

Abstract
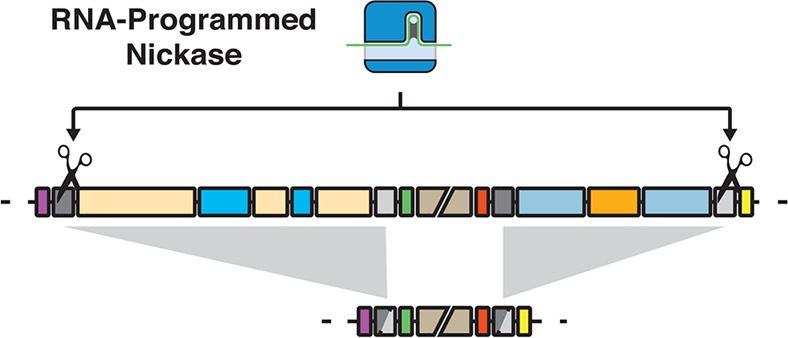
Programmable CRISPR-Cas systems have augmented our ability to produce precise genome manipulations. Here we demonstrate and characterize the ability of CRISPR-Cas derived nickases to direct targeted recombination of both small and large genomic regions flanked by repetitive elements in Escherichia coli. While CRISPR directed double-stranded DNA breaks are highly lethal in many bacteria, we show that CRISPR-guided nickase systems can be programmed to make precise, nonlethal, single-stranded incisions in targeted genomic regions. This induces recombination events and leads to targeted deletion. We demonstrate that dual-targeted nicking enables deletion of 36 and 97 Kb of the genome. Furthermore, multiplex targeting enables deletion of 133 Kb, accounting for approximately 3% of the entire E. coli genome. This technology provides a framework for methods to manipulate bacterial genomes using CRISPR-nickase systems. We envision this system working synergistically with preexisting bacterial genome engineering methods.
Keywords: CRISPR, Cas9 nickase, genome engineering, chromosome deletion, direct repeats, recombination
Large chromosomal rearrangements and deletions have been observed in both natural and laboratory bacterial evolution studies1−3 and shown to have profound impacts on bacterial physiology, such as improved bioproduct production,4 increased strain fitness,5 or changed tolerance to stress.6 However, such desired genotypes can only be produced in the lab by time-consuming and laborious directed evolution experiments. This is because bacterial genome rearrangement and deletion events only occur when a spontaneous but stochastic DNA break emerges between direct-repeat sequences.3 Efficient methods to target this process for large-scale genome manipulations would be beneficial to biotechnology. Current genome rearrangement strategies often rely on the insertion of exogenous recombinase sites into the bacterial genome to facilitate remodeling.7−12 The use of site-specific recombinases provide efficient tools for in vivo DNA manipulation, however, targeting recombination in a programmable and controllable fashion would broaden and simplify implementations of genome engineering for more applications.
Recent advances in using the clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR associated (Cas) systems for precise genome editing13−18 have paved the way of using it for large scale genome modifications. CRISPR functions as a prokaryotic and archaeal immune system protecting cells from foreign nucleic acids.19−21 CRISPR RNAs (crRNAs) direct degradation of target DNAs through simple Watson–Crick base pairing to recruit Cas9 nuclease to introduce double stranded DNA breaks (DSBs) to foreign nucleic acids.20−22 These systems have minimal sequence targeting constraints, requiring only a small proto-spacer adjacent motif (PAM) sequence,23,24 which makes it possible to program CRISPR to target almost any sequence. CRISPR induced chromosomal DSBs are highly lethal to many bacteria15,25−28 and lead to rapid target DNA degradation.26 This has been exploited to select for desired mutations15,29 and also enabled genome engineering methods in bacteria that are capable of DSB repair.30,31 However, current genomic DNA manipulations in Escherichia coli largely employ site-specific recombinases, which can facilitate recombination between defined recombinase sites. These methods have enabled large-scale manipulations of the bacterial genome including deletion,7,8 inversion,7 and insertion10,32 of DNA. Employing CRISPR-Cas systems to target recombination between arbitrary sequences would complement these approaches to increase the versatility of rationally designed genome manipulations.
Type II CRISPR systems containing Streptococcus pyogenes Cas9 nuclease have been shown to enable user designated single-stranded DNA cleavage (nicking). This is accomplished through mutation of the RuvCI or HNH nuclease domains.33 Methods employing nicking versions of Cas9 have been utilized in eukaryotic organisms to direct homologous recombination (HR) and reduce off-target mutations.13,34,35 As genomic DSBs are highly lethal to many bacterial species, we sought to investigate the potential application of nicking CRISPR systems to induce recombination on the bacterial chromosome.
Here, we demonstrate that nicking versions of Cas9 are nonlethal when targeting the wild type bacterial genome. Short guide RNAs (sgRNAs)33 can be programmed to direct single-stranded DNA breaks (SSBs) resulting in host cell recombination. We functionally validate this with nicking mutant Cas9D10A using a fluorescent reporter system. We show that dual-targeting sgRNAs to genomic repeat sequences leads to recombination across 36 to 97 Kilobases. The easily implementable and modular nature of our of CRISPR plasmid system enables multiplex targeting. Multitargeted CRISPR-nickase systems can remodel the E. coli genome resulting in 133 Kb of deleted DNA, leading to changes of multiple phenotypes.
To experimentally assess the lethality of CRISPR-directed SSBs in wild type E. coli, we constructed a two-plasmid system (Supplementary Figure S1): one plasmid expresses Cas9 and another expresses an sgRNA against a genome integrated green fluorescent protein (GFP) gene (Figure 1a). Multiple mutant versions of Cas9, including Cas9D10A, Cas9H840A, and a Cas9D10A,H840A double mutant,36 were tested. The D10A mutation abrogates function of the RuvCI nuclease domain, generating a Cas9 capable of only nicking the DNA strand complementary to the sgRNA. On the other hand, the H840A mutation removes function of the HNH nuclease domain resulting in a Cas9 that cleaves only the noncomplementary strand to the sgRNA (Figure 1a).33 Combination of both D10A and H840A leaves Cas9 catalytically dead (dCas9).36 Therefore, dCas9 can only bind to target DNA without any cleavage of the chromosome.
Figure 1.
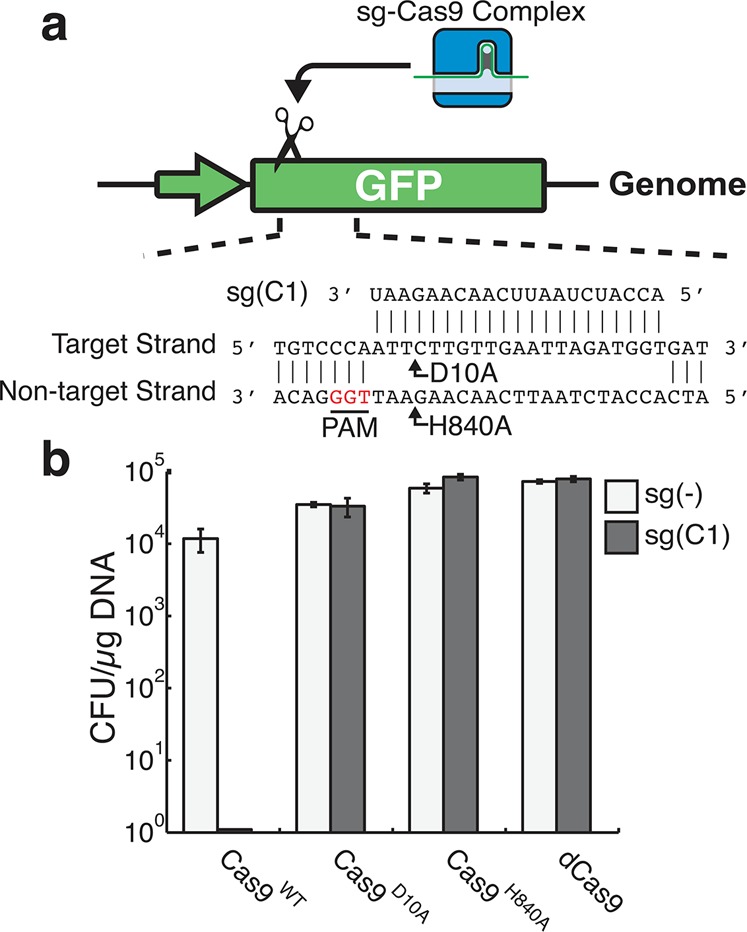
CRISPR-directed SSBs are not lethal to K12 E. coli. (a) A schematic representation of sgRNA guided Cas9s targeting a chromosomally integrated GFP reporter. The sgRNA guide sequence is visualized adjacent to its target sequence. The protospacer adjacent motif (PAM) sequence is highlighted in red. Arrows denote where DNA cleavage occurs by two Cas9 mutants, respectively. Cas9D10A cleaves the target DNA strand of the sgRNA and Cas9H840A cleaves the nontarget strand. (b) Transformation efficiencies of sg(C1) (gray) or untargeted sg(−) (white) in K12 E. coli containing Cas9WT, CasD10A, Cas9H840A or dCas9. Error bars represent ± standard deviation (n = 3).
We transformed GFP targeting sgRNA, sg(C1), into E. coli K12 coexpressing the aforementioned Cas9 variants and compared resulting colonies to a control nontargeting sgRNA, sg(−). As can be seen in Figure 1b, sgRNA targeting GFP showed no viable colony forming units when wildtype Cas9 (Cas9WT) was expressed, while nicking or catalytically inactive variants of Cas9 do not affect viability. This suggests that CRISPR induced DSBs are lethal to E. coli cells while CRISPR induced SSBs have no severe impact on viability. It is also demonstrated that such impacts are not cleavage position dependent (Supplementary Figure S2).
Although it has been reported that HR could be an inconvenience in building synthetic gene circuits with repetitive parts,37,38 a precise nick between repeat sequences could hypothetically induce rationally designed HR to achieve targeted genome deletion.39 To explore the potential of using CRISPR generated nicking to induce targeted HR, we constructed a genome integrated synthetic dual-reporter system (Figure 2a) to query gene deletion and viability. In this system, GFP was designed to be flanked by two 129 base pair direct repeats (R). We systematically targeted nicking sites between direct repeats to both strands of GFP with Cas9D10A and Cas9H840A. The fluorescent population distributions of sgRNA(T1) Cas9D10A indicate that GFP is phenotypically lost in 86% of the cells (Figure 2b, red). This target site is ∼110bp away from the left repeat. It is interesting to see that the percentage of GFP negative cells quickly decreases as the target site is moved further away from the left repeat (Figure 2c, blue triangles). However, Cas9H840A nicks result in less recombination (Figure 2c). This suggests a strong strand and position dependency of single-nick induced local-homologous recombination.
Figure 2.

Nick-induced recombination leads to single gene chromosomal deletion. (a) A diagram of chromosomally integrated dual-fluorescent reporter with internal homologous DNA sequences (R), shown as dark and light gray boxes. sgRNA-guided Cas9 nicking enzyme generates an SSB, illustrated by the scissor. CRISPR induced deletions of GFP results in the joining of two repeats, shown as a gray box with both dark and light shades. Primers (Pr1 and Pr4) flank the dual fluorescence reporter. (b) Flow cytometry screening of pooled sgRNA transformations for homologous recombination. Histograms show population fluorescence distributions for controls sg(−) (green), cells containing RFP with no GFP (gray), and sg(T1) coexpressed with Cas9D10A (red). Vertical dashed line represents threshold for GFP expression. 86% of sg(T1) cells fall to the left of the line, indicating the loss of GFP expression. (c) Percent of flow cytometry estimated GFP deletion for various sgRNAs targeting the noncoding strand using Cas9D10A (blue) and Cas9H840A (orange). Nicking sites within GFP are represented by blue and orange triangles and black squares representing sgRNA target sites. Cut positions are defined as bp from the left repeat (d) Fluorescence microscopy interrogation of dual-fluorescent reporter expression for sg(−) and sg(T1) transformants in E. coli expressing Cas9D10A. GFP expression is undetectable in sg(T1) while RFP expression is similar in both sg(−) and sg(T1). Phase contrast images show cells have normal morphology and RFP expression. (e) Gel-electrophoresis of amplicons using primers Pr1 and Pr4 from transformants for sg(T1) and sg(−). The primers generate a 4.6 kilobase (Kb) amplicon for the initial RFP:GFP site, and following HR the primers generate a 3.6 Kb amplicon. Six out 6 sg(T1) transformants amplify at sizes of approximately 3.6 kilobases, while a sg(−) control template results in banding at 4.6 Kb.
To verify HR as the cause of GFP signal loss, individual colonies were obtained from transformations for further analysis. Fluorescence microscope images show that, when targeted by sgRNA(T1) and Cas9D10A, cells have normal morphology and red fluorescent protein (RFP) expression while GFP fluorescence is undetectable (Figure 2d, bottom), (for microscopy controls see Supplementary Figure S3). This ruled out cell death as the cause of diminished GFP signal. Gel-electrophoresis results also verified a GFP knockout due to HR (Figure 2e). Sanger sequencing of the PCR products further confirmed that recombination indeed occurred between the two direct repeats, yielding the same GFP deletion product (Supplementary Figure S4).
Finally, without the presence of two direct repeats on each end of GFP, neither Cas9D10A nor Cas9H840A can induce targeted GFP fluorescence abolishment (Supplementary Figure S5). Furthermore, DSBs targeted between homologous sequences do not effectively lead to target deletion (Supplementary Figure S6). Taken together, our results show that chromosomally targeted Cas9D10A can induce targeted SSBs and enable recombination between homologous sequences.
To investigate how CRISPR-directed SSBs can be applied to delete large sections of genome in E. coli, we designed sgRNAs to target insertion sequences (IS) in direct repeat orientations and tested for genome deletion. Widespread distributions of transposable DNAs, such as IS elements, have been implicated to be responsible for large genomic changes,1,40 and also provide a rich source of repetitive sequences to apply our method. We identified a region with two 1.2 Kb IS5 elements 35 Kb apart. The DNA flanked by IS5 elements contains 33 genes in total, including the Histidine (His) biosynthesis operon (Figure 3a). The His operon provides us a convenient screenable marker for deletions. Cells containing the His operon are histidine prototrophs. When deleted, E. coli are converted to histidine auxotrophs (unable to grow without provision of the amino acid).
Figure 3.

Nickase directed deletion of a 36 kilobase genomic region. (a) Top half is a schematic representation of a 36.8 Kb genomic region with two IS5 direct-repeats (gray, R) on the K12 MG1655 chromosome. Colored boxes serve as visual aids to mark relative positions and sizes of various genomic regions. A series of 10 sgRNAs were designed to target various locations (black squares, sg(A–J)). The position of black squares indicate which strand is targeted by the sgRNA with I, A and H targeting the + strand, and J and B-G targeting the -. The distances between sgRNA targets are shown in panel C. Primers flanking each repeat (Ph1–Ph4) are used for PCR based genotyping of chromosomal deletions. The His-biosynthetic operon (His-Operon) (orange) located between the IS5 elements serves as a screenable marker for cells harboring deletions. Bottom half represents results of CRISPR induced deletion of genomic region between two repeats (shown gray box with mixed shades). (b) A schematic representation of multitargeting nicking systems around the left IS5, with 3′ Overhang (OH) and 5′ OH cut orientations highlighted. Bp apart is the base pair distance between cut sites. Blue triangles indicate cut sites for respective guides. (c) PCR screening and gel electrophoresis of pooled transformations to detect formation of recombined site (red, gray yellow icon) using primers Ph1 and Ph4 (d) Histidine auxotrophy screening of individual isolated transformants expressing Cas9D10A with sg(B:I) and sg(A:B). Differential growth on M9 minimal medium containing synthetic complete amino acids (M9SC) or M9 without histidine (M9-H) suggests deletions of the targeted region when no colonies are formed in M9-H. Colony positions on M9SC correspond to those on M9-H. Success rates are denoted as the number of colonies unable to grow in M9-H over the total number of colonies (e) PCR genotyping of 6 sg(B:I) histidine auxtrophs and a control sg(−) transformant using 3 different primer combinations confirming formation of the recombined site (bottom row).
To employ single-stranded lesions to direct recombination across the Histidine biosynthetic region, we systematically targeted a series of 10 sgRNAs (sgA-J) in the vicinity of the two IS5 sequences. These included targets directly adjacent to the repeats (R) on both strands along with multiple target sites within the 36.8 Kb region (Figure 3a,b). We transformed K12 E. coli expressing nicking Cas9D10A with plasmids expressing individual and paired sgRNA combinations and cultured in pooled transformations and determined paired nicking did not lead to a substantial reduction in viability (Supplementary Figure S7). We employed a PCR based approach to monitor recombination between repeats. Interestingly, the recombined site was undetectable in single targeting versions, unlike what’s shown above for GFP deletion. This indicates individual single-strand breaks are insufficient to direct HR across large regions (Figure 3c). Interestingly, when employing paired sgRNA, formation of the deletion product was detectable for multiple combinations (Figure 3c). We isolated individual colonies from pooled transformations and subjected them to random screening for histidine auxotrophy. We determined sgRNAs targeting adjacent to each other, sg(A:B), and across the repetitive sequences, sg(B:I), had prominently undergone recombination (Figure 3d). Our results show that sgRNAs generating a 5′ overhang across the repeat sequence, sg(B:I), and sgRNAs targeting in close proximity to each other, sg(A:B), have roughly 17 ± 1.6% and 11 ± 3.2% (± standard devation, 3 biological replicates) success rate of 36 Kb deletion, respectively (Figure 3b,d). Other paired-nicking transformations and sgRNA pair H:G were screened and confirm histidine operon deletions were rare (Supplementary Figure S8 and S9). Finally, we confirmed that histidine auxotrophs consistently harbor the expected deletion by using multiple primer sets querying the presence of starting and recombined junctions (Figure 3e). These results show that dual-targeted nicking systems can effectively induce recombination across 36 Kb with efficiencies approaching 20% of a bacterial population.
The high efficacy of sg(B:I) provided us with design principles for further implementation of nicking-directed recombination systems. sgRNAs should be targeted adjacent to both ends of a repetitive sequence with Cas9D10A cut sites within close proximity (approximately 22 nucleotides) of the homology. The left sgRNA targets the + strand and the right guide targets the – DNA strand. This results in a 5′ overhang cleavage orientation (see Figure 3b, 5′ OH). Interestingly, the requisite for a 5′ cut orientation is consistent with previously reported application of double nicking systems in mammalian cells.34
To further explore the potential of our method to direct multiplexed large-scale genomic recombination, we identified a 97-kilobase genomic region with the tryptophan biosynthesis operon (TRP) and the pyrF gene flanked by identical 1.2 Kb IS5 repeats (Figure 4a). This region enables screening for deletion by testing for tryptophan auxotrophy along with selection of deletion using 5-fluoroorotic acid (5-FOA).41 We created strain KSB1 with duplicate copies of essential genes found within the 97 Kb section of genome integrated into separate loci. This alleviated the essentiality of the region and enabled nickase-directed remodeling.
Figure 4.

Multiplex targeted genome remodeling achieves 133 Kb deletion. (a) A schematic view of two separate genomic regions surrounded by direct repeat sequences. Definitions of illustrations are the same as in Figure 3. The middle disjoint beige box represents 670 Kb distance between these two regions. The left region is 97 kilobases and contains the tryptophan biosynthesis operon (TRP) and the pyrF gene (labeled bright blue boxes). Recombination between these repeats results in deletion of 97 Kb (Δ97 Kb) and tryptophan auxotrophy. The remodeled site (purple, gray and green) brings primers Pt1 and Pt4 in into close proximity. sgRNAs K and L target the sequences directly adjacent to the left repeat to this region. In parallel, guides B and I target a separate region containing His (orange box). The position of black squares indicate which strand is targeted by the sgRNA with K and I targeting the + strand and L and B targeting the – strand (see Figure 3 also). Deletion of this region results in a loss of 36 Kb (Δ36 Kb) and histidine auxotrophy. The remodeled sequence (red, gray and yellow) results in close proximity of Ph1 and Ph4. (b) PCR monitoring of HR in Cas9D10A expressing cells. Primers Pt1 and Pt4 detect deletion of 97 Kb (purple, gray and green icon, resulting in a 1.5 Kb amplicon). Ph1 and Ph4 detect recombination across 36 Kb (red, gray and yellow icon, resulting in a 1.8 Kb amplicon). Different sgRNA targets are indicated beneath the gel photo. sg(−) is a guide sequence not matching the bacterial genome. Amplification indicates occurrence of recombination. (c) Resulting phenotypes of isolated E. coli containing different remodeling combinations. Cells isolated containing different deletions replica plated on M9 synthetic complete medium (M9SC), M9 lacking histidine (M9-H), and M9 without tryptophan (M9-T). Vertical columns correspond to different deletions. sg(−) with no genomic deletion (Δ0) serves as the control. sg(B:I:K:L) expressing cells can harbor 36 (Δ36), 97 (Δ97) or combined 133 kilobase (Δ133) deletions. Δ36, Δ97 and Δ133 are auxotrophic histidine, tryptophan and both, respectively.
To multiplex target the 97 Kb genomic region along with the 36 Kb region tested, we applied principles of dual sgRNA design from above experiments. sgRNAs K and L were designed and coupled to the previously employed guides B and I. This generated a four-target CRISPR system, sg(B:I:K:L), which should direct multiplex remodeling of the E. coli genome (Figure 4a). We validated the necessity of cooperative nicking to induce homologous recombination and determined that four-targeted nicking did not substantially reduce transformation efficiency (Supplementary Figure S10b,c). In pooled transformations of the multitargeted CRISPR device, we detected recombination at both His and Trp regions (Figure 4b). This indicated that HR at both sites was simultaneously occurring within the population of cells. To quantify the 97 Kb deletion we combined results for 5-FOA and TRP markers. 1.4 ± 0.8% of cells exhibit 5-FOA resistance where 47% of the 5-FOA resistant colonies exhibited clear tryptophan auxotrophy (Supplementary Figure S10d). Combining results for both markers, we estimated deletion efficiency to be 7 ± 4 deletions per 1000 cells. After initial 5-FOA/TRP screens we further tested for histidine deletion. Using stepwise screening (see Methods for details) we reliably obtained 36, 97 Kb single and 133 Kb dual-deletions from sg(B:I:K:L) transformations (Figure 4c), indicating successful deletion of 133 Kb of E. coli genome using our approach. These results verify the dual-targeted sgRNA design for directed recombination. Likewise, the results exemplify the potential of multiplex CRISPR-guided nicking systems to remove large genomic regions.
Here we demonstrate the ability for CRISPR targeted nicking systems to direct recombination across both small and large genomic regions. Furthermore, we systematically characterized various sgRNA target locations, choice of nicking Cas9, and overhang orientations to identify the optimal combination for efficient large-scale genome deletion. Targeting paired nicking across repeat elements in a 5′ overhang orientation with nick sites located within 40 bp of the homology sequence results in directed recombination. This enables a 3% reduction of the E. coli genome with only one plasmid transformation. Although the genome reductions reported here utilize repeat sequences to define the domain of remodeling, it is feasible to generate deletions of arbitrary position or to target genome integration by incorporation of plasmid based homologous sequences.30,31
Genome rearrangements are an important part of bacterial evolution, which can facilitate large-scale organismal changes beneficial to biotechnology.4−6 Using engineered SSBs, we enable recombination in bacterial cells resulting in multiplex genome rearrangements reaching 133 Kb in total, about 3% of the E. coli genome. In E. coli, genome reductions of such scale have been generated through multiple years of evolution1 and can be engineered through recombineering based methods.12,42−44 Our system can recapitulate this scale of reduction through a single plasmid transformation. More importantly, the removal of these genome segments is targeted and its execution is controlled. It is conceivable that this method could assist in rationally designed and multitargeted removal of pathways competing with the production of industrially useful chemicals and factors deleterious to growth. Collectively, our method provides a new tool for design and engineering of bacterial genomes.45,46
Our results also provide a framework for methods that can change the landscape of bacterial genomes. Currently, genome-remodeling strategies in many bacteria often utilize site-specific recombinases.7,47 These methods can direct high-efficiency recombination between recombinase sites. The current efficiency of CRISPR-nickase targeted deletion is relatively low, especially compared to efficiencies of site-specific recombinases, however these concerns may be addressed with improved sgRNA combinations and expression vectors. Nicking CRISPR systems, however, can be targeted with high versatility and leverage HR, which can utilize arbitrary sequences as substrates. Nicking CRISPR systems are also desirable to synthetic biology applications.48−55 For instance, systems employing retron mediated reverse transcription have enabled genome directed rewriting and memory storage,56 coupling CRISPR-nickase systems to the output of inducible promoters could enable remodeling of the genome in response to environmental inputs.
In summary, we present a strategy to overcome the cytotoxicity of genome targeting DSBs to make a platform for accurate large-scale bacterial genome manipulations. RNA-programmed SSBs enable recombination resulting in bacterial genome deletion. With the growing availability of full genome sequencing, it is foreseeable that laboratories could identify an organism’s repetitive sequences in silico to predictably and combinatorialy direct large-scale remodeling to test strain characteristics for a myriad of basic science and engineering applications. Further extension of this method would enable researchers to target genome integration of synthetic gene pathways;57 likewise, we envision this method working synergistically with preexisting genome engineering technologies.
Methods
Strains and Media
Molecular cloning was conducted using E. coli NEB-10-Beta (New England Biolabs, NEB). Maintenance and cloning of vectors containing an R6Kγ origin of replication was conducted in a Pir+ E. coli strain. Fluorescent reporter experiments were conducted in K12 MG1655 (ΔLacI ΔAraC) HK022 (J04450 and/or I13522) (Registry of Standard Biological Parts). Initial 36 kilobase deletion experiments were conducted in K12 MG1655 Δ(LacI) Δ(AraC). Experiments involving the 97 kilobase deletion were conducted in strain “KSB1”, which is K12 MG1655 (American Type Culture Collection, ATCC, #700926) HK022(topA,cysB,ribA,fabI), 186(lapAB). Integrations into HK022 attB and 186 attB sites are described in Strain Construction section. LB Miller Medium (Sigma-Aldrich, Sigma) was supplemented with appropriate antibiotics for plasmid maintenance: Ampicillin (100 μg/mL) and/or Kanamycin (30 μg/mL). Experiments involving fluorescent microscopy visualization of genome-integrated reporters used M9 minimal Medium containing 0.2% glycerol. Screening for histidine and tryptophan auxotrophs was done on M9 minimal Medium 0.4% glucose, 1.5% agar, 2 mg/mL Uracil with synthetic complete (SC) (i.e., Leucine deficient supplement (Clonetech, #630414) + 200 mg/L Leucine (Sigma, #L8000), without histidine (-H) or without tryptophan (-T) amino acid supplement (Clonetech, #630415, #630414 respectively). 5-Fluoroorotic Acid (5-FOA, Gold Biotechnology, #F-230-5) media was made by supplementing 5-FOA to a final concentration of 600 mg/L and boiling to dissolve in LB agar miller medium. All cell culture was conducted at 37 °C in either a stationary incubator for Petri dishes, or 250 rpm shaker for liquid media unless noted otherwise.
Strain Construction
Genome integration of reporters and other genes were conducted using methods described previously.32 Plasmids pOSIP-KH and pOSIP-KO were used to integrate into HK022 and 186 attB sites, respectively. Fluorescent reporters (BioBrick #) parts RFP (J04450) and GFP (I13522) were cloned into the EcoRI and PstI sites of pOSIP-KH. The strain used for 97 Kb remodeling experiments was derived from ATCC, #700926. Integration vectors containing essential genes43,58 were created by stepwise cloning of topA:cysB, ribA, and fabI into pOSIP-KH using primers 18 through 23 with corresponding restriction sites indicated in Supplementary Table S2 and lapAB in pOSIP-KO. The lapAB cassette was ligated and integrated in one step. Purified PCR products of primers 24 and 25 were digest with BamHI and SpeI and ligated into pOSIP-KO. Vectors were integrated by transforming ≤10 ng of plasmid into destination strains. Integrations were confirmed at HK022 att with primers 7 and 8, and 186 att with 26 and 27. R6Kγ vectors were propagated as plasmids at 30 °C and integrated at 37 °C.
Construction of Cas9 Mutations
pCas9WT was obtained from Addgene (plasmid #44520).36 pCas9WT expresses S. pyogenes wild type Cas9 from a pLTetO promoter. The plasmid contains a high copy ColE1 origin of replication (∼100 copies), the TetR repressor protein and an ampicillin resistance marker (Supplementary Figure S1a). Cas9 D10A and H840A mutations were generated by PCR amplifying 10 pg of pCas9WT using primer pairs 3,4 and 5,6 respectively (Supplementary Table S2). PCR reactions were purified using GenElute PCR Cleanup Kit (Sigma, #NA1020) following the manufacturer’s protocol. DNA was digested with SapI and self-ligated with <40 ng of DNA in 20 μL reactions containing T4 DNA ligase (NEB).
Design, Cloning and Multiplexing of sgRNAs
Short guide RNAs (sgRNAs) were designed by identifying the “NGG” Protospacer Adjacent Motif (PAM) on the noncomplementary strand of the DNA target. 20–21 nucleotides were used as the guide region. sgRNAs contained an Adenine on the 5′ end, since A nucleotides function as effective transcriptional start site. sgRNA guides were synthesized as a pair of 25 nmole oligonucleotides (Integrated DNA Technologies, IDT) in a format shown in Supplementary Figure S1b. 5′ phosphates were added to oligonucleotides by incubating 1ug of top/bottom oligonucleotides in 50 μL reactions containing 1X T4 DNA Ligase Buffer and 10 units of T4 Polynucleotide Kinase (NEB, #M0201) at 37 °C overnight. Oligonucleotides were duplexed by heating the kinase reactions to 90 °C on an aluminum heating block for 5 min followed by slowly returning the reaction to room temperature (25 °C) over approximately 1 h.
The sgRNA plasmid vector (pSG4K5) (Supplementary Figure S1a), derived from pSB4K5 (Registry of Standard Biological Parts) expresses sgRNAs from a constitutive J23119 promoter. To clone new sgRNAs, pSG4K5 was digested with SapI and dephosphorylated using Antarctic Phosphatase (NEB, #M0289) using the manufacture’s protocol. 40 ng of SapI digested and dephosphorylated pSG4K5 was ligated to an equimolar amount of sgRNA duplex in 20 μL reactions containing T4 DNA ligase (NEB, #M0202).
Multiplex sgRNA plasmids were constructed by PCR amplifying 10 pg of template plasmid with primers 1 and 2. PCR products were prepared by purifying with GenElute PCR Cleanup Kit, followed by digestion of 500 ng with EcorI and SpeI (NEB). Reactions were heat inactivated at 80 °C for 20 min followed by slowly returning to room temperature. pSG4K5 plasmids containing other guide sequences were digested with BsaI and deposphorylated using Antarctic Phosphatase. 40 ng of BsaI digested and dephosphorylated pSG4K5 was ligated to an equimolar amount of sgRNA expression cassette in 20 μL reactions containing T4 DNA ligase.
Transformation of sgRNAs
All strains were made chemically competent using the Z-competent E. coli Tranformation Kit (Zymo Research, #T3002) following the manufacturer’s instructions. For transformation of sgRNA expressing pSG4K5, 10 ng of DNA was incubated with 50 μL of competent cell solution for 30 min on ice. Samples were heat shocked for 30 s in a 42 °C water bath. Samples were then kept on ice for 3 min. 300 μL of prewarmed LB media was added and cells were shaken at 37 °C for 1 h. Following outgrowth, 100 to 300 μL of sample was plated on LB agar containing ampicillin and kanamycin. Plates were incubated overnight.
In the case of pooled transformations, following outgrowth for 1 h, 100 μL of the transformation was added to 5 mL of prewarmed LB liquid medium containing ampicillin and kanamycin. Samples were cultured for 12–14 h. For fluorescent reporter recombination experiments, samples were analyzed by flow cytometry. For histidine and tryptophan remodeling experiments samples were streak plated or serial diluted onto LB agar plates and/or 5-FOA medium containing appropriate antibiotics.
Flow Cytometry
All flow cytometry was conducted on an Accuri C6 Flow Cytometer (BD Biosciences, CA). Samples were gated by consistent forward scatter (FSC) and side scatter (SSC) and 10 000 events within the FSC/SSC gate were collected. A 488 nm laser excitation and a 530 ± 15 nm emission filter was used for GFP fluorescence determination. Flow cytometry files were analyzed in MatLab (The MathWorks).
Fluorescence Microscopy
Colonies picked from sgRNA transformations were cultured for roughly 48 h in M9 minimal media 0.2% glycerol. Cells were spun down at 5000 g for 2 min and washed with 1X Phosphate Buffer Solution (PBS). Two μL of concentrated cell solution was placed on glass microscope slides and visualized on a Nikon Ti-Eclipse inverted microscope with and LED-based Lumencor SOLA SE Light Engine with appropriate filter sets. GFP was visualized with an excitation at 472 nm and emission at 520/35 nm using a Semrock band-pass filter. RFP was visualized with excitation at 562 nm and emission at 641/75 nm. For exposure times and experimental controls see Supplementary Figure S3. Constant exposure times, LUT and image gain adjustments were applied to all microscopy data.
Remodeling of 36 Kilobase Histidine Genomic Region
The 36 kilobase region corresponds to NCBI U00096.3 Positions: 2066159–2102943. sgRNA expression plasmids targeting the IS5-histidine operon region were transformed as described above into K12 E. coli expressing Cas9D10A. Pooled transformations were cultured in 5 mL liquid medium containing ampicillin and kanamycin for 12–14 h. To isolate individual histidine deletions, 5 μL of cells were streak plated or serial diluted onto LB agar medium containing ampicillin and kanamycin. Plates were cultured overnight. Individual colonies were randomly selected and replica plated on M9SC and M9–H media. Deletion efficiency is the number of histidine auxotrophs over the total number of screened colonies. Genomic DNA was prepared from 2 mL of pooled transformation and used for PCR genotyping with primers Ph1-Ph4 (Supplementary Table S2 #10–13).
Remodeling of 97 Kilobase Tryptophan Genomic Region
The 97 kilobase region corresponds to NCBI U00096.3 Positions: 1299494–1397238. sgRNA expression plasmids targeting the IS5-tryptophan operon region were transformed as described above into strain KSB1 expressing Cas9D10A. Pooled transformations were grown for 12–14 h in liquid medium and plated in serial dilutions on LB agar and LB 5-FOA media with ampicillin and kanamycin. Individual 5-FOA resistant colonies were counted and selected at ≥24 h of growth, as colonies tended to grow slowly on 5-FOA plates, furthermore 97 Kb deletions exhibited a slow growth phenotype on LB medium. Selected colonies were replica plated on M9SC and M9-T plates. Deletion efficiency was determined by multiplying the fraction of 5-FOA resistant colonies by the fraction of tryptophan operon deletions (Supplementary Figure S10d). Genomic DNA was prepared from 2 mL of pooled transformation and used for PCR genotyping with primers Pt1-Pt4 (Supplementary Table S2 #14–17).
To Isolate 133 kilobase, 36 and 97 Kb dual-deletions, we conducted multiple step screening to isolate each deletion. Tryptophan deletions were picked and regrown overnight in 5 mL LB medium with antibiotics. Individual colonies were isolated through serial dilutions and replica plated on M9SC and M9-H medium. We also obtained dual-deletions through histidine deletion screening followed by regrowing overnight then 5-FOA and tryptophan screening.
PCR Genotyping
PCR was conducted on a Bio-Rad C1000 thermocycler with Dual 48/48 Fast Reaction Modules (Bio-Radiations, BioRad). Genomic DNA of cells was prepared with the GenElute Bacterial Genomic DNA Kit (Sigma, #NA2110). Genomic DNA preparations were diluted 100-fold and 1 μL of genomic template was used in 20 μL PCR reactions. PCR reactions contained Phusion DNA Polymerase (NEB, #E0553) and corresponding primers (Supplementary Table S2). Annealing temperatures and extension times were calculated using the manufacturer’s protocol. PCR products were visualized via 1% agarose gel-electrophoresis.
Acknowledgments
We thank members of Xiao Wang’s lab for helpful discussions and suggestions. K.S.B. is partially supported by ASU School of Life Science’s SOLUR Program and Waeso (S2012ur0006). This study was financially supported by National Science Foundation Grant DMS-1100309 and National Institutes of Health Grant GM106081 (to X.W.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.5b00132.
Figures S1–S10, Tables S1 and S2, and supplemental sequences. (PDF)
Author Contributions
K.S.B. and X.W. designed the study. K.S.B. and Q.Z. conducted experiments. K.S.B. and X.W. analyzed data and wrote the paper.
The authors declare the following competing financial interest(s): A patent application has been filed relating to this work.
Supplementary Material
References
- Raeside C.; Gaffé J.; Deatherage D. E.; Tenaillon O.; Briska A. M.; Ptashkin R. N.; Cruveiller S.; Médigue C.; Lenski R. E.; Barrick J. E.; Schneider D. (2014) Large chromosomal rearrangements during a long-term evolution experiment with Escherichia coli. mBio 5, e01377–01314 10.1128/mBio.01377-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick J. E.; Yu D. S.; Yoon S. H.; Jeong H.; Oh T. K.; Schneider D.; Lenski R. E.; Kim J. F. (2009) Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461, 1243–1247 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- Darling A. E.; Miklós I.; Ragan M. A. (2008) Dynamics of Genome Rearrangement in Bacterial Populations. PLoS Genet. 4, e1000128. 10.1371/journal.pgen.1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W., Dole S., Grabar T., Collard A. C., Pero J. G., and Yocum R. R. (2012) Engineering microbes for efficient production of chemicals, WO 2011063055 A3.
- Cooper V. S.; Schneider D.; Blot M.; Lenski R. E. (2001) Mechanisms causing rapid and parallel losses of ribose catabolism in evolving populations of Escherichia coli B. J. Bacteriol. 183, 2834–2841 10.1128/JB.183.9.2834-2841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle M. M.; Bennett A. F.; Long A. D. (2001) Genetic architecture of thermal adaptation in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 98, 525–530 10.1073/pnas.98.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyeart P. J.; Chirieleison S. M.; Dao M. N.; Perutka J.; Quandt E. M.; Yao J.; Whitt J. T.; Keatinge-Clay A. T.; Lambowitz A. M.; Ellington A. D. (2013) Generalized bacterial genome editing using mobile group II introns and Cre-lox. Mol. Syst. Biol. 9, 685. 10.1038/msb.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R.; Grose C.; Haft D. H.; Zaveri J.; Alperovich N.; Gibson D. G.; Merryman C.; Glass J. I. (2014) Simultaneous non-contiguous deletions using large synthetic DNA and site-specific recombinases. Nucleic Acids Res. 42, gku509. 10.1093/nar/gku509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos C. N. S.; Yoshikuni Y. (2014) Engineering complex biological systems in bacteria through recombinase-assisted genome engineering. Nat. Protoc. 9, 1320–1336 10.1038/nprot.2014.084. [DOI] [PubMed] [Google Scholar]
- Santos C. N. S.; Regitsky D. D.; Yoshikuni Y. (2013) Implementation of stable and complex biological systems through recombinase-assisted genome engineering. Nat. Commun. 10.1038/ncomms3503. [DOI] [PubMed] [Google Scholar]
- Kuhlman T. E.; Cox E. C. (2010) Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res. 38, e92. 10.1093/nar/gkp1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolisnychenko V.; Plunkett G.; Herring C. D.; Feher T.; Posfai J.; Blattner F. R.; Posfai G. (2002) Engineering a Reduced Escherichia coli Genome. Genome Res. 12, 640–647 10.1101/gr.217202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L.; Ran F. A.; Cox D.; Lin S.; Barretto R.; Habib N.; Hsu P. D.; Wu X.; Jiang W.; Marraffini L. A.; Zhang F. (2013) Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P.; Yang L.; Esvelt K. M.; Aach J.; Guell M.; DiCarlo J. E.; Norville J. E.; Church G. M. (2013) RNA-Guided Human Genome Engineering via Cas9. Science 339, 823–826 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W.; Bikard D.; Cox D.; Zhang F.; Marraffini L. A. (2013) RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo J. E.; Norville J. E.; Mali P.; Rios X.; Aach J.; Church G. M. (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41, 4336–4343 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz A. A.; Walter J. M.; Schubert M. G.; Kung S. H.; Hawkins K.; Platt D. M.; Hernday A. D.; Mahatdejkul-Meadows T.; Szeto W.; Chandran S. S.; Newman J. D. (2015) Efficient Multiplexed Integration of Synergistic Alleles and Metabolic Pathways in Yeasts via CRISPR-Cas. Cell Syst. 1, 1–9 10.1016/j.cels.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Bao Z.; Xiao H.; Liang J.; Zhang L.; Xiong X.; Sun N.; Si T.; Zhao H. (2015) Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth. Biol. 4, 585–594 10.1021/sb500255k. [DOI] [PubMed] [Google Scholar]
- Makarova K. S.; Haft D. H.; Barrangou R.; Brouns S. J. J.; Charpentier E.; Horvath P.; Moineau S.; Mojica F. J. M.; Wolf Y. I.; Yakunin A. F.; van der Oost J.; Koonin E. V. (2011) Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R.; Fremaux C.; Deveau H.; Richards M.; Boyaval P.; Moineau S.; Romero D. A.; Horvath P. (2007) CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 315, 1709–1712 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Marraffini L. A.; Sontheimer E. J. (2008) CRISPR Interference Limits Horizontal Gene Transfer in Staphylococci by Targeting DNA. Science 322, 1843–1845 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapranauskas R.; Gasiunas G.; Fremaux C.; Barrangou R.; Horvath P.; Siksnys V. (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 39, 9275–9282 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica F. J. M.; Diez-Villasenor C.; Garcia-Martinez J.; Almendros C. (2009) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- Marraffini L. A.; Sontheimer E. J. (2010) Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568–571 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citorik R. J.; Mimee M.; Lu T. K. (2014) Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 32, 1141–1145 10.1038/nbt.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliando B. J.; Voigt C. A. (2015) Targeted DNA degradation using a CRISPR device stably carried in the host genome. Nat. Commun. 6, 6989. 10.1038/ncomms7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.; Qimron U. (2010) The Escherichia coli CRISPR system protects from λ lysogenization, lysogens, and prophage induction. J. Bacteriol. 192, 6291–6294 10.1128/JB.00644-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercoe R. B.; Chang J. T.; Dy R. L.; Taylor C.; Gristwood T.; Clulow J. S.; Richter C.; Przybilski R.; Pitman A. R.; Fineran P. C. (2013) Cytotoxic Chromosomal Targeting by CRISPR/Cas Systems Can Reshape Bacterial Genomes and Expel or Remodel Pathogenicity Islands. PLoS Genet. 9, e1003454. 10.1371/journal.pgen.1003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J.-H.; Pijkeren J.-P. v. (2014) CRISPR–Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res. 42, gku623. 10.1093/nar/gku623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb R. E.; Wang Y.; Zhao H. (2015) High-Efficiency Multiplex Genome Editing of Streptomyces Species Using an Engineered CRISPR/Cas System. ACS Synth. Biol. 4, 723–728 10.1021/sb500351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y.; Charusanti P.; Zhang L.; Weber T.; Lee S. Y. (2015) CRISPR-Cas9 Based Engineering of Actinomycetal Genomes. ACS Synth. Biol. 4, 1020. 10.1021/acssynbio.5b00038. [DOI] [PubMed] [Google Scholar]
- St-Pierre F.; Cui L.; Priest D. G.; Endy D.; Dodd I. B.; Shearwin K. E. (2013) One-Step Cloning and Chromosomal Integration of DNA. ACS Synth. Biol. 2, 537–541 10.1021/sb400021j. [DOI] [PubMed] [Google Scholar]
- Jinek M.; Chylinski K.; Fonfara I.; Hauer M.; Doudna J. A.; Charpentier E. (2012) A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816–821 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A.; Hsu P. D.; Lin C.-Y.; Gootenberg J. S.; Konermann S.; Trevino A. E.; Scott D. A.; Inoue A.; Matoba S.; Zhang Y.; Zhang F. (2013) Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 155, 479–480 10.1016/j.cell.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P.; Aach J.; Stranges P. B.; Esvelt K. M.; Moosburner M.; Kosuri S.; Yang L.; Church G. M. (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L. S.; Larson M. H.; Gilbert L. A.; Doudna J. A.; Weissman J. S.; Arkin A. P.; Lim W. A. (2013) Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 152, 1173–1183 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-J.; Liu P.; Nielsen A. A. K.; Brophy J. A. N.; Clancy K.; Peterson T.; Voigt C. A. (2013) Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 10, 659–664 10.1038/nmeth.2515. [DOI] [PubMed] [Google Scholar]
- Jack B. R.; Leonard S. P.; Mishler D. M.; Renda B. A.; Leon D.; Suárez G. A.; Barrick J. E. (2015) Predicting the Genetic Stability of Engineered DNA Sequences with the EFM Calculator. ACS Synth. Biol. 4, 939–943 10.1021/acssynbio.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett S. T.; Drapkin P. T.; Sutera-Jr V. A.; Gluckman-Peskind T. J. (1993) A Sister-Strand Exchange Mechanism for Reca-Independent Deletion of Repeated DNA Sequences in Escherichia Coli. Genetics 135, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro J. A. (2009) Letting Escherichia coli Teach Me About Genome Engineering. Genetics 183, 1205–1214 10.1534/genetics.109.110007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redder P.; Linder P. (2012) New Range of Vectors with a Stringent 5-Fluoroorotic Acid-Based Counterselection System for Generating Mutants by Allelic Replacement in Staphylococcus aureus. Appl. Environ. Microbiol. 78, 3846–3854 10.1128/AEM.00202-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pósfai G.; Plunkett G.; Fehér T.; Frisch D.; Keil G. M.; Umenhoffer K.; Kolisnychenko V.; Stahl B.; Sharma S. S.; Arruda M.; de Burland V.; Harcum S. W.; Blattner F. R. (2006) Emergent Properties of Reduced-Genome Escherichia coli. Science 312, 1044–1046 10.1126/science.1126439. [DOI] [PubMed] [Google Scholar]
- Baba T.; Ara T.; Hasegawa M.; Takai Y.; Okumura Y.; Baba M.; Datsenko K. A.; Tomita M.; Wanner B. L.; Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan S. K.; Thomason L. C.; Kuznetsov S. G.; Court D. L. (2009) Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 4, 206–223 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way J. C.; Collins J. J.; Keasling J. D.; Silver P. A. (2014) Integrating Biological Redesign: Where Synthetic Biology Came From and Where It Needs to Go. Cell 157, 151–161 10.1016/j.cell.2014.02.039. [DOI] [PubMed] [Google Scholar]
- Pál C.; Papp B.; Pósfai G. (2014) The dawn of evolutionary genome engineering. Nat. Rev. Genet. 15, 504–512 10.1038/nrg3746. [DOI] [PubMed] [Google Scholar]
- Val M.-E.; Skovgaard O.; Ducos-Galand M.; Bland M. J.; Mazel D. (2012) Genome Engineering in Vibrio cholerae: A Feasible Approach to Address Biological Issues. PLoS Genet. 8, e1002472. 10.1371/journal.pgen.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs F. J.; Carr P. A.; Wang H. H.; Lajoie M. J.; Sterling B.; Kraal L.; Tolonen A. C.; Gianoulis T. A.; Goodman D. B.; Reppas N. B.; Emig C. J.; Bang D.; Hwang S. J.; Jewett M. C.; Jacobson J. M.; Church G. M. (2011) Precise Manipulation of Chromosomes in Vivo Enables Genome-Wide Codon Replacement. Science 333, 348–353 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T. K.; Khalil A. S.; Collins J. J. (2009) Next-generation synthetic gene networks. Nat. Biotechnol. 27, 1139–1150 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra D.; Rivera P. M.; Lin A.; Del Vecchio D.; Weiss R. (2014) A load driver device for engineering modularity in biological networks. Nat. Biotechnol. 32, 1268–1275 10.1038/nbt.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.; Su R.-Q.; Li X.; Ellis T.; Lai Y.-C.; Wang X. (2013) Engineering of regulated stochastic cell fate determination. Proc. Natl. Acad. Sci. U. S. A. 110, 201305423. 10.1073/pnas.1305423110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litcofsky K. D.; Afeyan R. B.; Krom R. J.; Khalil A. S.; Collins J. J. (2012) Iterative plug-and-play methodology for constructing and modifying synthetic gene networks. Nat. Methods 9, 1077–1080 10.1038/nmeth.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis T.; Wang X.; Collins J. J. (2009) Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat. Biotechnol. 27, 465–471 10.1038/nbt.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker J.; Cookson S.; Bennett M. R.; Mather W. H.; Tsimring L. S.; Hasty J. (2008) A fast, robust and tunable synthetic gene oscillator. Nature 456, 516–519 10.1038/nature07389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne S.; Li B.; Cao Y.; Schaeffer D.; Ryser M. D.; You L. (2013) Temporal control of self-organized pattern formation without morphogen gradients in bacteria. Mol. Syst. Biol. 9, 697. 10.1038/msb.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzadfard F.; Lu T. K. (2014) Genomically encoded analog memory with precise in vivo DNA writing in living cell populations. Science 346, 1256272. 10.1126/science.1256272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme K.; Zhao D.; Voigt C. A. (2012) Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc. Natl. Acad. Sci. U. S. A. 109, 7085–7090 10.1073/pnas.1120788109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalakshmi S.; Sunayana M. R.; SaiSree L.; Reddy M. (2014) yciM is an essential gene required for regulation of lipopolysaccharide synthesis in Escherichia coli. Mol. Microbiol. 91, 145–157 10.1111/mmi.12452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.