

Abstract
Interfering with anaplerotic utilization of glutamine (Gln) was recently reported to sensitize KRAS-driven cancer cells to the cytotoxic effects of capecitabine and paclitaxel. This effect was due to bypass of a Gln-dependent G1 cell cycle checkpoint in these cells. This study highlights therapeutic opportunities created by metabolic reprogramming in cancer cells.
Keywords: Glutamine, anaplerosis, TCA cycle, KRAS, cell cycle
Abbreviations
- Gln
glutamine
- TCA
tricarboxylic acid
Over the last decade there has been a resurgence of interest in metabolism stimulated largely by the observation of metabolic reprogramming in cancer cells.1 To meet the increased anabolic demand to accommodate high rates of cell growth and proliferation, cancer cells increase glucose uptake and reprogram the fate of glycolytic and tricarboxylic acid (TCA) cycle intermediates toward synthesis of the amino acids, nucleotides, and lipids needed for the cell to double its mass and divide. In dividing cells, citric acid, which is synthesized from the condensation reaction between acetyl-CoA and oxaloacetate in the first step of the TCA cycle, exits the mitochondria and regenerates acetyl-CoA, which is then used for the generation of lipids needed for membrane biosynthesis. The exit of citric acid from the mitochondria and the TCA cycle creates a need for anaplerotic replenishment of TCA cycle intermediates downstream of citric acid. The major source for anaplerotic replenishment of TCA cycle intermediates is the conditionally essential amino acid glutamine (Gln). Gln is first deaminated to glutamate, and then converted to α-ketoglutarate by glutamate dehydrogenase or during transamination reactions with α-keto-acids such as oxaloacetate to generate aspartate (Fig. 1). Up to 25% of the Gln is incorporated into membrane lipids,2 indicating that a substantial amount of Gln can be converted to citric acid for export to the cytosol for fatty acid synthesis. Gln-derived α-ketoglutarate is also critical for redox balance and the generation of NADPH via the conversion of malate to pyruvate (Fig. 1). These observations underscore the critical importance of Gln as a nutrient source in dividing and metabolically reprogrammed cancer cells.
Figure 1.
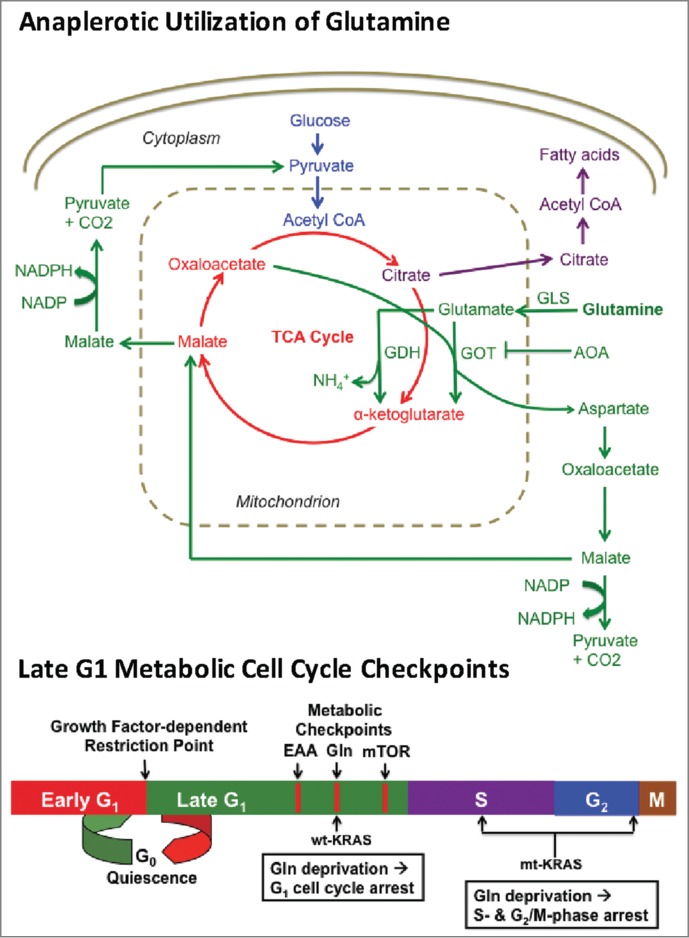
Schematic overview of anaplerotic glutamine (Gln) utilization and late G1 metabolic cell cycle checkpoints. Gln is first deaminated to glutamate by glutaminase (GLS). Glutamate is then converted to α-ketoglutarate via either glutamate dehydrogenase (GDH) or through transamination catalyzed by glutamate oxaloacetate transaminase (GOT), which uses oxaloacetate as the amino group acceptor to generate aspartate. Aminooxyacetate (AOA) inhibits GOT and therefore suppresses generation of α-ketoglutarate from Gln-derived glutamate. The aspartate generated by GOT is critical for redox balance and NAPDH production,7 as is the Gln-derived TCA cycle component malate, which can be converted to pyruvate and NADPH. The relative positions of the growth factor-dependent restriction point and late G1 metabolic checkpoints mediated by essential amino acids (EAA), Gln, and mTOR are depicted in the lower schematic.
Consistent with the importance of Gln as a nutrient for dividing cells, we recently identified a Gln-dependent late-G1 cell cycle checkpoint that could be distinguished from 2 other late-G1 checkpoints – one dependent on essential amino acids and the other dependent on mammalian/mechanistic target of rapamycin (mTOR).3 All 3 metabolic checkpoints were clearly distinguished from the mid-G1 growth factor-dependent restriction point.3 Thus, after the cell receives growth factor signals indicating that it is appropriate to divide, there appear to be several late G1 metabolic checkpoints that monitor whether there are sufficient nutrients available for the cell to double its mass and divide.4 This is shown schematically in Fig. 1. Importantly, cancer cells harboring KRAS mutations do not arrest in G1 upon Gln deprivation. Instead, KRAS-driven cancer cells progress into S and G2/M phases where they are arrested.5 Thus, mutant KRAS confers the ability to override the Gln-dependent late G1 checkpoint allowing progression from G1 into S phase in the absence of Gln. Suppression of the KRAS downstream effectors mTOR and Erk restores G1 arrest in response to Gln deprivation in KRAS-driven cancer cells indicating that override of the G1 Gln checkpoint is mediated by activation of mTOR and Erk. Cells that have committed to divide and progress into S and G2/M are, in general, more vulnerable to apoptotic insult. Thus, KRAS-driven cancer cells that override a Gln-dependent G1 cell cycle checkpoint and arrest in S and G2/M could be sensitive to therapeutic strategies that deprive cells of Gln and target cells in S and G2/M.
To test the hypothesis that Gln deprivation in KRAS-driven cancer cells could induce sensitivity to cell cycle phase-specific cytotoxic compounds, we deprived KRAS-driven cancer cells of Gln and examined their sensitivity to capecitabine, which interferes with DNA synthesis, and paclitaxel, which interferes with microtubule breakdown during mitosis. Both capecitabine and paclitaxel induced apoptosis in KRAS-driven cancer cells, but not in cancer cells lacking a KRAS mutation that arrested in G1 upon Gln depletion. Clearly, Gln deprivation is not a viable therapeutic option; however, interfering with anaplerotic utilization of Gln is a possible approach.6 Kimmelman and colleagues recently reported that Gln is utilized in KRAS-driven pancreatic cancer cells through a transamination reaction in which glutamate is deaminated to α-ketoglutarate with concomitant generation of aspartate from oxaloacetate7 (Fig. 1). Thus, in KRAS-driven cancer cells, the transaminase pathway is apparently preferred over the glutamate hydrogenase pathway, which is used when glucose levels are low.6 Consistent with findings reported by the Kimmelman group, we found that the transaminase inhibitor aminooxyacetate mimicked Gln depletion and induced sensitivity to capecitabine and paclitaxel in KRAS-driven cancer cells. Thus, a combination of transaminase inhibitors together with capecitabine and paclitaxel could be a viable strategy for the treatment of KRAS-driven cancers that exploits the ability of KRAS to stimulate override of the Gln-dependent G1 checkpoint. This approach represents a “synthetic lethal” situation8 whereby activating KRAS mutations in combination with suppressed Gln utilization sensitizes cells to the cytotoxic effects of the cell cycle phase-specific compounds capecitabine and paclitaxel.
KRAS-driven cancer cells have been notoriously resistant to therapeutic intervention and KRAS itself is considered undruggable.9 Thus, alternative strategies are needed to treat what may be as many of 30% of human cancers that are driven by KRAS mutations.10 The observation that KRAS-driven cancer cells override a late G1 Gln-dependent cell cycle checkpoint and arrest in a part of the cell cycle where they become sensitive to S- and G2/M-phase specific cytotoxic drugs represents a potentially exploitable vulnerability of KRAS-driven cancer cells. The ability to target Gln utilization in combination with capecitabine and paclitaxel could provide a basis for targeting the significant percentage of human cancers harboring KRAS mutations that have to date been resistant to therapeutic intervention.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
I would like to acknowledge Mahesh Saqcena and Suman Mukhopadhyay for comments on this manuscript and for their contributions as co-first authors of the article that stimulated this commentary.
Funding
This original study was supported by National Institutes of Health grant R01-CA046677 and a pilot project award from the Research Centers in Minority Institutions award RP-03037 from the National Center for Research Resources of the National Institutes of Health.
References
- 1. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/ 10.1016/j.ccr.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 2007; 104:19345-50; PMID:18032601; http://dx.doi.org/ 10.1073/pnas.0709747104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saqcena M, Menon D, Patel D, Mukhopadhyay S, Chow V, Foster DA. Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS One 2013; 8:e74157; PMID:23977397; http://dx.doi.org/ 10.1371/journal.pone.0074157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Foster DA, Yellen P, Xu L, Saqcena M. Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 2010; 1:1124-31; PMID:21779436; http://dx.doi.org/ 10.1177/1947601910392989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saqcena M, Mukhopadhyay S, Hosny C, Alhamed A, Chatterjee A, Foster DA. Blocking anaplerotic entry of glutamine into the TCA cycle sensitizes K-Ras mutant cancer cells to cytotoxic drugs. Oncogene 2014; PMID:25023699; http://dx.doi.org/ 10.1038/onc.2014.207; Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 2013; 123:3678-84; PMID: 23999442; http://dx.doi.org/ 10.1172/JCI69600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013; 496:101-5; PMID:23535601; http://dx.doi.org/ 10.1038/nature12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reinhardt HC, Jiang H, Hemann MT, Yaffe MB. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle 2009; 8:3112-9; http://dx.doi.org/ 10.4161/cc.8.19.9626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell 2014; 25:272-81; PMID:24651010; http://dx.doi.org/ 10.1016/j.ccr.2014.02.017 [DOI] [PubMed] [Google Scholar]
- 10. Bos JL. RAS oncogenes in human cancer: a review. Cancer Res 1989; 49:4682-9 [PubMed] [Google Scholar]