrol efficacy and plant growth promoting activity of bacillus altitudinis isolated from darjeeling hills, india. Journal of basic microbiology 55, 91–104 (2015). [DOI] [PubMed] [Google Scholar]
- Das K. & Mukherjee A. K. Crude petroleum-oil biodegradation efficiency of bacillus subtilis and pseudomonas aeruginosa strains isolated from a petroleum-oil contaminated soil from north-east india. Bioresource technology 98, 1339–45 (2007). [DOI] [PubMed] [Google Scholar]
- Dawkar V. V., Jadhav U. U., Jadhav S. U. & Govindwar S. P. Biodegradation of disperse textile dye brown 3rel by newly isolated bacillus sp. vus. Journal of Applied Microbiology 105, 14–24 (2008). [DOI] [PubMed] [Google Scholar]
- Rasko D. A., Altherr M. R., Han C. S. & Ravel J. Genomics of the bacillus cereus group of organisms. FEMS microbiology reviews 29, 303–29 (2005). [DOI] [PubMed] [Google Scholar]
- Jeyaram K. et al. Distinct differentiation of closely related species of bacillus subtilis group with industrial importance. Journal of Microbiological Methods 87, 161–164 (2011). [DOI] [PubMed] [Google Scholar]
- Satomi M., La Duc M. T. & Venkateswaran K. Bacillus safensis sp.nov., isolated from spacecraft and assembly-facility surfaces. International Journal of Systematic and Evolutionary Microbiology 56, 1735–1740 (2006). [DOI] [PubMed] [Google Scholar]
- Shivaji S. et al. Bacillus aerius sp. nov., bacillus aerophilus sp. nov., bacillus stratosphericus sp. nov. and bacillus altitudinis sp. nov., isolated from cryogenic tubes used for collecting air samples from high altitudes. International Journal of Systematic and Evolutionary Microbiology 56, 1465–1473 (2006). [DOI] [PubMed] [Google Scholar]
- Liu Y. et al. Phylogenetic diversity of the bacillus pumilus group and the marine ecotype revealed by multilocus sequence analysis. PLoS ONE 8, e80097 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins C., Lay J. (eds) Identification of microorganisms by mass spectrometry (John Wiley & Sons, Inc., 2006). [Google Scholar]
- Wang Z., Russon L., Li L., Roser D. C. & Long S. R. Investigation of spectral reproducibility in direct analysis of bacteria proteins by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid communications in mass spectrometry : RCM 12, 456–64 (1998). [DOI] [PubMed] [Google Scholar]
- Mellmann A. et al. Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to 16s rrna gene sequencing for species identification of nonfermenting bacteria. Journal of clinical microbiology 46, 1946–54 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellmann A. et al. High interlaboratory reproducibility of matrix-assisted laser desorption ionization-time of flight mass spectrometry-based species identification of nonfermenting bacteria. Journal of Clinical Microbiology 47, 3732–3734 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold E., Maier T., Kostrzewa M., Zeman E. & Splettstoesser W. Identification of francisella tularensis by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry: Fast, reliable, robust, and cost-effective differentiation on species and subspecies levels. Journal of Clinical Microbiology 48, 1061–1069 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiwald A. & Sauer S. Phylogenetic classification and identification of bacteria by mass spectrometry. Nature protocols 4, 732–42 (2009). [DOI] [PubMed] [Google Scholar]
- Šedo O., Várová A., Vad’urová M., Tvrzová L. & Zdráhal Z. The influence of growth conditions on strain differentiation within the lactobacillus acidophilus group using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry profiling. Rapid communications in mass spectrometry : RCM 27, 2729–36 (2013). [DOI] [PubMed] [Google Scholar]
- Kliem M. & Sauer S. The essence on mass spectrometry based microbial diagnostics. Current Opinion in Microbiology 15, 397–402 (2012). [DOI] [PubMed] [Google Scholar]
- Carbonnelle E. et al. Rapid identification of staphylococci isolated in clinical microbiology laboratories by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Journal of clinical microbiology 45, 2156–2161 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm R. et al. Detection and identification of protein isoforms using cluster analysis of maldi-ms mass spectra. Journal of Proteome Research 5, 785–792 (2006). [DOI] [PubMed] [Google Scholar]
- De Bruyne K. et al. Bacterial species identification from maldi-tof mass spectra through data analysis and machine learning. Systematic and applied microbiology 34, 20–9 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang L., Vranckx K., Janssens K. & Sandrin T. R. Use of maldi-tof mass spectrometry and a custom database to characterize bacteria indigenous to a unique cave environment (kartchner caverns, az, usa). Journal of Visualized Experiments e52064 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. et al. Comparison of feature selection and classification for maldi-ms data. BMC genomics 10 Suppl 1, S3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polunin D., Shtayger I. & Efimov V. Jacobi4 software for multivariate analysis of microarray data. Vestnik NSU 12, 90–98 (2014). [Google Scholar]
- Levandowsky M. & Winter D. Distance between sets. Nature 239, 174–174 (1972).4561969 [Google Scholar]
- Fernández-No I. C. et al. Characterisation and profiling of bacillus subtilis, bacillus cereus and bacillus licheniformis by maldi-tof mass fingerprinting. Food Microbiology 33, 235–242 (2013). [DOI] [PubMed] [Google Scholar]
- Hotta Y., Sato J., Sato H., Hosoda A. & Tamura H. Classification of the genus bacillus based on maldi-tof ms analysis of ribosomal proteins coded in s10 and spc operons. Journal of Agricultural and Food Chemistry 59, 5222–5230 (2011). [DOI] [PubMed] [Google Scholar]
- Lasch P. et al. Identification of bacillus anthracis by using matrix-assisted laser desorption ionization-time of flight mass spectrometry and artificial neural networks. Applied and Environmental Microbiology 75, 7229–7242 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branquinho R. et al. Differentiation of bacillus pumilus and bacillus safensis using maldi-tof-ms. PloS one 9, e110127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J. & Higgins D. G. Multiple sequence alignment using clustalw and clustalx. Current protocols in bioinformatics Chapter 2, Unit 2.3 (2002). [DOI] [PubMed] [Google Scholar]
- Dempster A., Laird N. & Rubin D. B. Maximum likelihood from incomplete data via the em algorithm. Journal of the Royal Statistical Society 39, 1–38 (1977). [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A. & Kumar S. Mega6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohalm M., Kavan D., Novák P., Volný M. & Havlícek V. mmass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Analytical chemistry 82, 4648–51 (2010). [DOI] [PubMed] [Google Scholar]
- Hammer Ø., Harper D. a. T. & Ryan P. D. Paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4, 9–18 (2001). [Google Scholar]
- Jaccard P. Nouvelles recherches sur la distribution. Bulletin de la Societe vaudoise des sciences Naturelles 44, 223–270 (1908). [Google Scholar]
- Deza M. M. & Deza E. Encyclopedia of Distances (Springer Berlin Heidelberg, 2009). [Google Scholar]