

Abstract
Terpenes are ubiquitous natural chemicals with diverse biological functions spanning all three domains of life. In specialized metabolism, the active sites of terpene synthases (TPSs) evolve in shape and reactivity to direct the biosynthesis of a myriad of chemotypes for organismal fitness. As most terpene biosynthesis mechanistically involves highly reactive carbocationic intermediates, the protein surfaces catalyzing these cascade reactions possess reactive regions possibly prone to premature carbocation capture and potentially enzyme inactivation. Here, we show using proteomic and X-ray crystallographic analyses that cationic intermediates undergo capture by conserved active site residues leading to inhibitory self-alkylation. Moreover, the level of cation-mediated inactivation increases with mutation of the active site, upon changes in the size and structure of isoprenoid diphosphate substrates, and alongside increases in reaction temperatures. TPSs that individually synthesize multiple products are less prone to self-alkylation then TPSs possessing relatively high product specificity. In total, the results presented suggest that mechanism-based alkylation represents an overlooked mechanistic pressure during the evolution of cation-derived terpene biosynthesis.
Diverse terpene-based natural products are widespread across all three domains of life. In the specialized metabolism of bacteria, plants, fungi, and animals, terpene natural products are used for defense and attraction, ecological communication, and physiologic regulation.1−4 Furthermore, cyclic terpene structures like sterols and hopanes serve as important membrane constituents in all life forms.5,6 Many cyclic terpenes are also commercially important compounds used as agrochemicals, pharmaceuticals, flavors, and fragrances.7−10
For terpene natural products such as 15-carbon sesquiterpenes, their functional complexity centers on the structural diversity of multicyclic, chiral hydrocarbon scaffolds most commonly biosynthesized starting with the primary metabolite, (E,E)-farnesyl diphosphate (FPP) (Figure S1).11 Plant class I TPSs consist of two α-helical domains, while bacterial and fungal class I TPSs contain a single α-helical catalytic domain.11 While the N-terminal domains in plant class I TPSs appear to lack catalytic activity, their C-terminal domains and the single helical domains of bacterial and fungal TPSs contain the active sites employing conserved Mg2+-binding motifs and variable surrounding residues. Together, these motifs catalyze the ionization of FPP and the ensuing structural diversity of cyclic sesquiterpene scaffolds through carbocation-based mechanisms.12,13
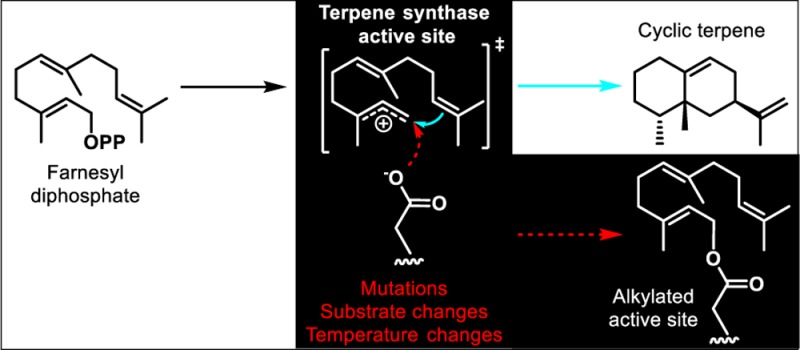
TPSs typically catalyze cationic mechanisms involving multiple highly reactive intermediates.14 The catalytic cycle of a sesquiterpene synthase begins with binding of the diphosphate moiety of FPP to positively charged residues and Mg2+ cofactors coordinated by two Mg2+-binding elements, the “DDxxD” motif and the “NSE/DTE” motif, both sitting at the entrance to the active site.15 The farnesyl chain tucks into the hydrophobic and aromatic-rich portion of the active site (Figure 1A). Subsequently, the carbon–phosphoester bond of FPP breaks through ionization, and an allylic farnesyl cation forms. The farnesyl cation then undergoes cationic reaction cascades consisting of electrophilic additions, deprotonation–reprotonation reactions, and hydride/alkyl rearrangements mediated by the shape and chemistry of the active site. The high reactivities of cationic intermediates are balanced in TPSs by hydrophobic active sites and several aromatic active site residues that stabilize cations through π-cation interactions.16 In addition, induced-fit closures of the active sites after substrate binding and ionization by C-terminal loops limit premature solvent quenching.17,18 Reaction termination occurs through cation neutralization. Three possible cation-based quenching scenarios exist: (1) cation neutralization through proton loss from a neighboring carbon,19,20 (2) quenching through nucleophilic attack by an exogenous species such as water,10 and (3) nucleophilic substitution by a reactive enzyme side chain (Figure 1B). Scenarios 1 and 2 yield sesquiterpene and sesquiterpenol products, respectively (Figure 1B). Scenario 3, however, lead to active site alkylations of TPSs possibly resulting in mechanism-based inhibition by covalently bound biosynthetic intermediates.
Figure 1.

Structural basis of cyclic sesquiterpene biosynthesis and carbocation quenching scenarios in sesquiterpene synthases. (a) Active site model of wild-type TEAS with bound substrate mimic 1-hydroxyfarnesyl phosphonate (colored yellow) based on Protein Data Bank entry 5EAT.19 Selected active site residues are displayed and numbered. The dynamic active site J/K loop is colored magenta. Three Mg2+ ions are highlighted as gray spheres and waters as red crosses. Active site mutation sites for alkylation studies (Table 1) are colored orange. (b) Carbocation quenching scenarios in sesquiterpene synthases. During enzyme-catalyzed sesquiterpene cyclization, cationic intermediates are neutralized by deprotonation as exemplified by the proposed last step in 5-epi-aristolochene biosynthesis,19 quenching with an exogenous nucleophile such as water exemplified by the proposed last step in patchoulol biosynthesis,9 or quenching with an endogenous nucleophile such as an active site residue to yield an alkylated active site. Biosynthetic steps in TPS active sites are highlighted in gray.
Mechanism-based enzyme inhibition includes inactivation of enzymes by turnover of substrate analogues, resulting in covalent attachment to amino acid residues or cofactors in enzyme active sites.21 Mechanism-based inhibition has been extensively studied for drug discovery.21 Although enzymatic self-inactivation by oxidative damage occurs in many heme-dependent enzymes such as prostaglandin endoperoxide H synthase,22,23 the occurrence of mechanism-based inhibition of TPSs has received little attention. Because sesquiterpene synthase active sites undergo changes in shape and reactivity during TPS evolution12,13 and upon protein engineering,24 self-alkylations may represent significant selective pressures on TPS evolution and engineering.25 In addition to mutational modulation of the TPS itself, self-alkylation may also result from the use of nonconventional or synthetic substrates, as recently demonstrated by the mechanism-based inhibition of aristolochene synthase of Penicillium roquefortii with vinyl-farnesyl diphosphate.26
Here, we investigate the occurrence, location, and chemistry of inhibitory self-alkylations in both specific and promiscuous sesquiterpene synthases from plants and fungi using proteomics and X-ray crystallography. Our in vitro characterizations of self-alkylations in sesquiterpene synthases in response to active site mutations, substrate changes, and reaction temperatures strongly suggest that mechanism-based alkylations during the course of natural evolution and synthetic engineering of these mechanistically complex, ecologically important, and commercially promising enzymes are additional constraints on TPS biosynthetic diversification.
Results
Self-Alkylation of Tobacco 5-epi-Aristolochene Synthase (TEAS) Mutants
To characterize self-alkylation in a sesquiterpene synthase, we chose tobacco 5-epi-aristolochene synthase (TEAS) as a model enzyme, as X-ray crystallography methods are well-established for facile determination of TEAS X-ray crystal structures (Figure 1A).19 Self-alkylations by active site changes were investigated for two groups of TEAS mutants. The first group of mutants possessed single active site changes via introduction of nucleophilic side chains such as cysteines or glutamates (Table 1). The second group of mutants focused on single active site changes causing subtle steric changes to the active site with minimal alterations in side chain chemistry (Table 1). The Mg2+-binding sites19 were not mutated to maintain Mg2+ coordination essential for TPS activity. To detect site-specific alkylation of TEAS, the reaction mixtures of TEAS mutants or wild-type TEAS and (E,E)-FPP were analyzed by bottom-up proteomics. The masses of nonalkylated and alkylated tryptic peptides were calculated and identified in the corresponding LC–MS/MS data sets. When charge states of detected peptide mass signals (usually z = 1–3) were taken into account, the detection of tryptic peptide mass signals shifted by 204.188 Da (C15H24+, i.e., m/z 204.2 for z = 1, m/z 102.1 for z = 2, and m/z 68.1 for z = 3) was indicative of alkylated tryptic peptides. A monoalkylation mass shift of 204.188 Da was detected for all mutants except I294L on the tryptic peptide V[442–453]K residing on the active site surface (Table 1, Figure 2A,B, and Figures S2 and S3). In some cases, TEAS mutants, including W273C, W273E, W273F, Y404F, Y404C, and L407I, showed an alkylation mass shift on the tryptic active site peptide I[515–532]K (Table 1, Figure 2B, and Figures S2 and S3). Quite surprisingly, no alkyl mass shifts were detected on peptides spanning the introduced cysteine or glutamate residues as putative cation traps (W273C, W273E, and Y404C) (Table 1 and Figure S3). V[442–453]K spans the Mg2+-binding “DTE” motif of TEAS, while I[515–532]K includes part of the J/K loop. This catalytic loop functions as a dynamic active site lid upon substrate binding and ionization.19
Table 1. Alkylation Analyses of Specific (dominant-product) and Promiscuous (multiproduct) Sesquiterpene Synthasesa.
| terpene synthase (organism) | product specificity (wt) | mutation | temp (°C) | substrate | alkylation on the “NSE/DTE” motif [peptide with a putative alkyation sites (*)] | alkylation on the active site lid [peptide with a putative alkylation site (*)] | main product |
|---|---|---|---|---|---|---|---|
| 5-epi-aristolochene synthase (Nicotiana tabacum) | dominant-product | wt | 25 | (E,E)-FPP | N/D | N/D | 5-epi-aristolochene |
| W273C | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | yes (I515VEVTY*IHNLDGYTHPEK532) | β-farnesene | ||
| W273E | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | yes (I515VEVTY*IHNLDGYTHPEK532) | β-farnesene | ||
| W273F | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | yes (I515VEVTY*IHNLDGYTHPEK532) | β-farnesene | ||
| V277L | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | N/D | 5-epi-aristolochene | ||
| Y404C | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | yes (I515VEVTY*IHNLDGYTHPEK532) | unknown | ||
| Y404F | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | yes (I515VEVTY*IHNLDGYTHPEK532) | 5-epi-aristolochene | ||
| L407I | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | yes (I515VEVTY*IHNLDGYTHPEK532) | 5-epi-aristolochene | ||
| L407P | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | N/D | 5-epi-aristolochene | ||
| L512I | 25 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | N/D | 5-epi-aristolochene | ||
| wt | 37 | (E,E)-FPP | N/D | N/D | 5-epi-aristolochene | ||
| wt | 42 | (E,E)-FPP | yes (V442ID*DTATYEVEK453) | N/D | 5-epi-aristolochene | ||
| wt | 25 | (Z,E)-FPP | N/D | yes (I515VEVTY*IHNLDGYTHPEK532) | 5-epi-prezizaene | ||
| wt | 25 | SPP | yes (V442ID*DTATYEVEK453) | yes (I515VEVTY*IHNLDGYTHPEK532) | sesquilavandulene | ||
| premnaspirodiene synthase (Hyoscyamus muticus) | dominant-product | wt | 25 | (E,E)-FPP | N/D | N/D | premnaspirodiene |
| W280E | 25 | (E,E)-FPP | yes (V449VD*DIATYEVEK460) | yes (I522IDVTY*K528) | premnaspirodiene | ||
| wt | 25 | SPP | yes (V449VD*DIATYEVEK460) | yes (I522IDVTY*K528) | sesquilavandulene | ||
| wt | 25 | (Z,E)-FPP | yes (V449VD*DIATYEVEK460) | yes (I522IDVTY*K528) | unknown | ||
| valencene synthase (Citrus sinensis) | dominant-product | wt | 25 | (E,E)-FPP | N/D | N/D | valencene |
| W273E | 25 | (E,E)-FPP | N/D | yes (A517IDFIY*KEDDGYTHSYLIK535) | unknown | ||
| wt | 25 | SPP | N/D | N/D | unknown | ||
| aristolochene synthase (Aspergillus terreus) | dominant-product | wt | 25 | (E,E)-FPP | N/D | N/D | aristolochene |
| Y61C | 25 | (E,E)-FPP | yes (H214LSVVN*DIYSYEK226) | N/D | unknown | ||
| patchoulol synthase (Pogostemon cablin) | multiproduct | wt | 25 | (E,E)-FPP | N/D | N/D | patchoulol |
| W276E | 25 | (E,E)-FPP | N/D | N/D | unknown | ||
| wt | 25 | SPP | yes (L447VN*DITGHEFEK458) | N/D | sesquilavandulene | ||
| santalene synthase (Santalum spicatum) | multiproduct | wt | 25 | (E,E)-FPP | N/D | N/D | α-santalene |
| W293E | 25 | (E,E)-FPP | N/D | N/D | β-bergamotene | ||
| wt | 25 | SPP | N/D | N/D | unknown | ||
| γ-humulene synthase (Abies grandis) | multiproduct | wt | 25 | (E,E)-FPP | N/D | N/D | γ-humulene |
| W315P | 25 | (E,E)-FPP | N/D | N/D | β-farnesene | ||
| wt | 25 | SPP | N/D | N/D | unknown |
For detailed MS and MS/MS analyses, see Figures S2, S3, S8, S12, and S14. For GC–MS analyses, see Figures S4, S9, and S13. Product specificities describe wild-type enzyme specificities. Abbreviations: N/D, not detected; wt, wild type.
Figure 2.

Self-alkylation analysis of TEAS active site mutant W273E with (E,E)-farnesyl diphosphate. (A) LC–MS analysis of a tryptic digest of TEAS W273E after reaction with (E,E)-farnesyl diphosphate. Two tryptic peptides encompassing active site residues, V[442–453]K and I[515–532]K, showed an alkylation mass shift (red chromatograms). (B) MS analysis of alkylated and nonalkylated tryptic peptides V[442–453]K and I[515–532]K. For detailed MS/MS analysis, see Figure S2. (C) X-ray crystallographic analysis of alkylated TEAS W273E reveals a C1-farnesyl-γO-aspartate ester at Asp444. (D) X-ray crystallographic analysis of nonalkylated TEAS W273E. The electron density from unbiased (i.e., simulated-annealing omit) sigma-A-weighted 2Fo – Fc maps is shown at contour level 0.8σ with refined active site models in panels C and D. (E) Characterization of alkylation site Asp444 by EThcD-MS/MS analysis of alkylated tryptic peptide V[442–453]K. See also Figure S5. Abbreviations: BPC, base peak chromatogram; m(obs), observed mass; m(calc), calculated mass; ESI, electrospray ionization; Δm, mass error; EThcD, electron transfer and high-energy collision dissociation; FTMS, Fourier transform ion cyclotron resonance mass spectrometry.
In general, self-alkylation occurred most readily for active site mutants involving cation-stabilizing aromatic residues (Trp273 and Tyr404) (Figure 2A and Figures S2 and S3). The most predominant self-alkylation occurred in the TEAS W273E mutant on the V[442–453]K tryptic peptide (Figure 2A). This mutant did not produce any 5-epi-aristolochene but, instead, produced β-farnesene and farnesol as the major products (Figure S4). TEAS mutants with more subtle active site changes, including V277L, Y404F, L407I, and L512I, showed alkylation on the V[442–453]K peptide, as well (Table 1 and Figure S3). Unlike the W273E mutant, these mutants still produced some 5-epi-aristolochene during the time course of alkylation and inactivation (Figure S4). These results demonstrate that active site mutations in a relatively product-specific sesquiterpene synthase such as TEAS elicit self-alkylation of a catalytically essential and highly conserved TPS motif. Unexpectedly, alkylation did not occur on the nucleophilic mutations originally designed to trap specific carbocations.
Quantitative Self-Alkylation of TEAS W273E Yields a Farnesyl Ester of a Mg2+-Coordinating Aspartate
We used protein X-ray crystallography to characterize both the site of modification and the chemical structure of the alkylated residue in TEAS. We focused on the W273E mutant of TEAS, which yielded the highest level of farnesylation [∼50% for tryptic peptide V[442–453]K (see Figure 2A)]. For comparison, we obtained a crystal structure of nonalkylated TEAS W273E refined to 2.3 Å resolution (PDB-entry: 5DHI, Table S1). This “apo” TEAS crystal structure revealed several pertinent chemical features, including the extended conformation of the Glu273 side chain and a Mg2+ ion coordinated to the “DTE” motif, Asp444, Thr448, and Glu452 (Figure 2D). In marked contrast, the crystal structure of alkylated TEAS W273E refined to 2.43 Å resolution (PDB-entry: 5DHK, Table S1) unmistakably revealed a bent conformation of Glu273, the absence of Mg2+ coordinated to the “DTE” motif, and, most notably, an additional elongated stretch of electron density contiguous with Asp444. This latter electron density is most consistent with a linear farnesyl group covalently bound as an ester (Figure 2C). The covalent linkage refined and displayed unbiased electron density most reliably as a C1–farnesyl ester bond linked to the carboxyl group of the Asp444 side chain in phenix.refine.27
This site of alkylation, Asp444, within tryptic peptide V[442–453]K, was further verified by electron transfer and higher-energy collision dissociation tandem mass spectrometry (EThcD-MS/MS) (Figure 2E and Figure S5).28 Alkylation within tryptic peptide I[515–532]K was not detected by EThcD-MS/MS. However, the W280E mutant (equivalent to TEAS W273E) of the orthologous TEAS-like TPS, premnaspirodiene synthase from Hyoscyamus muticus (HPS),29 yields an alkylated tryptic peptide I[522–528]K consistent with the alkylation of the tyrosine corresponding to TEAS-Tyr520 (Figure S6). Thus, on the basis of the high degree of sequence and active site homology of these two sesquiterpene synthases,12,13 TEAS-Tyr520 and HPS-Tyr527 most likely represent additional alkylation sites in TEAS and HPS, respectively, via a tyrosine farnesyl ether. In addition, farnesylation of tyrosine via an electrophilic aromatic substitution is possible. However, the tyrosine–farnesyl linkage appears to be labile in the MS gas phase. There is a significant mass loss attributed to the farnesyl moiety in the MS spectrum (Figure 2B, spectrum 4). This experimental observation is most consistent with the presence of a farnesyl–tyrosine ether linkage rather than a farnesyl–tyrosine carbon–carbon bond.
Self-Alkylation of a Natural Mutation in a TEAS Homologue Revealed by Genome Mining
Gene duplication in plants often leads to the evolution of new chemotypes particularly in polyploid plants.30 To assess whether self-alkylation-causing mutations can naturally occur in sesquiterpene synthases, we searched the recently published genome of tobacco strain Nicotiana tabacum Basma Xanthi31 for TEAS homologues possessing active site changes (Figure S7). Among six homologous TEAS genes identified, including three genes with conserved active site residues and a putative premnaspirodiene synthase (TEAS M9 mutant),12,13 we uncovered one highly homologous TEAS pseudogene with a L407P mutation, a truncated N-terminus, and two internal stop codons at positions 376 and 520. Because of the N-terminal truncation and the two additional stop codons, the gene represents a loss-of-function TEAS homologue, but nonetheless, this sequence reveals a mutation at an active site position we showed to cause self-alkylation upon even subtle structural substitutions (Figure S3). The corresponding TEAS L407P mutant, when expressed as a full length protein, showed pronounced self-alkylation at Asp444 (Figure S7). This loss-of-function TEAS gene homologue with a self-alkylation-promoting mutation identified in the N. tabacum genome suggests that self-alkylation can occur in the evolution of plant sesquiterpene synthases and represents a significant constraint during the natural evolution of TPSs.
Carbocation-Mediated Alkylation Results in Mechanism-Based Enzyme Inhibition
To investigate the catalytic consequences of inactivation of TEAS mutants by alkylation at a Mg2+-binding site, the sesquiterpene products of TEAS W273E and TEAS Y404F were characterized after incubation with FPP. TEAS W273E was chosen because of its near stoichiometric level of C15 alkylation (Figure 2A), and TEAS Y404F was selected because it still produces the wild-type product 5-epi-aristolochene (Figure S4). After incubation (consistent with the physiological time course of TPS activity in planta) with FPP for 12 h and protein purification, TEAS W273E showed almost no sesquiterpene production compared to the positive control (Table 2). Similarly, TEAS Y404F showed considerably reduced 5-epi-aristolochene production. The reduction of product formation over a 12 h incubation with FPP was ∼99% for TEAS W273E (β-farnesene as the main product detected) and 7–21% for TEAS Y404F (5-epi-aristolochene as the main product detected) (Table 2).
Table 2. GC–MS Analyses of Product Yields of TEAS Reactions with (E,E)-FPP Depending on the Alkylation Statea.
| reaction time |
|||
|---|---|---|---|
| protein | 0–12 h | 12–24 h | product yield (%)b (main product) |
| TEAS W273E | no FPP | FPP | 100 (β-farnesene) |
| FPP | FPP | 1 ± 0.3 (β-farnesene) | |
| FPP | no FPP | N/D | |
| TEAS Y404F | no FPP | FPP | 100 (5EA) |
| FPP | FPP | 86 ± 7 (5EA) | |
| FPP | no FPP | N/D | |
| TEAS wild type | no FPP | FPP | 100 (5EA) |
| FPP | FPP | 105 ± 11 (5EA) | |
| FPP | no FPP | N/D | |
Analyzed TEAS constructs were TEAS W273E, TEAS Y404F, and wild-type TEAS. Abbreviations: SD, standard deviation; 5EA, 5-epi-aristolochene; N/D, not detected.
Means ± the standard deviation, reactions in triplicate (2.5 μM enzyme and 250 μM FPP).
To test whether substrate-mediated alkylation of TEAS was reversible, TEAS W273E and TEAS Y404F were incubated for 12 h with FPP, buffer exchanged to remove FPP and reaction products, and incubated for an additional 12 h without FPP. The corresponding hexane extracts of TEAS W273E and TEAS Y404F incubated with FPP substrate showed no β-farnesene, farnesol, or other putative hydrolyzed alkylation products, indicating that the alkylation modification is stable and resistant to recovery of catalytic activity. Additional tests with wild-type TEAS demonstrated that the reduction in major product production over time was not a result of enzyme degradation (Table 2). The estimated alkylation levels of TEAS W273E (∼50%) and TEAS Y404F (∼5%) after FPP reaction for 12 h (Figure 2A and Figure S3) are not sufficiently high to account for the corresponding product yield reductions. This discrepancy is likely due to both loss of alkylated peptides during proteomic analysis and protein aggregation precipitated by alkylated TEAS mutants. Although inactivation rates seem low at first blush (Table S3), they result in a significant loss of product yields as shown in Table 2 and, in the context of multiple mutations, likely influence TPS active site evolution during diversification and natural selection.
Increased Temperatures and Introduction of Substrate Analogues Lead to Alkylation in Wild-Type TEAS
Mechanism-based inhibition has been characterized in TPSs with unnatural substrate analogues such as vinyl-farnesyl diphosphate.26 To show that natural substrate analogues can also cause self-alkylation in TPSs, wild-type TEAS was incubated for 12 h with sesquilavandulyl diphosphate (SPP), an irregular sesquiterpene substrate employed during secondary metabolism of plants and marine microbes (Figure 3A).32−34 Proteomic LC–MS analysis of wild-type TEAS after SPP incubation detected alkylation on tryptic peptides V[442–453]K and I[515–532]K (Table 1, Figure 3B, and Figure S8). The GC–MS product profile of wild-type TEAS with SPP substrate showed sesquilavandulene as the main product and sesquilavandulol and cyclized sesquilavandulenes as minor products based on GC–MS spectra (Figure S9). The natural substrate (Z,E)-FPP (Figure 3A), with altered double bond stereochemistry, resulted in alkylation of Tyr520 (Table 1 and Figure S8). The characterized alkylation of wild-type TEAS with SPP and (Z,E)-FPP shows that natural substrates of TPSs can also induce alkylation in sesquiterpene synthases.
Figure 3.

Self-alkylation analysis of wild-type TEAS with (E,E)-farnesyl diphosphate analogues at different reaction temperatures. (A) Sesquiterpene substrates tested in self-alkylation studies (Table 1). (B) Self-alkylation analysis of wild-type TEAS with sesquilavandulyl diphosphate (SPP). (C) Self-alkylation analysis of wild-type TEAS with (E,E)-FPP at 25 and 42 °C. For detailed MS and MS/MS analysis, see Figure S8.
Because plants experience wide fluctuations in temperature during day–night cycles, we tested whether increased reaction temperatures can cause alkylation of wild-type TEAS during substrate turnover. Alkylation levels were measured using the conventional substrate (E,E)-FPP at 25, 37, and 42 °C. Wild-type TEAS produced 5-epi-aristolochene as a main product at all reaction temperatures (Figure S9). Alkylation was not detected on Asp444 or on Tyr520 of TEAS at 25 or 37 °C. However, at 42 °C, alkylation was readily detected on Asp444 (V[442–453]K) (Table 1, Figure 3C, and Figure S8), demonstrating that self-alkylation can also be promoted by increased reaction temperatures.
Alkylation Analysis of Catalytically Specific and Catalytically Promiscuous Sesquiterpene Synthases
The alkylation resulting from mutational replacement of active site residues or substitution of structurally diverse substrates was investigated in other sesquiterpene synthases possessing varying levels of product complexity. Three dominant-product sesquiterpene synthases were employed: H. muticus premnaspirodiene synthase (HPS),28 citrus valencene synthase (CVS),35 and Aspergillus terreus aristolochene synthase (ATAS).36 In addition, three multiproduct sesquiterpene synthases were tested: Santalum spicatum santalene synthase (SspiSSy),13Abies grandis γ-humulene synthase (γHS),24 and Pogostemon cablin patchoulol synthase (PAS).9 Alkylations by active site changes were tested in all TPSs by mutations of tryptophan or tyrosine residues structurally homologous to the active site Trp273 in TEAS to glutamate or cysteine residues (Figure 1A and Figures S10 and S11). Mutation of this site in TEAS resulted in appreciable substrate-dependent alkylation. Alkylations by natural (E,E)-FPP analogues were tested in selected TPSs by incubation with SPP or (Z,E)-FPP (Table 1).
All dominant-product sesquiterpene synthases showed self-alkylation with their conventional substrate FPP after active site mutations (Table 1 and Figures S12 and S13). Incubation with nonconventional substrates resulted in alkylation in wild-type HPS but not in CVS (Table 1 and Figure S12). Multiproduct sesquiterpene synthases showed little to no alkylation with FPP upon active site mutations, and incubation with nonconventional FPP substrates resulted in alkylation in only wild-type PAS (Table 1 and Figures S13 and S14). Our alkylation analyses of specific and promiscuous sesquiterpene synthases suggest that dominant-product synthases are more prone to self-alkylation upon active site mutations. In addition, the detected alkylation of PAS after SPP incubation showed that alkylation can also occur in multiproduct sesquiterpene synthases with nonconventional substrates, although to a lesser degree than observed in dominant-product synthases. Finally, the alkylation of ATAS Y61C after (E,E)-FPP turnover, and the alkylation of wild-type PAS with SPP, showed that sesquiterpene synthases with an asparagine instead of an aspartate in the “NSE/DTE” motif for Mg2+ binding can also undergo alkylation, implying alkyl asparagine amides as putative alkylation products.
Discussion
In this study, the occurrences, locations, chemistries, and inhibitory consequences of mechanism-based alkylations of specific and promiscuous sesquiterpene synthases were investigated. Alkylation assays relied on bottom-up proteomic LC–MS analyses of sesquiterpene synthases by monitoring the mass shifts of covalently modified enzyme side chains on tryptic peptides by C15 carbocations (C15H24+; Δm = 204.188 Da). Initial complications associated with the detection of alkylated peptides with protected proteolytic cleavage sites, e.g., CVS, γHS, and PAS, and with cleavage sites residing in the “NSE/DTE” motif or on the active site lid as seen in γHS (Figure S13) were addressed using contrasting solid phases for LC–MS separation and analyses. Accurate quantification of substrate-mediated TPS alkylation in this study by LC–MS was not possible because of differences in tryptic cleavage efficiencies between nonalkylated and alkylated TPSs, loss of alkylated peptides during sample preparation and separation, and variation in ionization efficiencies between nonalkylated and alkylated peptides. However, our LC–MS method is a relatively facile starting point for estimating the extent of substrate-mediated alkylation levels in more complex samples such as those found in elicited plant proteomes during the course of terpene biosynthesis. Ongoing studies are aimed at quantitation of alkylation using 14C-labeled substrates37 and quantitative proteomics.38 Nevertheless, ETD-MS/MS yielded a low-abundance z-ion series that confirmed the Asp444 alkylation site (Figure S5).
The mechanism-based alkylations of sesquiterpene synthases at Mg2+-binding “NSE/DTE” motifs and along active site catalytic loops result from early linear farnesyl cation captures instead of alkylation by cyclic carbocations formed during latter parts of the cation-based cascade mechanisms. For example, TEAS W273E still binds and ionizes FPP, but the active site mutation disfavors formation of the germacryl cation (Figure S1) because of the loss of the tryptophan−π-cation interaction. The result is reduced production of cyclic products in TEAS W273E compared to that in wild-type TEAS. Instead of intramolecular cyclization, the farnesyl cation can first undergo rapid proton loss to yield the main product β-farnesene. Second, the farnesyl cation can be quenched by water to yield farnesol as a minor product. Third, the farnesyl cation can be captured by attack of active site nucleophiles, including the side chains of Asp444 and Tyr520 buttressing the farnesyl–C1 cationic centers. The latter covalent modification of the TEAS active site leads to enzyme inhibition.
These results suggest that enzymes employing chemically reactive carbocation chemistry must balance the presence of nucleophilic residues used for substrate turnover with the unintended consequences of mechanism-based inactivation by covalent modification of the active site. Given a recent study of the evolutionary trajectory of sesquiterpene cyclization promoted by epistatic active site mutations,39 it will be interesting to assess the degree to which cyclization-inducing mutations favored by natural selection also limit self-alkylation in comparison to mutations suggested to favor more ancient noncyclizing activity.
Previous studies hypothesized that cationic intermediates of the catalytic cascade reactions of TPSs might be trapped in active sites by nucleophilic residues.25 Our investigation of self-alkylation in TEAS is, to the best of our knowledge, the first study to confirm this hypothesis and to identify two conserved catalytic motifs acutely sensitive to farnesyl cation-based covalent modifications. Moreover, multiple alkylated species were detected at two active site motifs. The J/K loop lid peptide, in particular, showed multiple species based on different retention times of alkylated peptides during LC-based separation (Figure 2A and Figure S2). In several TEAS point mutants undergoing alkylation, including V277L, Y404F, L407I, and L512I, 5-epi-aristolochene is formed, suggesting that structurally distinct carbocations may also undergo quenching by other nucleophiles positioned proximal to cationic centers, including Thr448 and Asp525. Ongoing studies will address the structural identity of captured isoprenoid intermediates by hydrolytic release of these trapped moieties from alkylated TPSs.
Substrate-mediated alkylations were readily detected in active site mutants of dominant-product TPSs but were rarely detected in multiproduct TPSs. Multiproduct TPSs may direct mechanisms possessing more conformational freedom for initially formed farnesyl cations. In turn, these dynamic TPS scaffolds provide more alternative pathways during the cationic rearrangements leading to additional sesquiterpene products.9,24 This level of catalytic promiscuity in cationic reaction pathways mediated by the active site of multiproduct TPSs may have evolved, in some instances, not only to produce a mixture of advantageous products, but to also avoid covalent cation capture. Avoidances of premature enzyme inhibitions during the transformations of isoprenoid diphosphates in TPSs likely serves as additional hitherto unappreciated constraints during the evolution of TPS-based natural product biosyntheses. The large structural diversity of cyclic scaffolds found in sesquiterpene natural products is principally determined by the stereo- and regiochemistry of cationic transformations catalyzed by class I TPSs.40 To facilitate specific cationic transformations leading to bioactive terpene structures, TPSs must evolve to promote cationic reaction cascades while avoiding the premature enzyme-based capture of these cations and ensuing inactivation. These hypotheses are supported by the observations that none of the investigated wild-type TPSs showed significant levels of self-alkylation with the conventional substrate (E,E)-FPP at room temperature (Table 1, Figure 3C, and Figures S11 and S13). During the evolution of terpene biosynthesis, enzyme inhibition by self-alkylation may be limited by maintaining large active site volumes, such as those found in multiproduct sesquiterpene synthases. In contrast, the evolution of increased product specificity often results in smaller and more specialized active sites that appear to be more susceptible to substrate-based alkylation upon subtle mutational changes. In short, alkylations may serve as constraints during the evolution of other enzymes employing cationic mechanisms, Mg2+ ions and carboxyl-bearing side chains for isoprenoid diphosphate ionization.11 Finally, mechanism-based self-alkylation may underlie metabolic diseases associated with mutations in pervasive terpene biosynthetic genes found across all three domains of life.41
The propensity for cation-based alkylation of TPSs is also an important consideration for the metabolic engineering of high-value terpene production in heterologous hosts. Metabolic engineering of host organisms for biotechnological production of sesquiterpenes has advanced considerably.42 However, the optimization of TPSs or engineering of new TPS activities has failed to account for the possibility of mechanism-based TPS inactivation through active site alkylation.43,44 Understanding the correlations between the extent and regiochemistry of alkylations of TPSs with mutations and substrate alterations should facilitate TPS optimizations through rapid alkylation analyses of mutant libraries. Future studies will focus on in vivo characterization of self-alkylation of sesquiterpene synthases in plant cells and expression hosts used commercially, and additional enzymes exposed to reactive carbocations that biosynthesize terpene natural products.
Methods
Chemicals and Primers
Chemicals were obtained from Sigma-Aldrich, if not noted otherwise. LC–MS solvents were Honeywell LabReady blends for LC–MS application. All DNA primers were obtained from IDT DNA Technologies. (E,E)-FPP and (Z,E)-FPP were obtained from Isoprenoids, LLC. SPP was obtained by synthesis as described previously45 and provided by the Moore lab (University of California at San Diego, La Jolla, CA).
Sesquiterpene Synthase Constructs
TPS-encoding genes (TEAS, TOBSQPC; HPS, XXU20187; CVS, GQ988384; SspiSSy, HQ343278; γHS, U92267; PAS, AY508730) were expressed and mutated from pET28a constructs harboring the corresponding genes with an N-terminal His tag (nine-His tag, PAS; eight-His tag, TEAS and HPS; six-His tag, SspiSSy, γHS, and CVS) and N-terminal thrombin cleavage site before the start codon of the terpene synthase gene. Terpene synthase mutants were constructed by site-directed mutagenesis based on the QuikChange site-directed mutagenesis kit (Agilent) using designed mutagenesis primers (Table S2).
Protein Expression and Purification
See the text of the Supporting Information.
Sesquiterpene Synthase Assays and Product Profiling by GC–MS
Sesquiterpene synthases (2.5 μM) were incubated with substrates [250 μM, (E,E)-FPP, (Z,E)-FPP, or SPP] in reaction buffer [50 mM Bis-Tris propane and 15 mM MgCl2 (pH 7.5)] for 12 h at 25 °C. The reaction mixtures were subsequently extracted with hexane by being vortexed for 30 s and the hexane layers were analyzed by GC–MS as described previously.45 For investigation of reaction temperature effects, wild-type TEAS was also assayed at 37 and 42 °C.
For inhibition analysis, wild-type TEAS, TEAS W273E, or TEAS Y404F (2.5 μM) was incubated with (E,E)-FPP (250 μM) in reaction buffer for 12 h at 25 °C (reaction time of 0–12 h). The reaction mixtures were then concentrated using 30 kDa cutoff protein concentrators (Millipore) and washed three times with reaction buffer. Subsequently, buffer-exchanged wild-type TEAS, TEAS W273E, or TEAS Y404F (2.5 μM) was incubated again with (E,E)-FPP (250 μM) in reaction buffer for 12 h at 25 °C (reaction time of 12–24 h). The reaction mixtures were then extracted with hexane and analyzed by GC–MS as described previously.46 For comparison of enzyme inhibition by the first reaction period, wild-type TEAS, TEAS W273E, or TEAS Y404F (2.5 μM) was incubated without substrate for 12 h at 25 °C (reaction time of 0–12 h), buffer exchanged as described above, and then incubated with (E,E)-FPP (250 μM) in reaction buffer for 12 h at 25 °C (reaction time of 12–24 h, 2.5 μM enzyme). The reaction mixtures were then extracted with hexane and analyzed by GC–MS as described previously.46 For analysis of putative hydrolysis of enzyme alkylation modifications, wild-type TEAS, TEAS W273E, or TEAS Y404F was incubated with (E,E)-FPP for 12 h at 25 °C (reaction time of 0–12 h), buffer exchanged as described above, and then incubated without substrate in reaction buffer for 12 h at 25 °C (reaction time of 12–24 h, 2.5 μM). The reaction mixtures were then extracted with hexane and analyzed by GC–MS as described previously.46 Assays for inhibition analyses were conducted in triplicate with 4-chlorotoluene (Sigma-Aldrich) as an internal standard. Sesquiterpene products were quantified by autointegration of product peaks in GC–MS total ion chromatograms with Agilent Chemstation software (G1701DA). Kinetic analyses of TEAS W273E and TEAS Y404F were conducted as described previously.46
Alkylation Analysis of Sesquiterpene Synthases by Bottom-Up Proteomics
Sesquiterpene synthases (2.5 μM) were incubated with substrate [250 μM, (E,E)-FPP, (Z,E)-FPP, or SPP] in reaction buffer [50 mM Bis-Tris propane and 15 mM MgCl2 (pH 7.5)] for 12 h at 25 °C. For investigation of reaction temperature effects, wild-type TEAS was also assayed at 37 and 42 °C. The reaction mixtures were adjusted to pH 8 with 1 M Tris-HCl (pH 8), with 1 μg of trypsin (Trypsin singlets, Sigma-Aldrich) added, and incubated for 2–6 h at 25 °C. The tryptic digests were analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS) on a PROTO300 column (C4, 5 μm, 250 mm × 4.6 mm, Higgins Analytical, Inc.) connected to an Agilent 6530 Accurate-Mass Q-TOF LC/MS instrument in positive ion mode. The LC gradient of solvent A (acetonitrile and 0.1% formic acid) and solvent B (0.1% formic acid) was as follows: 10% A (0 min), 10% A (10 min), 30% A (15 min), 70% A (55 min), 100% A (60 min), 100% A (63 min), 10% A (64 min), and 10% A (70 min) at a flow rate of 0.7 mL min–1. MS parameters were as follows: time segment 1, 0–10 min (LC flow to waste); time segment 2, 10–70 min (LC flow to MS). Source parameters were as follows: gas temperature, 350 °C; gas flow, 8 L min–1; nebulizer, 35 psig; positive ion polarity. Scan source parameters were as follows: VCap, 3500 V; Fragmentor, 175 V; Skimmer1, 65 V; OctopoleRFPeak, 750 V; Acquisition mode AutoMS2, MS range of m/z 400–1700, MS scan range of 2 spectra/s, MS/MS scan range of 3 spectra/s, MS/MS isolation width of m/z 4, collision energy 30 eV, Precursor Selection: Static Exclusion Range 100–500 m/z, MS Abs. threshold 200, MS Rel. threshold 0.01%, MS/MS Abs. threshold 500, MS/MS Rel. threshold 0.01%. Q-TOF MS data were analyzed with Agilent MassHunter software (B.05.00).
Alkylation Analysis of Sesquiterpene Synthases by EThcD-MS/MS
Samples were precipitated with methanol and chloroform. Dried pellets were dissolved in an 8 M urea/100 mM Tris-HCl (pH 8.5) mixture. Proteins were reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (Sigma-Aldrich) and alkylated with 10 mM iodoacetamide (Sigma-Aldrich). Proteins were digested overnight at 37 °C in a 2 M urea/100 mM Tris-HCl (pH 8.5) mixture with trypsin (Promega). Digests were quenched with formic acid, at a final concentration of 5%. The digested samples were analyzed on a Fusion Orbitrap tribrid mass spectrometer (Thermo). The digests were injected directly onto a 20 cm, 100 μm inside diameter column packed with 5 μm ODS-AQ C4, C8, or C18 resin (YMC). Samples were separated at a flow rate of 400 nL min–1 on an Easy nLC2 instrument (Thermo). Buffers A and B were 0.1% formic acid in water and acetonitrile, respectively. A gradient of 1 to 10% B over 10 min, an increase to 45% B over 70 min, an increase to 100% B over an additional 50 min, and a hold at 90% B for a final 10 min of washing was used before returning to 1% B. The column was re-equilibrated with 10 μL of buffer A prior to the injection of samples. Peptides were eluted directly from the tip of the column and nanosprayed directly into the mass spectrometer by application of a voltage of 2.5 kV at the back of the column. The Orbitrap Fusion was operated in a data-dependent mode. Full MS1 scans were collected in the Orbitrap at 120K resolution with a mass range of m/z 400–1600 and an AGC target of 5e5. The cycle times were set to 5 s, and within this 5 s interval, the most abundant ions per scan were selected for either CID or EThcD and detected in the orbitrap with a resolution of 15K with an AGC target of 5e5 and a minimal intensity of 5000. Maximal fill times were set to 250 and 100 ms for MS and MS/MS scans, respectively. Quadrupole isolation at m/z 3 was used; monoisotopic precursor selection was disabled, and dynamic exclusion was used with an exclusion duration of 30 s. Orbitrap MS data were analyzed with QualBrowser software from the Thermo Xcalibur package (version 2.1).
Genome Mining of N. tabacum for Tobacco 5-epi-Aristolochene Synthase Homologues
The genome of N. tabacum str. Basma Xanthi (GenBank entry AWOK01000000)31 was searched for gene homologues of tobacco 5-epi-aristolochene synthase (GenBank entry TOBSQPC)19 with blastn. Highest blastn hits (>80% identity) were translated and analyzed for completeness of the catalytic C-terminal domain sequence and for active site residues and M9 residues.12,13
Protein Crystallization
For preparation of farnesylated protein for crystallization trials, TEAS W273E (2.5 μM) was incubated with FPP (250 μM) for 18 h at 25 °C in reaction buffer [50 mM Bis-Tris propane and 15 mM MgCl2 (pH 7.5)], and the treated protein was then transferred to a 30 kDa protein concentrator (Millipore), washed three times with protein buffer [50 mM Tris-HCl, 100 mM NaCl, and 2 mM DTT (pH 8)], and finally brought to a concentration suitable for crystallization. Alkylated and untreated TEAS W273E samples were crystallized by the hanging-drop vapor-diffusion method with drops containing a 1:1 ratio of a protein solution (10–12 mg mL–1) and a crystallization solution [0.1 M MOPSO (pH 7), 100 mM MgOAc, and 14% (w/v) PEG 8000]. Tetragonal crystals were observed within 1 day. For X-ray data collection, crystals were flash-frozen in liquid nitrogen in a crystallization solution containing 20% (v/v) glycerol.
X-ray Crystallographic Analyses of TEAS Active Site Mutant W273E
X-ray diffraction data were obtained from frozen crystals at beamline 8.2.1 of the Advanced Light Source, Lawrence Berkeley National Laboratory (Berkeley, CA). Diffraction images were collected on an ADSC Quantum 315R CCD detector and indexed and integrated with iMOSFLM,47 and measured diffraction intensities were scaled with AIMLESS48 from the CCP4 suite.49 For the crystallographic analyses of TEAS W273E and farnesylated TEAS W273E, the structure of isomorphously crystallized, wild-type TEAS (with residue 273 truncated to CB) provided a template for the initial atomic model, which was further refined (phenix.refine) in Phenix.27 Coot50 was used for graphical map inspection and manual rebuilding of atomic models. C1-Farnesyl-γO-aspartate was created in JLigand51 from the CCP4 suite.49 For the generation of unbiased electron density maps of alkylated and nonalkylated TEAS W273E, the side chain atoms of all active site residues (E273, I294, D301, D305, T401, Y404, L407, D444, T448, E452, L512, Y520, D525, and Y527), Mg2+ cofactors, active site waters, and additional ligand atoms (if present) were omitted from the final refined models and a simulated annealing (Cartesian) refinement was conducted with phenix.refine.27 For graphical display in PyMOL,52 electron density maps covering the entire asymmetric unit were calculated with unbiased map coefficients in Phenix.27
Acknowledgments
We thank G. Louie for training in X-ray crystallography and H.-J. Koo for GC–MS training and providing a TEAS model for initial molecular refinement. We thank B. Moore (University of California at San Diego) for mass spectrometry instrument access, S. Diethelm (University of California at San Diego) for providing synthetic SPP, and J. Bohlmann and J. Chappell for discussions and TPS genes. J.P.N. is an Investigator of the Howard Hughes Medical Institute. R.D.K. is a Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation. This research was funded by the National Science Foundation (EEC-0813570 to J.P.N.) and the National Institute of General Medical Sciences (8 P41 GM103533 to J.R.Y.III). The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00539.
Supporting methods, X-ray crystallographic data collection and refinement statistics, mutagenesis primer sequences, kinetic data and estimation of alkylation rates of TEAS mutants, proteomic alkylation analyses of TPSs, GC–MS product profiling of TPSs, genome mining of N. tabacum Basma Xanthi for TEAS gene homologues, and sequence alignment of analyzed TPSs (PDF)
Structure factor and coordinate files of the reported structures are available from the Protein Data Bank (5DHI and 5DHK).
The authors declare no competing financial interest.
Supplementary Material
References
- Jiang J.; He X.; Cane D. E. (2007) Biosynthesis of the earthy odorant geosmin by a bifunctional Streptomyces coelicolor enzyme. Nat. Chem. Biol. 3, 711–715 10.1038/nchembio.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicke M.; Baldwin I. T. (2010) The evolutionary context for herbivore-induced plant volatiles: beyond the ‘cry for help’. Trends Plant Sci. 15, 167–175 10.1016/j.tplants.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Weng J. K.; Philippe R. N.; Noel J. P. (2012) The rise of chemodiversity in plants. Science 336, 1667–1670 10.1126/science.1217411. [DOI] [PubMed] [Google Scholar]
- Gershenzon J.; Dudareva N. (2007) The function of terpene natural products in the natural world. Nat. Chem. Biol. 3, 408–414 10.1038/nchembio.2007.5. [DOI] [PubMed] [Google Scholar]
- Mellon S. H.; Griffin L. D.; Compagnone N. A. (2001) Biosynthesis and action of neurosteroids. Brain Res. Rev. 37, 3–12 10.1016/S0165-0173(01)00109-6. [DOI] [PubMed] [Google Scholar]
- Dufourc E.J. J. (2008) Sterols and membrane dynamics. Chem. Biol. 1, 63–77 10.1007/s12154-008-0010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitmaier E. (2006) Terpenes: Flavors, Fragrances, Pharmaca, Pheromones, Wiley-VCH, Weinheim, Germany. [Google Scholar]
- Eckstein-Ludwig U.; Webb R. J.; Van Goethem I. D. A.; East J. M.; Lee A. G.; Kimura M.; O’Neill P. M.; Bray P. G.; Ward S. A.; Krishna S. (2003) Artemisinins target the SERCA of Plasmodium falciparum. Nature 424, 957–961 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- Jones C. G.; Moniodis J.; Zulak K. G.; Scaffidi A.; Plum-mer J. A.; Ghisalberti E. L.; Barbour E. L.; Bohlmann J. (2011) Sandalwood fragrance biosynthesis involves sesquiterpene synthases of both the terpene synthase (TPS)-a and TPS-b subfamilies, including santalene synthases. J. Biol. Chem. 286, 17445–17454 10.1074/jbc.M111.231787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munck S.; Croteau R. (1990) Purification and characterization of the sesquiterpene cyclase patchoulol synthase from Pogostemon cablin. Arch. Biochem. Biophys. 282, 58–64 10.1016/0003-9861(90)90086-E. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Honzatko R. B.; Peters R. J. (2012) Terpenoid synthase structures: a so far incomplete view of complex catalysis. Nat. Prod. Rep. 29, 1153–1175 10.1039/c2np20059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhagen B. T.; O’Maille P. E.; Noel J. P.; Chappell J. (2006) Identifying and manipulating structural determinates linking catalytic specificities in terpene synthases. Proc. Natl. Acad. Sci. U. S. A. 103, 9826–9831 10.1073/pnas.0601605103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Maille P. E.; Malone A.; Dellas N.; Andes Hess B.; Smentek L.; Sheehan I.; Greenhagen B. T.; Chappell J.; Manning G.; Noel J. P. (2008) Quantitative exploration of the catalytic landscape separating divergent plant sesquiterpene synthases. Nat. Chem. Biol. 4, 617–623 10.1038/nchembio.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantillo D. J. (2011) Biosynthesis via carbocations: Theoretical studies on terpene formation. Nat. Prod. Rep. 28, 1035–1053 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- Aaron J. A.; Christianson D. W. (2010) Trinuclear metal clusters in catalysis by terpenoid synthases. Pure Appl. Chem. 82, 1585–1597 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty D. A. (1996) Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 271, 163–168 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- Baer P.; Rabe P.; Fischer K.; Citron C. A.; Klapschinski T. A.; Groll M.; Dickschat J. S. (2014) Induced-Fit Mechanism in Class I Terpene Cyclases. Angew. Chem., Int. Ed. 53, 7652–7656 10.1002/anie.201403648. [DOI] [PubMed] [Google Scholar]
- Van der Kamp M. W.; Sirirak J.; Zurek J.; Allemann R. K.; Mulholland A. J. (2013) Conformational change and ligand binding in the aristolochene synthase catalytic cycle. Biochemistry 52, 8094–8105 10.1021/bi400898k. [DOI] [PubMed] [Google Scholar]
- Starks C. M.; Back K.; Chappell J.; Noel J. P. (1997) Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science 277, 1815–1820 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- Lesburg C. A.; Zhai G.; Cane D. E.; Christianson D. W. (1997) Crystal structure of pentalenene synthase: mechanistic insights on terpenoid cyclization reactions in biology. Science 277, 1820–1824 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- Walsh C. (1982) Suicide substrates: mechanism-based enzyme inactivators. Tetrahedron 38, 871–909 10.1016/0040-4020(82)85068-0. [DOI] [PubMed] [Google Scholar]
- Smith W. L.; Lands W. E. (1972) Oxygenation of polyunsaturated fatty acids during prostaglandin biosynthesis by sheep vesicular glands. Biochemistry 11, 3276–3285 10.1021/bi00767a024. [DOI] [PubMed] [Google Scholar]
- Smith W. L.; Urade Y.; Jakobsson P. J. (2011) Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 111, 5821–5865 10.1021/cr2002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikuni Y.; Ferrin T. E.; Keasling J. D. (2006) Designed divergent evolution of enzyme function. Nature 440, 1078–1082 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- Allemann R. K. (2008) Chemical wizardry? The generation of diversity in terpenoid biosynthesis. Pure Appl. Chem. 80, 1791–1798 10.1351/pac200880081791. [DOI] [Google Scholar]
- Cane D. E.; Bryant C. (1994) J. Am. Chem. Soc. 116, 12063–12064 10.1021/ja00105a061. [DOI] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Da-vis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. D66, 213–221 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. M.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J.; Mohammed S. (2012) Toward Full Peptide Sequence Coverage by Dual Fragmentation Combining Electron-Transfer and Higher-Energy Collision Dissociation Tandem Mass. Anal. Chem. 84, 9668–9673 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Back K.; Chappell J. (1995) Aristolochene synthase. Mechanism-based inhibition of a terpenoid cyclase. J. Biol. Chem. 270, 7375–7381. [DOI] [PubMed] [Google Scholar]
- Roulin A.; Auer P. L.; Libault M.; Schlueter J.; Farmer A.; May G.; Stacey G.; Doerge R. W.; Jackson S. A. (2013) The fate of duplicated genes in a polyploid plant genome. Plant J. 73, 143–153 10.1111/tpj.12026. [DOI] [PubMed] [Google Scholar]
- Sierro N.; Battey J. N.; Ouadi S.; Bakaher N.; Bovet L.; Willig A.; Goepfert S.; Peitsch M. C.; Ivanov N. V. (2014) The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 5, 3833. 10.1038/ncomms4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath G.; Potterat O.; Mavi S.; Hostettmann K. (1996) Xanthones from Hypericum roeperanum. Phytochemistry 43, 513–520 10.1016/0031-9422(96)00284-1. [DOI] [PubMed] [Google Scholar]
- Kaysser L.; Bernhardt P.; Nam S. J.; Loesgen S.; Ruby J. G.; Skewes-Cox P.; Jensen P. R.; Fenical W.; Moore B. S. (2012) Merochlorins A–D, cyclic meroterpenoid antibiotics biosynthesized in divergent pathways with vanadium-dependent chloroperoxidases. J. Am. Chem. Soc. 134, 11988–11991 10.1021/ja305665f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diethelm S.; Teufel R.; Kaysser L.; Moore B. S. (2014) A Multitasking Vanadium-Dependent Chloroperoxidase as an Inspiration for the Chemical Synthesis of the Merochlorins. Angew. Chem. 126, 11203–11206 10.1002/ange.201405696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon-Asa L.; Shalit M.; Frydman A.; Bar E.; Holland D.; Or E.; Lavi U.; Lewinsohn E.; Eyal Y. (2003) Citrus fruit flavor and aroma biosynthesis: isolation, functional characterization, and developmental regulation of Cstps1, a key gene in the production of the sesquiterpene aroma compound valencene. Plant J. 36, 664–674 10.1046/j.1365-313X.2003.01910.x. [DOI] [PubMed] [Google Scholar]
- Chen M.; Al-Lami N.; Janvier M.; D’Antonio E. L.; Faral-dos J. A.; Cane D. E.; Allemann R. K.; Christianson D. W. (2013) Mechanistic Insights from the Binding of Substrate and Carbocation Intermediate Analogues to Aristolochene Synthase. Biochemistry 52, 5441–5453 10.1021/bi400691v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong S. A.; Hannah V. C.; Goldstein J. L.; Brown M. S. (1995) CAAX geranylgeranyl transferase transfers farnesyl as efficiently as geranylgeranyl to RhoB. J. Biol. Chem. 270, 7864–7868 10.1074/jbc.270.14.7864. [DOI] [PubMed] [Google Scholar]
- Lewandowska D.; ten Have S.; Hodge K.; Tillemans V.; Lamond A. I.; Brown J. W. (2013) Plant SILAC: stable-isotope labelling with amino acids of Arabidopsis seedlings for quantitative proteomics. PLoS One 8, e72207. 10.1371/journal.pone.0072207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon M.; Laurendon C.; Vardakou M.; Cheema J.; Defernez M.; Green S.; Faraldos J. A.; O’Maille P. E. (2015) Emergence of terpene cyclization in Artemisia annua. Nat. Commun. 6, 6143. 10.1038/ncomms7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin M. B.; O’Maille P. E.; Noel J. P. (2008) Evolving biosynthetic tangos negotiate mechanistic landscapes. Nat. Chem. Biol. 4, 217–222 10.1038/nchembio0408-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinzii C.; Naini A.; Salviati L.; Trevisson E.; Navas P.; Dimauro S.; Hirano M. (2006) A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q 10 deficiency. Am. J. Hum. Genet. 78, 345–349 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez S.; Kirby J.; Denby C. M.; Keasling J. D. (2014) Production and quantification of sesquiterpenes in Saccharomyces cerevisiae, including extraction, detection and quantification of terpene products and key related metabolites. Nat. Protoc. 9, 1980–1996 10.1038/nprot.2014.132. [DOI] [PubMed] [Google Scholar]
- Lauchli R.; Rabe K. S.; Kalbarczyk K. Z.; Tata A.; Heel T.; Kitto R. Z.; Arnold F. H. (2013) High-Throughput Screening for Terpene-Synthase-Cyclization Activity and Directed Evolution of a Terpene Synthase. Angew. Chem. 125, 5681–5684 10.1002/ange.201301362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furubayashi M.; Ikezumi M.; Kajiwara J.; Iwasaki M.; Fujii A.; Li L.; Saito K.; Umeno D. A. (2014) High-Throughput Colorimetric Screening Assay for Terpene Synthase Activity Based on Substrate Consumption. PLoS One 9, e93317. 10.1371/journal.pone.0093317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel R.; Kaysser L.; Villaume M. T.; Diethelm S.; Carbullido M. K.; Baran P. S.; Moore B. S. (2014) One-Pot Enzymatic Synthesis of Merochlorin A and B. Angew. Chem., Int. Ed. 53, 11019–11022 10.1002/anie.201405694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Maille P. E.; Chappell J.; Noel J. P. (2004) A single-vial analytical and quantitative gas chromatography–mass spectrometry assay for terpene synthases. Anal. Biochem. 335, 210–217 10.1016/j.ab.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Battye T. G. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr. D67, 271–281 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. D62, 72–82 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. D67, 235–242 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. D66, 486–501 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev A. A.; Young P.; Isupov M. N.; Moroz O. V.; Vagin A. A.; Murshudov G. N. (2012) JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr., Sect. D: Biol. Crystallogr. D68, 431–440 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. (2002) PyMOL molecular graphics system, DeLano Scientific, San Carlos, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.